

Abstract
The formal first step in in vitamin A metabolism is the conversion of its natural precursor β,β-carotene (C40) to retinaldehyde (C20) This reaction is catalyzed by the enzyme β,β-carotene-15,15′-monooxygenase (BCMO1). BCMO1 has been cloned from several vertebrate species, including humans. However, knowledge about this protein’s enzymatic and structural properties is scant. Here we expressed human BCMO1 in Spodoptera frugiperda 9 insect cells. Recombinant BCMO1 is a soluble protein that displayed Michaelis-Menten kinetics with a KM of 14 μM for β,β-carotene. Though addition of detergents failed to increase BCMO1 enzymatic activity, short chain aliphatic detergents such as C8E4 and C8E6 decreased enzymatic activity probably by interacting with the substrate binding site. Thus we purified BCMO1 in the absence of detergent. Purified BCMO1 was a monomeric enzymatically active soluble protein that did not require cofactors and displayed a turnover rate of about 8 molecules of β,β-carotene per second. The aqueous solubility of BCMO1 was confirmed in mouse liver and mammalian cells. Establishment of a protocol that yields highly active homogenous BCMO1 is an important step towards clarifying the lipophilic substrate interaction, reaction mechanism and structure of this vitamin A forming enzyme.
Keywords: β,β-carotene; all-trans-retinal; vitamin A; β,β-carotene-15; 15′-monooxygenase; cytosolic; symmetric carotenoid cleavage; non-heme iron oxygenase
Introduction
Vitamin A (all-trans-retinol, ROL) is critical for vision, embryonic development, cellular homeostasis and immunity [1–4]. Plant carotenoids such as β,β-carotene are the main dietary source of vitamin A for most of the world’s population [5, 6]. Deficiency of this vitamin, especially in developing countries, leads to blindness in hundreds of thousands of children annually, as well as great increases in childhood morbidity [7]. The amount of vitamin A obtainable from dietary β,β-carotene depends mainly on two factors: the bioavailability of the ingested carotenoids and their conversion to vitamin A by endogenous enzymes [8, 9]. This conversion is catalyzed by the enzyme β,β-carotene-15,15′-monooxygenase (BCMO1) located in intestinal enterocytes [10, 11]. The reaction yields two molecules of retinaldehyde (RAL) which can be converted to ROL and retinoic acid (Fig. 1). BCMO1 has been cloned from several vertebrate species, including chicken, mouse, human and zebrafish [12–16]. A rare missense mutation in human BCMO1, as well as genetic disruption of BCMO1 in mice, result in highly elevated β,β-carotene blood levels and cause hypovitaminosis A [17, 18], indicating that BCMO1 is the major enzyme for vitamin A production. BCMO1 only cleaves carotenoids with a non-substituted β-ionone ring and thus has limited substrate specificity for provitamin A carotenoids [15, 19]. The enzyme has a slightly alkaline pH optimum [15, 20] and can be inhibited by various ferrous iron chelators and sulfhydryl alkylating compounds [11, 15, 20] as well as activated or protected by sulfhydryl reducing compounds [11, 15, 21–24]. Because BCMO1 activity could be inhibited by iron chelating agents but not by cyanide, an inhibitor of ferric protoporphyrin enzymes, this carotenoid oxygenase was classified as a non-heme iron oxygenase [15, 25].
Figure 1. BCMO1 catalyzes the oxidative conversion of b,b-carotene to retinoids.

Oxidative cleavage of β,β-carotene yields all-trans-retinal that can be either further oxidized to all-trans-retinoic acid or reduced to all-trans-retinol (vitamin A) (30).
