

Abstract
Blood-borne neutrophils are excluded from entering lymph nodes across vascular portals termed high endothelial venules (HEVs) due to lack of expression of the CCR7 homeostatic chemokine receptor. Induction of sterile inflammation increases neutrophil entry in tumor draining lymph nodes (TDLNs), which is critical for induction of anti-tumor adaptive immunity following treatments such as photodynamic therapy (PDT). However, the mechanisms controlling neutrophil entry in TDLNs remain unclear. Prior evidence that IL-17 promotes neutrophil emigration to sites of infection via induction of CXCL2 and CXCL1 inflammatory chemokines raised the question of whether IL-17 contributes to chemokine-dependent trafficking in TDLNs. Here we demonstrate rapid accumulation of IL-17 producing Th17 cells in the TDLN following induction of sterile inflammation by PDT. We further report that non-hematopoietic expression of IL-17RA regulates neutrophil accumulation in TDLNs following induction of sterile inflammation by PDT. We show that HEVs are the major route of entry of blood-borne neutrophils into TDLN through interactions of L-selectin with HEV-expressed peripheral lymph node addressin (PNAd) and by preferential interactions between CXCR2 and CXCL2, but not CXCL1. CXCL2 induction in TDLNs was mapped in a linear pathway downstream of IL-17RA-dependent induction of IL-1β. These results define a novel IL-17-dependent mechanism promoting neutrophil delivery across HEVs in TDLNs during acute inflammatory responses.
Keywords: sterile inflammation, neutrophils, IL-17, HEV, IL-1β
Introduction
Sterile inflammation refers to inflammation that occurs in the absence of pathogens and is observed following ischemia-reperfusion, chemotherapy, radiation, and other anti-cancer modalities (1). Like pathogen-induced inflammation, sterile inflammation is characterized by a rapid infiltration of neutrophils into the site of damage. Recent studies have shown that neutrophils can also accumulate in inflamed lymph nodes (LNs) following induction of both pathogen-mediated (2–5) and sterile inflammation (6). However, the mechanisms controlling neutrophil entry in inflamed LNs remain unclear.
Neutrophil migration into inflamed extralymphoid tissue is mediated by the chemokines CXCL2 (MIP-2) and CXCL1 (KC) through their interaction with CXCR2 (7). CXCL2 and CXCL1 expression is regulated by the pro-inflammatory cytokine interleukin-17 (IL-17) (8). The IL-17 family of cytokines consists of six members, IL-17A-F (9). The biological effects of IL-17A, hereafter referred to as IL-17, are mediated through IL-17RA, the receptor subunit necessary for signal transduction (10). IL-17 signaling leads to enhanced stability of CXCL1 mRNA (11), expression of CXCL2 through mRNA transcription and translation in mesangial cells (12) and production of IL-8 (human homolog of murine CXCL2) in human lung endothelial cells (13).
IL-17 induction of CXCL2 expression is augmented by interleukin-1β (IL-1β) (14). IL-1β is a potent pro-inflammatory cytokine that has also been shown to play a critical role in controlling neutrophil recruitment (1). IL-1β regulates endothelial cell expression of adhesion molecules that support recruitment of neutrophils (15) and enhances CXCL2 expression (16). Recruitment of neutrophils to sterile inflammation of the liver is dependent on IL-1β and CXCL2 (17).
Migration of naïve and central memory lymphocytes in LNs is orchestrated via a multi-step adhesion cascade that begins with tethering/rolling along specialized post-capillary venules termed high endothelial venules (HEVs) (18). Tethering and rolling is mediated by HEV-expressed peripheral lymph node addressin (PNAd) and L-selectin expressed on the lymphocyte (19,20). Transition to firm arrest during homeostatic recirculation is mediated by the interaction of lymphocyte CCR7 with the chemokine CCL21 and subsequent LFA-1 binding to ICAM-1 and ICAM-2 (21), which have redundant functions during steady-state lymphocyte trafficking across HEVs (22).
Neutrophils were initially thought to be excluded from LNs because, while they can engage HEV-borne PNAd through interaction with L-selectin expressed on their cell surface, they do not express CCR7 (18,19). Recent studies showed that neutrophils could enter inflamed LNs via the afferent lymphatics (2,3) following infection and that entry is mediated by pathogen-induced up-regulation of CCR7 on neutrophils (5). It is unclear whether neutrophils use a similar mechanism to gain entry into LNs following sterile inflammation.
Previous studies by our group have shown that neutrophils play a pivotal role in the induction of anti-tumor immunity following treatment of tumors with photodynamic therapy (PDT) (6). PDT is a Food and Drug Administration (FDA) approved anti-cancer therapy used to cure early stage disease and achieve palliation of advanced disease (23). PDT of tumors induces a sterile inflammatory response that is characterized by systemic release of pro-inflammatory cytokines (24), increased systemic neutrophilia (25), and rapid migration of neutrophils into the tumor (26) and tumor draining lymph nodes (TDLNs) (6). The inflammatory response generated by PDT contributes to the overall efficacy of treatment (26). Induction of neutrophils and migration of neutrophils into tumor and TDLNs is critical for generation of adaptive immunity following PDT (6). Long-term tumor control by PDT in patients is dependent upon induction of anti-tumor immunity (27).
Our results show that increased entry of neutrophils in TDLNs following induction of sterile inflammation by PDT is regulated by IL-17:IL-17RA. We also report that expression of IL-17RA by the non-hematopoietic compartment is required for the migration of neutrophils in TDLNs following induction of sterile inflammation. Neutrophil entry in TDLNs preferentially occurs across HEVs and is supported by IL-17-induced expression of CXCL2, but not CXCL1. Our data further provides evidence that IL-17 increases the production of IL-1β, which in turn enhances expression of CXCL2 that provides a molecular switch for increased entry of neutrophils in TDLN HEVs. These findings describe critical differences in the regulation of neutrophils in inflamed LNs via pathogen-mediated and sterile induction of acute inflammation. Whereas pathogen-mediated inflammation increases neutrophil entry in inflamed LNs through afferent lymphatics, sterile inflammation modifies the chemokine availability allowing neutrophil recruitment in TDLNs through the vascular gateways.
Materials and Methods
Animals and tumor system
Pathogen-free BALB/c mice were obtained from the National Cancer Institute; C.129S2(B6)-Cmkar2tm1Mwm (Cxcr2−/−) mice were obtained from The Jackson Laboratory. Il17ra−/− mice were a generous gift from Sarah Gaffen and bred in the Roswell Park Department of Laboratory Animal Resources. All mice were female, of BALB/c background, and were housed in microisolator cages in a laminar flow unit under ambient light. Six- to 10-week-old animals were inoculated intra-dermally on the right shoulder with 106 Colo26 (murine colon carcinoma) cells transfected with hemagglutinin (HA) cDNA (Colo26-HA). The RPCI Institutional Animal Care and Use Committee approved all procedures carried out in this study.
Reagents and antibodies
Clinical-grade, pyrogen-free 2-[1-hexyloxyethyl]-2-devinyl pyropheophorbide-a (HPPH) was obtained from the Roswell Park Pharmacy and reconstituted to 0.4 mmol/L in pyrogen-free 5% dextrose in water (D5W; Baxter Corp.). Purified monoclonal antibody specific for murine IL-17A was a generous gift from Amgen. Purified antibodies against murine L-selectin (MEL-14) and PNAd (MECA-79) were purchased from PharMingen (San Diego, CA) and murine CXCL1 (clone 48415), CXCL2 (clone 40605), and IL-1β (clone 30311) were purchased from R&D Systems (Minneapolis, MN).
In vivo PDT treatment
Tumor-bearing mice were injected in the tail vein with 0.4 μmol/kg HPPH followed 24 h later by illumination as previously described (26). A spot of 1.1 cm2 that contained the tumor was illuminated with 665-nm light produced by a dye laser (375; Spectra Physics) pumped by an argon dye laser (2080; Spectra Physics) to a total dose of 88 J/cm2 or 48 J/cm2 given at 14 mW/cm2. Control mice were treated with photosensitizer alone.
Flow cytometry
Axillary and brachial TDLNs were harvested at the indicated time points and single-cell suspensions were generated. Cells were stained with monoclonal antibodies (mAb) against CD11b (clone M1/70), CD11c (clone HL3), Ly-6G (clone 1A8), Ly-6C (clone AL-21) (all purchased from PharMingen), and F4/80 (clone BM8) (e-bioscience). Intracellular staining was performed according to the manufacturer's suggestion using anti-IL-17 (BD Biosciences, San Jose, CA; clone TC11-18H10) and anti-RORγt (BD Biosciences, clone 31–378). The mAbs were directly conjugated with the fluorochromes FITC, PE, PerCPCy5.5, and allophycocyanin (APC). A BD FACSCalibur was used for flow cytometric analysis; data was acquired from 200,000 TDLN cells and analyzed using the WinList processing program (Verity Software House, Inc.).