BCMO1 is a member of an evolutionary well-conserved family of carotenoid cleavage enzymes (CCOs) [26]. Besides BCMO1, mammalian genomes encode the enzymes β,β-carotene- 9′,10′-dioxgenase (BCDO2) [27] and retinal pigment epithelium (RPE)-specific 65 kDa protein (RPE65) [28]. In contrast to BCMO1, BCMO2 cleaves carotenoids eccentrically at the C9,C10 double bond and shows a wide substrate specificity for carotenoids, including compounds with 3-hydroxy and 4-oxo-ionone ring substitutions [19, 29, 30]. As a consequence, BCDO2 can interact with both β and ε-3-OH ring sites of carotenoids [19, 30] and even with noncyclic carotenoids such as lycopene [19, 31]. Studies in animals indicate that BCDO2 plays a critical role in carotenoid homeostasis and in the prevention of oxidative stress caused by excess carotenoids [19, 29]. BCDO2 is localized in mitochondria [19, 29] whereas BCMO1 is a cytosolic enzyme [11, 15, 20], The differential localization of these carotenoid oxygenases in two different cell compartments appears logical because both enzymes are expressed in the same cell types and share β,β-carotene as a common substrate. Consequently, if both enzymes were expressed in the same cell compartment they would compete for β,β-carotene which then could decrease vitamin A production [32]. RPE65 is a monotopic membrane protein present in the RPE of vertebrates [28, 33]. Mutations in its gene can cause visual chromophore deficiency and thus blindness in humans [34] and homologous mouse models [35]. But RPE65, unlike other members of the carotenoid cleavage oxygenase family, does not cleave carotenoids oxidatively. Instead, it simultaneously cleaves and isomerizes all-trans-retinyl esters to 11-cis-retinol [28, 33, 36–40]. This isomerase may use the Lewis acidity of Fe2+ which leads to a polarization of the ester moiety to facilitate ester cleavage; the Lewis acidity of the iron is possibly strengthened by its uncharged 4-His ligand environment [33].
To date X-ray structures of three members of the CCO family have been determined. The first was apocarotenoid-15,15′-oxygenase (ACO) from Synechocystis sp. [41] followed by RPE65 [28] and Viviparous14 from plants [42]. Their common structural motifs are a seven bladed β-propeller, an active site with the catalytic iron coordinated by four completely conserved His residues, and a hydrophobic tunnel which leads from the active site with its catalytic iron to the protein exterior [28, 41–43]. It has been proposed that nonpolar patches surrounding the active site tunnels of these enzymes interact with membranes to allow the transfer of substrate which then can be transported to the active site [28, 41]. Superposition of RPE65 with ACO gives a rmsd of 2.5 Å for 443 Cαs [28] indicating a marked overall similarity between the two structures.
Though significant progress has been made towards characterizing this disease-relevant family of non-heme iron oxygenases, several structural and functional aspects of BCMO1, the key enzyme for vitamin A formation, have not yet been characterized in detail. Critical questions regarding the catalytic mechanism and the interaction with its lipophilic substrate remain to be answered. A prerequisite for such research is a protocol that provides the protein in high amounts in a homogenous and enzymatically active state. Here we report the recombinant expression of human BCMO1 in Spodoptera frugiperda 9 (Sf9) insect cells, its purification, its enzymatic properties in the presence of several detergents and its oligomeric state. Additionally, we show that native BCMO1 displayed similar properties to the purified enzyme in mammalian cell culture and its natural environment in mouse tissue.
Materials and Methods
Chemicals
All-trans-β,β-carotene was purchased from Calbiochem (San Diego, CA). Tetraethylene glycol monooctyl ether (C8E4), hexaethylene glycol monooctyl ether (C8E6), n-octyl-β-D-thioglucopyranoside (OTG), n-dodecyl-β-D-maltopyranoside (DDM) and 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) were obtained from Affymetrix. Talon Co2+-resin was from Clontech (Mountain View, CA).
Plasmid generation and cloning
The N-terminal TEV-cleavage site and 6x His-tag of the insect cell expression vector pFastBac HTa (Invitrogen) were deleted by site directed mutagenesis (QuikChange II XL Site-Directed Mutagenesis Kit, Agilent Technologies) by using the primer pair 5′-GCGCGGATCTCGGTCCGAAACCGCCATGGATCCGTTCAAAG-3′ and 5′-CTTTGAATTCCGGATCCATGGCGGTTTCGGACCGAGATCCGCGC-3′ (Integrated DNA Technologies) to generate the vector, pFastBac HTa-del. The gene sequences for a TEV-cleavage site, 6x His- and 1D4-tags [44] were attached at the 3′ end of the human BCMO1 gene by carrying out two PCR reactions. In the first reaction 5′-CTGAATTCATGGATATAATATTTGGCAGGAATAGG-3′ was used as a forward primer and 5′-CTGTCGACTCAGGCTGGAGCCACCTGGCTGGTCTCCGTATGATGATGATGATGATGG-3 (Invitrogen) as reverse primer, whereas in the second reaction the primers were 5′-CTGAATTCATGGATATAATATTTGGCAGGAATAGG-3′ forward and 5′-ATGATGATGATGATGATGGCCCTGGAAATACAAGTTTTCGGTCAGAGGAGCCCCGTGGCA G-3′ reverse (Invitrogen). The obtained insert was subcloned into pFastBac HTa-del with Eco RI and SalI (Roche) used as restriction enzymes to generate the construct, pFastBac HTa-del-BCMO1. MAX EfficiencyR DH10Bac™ Competent Cells (Invitrogen) were then transformed withthe obtained construct to generate the bacmid DNA.
Insect cell transfection, protein expression and purification
Sf9 cells were transfected with bacmid DNA with FuGENER 6 (Promega, Madison, WI) used as the transfection reagent. Recombinant baculovirus was produced with the Bac-to-BacR Baculovirus Expression System (Life Technologies, Grand Island, NY). Expression of carboxyl terminal tagged recombinant human BCMO1 was carried out by adding either 5 mL, 1 mL, 0.2 mL, or 0.04 mL of baculovirus suspension to 60 mL Sf9 starter cultures (cell number 1.5 × 106) in 250 mL baffled flasks. Serum-free Sf-900™ III SFM (Gibco) was employed as growth media. Cells grown at 27°C and rotated at 115 rpm were harvested after 3 days and the pellets were frozen at −80°C until further use.
The same conditions for soluble expression were reproduced at a larger scale for purification (20 mL baculovirus was added to 800 mL Sf9 cell culture in a 2 L baffled flask). Cell pellets (20–30 g) were re-suspended in 50 mL of sample buffer containing 20 mM Tricine, pH 7.5, 1 mM Tris(2-carboxyethyl)phosphine hydrochloride (TCEP/HCl), (Hampton Research) and one Complete EDTA free Protease Inhibitor Cocktail Tablet (Roche). Lysis of cells was performed by 30 strokes of homogenization in a glass tissue grinder. The lysate was centrifuged at 40,000 rpm for 1 h at 4°C (Beckman Coulter Optima™L-90K Ultracentrifuge). The supernatant was collected, transferred to 50 ml tubes and kept on ice. Then it was loaded onto a 10 mL column containing 1.5 mL of Talon Co2+-resin suspension (Clontech) pre-equilibrated with 5 column volumes of ice cold buffer containing 250 mM NaCl, 20 mM Tricine, pH 7.5, and 1 mM TCEP. After flow-through collection, the Talon column was first washed with 5 column volumes of ice cold buffer containing 250 mM NaCl, 20 mM Tricine, pH 7.5, and 1 mM TCEP and then with 5 column volumes of buffer containing 250 mM NaCl, 20 mM Tricine, pH 7.5, 1 mM TCEP and 1 mM imidazole. Finally BCMO1 was eluted in ice cold buffer containing 250 mM NaCl, 20 mM Tricine, pH 7.5, 1 mM TCEP and either 5 or 50 mM imidazole. Eluted BCMO1 fractions were pooled and concentrated in a 30K AmiconR Ultra Centrifugal Filter (Millipore) before being loaded onto a Superdex™ 200 10/300 GL size exclusion column (GE Healthcare Life Sciences). BCMO1 fractions were eluted from the column in 0.5 mL fractions at a flow rate of 0.4 mL/min. with buffer containing 100 mM NaCl, 20 mM Tricine, pH 7.5, and 1 mM TCEP. Fractions containing purified enzyme were then pooled, concentrated in a 30 K AmiconR Ultra Centrifugal Filter and stored on ice until further use. To determine the monomeric state of purified human BCMO1 a mix of proteins (Bio-Rad Gel filtration Standard) with known molecular weights were run simultaneously on the size exclusion column.