Generation of bone marrow chimeras
Il17ra−/− and BALB/c mice were given two doses of 6 Gy irradiation 24 hours apart. Six hours after the second dose of irradiation the mice were rescued with 4 million bone marrow cells injected i.v. The mice were allowed to reconstitute the hematopoietic compartment for nine weeks prior to use.
In vivo antibody treatments
For chemokine and cytokine neutralization studies 100 μg of the following antibodies were administered i.v. immediately before PDT: anti-CXCL1 R&D Systems clone 48415), anti-CXCL2 (R&D Systems clone 40605), anti-IL-1β (R&D Systems clone 30311), or anti-IL-17A (24). For L-selectin (Pharmingen clone MEL-14) and PNAd (Pharmingen clone MECA-79) neutralization, 100 μg of antibody was injected i.v. immediately prior to PDT.
Total RNA extraction
TDLNs were harvested at the indicated time points and frozen in TRIzol® Reagent. Total mRNA was extracted according to manufacturer's protocol (Invitrogen, Carlsbad, CA). Briefly, tissue was homogenized in TRIzol and extracted with chloroform. The RNA was then precipitated using isopropanol and washed once with ethanol. The RNA pellet was then dried and reconstituted in Molecular Grade Water.
Short-term homing assays and immunofluorescence histology
Quantification of neutrophil trafficking was performed as essentially as previously described (28,29). Briefly, an enriched population of neutrophils (~70% Ly-6G+) was isolated from bone marrow by FACS-based flow sorting based on their characteristic size and granularity. These cells were labeled with CellTracker Orange CMTMR (CTO, Molecular Probes) and adoptively transferred i.v. into tumor-bearing mice 3 hours after PDT treatment. TDLNs (axillary and brachial) and peripheral lymph nodes (PLN, inguinal and popliteal) were harvested 30 minutes later from either PDT-treated or HPPH-only treated mice and embedded in optimum cutting temperature compound (OTC; Sakura Fineteck). Both endogenous and adoptively transferred neutrophils were identified by immunofluorescence staining of 9 μm cryosections that were fixed in methanol:acetone (3:1) for Ly-6G (clone 1A8, 68 μg/mL, PharMingen). Expression of adhesion molecules was determined by performing intravascular staining by injecting 20 μg purified mAb specific for ICAM-1 (clone 3E2, PharMingen) in the tail vein. TDLNs were harvested twenty minutes later and embedded in OTC. Tissues were counterstained for PNAd (clone MECA-79, 20 μg/mL, PharMingen) and CD31 (clone 2H8, 5 μg/mL, AbD Serotec). After washing, the primary antibodies were detected by fluorochrome-conjugated (fluorescein, rhodamine, or AmCa) secondary antibodies from Jackson ImmunoResearch (West Grove, PA). Digital images were captured by investigators blinded to sample identity with an Olympus BX50 upright fluorescence microscope equipped with a SPOT RT camera (Spectra Services); all images were captured with identical exposure times and settings. The number of Ly-6G+ (± CTO) cells associated with CD31+ vessels were quantified in at least 9 fields (area of field is 0.34 mm2) unless the size of the TDLNs restricted the number of fields. Images were analyzed with ImageJ software to determine the relative fluorescence staining intensity of hundreds of vessels across 10 fields for PNAd, CD31, ICAM-1, and ICAM-2.
Quantitative real-time PCR (qRT-PCR)
The SuperScript™ III Platinum® SYBR® green one-step qRT-PCR kit was purchased from Invitrogen (Carlsbad, CA) and used according to manufacturer's protocol. For each reaction, 0.5 μg total RNA was used and primers specific for murine IL-17 (100 nM), CXCL2 (200 nM), and GAPDH (100 nM) were utilized. Primer sequences are as follows: IL-17A forward 5'-CTC CAG AAG GCC CTC AGA CTA C-3' and reverse 5'-AGC TTT CCC TCC GCA TTG ACA CAG-3'; CXCL2 forward 5'-GAA CAA AGG CAA GGC TAA CTG A-3' and reverse 5'-AAT CCA TGG CTC AAC TTT CTG CCC-3'; GAPDH forward 5'-ACG GCA AAT TCA ACG GCA CAG TCA-3' and reverse 5'-TGG GGG CAT CGG CAG AAG G-3'. The samples were run on an ABI (Applied Biosystems) 7900 real-time instrument (Carlsbad, CA) with a dissociation curve. The analysis was performed using SDS 2.3 software (Applied Biosystems) to obtain the Ct values for IL-17, CXCL2, and GAPDH. The ΔCt was determined by subtracting the GAPDH Ct value from either the IL-17 or CXCL2 Ct value. The relative amount of IL-17 or CXCL2 mRNA is reported and was calculated by using the equation 2−ΔCt.
Determination of CXCL1, CXCL2, and IL-1β protein expression
TDLNs were harvested at the indicated time points and flash frozen on dry ice. Total protein lysate was generated as previously described (30). Briefly, tissue was homogenized in CelLytic™MT (Sigma Saint Louis, MO) containing protease inhibitor cocktail (Sigma). Cellular debris was removed by centrifugation. The protein concentration of the supernatant was determined using the Bio-Rad Protein Assay (Hercules, CA). ELISA kits specific for murine CXCL1, CXCL2, and IL-1β were purchased from R&D systems (Minneapolis, MN) and used according to the manufacturer's instructions. The results were normalized to per μg total protein.
Statistical evaluation
All measured vales are presented as mean ± SEM. The non-paired Student's t test with Welch's correction was used for comparison between groups in all experiments except for tumor response experiments. In all cases, significance was defined as P ≤ 0.05.
Results
IL-17 regulates accumulation of neutrophils in TDLNs following induction of sterile inflammation by PDT
To determine whether the induction of sterile inflammation by PDT modulates TDLN expression of IL-17, quantitative real-time PCR with IL-17 specific primers was performed using RNA collected from TDLNs of Colo26-HA tumor-bearing mice. IL-17 message levels significantly increased within 4h following PDT and returned to baseline levels by 8h (Figure 1A).
Figure 1. Increased expression of IL-17 by Th17 cells regulates entry of neutrophils to TDLNs following induction of sterile inflammation by PDT.

(A) BALB/c Colo26-HA tumor-bearing mice were subjected to PDT. TDLNs were harvested at the indicated time points post PDT to measure IL-17 mRNA by quantitative RT-PCR. The amount of IL-17 message was normalized to GAPDH and is reported as IL-17 mRNA copies. Each group contains a total of 6 mice and the experiment was repeated once. (B) BALB/c Colo26-HA tumor-bearing mice were subjected to PDT. TDLNs were harvested at the indicated time points post PDT and a single-cell suspension was generated. The absolute number of Th17 in the TDLN was analyzed by flow cytometry and is defined by CD3+CD4+RORγt+iIL-17+. Each group contains a total of 6 mice and the experiment was repeated once. (C) Il17ra−/− or BALB/c mice bearing Colo26-HA tumors were subjected to PDT. (D) BALB/c mice bearing Colo26-HA tumors were treated with 100 μg anti-IL-17A antibodies or isotype control intravenously immediately before PDT. (B – C) TDLNs were harvested at the indicated time points and a single-cell suspension was generated. The absolute number of neutrophils in the TDLN was analyzed by flow cytometry and is defined by CD11b+Ly-6GhiLy-6C+F4/80−CD11c−. Each group contains a total of at least 9 mice and the experiment was repeated twice. Error bars represent mean ± SEM. *P<0.05 to 0h; #P<0.05 to BALB/c.
Potential sources of IL-17 within the TDLN were examined using cell type specific markers and intracellular staining for IL-17. The major IL-17 producing cells in the TDLN following PDT appear to be Th17 cells (CD3+CD4+RORγt+iIL-17+). Th17 cells increased significantly within 2h of PDT (Figure 1B). Innate immune cells such as macrophages and γδ T cells, which are resident populations of LNs can produce IL-17 in response to inflammation (31). IL-17 producing γδ T cells were not detected with in the TDLN before or after PDT. A minor population of IL-17 producing macrophages (CD11b+Ly6C+) was present prior to PDT, but this population did not change following PDT (data not shown). Based on these results, we characterized the regulation of neutrophil entry to TDLNs by IL-17 4-8h post the induction of sterile inflammation by PDT.
To determine whether IL-17 regulates increased neutrophil accumulation in TDLNs following induction of sterile inflammation by PDT, the presence of these leukocytes was measured by flow cytometry in TDLNs of BALB/c and Il17ra−/− mice. Neutrophils were defined as cells expressing CD11b, high levels of Ly-6G, and Ly-6C but lacking expression of F4/80 and CD11c (32); the identity of these cells as neutrophils was confirmed morphologically [(6) and data not shown]. The number of neutrophils in TDLNs of BALB/c mice significantly increased within 4h of treatment (Figure 1C). The accumulation of neutrophils in TDLNs of Il17ra−/− mice following PDT was substantially reduced compared to accumulation in BALB/c TDLNs. Similar results were obtained when IL-17A was neutralized immediately prior to PDT by antibodies (Figure 1D), indicating that IL-17 activity was required during the acute phase of sterile inflammation induced following PDT. IL-17 also regulated accumulation of neutrophils in TDLNs following treatment of a second mouse model, EMT6 (Supplementary Figure 1A). PDT induces systemic neutrophilia (23), which could account for the presence of neutrophils in the TDLN. However equivalent increases in the number of systemic neutrophils were detected following PDT in both BALB/c and il17ra−/− mice, suggesting that improved entry of neutrophils into the TDLN involves an active mechanism of entry regulated by IL-17 rather than passive mechanisms due solely to neutrophilia (Supplementary Figure 1B).
To ascertain whether the contribution of IL-17 to neutrophil accumulation in TDLNs is unique to PDT or occurs in other sterile inflammatory settings, turpentine was injected intra-tumorally into Colo26-HA tumors grown in BALB/c and Il17ra−/− mice. Like PDT, intra-tumoral injection of turpentine results in tumor cell necrosis and acute inflammation characterized by rapid neutrophil infiltration and release of IL-6 (6,26). There is no difference between the acute tumor response of BALB/c and Il17ra−/− mice (data not shown). The number of neutrophils markedly increased in TDLNs within an hour of turpentine injection (Supplementary Figure 1C). Significantly fewer total neutrophils were detected 2h after turpentine injection in TDLNs of Il17ra−/− mice when compared to BALB/c mice. Thus, it appears that secretion of IL-17 by Th17 cells in TDLN contributes to neutrophil accumulation in TDLNs following induction of sterile inflammation.
Non-hematopoietic expression of IL-17RA regulates neutrophil accumulation in TDLNs post PDT
Both non-hematopoietic and hematopoietic cells express IL-17RA (33). Bone marrow chimeras were generated to investigate whether expression of IL-17RA by either hematopoietic or non-hematopoietic cells regulates accumulation of neutrophils in TDLNs following sterile inflammation, (Figure 2). Colo26-HA tumor-bearing chimeras were treated with PDT and neutrophil accumulation in TDLNs was examined. Neutrophil accumulation was similar in TDLNs of chimeras expressing IL-17RA in only non-hematopoietic cells (Il17ra−/− → BALB/c) and control chimeras (BALB/c → BALB/c). In contrast, significantly fewer neutrophils accumulated in TDLNs of chimeras lacking IL-17RA expression in non-hematopoietic cells (BALB/c → Il17ra−/−), which was similar to accumulation observed in Il17ra−/− → Il17ra−/− chimeras. These results demonstrate that expression of IL-17RA by non-hematopoietic stromal cells regulates accumulation of neutrophils in TDLNs following induction of sterile inflammation.
Figure 2. IL-17RA expression by non-hematopoietic cells regulates neutrophil accumulation in TDLNs post PDT.

Il17ra−/− chimera mice were generated as described in Methods. Colo26-HA tumor-bearing mice were subjected to PDT. TDLNs were harvested 8h post treatment; a single-cell suspension was generated and analyzed by flow cytometry for neutrophils. The results are expressed as absolute number of neutrophils per TDLN. Error bars represent mean ± SEM. Each group contains a minimum of 8 mice and the experiment was repeated twice. *P<0.03 to 0h control; #P<0.003 to 8h BALB/c → BALB/c.
HEVs of TDLNs are the primary portal of entry for neutrophils following PDT treatment
Neutrophil entry in TDLNs following PDT-induced sterile inflammation could potentially occur through three vascular ports: afferent lymphatics functioning as conduits for migration from tumor sites, as reported during infection (5); inflamed blood vessels within TDLNs; and post-capillary HEVs in TDLNs. To distinguish between these possibilities, we took a two-pronged approach as outlined in Figure 3A. First, the spatial distribution of endogenous Ly-6G+ neutrophils relative to vessels was quantified 3.5 hours post-PDT in TDLN cryosections. Vessels were counterstained for the pan-endothelial CD31 adhesion molecule which demarks flat-walled vessels (i.e., afferent and efferent lymphatics, and inflamed blood vessels) and HEVs (34). HEVs were distinguished morphometrically based on their cuboidal endothelium and confirmed, in all cases, to be PNAd+ in adjacent serial sections. Additionally, we tracked the destination of an enriched population of cell tracker orange (CTO)-labeled neutrophils following intravenous transfer in PDT-treated mice. Quantification at 30 minutes post-adoptive transfer allowed for definitive evaluation of the frequency of CTO/Ly-6G+ neutrophil interactions exclusively with blood vessels (CD31+ flat-walled or HEVs) since this time period is too short for leukocytes to emigrate from peripheral tissues via afferent lymphatics (29).
Figure 3. TDLN HEVs support increased neutrophil accumulation following PDT treatment.

(A) A single-cell suspension of neutrophils was fluorescently labeled with CellTracker Orange CMTMR (red) and adoptively transferred into treated mice as shown in the schematic. (B) Lymph node tissue sections were stained with anti-CD31 (green) and anti-Ly6G (blue). Endogenous (blue) and adoptively transferred Ly6G+ cells (pink) were counted following short-term (0 – 30 minute) homing assays. Representative photomicrographs of control (HPPH only) and PDT treated lymph nodes are shown. Quantification of endogenous Ly6G+ cells and adoptively transferred Ly6G+ neutrophils is presented in (C) and (D), respectively. Serial sections were stained to confirm that CD31-expressing cuboidal cells also expressed the HEV-specific marker PNAd. Data (mean ± SEM) are of ≥ 9 fields, unless the size of the TDLN restricted the number of fields, in individual mice and are representative of 3 independent experiments. *P<0.05 Scale bars represent 50 μm; parenchyma, TDLN stroma; PLN, peripheral LN (inguinal and popliteal).
The data shown in Figures 3B and C indicate that constitutive accumulation of endogenous neutrophils within the parenchyma of TDLNs or peripheral LNs (PLNs) is low following administration of the HPPH photosensitizer alone, consistent with reports of minimal homeostatic trafficking of neutrophils to LNs (29). A marked increase in the localization of endogenous neutrophils upon PDT treatment occurred in the HEV-rich paracortical region exclusively in TDLNs while homing in distal PLN remained low. Improved trafficking in TDLNs following PDT was accompanied by a modest increase (p<0.07) in the association of endogenous neutrophils with flat-walled vessels including the subcapsular sinus (Figures 3B, C and Supplementary Figure 2) that is indicative of influx via afferent lymphatics and/or egress through efferent lymphatics (35,36). However, these cells represented a relatively minor proportion (< 10%) of total vessel-associated neutrophils.
In sharp contrast, PDT-induced sterile inflammation caused a profound increase in the number of neutrophils associated with HEVs. Notably, > 90% of vessel-associated endogenous neutrophils interacted with HEVs. These findings were substantiated in short-term (30 minute) homing assays where > 95% of adoptively transferred neutrophils detected in TDLNs interacted selectively with HEVs (Figures 3B, D). Moreover, trafficking of endogenous neutrophils in TDLN was strongly inhibited by antibody neutralization of prototypical homing receptors required for binding to HEVs (i.e., leukocyte-expressed L-selectin and HEV-presented PNAd) (Figures 4A and B). The density of TDLN HEVs remained constant following PDT (average number of HEVs/field in HPPH-control and PDT-treated mice was 5.5 ± 0.95 and 5.4 ± 0.41, respectively; n = 2 mice) indicating that the intrinsic binding activity of endothelial cells lining HEVs was enhanced. The increase in HEV adhesion could not be explained by changes in the intravascular density of molecules required for transient tethering/rolling interactions (PNAd) or firm arrest (ICAM-1, ICAM-2), or transendothelial migration (CD31) as determined by quantitative immunofluorescence histology (Figure 4C, Supplementary Figure 3). Additionally L-selectin expression was not increased on neutrophils recovered from TDLNs after PDT (Supplementary Figure 3). Taken together, these data strongly implicate HEVs as the major route of neutrophil entry in TDLNs following induction of sterile inflammation by PDT and point to chemokine-dependent adhesion as the molecular switch that converts HEVs to an active site of neutrophil recruitment.
Figure 4. Neutrophils gain entry to TDLNs following induction of sterile inflammation through HEV interactions.

TDLNs were harvested at the indicated time points post PDT. A single-cell suspension was generated and analyzed by flow cytometry for the number of neutrophils per TDLN in the presence of (A) 100 μg isotype control or 100 μg anti-L-selectin antibodies or (B) 100 μg isotype control or 100 μg anti-PNAd antibodies. (C) Immunofluorescence staining of TDLNs for PNAd and ICAM-1 was performed as described in Methods. The numerical value represents the mean fluorescence intensity (MFI). Each group contains a minimum of 9 mice and the experiment was repeated twice. Error bars represent mean ± SEM. *P<0.05 to 0h; #P<0.007 to Isotype.