Enzymatic assays
Frozen Sf9 cell pellets (3–4 g) infected with human BCMO1 baculovirus were re-suspended in 13 mL sample buffer containing 200 mM NaCl, 20 mM Tricine, pH 7.5, 1 mM dithiothreitol (DTT, Roche) and one Complete EDTA free Protease Inhibitor Cocktail Tablet (Roche). Lysis of cells was performed by a 30 stroke homogenization in a glass tissue grinder. The lysate was centrifuged at 40,000 rpm for 1 h at 4°C (Beckman Coulter Optima™L-90K Ultracentrifuge). The supernatant (10 mL) was collected and the remaining pellet was dissolved in 10 mL of sample buffer. To monitor enzymatic activity in the presence of different concentrations of various detergents (tetraethylene glycol mono-octyl ether (C8E4), hexaethylene glycol mono-octyl ether (C8E6), n-octyl-β-D-thioglucopyranoside (OTG), n-dodecyl-β-D-maltopyranoside (DDM) and 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS)) detergent was added to the supernatant at the appropriate concentration and samples were kept on ice for 10 min. Enzymatic assays were carried out as previously described [25, 45].
Immunoblotting
Frozen 1 g Sf9 cell pellets (from 60 mL Sf9 cultures supplemented with either 5, 1, 0.2 or 0.04 mL BCMO1 baculovirus) were each re-suspended in 10 mL of sample buffer containing 200 mM NaCl, 20 mM Tricine, pH 7.5, 1 mM dithiothreitol (DTT, Roche) and one Complete EDTA free Protease Inhibitor Cocktail Tablet (Roche). Lysis of cells was performed by a 30 stroke homogenization in a glass tissue grinder. The lysates were centrifuged at 40,000 rpm (Beckman Coulter 50.2 Ti) for 1 h at 4°C (Beckman Coulter Optima™L-90K Ultracentrifuge). Supernatants (10 mL) were collected and the remaining pellets were each dissolved in 10 mL sample buffer. Equal amounts of supernatants and pellets were loaded on SDS-polyacrylamide gels and transferred to PVDF membranes. Blots were probed with an alkaline phosphatase conjugated monoclonal 1D4 antibody (Polgenix Inc.) diluted 1:10,000 in 137 mM NaCl, 2.7 mM KCl, 10 mM dibasic sodium phosphate, 2 mM monobasic potassium phosphate at a pH of 7.4 (PBS) and 5% milk powder for 1 h and developed colorimetrically with Western BlueR Stabilized Substrate for Alkaline Phosphatase. Protocols for detection of BCMO1 and lecithin: retinol acyl transferase (LRAT) were previously described [16, 46].
Triton X-114 phase separation experiments
RPE microsomes were prepared from bovine RPE as previously described [47]. Purified bovine serum albumin (BSA) was commercially obtained from Thermo-Scientific (Rockford, IL) and Talon-purified BCMO1 was prepared as described above. All protein samples (0.5 to 1.0 mg/mL) were ultimately suspended in 200 μl of 10 mM Tricine, pH 7.4, 150 mM NaCl, and 1.0% Triton X-114 at 0°C. The phase separation experiments were performed essentially as described by Bordier [48] except that 1.5% w/v of Triton X-114 was used to solubilize proteins instead of 1%. Also, after initial separation, the upper aqueous phase received 1% fresh Triton X-114 and after separation, the aqueous and detergent phases were equalized in volume by 10 % w/v Triton X-114 and the solubilization buffer, respectively. Equal volumes of each fraction were analyzed by SDS-PAGE followed by Coomassie staining.