Increased neutrophil entry in TDLNs depends on CXCR2 and CXCL2, but not CXCL1
Neutrophils are equipped with trafficking molecules necessary for binding PNAd and ICAM-1/2 (i.e., L-selectin and LFA-1, respectively) although they normally fail to extravasate across HEVs because they cannot initiate CCR7/CCL21-dependent transition from rolling to firm arrest within vessel walls (35). Beauvillain et al. (5) previously demonstrated that neutrophils acquire CCR7 and gain access to LNs through the afferent lymphatics in response to pathogen-mediated inflammation. However, neutrophils that accumulate in TDLNs following induction of sterile inflammation by PDT express CXCR2 but only minimal levels of CCR7 (Supplementary Figure 3). Furthermore there is no significant change in the levels of homeostatic chemokines, CCL19/21, expressed in the TDLNs post PDT (data not shown). These observations suggested that chemokines other than CCL19/21 are responsible for neutrophil entry across HEVs in TDLNs following induction of sterile inflammation.
CXCR2 mediates neutrophil migration into non-lymphoid tissue at sites of inflammation (7). To determine if neutrophils were using CXCR2 to enter TDLNs, neutrophil accumulation in TDLNs was measured in Cxcr2−/− mice following induction of sterile inflammation. The lack of CXCR2 expression significantly reduced the number of neutrophils in TDLNs following PDT (Figure 5A). The chemokines CXCL2 and CXCL1 are ligands for CXCR2 (37). Injection of antibodies specific to CXCL2 immediately prior to induction of sterile inflammation by PDT significantly reduced the number of neutrophils in TDLNs at 4h and 8h post PDT (Figure 5B). In contrast, neutralizing CXCL1 reduced the accumulation of neutrophils in TDLNs only at 8h post PDT (Figure 5C). Collectively, these results suggest that entry of neutrophils in TDLNs following induction of sterile inflammation is mediated by ligation of CXCR2 with CXCL2.
Figure 5. Increased neutrophil entry to TDLNs is supported by CXCR2 and CXCL2, but not CXCL1.

(A) Colo26-HA tumor-bearing Cxcr2−/− or BALB/c mice were subjected to PDT and the number of neutrophils per TDLN was determined by flow cytometry at the indicated time points. (B – C) TDLNs were harvested at the indicated time points post PDT and a single-cell suspension was generated. The number of neutrophils per TDLN was analyzed by flow cytometry in the presence of (B) 100 μg anti-CXCL2 antibodies or (C) 100 μg anti-CXCL1 antibodies given intravenously immediately prior to PDT. Each group contains a minimum of 9 mice and the experiment was repeated twice. Error bars represent mean ± SEM. *P<0.02 to 0h; #P<0.02 to Isotype or BALB/c.
IL-17 regulates CXCL2 expression in TDLNs post-PDT
IL-17:IL-17RA interaction is critical for CXCL2/CXCL1 expression following pathogen infection (8). To determine whether IL-17 also controls CXCL2/CXCL1 expression following induction of sterile inflammation, expression of CXCL2/CXCL1 in TDLNs following PDT was assessed. The expression of CXCL2 mRNA (Figure 6A) and protein (Figure 6B) profoundly increased in TDLNs of Colo26-HA tumor-bearing BALB/c mice post PDT. In sharp contrast, expression of CXCL2 mRNA and protein in TDLNs of Il17ra−/− mice was largely unchanged post PDT.
Figure 6. IL-17 regulates induction of CXCL2 in TDLNs post PDT.

Il17ra−/− or BALB/c Colo26-HA tumor-bearing mice were subjected to PDT. TDLNs were harvested at the indicated time points post PDT to measure expression of CXCL2 mRNA or protein. (A) Quantitative RTPCR was performed and the amount of CXCL2 message was normalized to GAPDH and is reported as CXCL2 mRNA copies. (B) An ELISA kit (R&D Systems) specific for murine CXCL2 was used and the amount of CXCL2 protein per μg total protein is reported. Each group contains a minimum of 9 mice and the experiment was repeated twice. Error bars represent mean ± SEM. *P<0.05 to 0h; #P<0.05 to BALB/c.
In contrast to the regulation of CXCL2 mRNA in TDLNs by IL-17 post PDT, minimal differences were observed in CXCL1 mRNA and protein expression in TDLNs of BALB/c and Il17ra−/− mice following PDT (Supplementary Figure 4). CXCL1 protein levels in Il17ra−/− TDLNs were 1.4 fold lower than in BALB/c mice at the 4h time point. In comparison, CXCL2 levels were 4-fold lower 4h post PDT. These results demonstrate that IL-17 regulates expression of CXCL2 mRNA and protein in TDLNs post the induction of sterile inflammation by PDT.
IL-17 regulated IL-1β expression enhances CXCL2 expression and accumulation of neutrophils in TDLNs post induction of sterile inflammation by PDT
IL-1β synergizes with IL-17 to augment the expression of CXCL2 (14). We therefore sought to determine the contribution of IL-1β to the regulation of CXCL2 expression and neutrophil entry in TDLNs by IL-17 post induction of sterile inflammation by PDT. Although the expression of IL-1β in TDLNs of tumor-bearing BALB/c and Il17ra−/− mice significantly increased following PDT (Figure 7A), induced levels of IL-1β were significantly less in Il17ra−/− than those observed in BALB/c mice. This result suggests that IL-17 contributes to the regulation of IL-1β in the TDLNs post PDT. To determine whether IL-1β regulates CXCL2 expression in TDLNs, neutralizing antibodies against IL-1β were administered prior to the induction of sterile inflammation by PDT (Figure 7B). Neutralizing IL-1β partially inhibited CXCL2 expression in TDLNs, suggesting that IL-1β regulates CXCL2 expression. To ascertain the contribution of IL-17 and IL-1β to the accumulation of neutrophils in TDLNs following induction of sterile inflammation, IL-17 or IL-1β either alone or in combination were neutralized with antibodies immediately before PDT treatment (Figure 7C). Blocking either IL-17 or IL-1β significantly reduced the entry of neutrophils in TDLNs post PDT. Although blocking IL-1β had a greater effect on the entry of neutrophils into TDLNs than blocking IL-17, the difference was not significant. Blocking both IL-17 and IL-1β resulted in no further reduction in the entry of neutrophils in TDLNs than blocking either alone. Taken together, these results suggest that rather than acting in synergy, IL-17 acts upstream of IL-1β to enhance CXCL2 expression and support entry of neutrophils to TDLNs following the induction of sterile inflammation.
Figure 7. Regulation of IL-1β by IL-17RA enhances CXCL2 expression and neutrophil access to TDLNs post induction of sterile inflammation.

(A) Il17ra−/− or BALB/c Colo26-HA tumor-bearing mice were subjected to PDT. TDLNs were harvested at the indicated time points post PDT and IL-1β protein expression was measured by ELISA (R&D Systems). The amount of IL-1β protein per μg total protein is reported. (B) Colo26-HA tumor-bearing BALB/c mice were treated with 100 μg isotype control or 100 μg anti-IL-1β antibodies intravenously immediately prior to PDT. TDLNs were harvested at the indicated time points to measure CXCL2 protein expression by ELISA (R&D Systems). The amount of CXCL2 protein per μg total protein is reported. (C) Colo26-HA tumor-bearing BALB/c mice were treated with 100 μg isotype control or 100 μg anti-IL-17A or 100 μg anti-IL-1β antibodies either alone or in combination intravenously immediately prior to PDT. TDLNs were harvested at the indicated time points post PDT and a single-cell suspension was generated. The number of neutrophils was determined by flow cytometry. Each group contains a minimum of 9 mice and the experiment was repeated twice. Error bars represent mean ± SEM. *P<0.04 to 0h; #P<0.04 to Isotype or BALB/c.
Discussion
The paradigm has been that neutrophils are normally excluded from gaining entry to LNs via HEVs (18) and thus, their contribution to controlling adaptive immune responses has been largely overlooked. Here we report that the induction of sterile inflammation by PDT increases the entry of neutrophils to TDLNs. Our results demonstrate that entry of neutrophils into TDLNs during sterile inflammation occurs via HEV portals and is dependent upon IL-17 mediated induction of CXCL2. The effects of IL-17 on CXCL2 are dependent upon IL-1β, which is enhanced by IL-17. To our knowledge, this is the first demonstration that IL-17 regulates IL-1β expression in TDLNs post induction of sterile inflammation. These findings suggest a model for neutrophil entry to TDLNs following induction of sterile inflammation whereby increased expression of IL-1β by IL-17 leads to the induction of CXCL2, which acts as a molecular switch that converts HEVs to active sites of neutrophil recruitment (Figure 8).
Figure 8. Blood-borne neutrophils enter TDLNs across HEVs following induction of sterile inflammation.
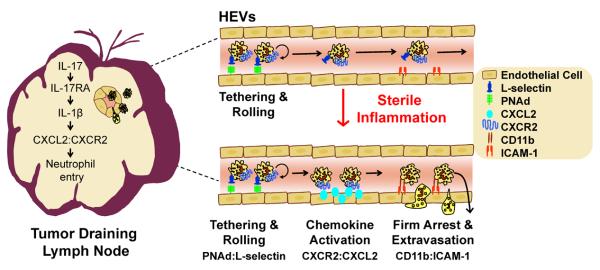
Our findings indicate that neutrophils gain entry to TDLNs through interactions of L-selectin with HEV-borne PNAd and is supported by preferential interactions between CXCR2 and CXCL2. Induction of CXCL2 in TDLNs is in a linear pathway downstream of IL-17:IL-17RA dependent regulation of IL-1β expression.
Immunohistochemical analysis of PDT-treated TDLNs revealed an extremely high density of neutrophils within the paracortex region, a HEV-rich region of LNs. PDT leads to an increase in the overall size of TDLNs (6), which results in an overall increase in the number of HEVs available for trafficking. PDT also increases the intrinsic binding properties of HEVs as evident by the findings that endogenous and adoptively transferred neutrophils exhibit increased association with individual HEVs following treatment. Thus, it appears that modulation of neutrophil numbers found in TDLNs following induction of sterile inflammation is due to a combination of increased LN size and enhanced adhesion within the HEVs.
The majority of studies demonstrating that neutrophils can access inflamed LNs during pathogen infections have suggested that migration occurs via the lymphatics (2,4,5) because while neutrophils can use L-selectin to roll along HEVs, firm adhesion and extravasation is prevented due to the lack of activating chemokines (19). Recent work by Beauvillain et al. (5) showed that neutrophils acquire CCR7 and access inflamed LNs through the afferent lymphatics during pathogen induced inflammation. Our study indicates that the majority of neutrophils that enter TDLNs through HEV portals following induction of sterile inflammation are CCR7low. Mechanistic studies reveal that during sterile inflammation HEVs undergo a molecular switch involving CXCR2 engagement by CXCL2 that enables blood-borne neutrophils to gain access to TDLNs. Blocking of CXCL2 or the use of CXCR2 deficient mice virtually eliminated neutrophil accumulation in the TDLNs. These results support the conclusion that neutrophil migration to TDLNs following induction of sterile inflammation by PDT is unique. The distinction between the mechanisms of neutrophil entry in LNs triggered by sterile and pathogen-induced inflammation is further supported by the lack of increase in neutrophil expression of CCR7 or LN expression of CCL19/21 following induction of sterile inflammation. Furthermore previous studies have shown that the lymphatic endothelium does not express either PNAd or ICAM-1 (19). There has been one report demonstrating that the lymphatic endothelium expresses CXCL2 mRNA (38) and additional support for the lack of involvement of CCR7 in neutrophil migration during sterile inflammation could be obtained by examining migration in CCR7-deficient mice. However, these mice are only available on the B6 background, which is incompatible with the CT26 tumor model.
IL-17 has no effect on the expression of CCR7 by neutrophils (5). However IL-17 has been shown to augment the migration of neutrophils in response to CCL19/21 in the presence of GMCSF (5). Systemic levels of GM-CSF increase following PDT (23,39) and neutrophils can enter LNs via CCR7 mediated migration through the lymphatics (5). Additionally sphingosine 1-phosphate (S1P) can enhance neutrophil transendothelial migration (40) and inhibition of S1P blocks the IL-23/IL-17/G-CSF mediated neutrophil migration from the blood into tissue (41). S1P is expressed in LNs and its expression increases during inflammation (41). We do observe a minor population of neutrophils that appears to enter TDLNs via afferent lymphatics or an alternative manner. Thus, it is possible that while our data indicates that neutrophils primarily gain access to TDLNs following induction of sterile inflammation via IL-17/CXCL2 mediated migration across HEV portals, minor populations of neutrophils enter the TDLN via the lymphatics through IL-17 mediated increases in CCR7 or via S1P mediated transendothelial migration.
The lack of L-selectin down regulation post migration of neutrophils through HEVs is somewhat surprising. Previous reports have shown that neutrophils shed L-selectin upon activation and that rolling neutrophils have reduced expression of L-selectin (42–45). L-selectin shedding is initiated by cell activation and the subsequent induction of apoptosis (44,46). However, activated neutrophils can re-express L-selectin following activation induced down-regulation and prior to cell death (47–49). Therefore it is possible that the L-selectin expression observed in our study is a result of the molecule being re-expressed following activation induced down regulation.
Neutrophil accumulation in TDLNs post PDT was not completely suppressed in Il17ra−/− mice or by blocking antibodies to IL-17A suggesting that other IL-17R subunits or isoforms of IL-17 may be involved. IL-17RC, which forms a hetermeric complex with IL-17RA, may also regulate neutrophil entry to TDLNs post PDT. Although the relative contributions of each cytokine receptor subunit to downstream signaling have remained elusive, IL-17RC has a higher affinity for IL-17F than IL-17A (9). Work by Ho et al. (50) demonstrated that in vitro IL-17RC does not require IL-17RA for IL-17-dependent signaling in HEK293T cells and in vivo IL-17RC acts indistinguishable from IL-17RA for IL-17-dependent responses. Therefore, it is possible that IL-17F may signal through IL-17RC expressed on the stromal elements of the TDLNs independently of IL-17RA and contribute to CXCL2 expression and neutrophil accumulation in TDLNs post PDT.
PDT induces a sterile inflammatory response within the treated tumor (23). Treatment results in rapid apoptosis of tumor cells induced by the generation of singlet oxygen. The primary apoptotic response is replaced by secondary necrosis due to an overwhelming of the clearance response. Direct tumor cell death is accompanied by vascular damage and induction of acute inflammation that contribute to overall tumor destruction. The acute inflammatory response within tumors is characterized by rapid neutrophil infiltration and systemic release of pro-inflammatory cytokines, including IL-6. A similar response is observed within the tumor bed upon injection of turpentine (6,24,26).
Our studies suggest that Th17 cells are the primary source of IL-17 following PDT, with significant increases in these cells detected in TDLN within 2h of PDT. The rapid accumulation of IL-17 expressing CD3+CD4+RORγT+ cells following PDT suggests that these cells may be natural Th17 cells. Natural Th17 (nTh17) cells acquire the ability to produce IL-17 in the thymus; conventional Th17 cells require further stimulation in peripheral tissue to produce IL-17 (51,52). nTh17 and conventional Th17 cells share similar phenotypes (CD3+, CD4+, RORγT+) and express similar cytokines (IL-17 and IL-122). Both cell types mediate host protection during inflammation through recruitment of neutrophils.
The distinct mechanisms by which neutrophils access the LNs following sterile inflammation and pathogen induced inflammation indicate that neutrophil migration into LNs depends on the inflammatory stimuli. Both PDT and infection result in acute inflammation. PDT generated sterile inflammation is triggered by release of danger associated molecular patterns (DAMPs), endogenous factors that are normally sequestered intracellularly from dying cells. In contrast, pathogen-generated acute inflammation is initiated by small molecular sequences conserved by pathogens called pathogen associated molecular patterns (PAMPs). The release of DAMPs from necrotic cells following PDT induces a sterile inflammatory response characterized by an increase in expression of IL-1β (53), a known regulator of CXCL2 expression (54), and marked enhancement of neutrophil recruitment in TDLNs. Thus, the sterile inflammatory response generated by PDT is poised to support the entry of neutrophils to TDLNs.
The mechanism by which IL-17 regulates IL-1β is unclear. Secretion of IL-1β is a two-step process that involves production of pro-IL-1β and cleavage of pro-IL-1β into its active form (1). The production of pro-IL-1β is dependent upon NF-κB activation (55). IL-17RA (9) and Toll-like receptor signaling (56) both activate NF-κB. Thus, the production of pro-IL-1β may be due to IL-17-dependent and independent mechanisms. Cleavage of pro-IL-1β into its biologically active form occurs following activation of the NALP3 inflammasome (55). NALP3 can be activated by a variety of endogenous mediators including reactive oxygen and extracellular ATP, which are released during sterile inflammation (57,58). Pro-IL-1β can also be processed by neutrophil serine proteases (59). Both NALP3 and elastase expression/activity can be increased by IL-17 (60,61).