BCMO1 plasmid construction for expression in Cos7 cells
The full-length BCMO1 open reading frame (ORF) was amplified with the Expand High Fidelity PCR system (Roche, Indianapolis, IN). The amplified BCMO1 cDNA product was then cloned in frame into the pCDNA 3.1 V5/His TOPO (Invitrogen, Carlsbad, CA). Appropriate construction of the plasmid was verified by sequence analysis of both strands (Genomics Core Sequencing Facility, Case Western Reserve University, Cleveland, OH). Monkey kidney COS7 cells were maintained in high-glucose DMEM supplemented with 10% fetal bovine serum (FBS), 1% penicillin-streptomycin sulfate, and cultured at 37°C with 5% CO2. For BCMO1 subcellular localization studies, COS7 cells were seeded at 50–70% confluence on glass coverslips in 6-well plates. The next day cells were transfected with 4–6 μg of purified plasmid DNA by using LipofectAMINE 2000 (L2000) and OptiMEM as described previously [49]. About 40–48 h post transfection, cells grown on coverslips were fixed in a freshly prepared mixture of 4% formalin in PBS for 20 min at room temperature and processed as previously described [49]. Subcellular localization of BCMO1 in COS7 cells was achieved by exposure to the anti-V5 primary antibody followed by the anti-rabbit conjugated Alexa 594 secondary antibody. Cells were examined under a Zeiss LSM 510 UVMETA confocal microscope with an HCX Plan 40X numerical aperture 1.4 oil immersion objective lens. Images were acquired with Zeiss confocal software version 2.0 (Zeiss, Jena, Germany).
Determination of BCMO1 subcellular localization in mouse liver
Three 12-week-old wild type mice with a C57/BL6;129Sv mixed genetic background were used for the described experiments. Mice were maintained at 24°C in a 12:12-h light-dark cycle with ad libitum access to food and water. Animal procedures and experiments were approved by the Case Western Reserve University Animal Care Committee and conformed to recommendations of both the American Veterinary Medical Association Panel on Euthanasia and the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. Mice were anesthetized by intraperitoneal injection of a mixture containing ketamine (80 mg/kg body weight) and xylazine (20 mg/kg body weight) in 10 mM sodium phosphate, pH 7.2, with 100 mM NaCl and blood was drawn directly from the heart after snipping the right atrium. Then mice were perfused with 10 ml of PBS and killed by cervical dislocation. The liver was dissected out and homogenized with a polytron tissue homogenizer blender in 3 ml of buffer (750 mM sucrose, 1 mM DTT, MOPS, pH 7.0). For cytoplasm isolation, the homogenate was subjected to centrifugation at 16,000 rpm in a Sorvall rotor 7017 at 4 °C. The supernatant contained the cytoplasmic fraction. Membranes were resuspended in PBS and subjected to centrifugation at 40,000 rpm in a Ti50 in a Beckman ultra-centrifuge. The pellet contained the microsomal fraction. Equal amounts of protein from the cytoplasmic and microsomal fractions were used for immunoblot analyses.
Results
Expression and enzymatic activity of human recombinant BCMO1
Two sequential PCR reactions were carried out to attach the gene sequences for a TEV-cleavage site, namely 6x His- and 1D4-tags at the 3′ end of the human BCMO1 open reading frame. The N-terminal TEV-cleavage site and 6x His tag, intrinsic in the pFastBac HTa commercial vector, were deleted by site directed mutagenesis. N-terminal fusions were avoided because such modifications of Nostoc. Sp. CCOs result in enzyme inactivation, possibly indicating a role for this region in protein folding and stability or substrate binding and/or cleavage. The BCMO1 PCR product was finally cloned into the modified insect cell expression vector. MAX EfficiencyR DH10Bac™ Competent Cells were then transformed with the obtained construct to generate the bacmid DNA.
After generation of the BCMO1 baculovirus vector, expression of C-terminally tagged recombinant human BCMO1 was tested over 3 days by adding baculovirus to Sf9 starter cultures. After cells were harvested, the four pellets were dissolved in equal amounts of sample buffer and samples were homogenized in a glass homogenizer. A centrifugation step at 40,000 rpm and 4°C for 1 h was carried out to separate the soluble fraction (supernatant) from the inclusion bodies (pellet). Then the volume of the supernatants was measured and the pellets were re-suspended in the same volume of sample buffer. Equal amounts of protein in all four supernatant and pellet fractions were subjected to immunoblot analyses with 1D4 antibody and developed colorimetrically with a substrate for alkaline phosphatase. Fig. 2A illustrates successful expression of recombinant human BCMO1 under all tested conditions. A stepwise increase of the amount of baculovirus did not increase BCMO1 expression. Also, a significant amount of BCMO1 was present in both the soluble and pellet fractions (Fig. 2A). To determine which fraction was enzymatically active, we used equal protein amounts from both fractions for activity assays with β,β-carotene. This experiment was performed because BCMO1 contains the same conserved hydrophobic patch of non-heme iron oxygenases which is proposed to dip into the membrane or micelles to enable transfer of the substrate so it can reach the catalytic iron in the active site [28, 41, 42]. Furthermore, it was recently reported that RPE65 requires a membrane-like environment to be active [33]. Consequently, we assumed that BCMO1 also could require a membrane-like environment for its carotenoid cleavage activity. However, robust activity of BCMO1 was only detected in the supernatant (Fig. 2B–F), in agreement with previously published data showing that soluble recombinant human BCMO1 cleaves β,β-carotene [15]. This result indicates that the insoluble fraction of the BCMO1 preparation represents a pool of misfolded protein rather than its membrane bound active form.
Figure 2. Baculovirus expression and enzymatic activity of human BCMO1.

A, Immunoblot of the soluble and pelleted fractions of human BCMO1 baculovirus-infected Sf9 insect cell extracts. Blots were probed as described in “Materials and Methods”. Precision Plus Protein™ Standard (lane 1); supernatant (lane 2) and pellet (lane 3) from Sf9 cells infected with 5 mL BCMO1 P3 baculovirus; supernatant (lane 4) and pellet (lane 5) from Sf9 cells infected with 1 mL BCMO1 P3 baculovirus; supernatant (lane 6) and pellet (lane 7) from Sf9 cells infected with 0.2 mL BCMO1 P3 baculovirus. Products of active recombinant human BCMO1. Soluble (supernatant) and pellet fractions of Sf9 cell extracts infected with human BCMO1 baculovirus were incubated with 20 μM all-trans-β,β-carotene at 28°C. Lipids were extracted after 8 min and their separation was achieved by normal-phase HPLC. B, When only buffer was incubated with all-trans-β,β-carotene, no products were detected by HPLC monitored at 360 nm. C, When the supernatant was incubated with all-trans-β,β-carotene, significant amounts of all-trans-retinal were formed. During extraction all-trans-retinal was converted to the corresponding syn- and anti-oximes separated by HPLC and detected at 360 nm. D, When the pelleted fraction was incubated with all-trans-β,β-carotene, no product was observed after HPLC monitored at 360 nm. E, Spectrum of all-trans-retinal oxime (syn). F, Spectrum of all-trans-retinal oxime (anti). 1, all-trans-β,β-carotene. 2, all-trans-retinal oxime (syn). 3, all-trans-retinal oxime (anti).