Intriguingly, our findings demonstrate that whereas IL-17 regulates expression of CXCL2 in the TDLNs post PDT, expression of CXCL1 is largely independent of IL-17. The mechanism of regulation for these chemokines in the TDLNs post induction of sterile inflammation remains unclear. IL-17 activates NF-κB, a hallmark transcription factor associated with the induction of inflammation (62). DNA elements that bind NF-κB in the promoters of IL-17 target genes are required for IL-17-induced gene expression (63). Sequence analysis identified three NF-κB DNA binding sites within the CXCL1 promoter region whereas the CXCL2 promoter region contains an IRF DNA binding site upstream of two NF-κB sites (64). This critical difference in the promoter regions suggests that in addition to the NF-kB signaling pathway, CXCL2 gene expression is also controlled by the TIR-domain-containing adapter-inducing interferon-β (TRIF) pathway. TRIF is an adapter protein that mediates signal transduction downstream of TLR3 and TLR4. TLRs have been shown to affect the efficacy of chemotherapy (65) and PDT (66). It has been reported that IL-17 additively cooperates with TLR4 ligands to augment the expression of neutrophilic chemokines (67). Therefore, cancer therapies that result in tumor cell death may initiate the release of DAMPs that bind TLR4 and lead to downstream signaling through TRIF resulting in binding of IRF to the CXCL2 promoter. Activated NF-κB by IL-17 would also bind to the CXCL2 promoter. Cooperation between the binding of IRF and NF-κB to their respective DNA binding elements in the CXCL2 promoter would enhance CXCL2 gene transcription.
Our results show that although CXCL2 and CXCL1 are both induced in TDLNs post sterile inflammation, only CXCL2 is required for the entry of neutrophils to TDLNs. Call et al. (68) reported that although acute inflammation increased expression of both CXCL2 and CXCL1 in the inflamed tissue, CXCL2 was limited to the inflamed tissue whereas CXCL1 was found systemically in the circulation. CXCL2 is considered an inducible inflammatory chemokine that is thought to mediate stress- or injury-induced neutrophil migration (69). The sequestration of CXCL2 in inflamed TDLNs may be explained by its ability to be presented on the HEVs; however, it remains unclear whether this occurs. A recent report demonstrated an intravascular chemokine gradient of CXCL2 on the venular endothelium of the cremaster muscle, which directed neutrophil crawling (70). This finding supports the possibility that CXCL2 is capable of being presented on the HEVs through a process termed transcytosis, leading to chemokine activation of neutrophils and transendothelial migration across HEVs into TDLNs. This scenario would not be unprecedented as Palframm et al. (71) has previously reported that inflammatory chemokines are capable of undergoing transcytosis and becoming expressed on HEVs.
The TDLNs is the site of T cell activation. Neutrophils have been shown to influence T cell activation through secretion of chemokines and granule proteins that recruit monocytes and dendritic cells (72), activation of DCs via cell-to-cell contact and secretion of TNF-α (73,74), and secretion of IFN-γ that stimulates monocytes and T cell differentiation (72). We have shown that neutrophils are required for PDT efficacy (26) and that migration of neutrophils to TDLNs enhances the generation of anti-tumor immunity (6). The precise mechanism by which neutrophils contribute to PDT efficacy and the generation of anti-tumor adaptive immunity is under investigation. However, the findings reported here suggest that sterile inflammation alters the microenvironment to permit neutrophil entry in TDLNs via a previously unrecognized delivery mechanism across HEV portals of entry.
Supplementary Material
Acknowledgements
We thank Dr. Sarah Gaffen (University of Pittsburgh) for the generous gift of the Il17ra−/− mice bred on the BALB/c background. The authors acknowledge Debbie Tabaczynski and Kimberley Ramsey (Roswell Park Cancer Institute, Buffalo NY) for their excellent breeding and genotyping of the Il17ra−/− mice. We thank Amgen (Thousand Oaks, California) for their generous gift of the anti-mouse IL-17A antibodies.
Abbreviations
- HA
hemagglutinin
- HEV
high endothelial venule
- HPPH
2-[1-hexyloxyethyl]-2-devinyl pyropheophorbide-a
- LN
lymph node
- PDT
photodynamic therapy
- TDLN
tumor draining lymph node
Footnotes
This work was supported by National Cancer Institute grants (CA109480 & CA98156, S.O.G.) and (CA79765 & CA094045, S.S.E.). C.M.B. was supported by National Cancer Institute T32 grant (CA085183) and in part by the Roswell Park Cancer Center Support Grant CA16056.
References
- 1.Rock KL, Latz E, Ontiveros F, Kono H. The sterile inflammatory response. Annu. Rev. Immunol. 2010;28:321–342. doi: 10.1146/annurev-immunol-030409-101311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abadie V, Badell E, Douillard P, Ensergueix D, Leenen PJ, Tanguy M, Fiette L, Saeland S, Gicquel B, Winter N. Neutrophils rapidly migrate via lymphatics after Mycobacterium bovis BCG intradermal vaccination and shuttle live bacilli to the draining lymph nodes. Blood. 2005;106:1843–1850. doi: 10.1182/blood-2005-03-1281. [DOI] [PubMed] [Google Scholar]
- 3.Maletto BA, Ropolo AS, Alignani DO, Liscovsky MV, Ranocchia RP, Moron VG, Pistoresi-Palencia MC. Presence of neutrophil-bearing antigen in lymphoid organs of immune mice. Blood. 2006;108:3094–3102. doi: 10.1182/blood-2006-04-016659. [DOI] [PubMed] [Google Scholar]
- 4.Chtanova T, Schaeffer M, Han SJ, van Dooren GG, Nollmann M, Herzmark P, Chan SW, Satija H, Camfield K, Aaron H, Striepen B, Robey EA. Dynamics of neutrophil migration in lymph nodes during infection. Immunity. 2008;29:487–496. doi: 10.1016/j.immuni.2008.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beauvillain C, Cunin P, Doni A, Scotet M, Jaillon S, Loiry ML, Magistrelli G, Masternak K, Chevailler A, Delneste Y, Jeannin P. CCR7 is involved in the migration of neutrophils to lymph nodes. Blood. 2011;117:1196–1204. doi: 10.1182/blood-2009-11-254490. [DOI] [PubMed] [Google Scholar]
- 6.Kousis PC, Henderson BW, Maier PG, Gollnick SO. Photodynamic therapy (PDT) enhancement of anti-tumor immunity is regulated by neutrophils. Can. Res. 2007;67:10501–10510. doi: 10.1158/0008-5472.CAN-07-1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cacalano G, Lee J, Kikly K, Ryan AM, Pitts-Meek S, Hultgren B, Wood WI, Moore MW. Neutrophil and B cell expansion in mice that lack the murine IL-8 receptor homolog. Science. 1994;265:682–684. doi: 10.1126/science.8036519. [DOI] [PubMed] [Google Scholar]
- 8.Ye P, Garvey PB, Zhang P, Nelson S, Bagby G, Summer WR, Schwarzenberger P, Shellito JE, Kolls JK. Interleukin-17 and lung host defense against Klebsiella pneumoniae infection. Am. J. Respir. Cell Mol. Biol. 2001;25:335–340. doi: 10.1165/ajrcmb.25.3.4424. [DOI] [PubMed] [Google Scholar]
- 9.Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat. Rev. Immunol. 2009;9:556–567. doi: 10.1038/nri2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toy D, Kugler D, Wolfson M, Vanden BT, Gurgel J, Derry J, Tocker J, Peschon J. Cutting edge: interleukin 17 signals through a heteromeric receptor complex. J. Immunol. 2006;177:36–39. doi: 10.4049/jimmunol.177.1.36. [DOI] [PubMed] [Google Scholar]
- 11.Datta S, Biswas R, Novotny M, Pavicic PG, Jr., Herjan T, Mandal P, Hamilton TA. Tristetraprolin regulates CXCL1 (KC) mRNA stability. J. Immunol. 2008;180:2545–2552. doi: 10.4049/jimmunol.180.4.2545. [DOI] [PubMed] [Google Scholar]
- 12.Iyoda M, Shibata T, Kawaguchi M, Hizawa N, Yamaoka T, Kokubu F, Akizawa T. IL-17A and IL-17F stimulate chemokines via MAPK pathways (ERK1/2 and p38 but not JNK) in mouse cultured mesangial cells: synergy with TNF-alpha and IL-1beta. Am. J. Physiol Renal Physiol. 2010;298:F779–F787. doi: 10.1152/ajprenal.00198.2009. [DOI] [PubMed] [Google Scholar]
- 13.Roussel L, Houle F, Chan C, Yao Y, Berube J, Olivenstein R, Martin JG, Huot J, Hamid Q, Ferri L, Rousseau S. IL-17 promotes p38 MAPK-dependent endothelial activation enhancing neutrophil recruitment to sites of inflammation. J. Immunol. 2010;184:4531–4537. doi: 10.4049/jimmunol.0903162. [DOI] [PubMed] [Google Scholar]
- 14.Dragon S, Rahman MS, Yang J, Unruh H, Halayko AJ, Gounni AS. IL-17 enhances IL-1beta-mediated CXCL-8 release from human airway smooth muscle cells. Am. J. Physiol Lung Cell Mol. Physiol. 2007;292:L1023–L1029. doi: 10.1152/ajplung.00306.2006. [DOI] [PubMed] [Google Scholar]
- 15.Wang X, Feuerstein GZ, Gu JL, Lysko PG, Yue TL. Interleukin-1 beta induces expression of adhesion molecules in human vascular smooth muscle cells and enhances adhesion of leukocytes to smooth muscle cells. Atherosclerosis. 1995;115:89–98. doi: 10.1016/0021-9150(94)05503-b. [DOI] [PubMed] [Google Scholar]
- 16.Armstrong DA, Major JA, Chudyk A, Hamilton TA. Neutrophil chemoattractant genes KC and MIP-2 are expressed in different cell populations at sites of surgical injury. J. Leukoc. Biol. 2004;75:641–648. doi: 10.1189/jlb.0803370. [DOI] [PubMed] [Google Scholar]
- 17.McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, Beck PL, Muruve DA, Kubes P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330:362–366. doi: 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
- 18.Warnock RA, Askari S, Butcher EC, von Andrian UH. Molecular mechanisms of lymphocyte homing to peripheral lymph nodes. J. Exp. Med. 1998;187:205–216. doi: 10.1084/jem.187.2.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.von Andrian UH, Mempel TR. Homing and cellular traffic in lymph nodes. Nat. Rev. Immunol. 2003;3:867–878. doi: 10.1038/nri1222. [DOI] [PubMed] [Google Scholar]
- 20.Vardam TD, Zhou L, Appenheimer MM, Chen Q, Wang WC, Baumann H, Evans SS. Regulation of a lymphocyte-endothelial-IL-6 trans-signaling axis by fever-range thermal stress: hot spot of immune surveillance. Cytokine. 2007;39:84–96. doi: 10.1016/j.cyto.2007.07.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shamri R, Grabovsky V, Gauguet JM, Feigelson S, Manevich E, Kolanus W, Robinson MK, Staunton DE, von Andrian UH, Alon R. Lymphocyte arrest requires instantaneous induction of an extended LFA-1 conformation mediated by endothelium-bound chemokines. Nat. Immunol. 2005;6:497–506. doi: 10.1038/ni1194. [DOI] [PubMed] [Google Scholar]
- 22.Chen Q, Fisher DT, Clancy KA, Gauguet JM, Wang WC, Unger E, Rose-John S, von Andrian UH, Baumann H, Evans SS. Fever-range thermal stress promotes lymphocyte trafficking across high endothelial venules via an interleukin 6 trans-signaling mechanism. Nat. Immunol. 2006;7:1299–1308. doi: 10.1038/ni1406. [DOI] [PubMed] [Google Scholar]
- 23.Agostinis P, Berg K, Cengel KA, Foster TH, Girotti AW, Gollnick SO, Hahn SM, Hamblin MR, Juzeniene A, Kessel D, Korbelik M, Moan J, Mroz P, Nowis D, Piette J, Wilson BC, Golab J. Photodynamic therapy of cancer: An update. CA Cancer J. Clin. 2011 doi: 10.3322/caac.20114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gollnick SO, Evans SE, Baumann H, Owczarczak B, Maier P, Vaughan L, Wang WC, Unger E, Henderson BW. Role of cytokines in photodynamic therapy-induced local and systemic inflammation. Br. J. Cancer. 2003;88:1772–1779. doi: 10.1038/sj.bjc.6600864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cecic I, Parkins CS, Korbelik M. Induction of systemic neutrophil response in mice by photodynamic therapy of solid tumors. Photochem. Photobiol. 2001;74:712–720. doi: 10.1562/0031-8655(2001)074<0712:iosnri>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 26.Henderson BW, Gollnick SO, Snyder JW, Busch TM, Kousis PC, Cheney RT, Morgan J. Choice of oxygen-conserving treatment regimen determines the inflammatory response and outcome of photodynamic therapy of tumors. Cancer Res. 2004;64:2120–2126. doi: 10.1158/0008-5472.can-03-3513. [DOI] [PubMed] [Google Scholar]
- 27.Dragieva G, Hafner J, Dummer R, Schmid-Grendelmeier P, Roos M, Prinz BM, Burg G, Binswanger U, Kempf W. Topical photodynamic therapy in the treatment of actinic keratoses and Bowen's disease in transplant recipients. Transplantation. 2004;77:115–121. doi: 10.1097/01.TP.0000107284.04969.5C. [DOI] [PubMed] [Google Scholar]
- 28.Chen Q, Fisher DT, Kucinska SA, Wang WC, Evans SS. Dynamic control of lymphocyte trafficking by fever-range thermal stress. Cancer Immunol. Immunother. 2006;55:299–311. doi: 10.1007/s00262-005-0022-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fisher DT, Chen Q, Skitzki JJ, Muhitch JB, Zhou L, Appenheimer MM, Vardam TD, Weis EL, Passanese J, Wang WC, Gollnick SO, Dewhirst MW, Rose-John S, Repasky EA, Baumann H, Evans SS. IL-6 trans-signaling licenses mouse and human tumor microvascular gateways for trafficking of cytotoxic T cells. J. Clin. Invest. 2011;121:3846–3859. doi: 10.1172/JCI44952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gollnick SO, Liu X, Owczarczak B, Musser DA, Henderson BW. Altered expression of interleukin 6 and interleukin 10 as a result of photodynamic therapy in vivo. Cancer Res. 1997;57:3904–3909. [PubMed] [Google Scholar]
- 31.Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat. Rev. Immunol. 2010;10:479–489. doi: 10.1038/nri2800. [DOI] [PubMed] [Google Scholar]
- 32.Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 2011;11:519–531. doi: 10.1038/nri3024. [DOI] [PubMed] [Google Scholar]
- 33.Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, Fujikado N, Tanahashi Y, Akitsu A, Kotaki H, Sudo K, Nakae S, Sasakawa C, Iwakura Y. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity. 2009;30:108–119. doi: 10.1016/j.immuni.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 34.Turley SJ, Fletcher AL, Elpek KG. The stromal and haematopoietic antigen-presenting cells that reside in secondary lymphoid organs. Nat. Rev. Immunol. 2010;10:813–825. doi: 10.1038/nri2886. [DOI] [PubMed] [Google Scholar]
- 35.Tewalt EF, Cohen JN, Rouhani SJ, Engelhard VH. Lymphatic endothelial cells - key players in regulation of tolerance and immunity. Front Immunol. 2012;3:305. doi: 10.3389/fimmu.2012.00305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Girard JP, Moussion C, Forster R. HEVs, lymphatics and homeostatic immune cell trafficking in lymph nodes. Nat. Rev. Immunol. 2012;12:762–773. doi: 10.1038/nri3298. [DOI] [PubMed] [Google Scholar]
- 37.Bargatze RF, Kurk S, Butcher EC, Jutila MA. Neutrophils roll on adherent neutrophils bound to cytokine-induced endothelial cells via L-selectin on the rolling cells. J. Exp. Med. 1994;180:1785–1792. doi: 10.1084/jem.180.5.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mancardi S, Vecile E, Dusetti N, Calvo E, Stanta G, Burrone OR, Dobrina A. Evidence of CXC, CC and C chemokine production by lymphatic endothelial cells. Immunology. 2003;108:523–530. doi: 10.1046/j.1365-2567.2003.01613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Henderson BW, Gollnick SO. Mechanistic principles of photodynamic therapy. In: Vo-Dinh T, editor. Biomedical Photonics Handbook. CRC Press; Boca Raton, FL: 2003. pp. 36-1–36-27. [Google Scholar]
- 40.Florey O, Haskard DO. Sphingosine 1-phosphate enhances Fc gamma receptor-mediated neutrophil activation and recruitment under flow conditions. J. Immunol. 2009;183:2330–2336. doi: 10.4049/jimmunol.0901019. [DOI] [PubMed] [Google Scholar]
- 41.Allende ML, Bektas M, Lee BG, Bonifacino E, Kang J, Tuymetova G, Chen W, Saba JD, Proia RL. Sphingosine-1-phosphate lyase deficiency produces a pro-inflammatory response while impairing neutrophil trafficking. J. Biol. Chem. 2011;286:7348–7358. doi: 10.1074/jbc.M110.171819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kishimoto TK, Jutila MA, Berg EL, Butcher EC. Neutrophil Mac-1 and MEL-14 adhesion proteins inversely regulated by chemotactic factors. Science. 1989;245:1238–1241. doi: 10.1126/science.2551036. [DOI] [PubMed] [Google Scholar]
- 43.Jutila MA, Rott L, Berg EL, Butcher EC. Function and regulation of the neutrophil MEL-14 antigen in vivo: comparison with LFA-1 and MAC-1. J. Immunol. 1989;143:3318–3324. [PubMed] [Google Scholar]
- 44.Fan H, Derynck R. Ectodomain shedding of TGF-alpha and other transmembrane proteins is induced by receptor tyrosine kinase activation and MAP kinase signaling cascades. EMBO J. 1999;18:6962–6972. doi: 10.1093/emboj/18.24.6962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee D, Schultz JB, Knauf PA, King MR. Mechanical shedding of L-selectin from the neutrophil surface during rolling on sialyl Lewis x under flow. J. Biol. Chem. 2007;282:4812–4820. doi: 10.1074/jbc.M609994200. [DOI] [PubMed] [Google Scholar]
- 46.Wang Y, Zhang AC, Ni Z, Herrera A, Walcheck B. ADAM17 activity and other mechanisms of soluble L-selectin production during death receptor-induced leukocyte apoptosis. J. Immunol. 2010;184:4447–4454. doi: 10.4049/jimmunol.0902925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walcheck B, Herrera AH, St HC, Mattila PE, Whitney AR, Deleo FR. ADAM17 activity during human neutrophil activation and apoptosis. Eur. J. Immunol. 2006;36:968–976. doi: 10.1002/eji.200535257. [DOI] [PubMed] [Google Scholar]
- 48.Ord DC, Ernst TJ, Zhou LJ, Rambaldi A, Spertini O, Griffin J, Tedder TF. Structure of the gene encoding the human leukocyte adhesion molecule-1 (TQ1, Leu-8) of lymphocytes and neutrophils. J. Biol. Chem. 1990;265:7760–7767. [PubMed] [Google Scholar]
- 49.Kahn J, Ingraham RH, Shirley F, Migaki GI, Kishimoto TK. Membrane proximal cleavage of L-selectin: identification of the cleavage site and a 6-kD transmembrane peptide fragment of L-selectin. J. Cell Biol. 1994;125:461–470. doi: 10.1083/jcb.125.2.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ho AW, Shen F, Conti HR, Patel N, Childs EE, Peterson AC, Hernandez-Santos N, Kolls JK, Kane LP, Ouyang W, Gaffen SL. IL-17RC is required for immune signaling via an extended SEF/IL-17R signaling domain in the cytoplasmic tail. J. Immunol. 2010;185:1063–1070. doi: 10.4049/jimmunol.0903739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim JS, Smith-Garvin JE, Koretzky GA, Jordan MS. The requirements for natural Th17 cell development are distinct from those of conventional Th17 cells. J. Exp. Med. 2011;208:2201–2207. doi: 10.1084/jem.20110680. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52.Marks BR, Nowyhed HN, Choi J-Y, Poholek AC, Odegard JM, Flavell RA, Craft J. Thymic self-reactivity selects natural interleukin 17-producing T cells that can regulae peripheral inflammation. Nat. Immunol. 2009;10:1125–1133. doi: 10.1038/ni.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garg AD, Krysko DV, Vandenabeele P, Agostinis P. DAMPs and PDT-mediated photo-oxidative stress: exploring the unknown. Photochem. Photobiol. Sci. 2011;10:670–680. doi: 10.1039/c0pp00294a. [DOI] [PubMed] [Google Scholar]
- 54.Hoffmann E, Dittrich-Breiholz O, Holtmann H, Kracht M. Multiple control of interleukin-8 gene expression. J. Leukoc. Biol. 2002;72:847–855. [PubMed] [Google Scholar]
- 55.Mariathasan S, Monack DM. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat. Rev. Immunol. 2007;7:31–40. doi: 10.1038/nri1997. [DOI] [PubMed] [Google Scholar]
- 56.Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nature Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 57.Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat. Rev. Immunol. 2010;10:826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cassel SL, Sutterwala FS. Sterile inflammatory responses mediated by the NLRP3 inflammasome. Eur. J. Immunol. 2010;40:607–611. doi: 10.1002/eji.200940207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Karmakar M, Sun Y, Hise AG, Rietsch A, Pearlman E. Cutting Edge: IL-1beta Processing during Pseudomonas aeruginosa Infection Is Mediated by Neutrophil Serine Proteases and Is Independent of NLRC4 and Caspase-1. J. Immunol. 2012 doi: 10.4049/jimmunol.1201447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hoshino H, Laan M, Sjostrand M, Lotvall J, Skoogh BE, Linden A. Increased elastase and myeloperoxidase activity associated with neutrophil recruitment by IL-17 in airways in vivo. J. Allergy Clin. Immunol. 2000;105:143–149. doi: 10.1016/s0091-6749(00)90189-1. [DOI] [PubMed] [Google Scholar]
- 61.Cho KA, Suh JW, Lee KH, Kang JL, Woo SY. IL-17 and IL-22 enhance skin inflammation by stimulating the secretion of IL-1beta by keratinocytes via the ROS-NLRP3-caspase-1 pathway. Int. Immunol. 2012;24:147–158. doi: 10.1093/intimm/dxr110. [DOI] [PubMed] [Google Scholar]
- 62.Yao Z, Fanslow WC, Seldin MF, Rousseau AM, Painter SL, Comeau MR, Cohen JI, Spriggs MK. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity. 1995;3:811–821. doi: 10.1016/1074-7613(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 63.Shen F, Gaffen SL. Structure-function relationships in the IL-17 receptor: implications for signal transduction and therapy. Cytokine. 2008;41:92–104. doi: 10.1016/j.cyto.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.De FK, Henderson RB, Laschinger M, Hogg N. Neutrophil chemokines KC and macrophage-inflammatory protein-2 are newly synthesized by tissue macrophages using distinct TLR signaling pathways. J. Immunol. 2008;180:4308–4315. doi: 10.4049/jimmunol.180.6.4308. [DOI] [PubMed] [Google Scholar]
- 65.Apetoh L, Ghiringhelli F, Tesniere A, Criollo A, Ortiz C, Lidereau R, Mariette C, Chaput N, Mira JP, Delaloge S, Andre F, Tursz T, Kroemer G, Zitvogel L. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol. Rev. 2007;220:47–59. doi: 10.1111/j.1600-065X.2007.00573.x. [DOI] [PubMed] [Google Scholar]
- 66.Lee S, Chen TT, Barber CL, Jordan MC, Murdock J, Desai S, Ferrara N, Nagy A, Roos KP, Iruela-Arispe ML. Autocrine VEGF signaling is required for vascular homeostasis. Cell. 2007;130:691–703. doi: 10.1016/j.cell.2007.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ruddy MJ, Shen F, Smith JB, Sharma A, Gaffen SL. Interleukin-17 regulates expression of the CXC chemokine LIX/CXCL5 in osteoblasts: implications for inflammation and neutrophil recruitment. J. Leukoc. Biol. 2004;76:135–144. doi: 10.1189/jlb.0204065. [DOI] [PubMed] [Google Scholar]
- 68.Call DR, Nemzek JA, Ebong SJ, Bolgos GR, Newcomb DE, Wollenberg GK, Remick DG. Differential local and systemic regulation of the murine chemokines KC and MIP2. Shock. 2001;15:278–284. doi: 10.1097/00024382-200115040-00005. [DOI] [PubMed] [Google Scholar]
- 69.Biedermann T, Kneilling M, Mailhammer R, Maier K, Sander CA, Kollias G, Kunkel SL, Hültner L, Röcken M. Mast cells control neutrophil recruitment during T cell-mediated delayed-type hypersensitivity reactions through tumor necrosis factor and macrophage inflammatory protein 2. J. Exp. Med. 2000;192:1441–1451. doi: 10.1084/jem.192.10.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Massena S, Christoffersson G, Hjertstrom E, Zcharia E, Vlodavsky I, Ausmees N, Rolny C, Li JP, Phillipson M. A chemotactic gradient sequestered on endothelial heparan sulfate induces directional intraluminal crawling of neutrophils. Blood. 2010;116:1924–1931. doi: 10.1182/blood-2010-01-266072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Palframan RT, Jung S, Cheng G, Weninger W, Luo Y, Dorf M, Littman DR, Rollins BJ, Zweerink H, Rot A, von Andrian UH. Inflammatory chemokine transport and presentation in HEV: a remote control mechanism for monocyte recruitment to lymph nodes in inflamed tissues. J. Exp. Med. 2001;194:1361–1373. doi: 10.1084/jem.194.9.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nathan C. Neutrophils and immunity: challenges and opportunities. Nat. Rev. Immunol. 2006;6:173–182. doi: 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- 73.van Gisbergen KPJM, Sanchez-Hernandez M, Geijtenbeek TBH, van Kooyk Y. Neutrophils mediate immune modulation of dendritic cells through glycosylation-dependent interactions between Mac-1 and DC-SIGN. J. Exp. Med. 2005;201:1281–1292. doi: 10.1084/jem.20041276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bennouna S, Denkers EY. Microbial antigen triggers rapid mobilization of TNF-alpha to the surface of mouse neutrophils transforming them into inducers of high-level dendritic cell TNF-alpha production. J Immunol. 2005;174:4845–4851. doi: 10.4049/jimmunol.174.8.4845. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.