Enzymatic activity of the BCMO1 soluble fraction in different detergents
Because other members of the non-heme iron oxygenase family require detergents for either their solubilization (RPE65, [28]) or crystallization (ACO, [41]), human recombinant BCMO1 activity was tested in the presence of several different detergents. For each individual trial, a cell pellet of Sf9 infected with human BCMO1 baculovirus was re-suspended in sample buffer. Cells were lysed, the lysate was centrifuged, and the supernatant was collected. To monitor enzymatic activity in the presence of various nonionic detergents (tetraethylene glycol monooctyl ether (C8E4), hexaethylene glycol monooctyl ether (C8E6), n-octyl-β-D-thioglucopyranoside (OTG), n-dodecyl-β-D-maltopyranoside (DDM)) and the zwitterionic detergent, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS)), tested detergents were added to the supernatant at three different concentrations related to their CMCs. Results of these enzymatic assays are listed in Table 1. When the supernatant was incubated with increasing concentrations (0.5 x, 1 x and 2 x CMC) of the aliphatic detergents C8E4 and C8E6, enzymatic activity was significantly decreased as compared to the activity of the detergent-free supernatant (Table 1). Only 12 % and 19 % of the original activity was retained when C8E4 or C8E6 were added at 2 x their CMCs. Addition of the sugar-containing detergents, OTG (one glucose molecule) or DDM (two mannose molecules) had little negative effect on BCMO1 activity relative to the untreated supernatant (Table 1). Thus 93 % of activity was retained when DDM was added at 2 x CMC, and 83 % of activity still was retained when OTG was added at a concentration of 1 x CMC. Only when OTG was added at a concentration of 2 x CMC did activity drop to 42 % (Table 1). This last result could be explained by the fact that OTG when dissolved in water lost its solubility when added to cold (0–4°C) solutions. Thus, when increasing concentrations of OTG were added to the supernatant, more precipitation of protein was observed. When the recombinant BCMO1-containing supernatant was incubated with increasing concentrations of the steroid detergent CHAPS, most of the enzymatic activity was retained (91% at 1 x CMC, 83 % at 0.5 x CMC (Table 1). That lower activity was observed at 0.5 x CMC of CHAPS (Table 1) could be explained by compromised substrate accessibility. Other studies of purified recombinant human BCMO1 indicated that the enzyme is most active in the presence of OTG at concentrations higher than 0.5 mM. However, when CHAPS was used in these assays, only 3% of the initial activity was retained [15]. This discrepancy could arise because purified human enzyme was used in the present study. Compromised enzymatic activity after adding the aliphatic detergents C8E4 and C8E6 could result because these detergents are much smaller than OTG, DDM and CHAPS and therefore could reach and inhibit the active site of BCMO1. The available crystal structure of the Synechocystis sp. apocarotenoid oxygenase ACO in complex with an active site bound C8E4 detergent molecule supports this idea [41]. Interestingly, RPE65 also did not display activity after solubilization and purification with C8E4 [28, 33].
Table 1.
Activity of human BCMO1 soluble fraction in the presence of different detergents (equal amounts of soluble fraction from the same preparation were used).
| Detergent | Concentration [%], (x of critical micelle concentration (CMC)) | Activity (pmols all-trans-retinal per min) |
|---|---|---|
| ------- | ------- | 150 |
| C8E4 | 0.125 (0.5 x CMC) | 27.6 |
| C8E4 | 0.25 (1 x CMC) | 25.9 |
| C8E4 | 0.5 (2 x CMC) | 17.4 |
| C8E6 | 0.195 (0.5 x CMC) | 58.4 |
| C8E6 | 0.39(1 x CMC) | 31.4 |
| C8E6 | 0.78 (2 x CMC) | 28.6 |
| OTG | 0.14 (0.5 x CMC) | 132.7 |
| OTG | 0.28 (1 x CMC) | 127.3 |
| OTG | 0.56 (2 x CMC) | 65.3 |
| DDM | 0.00435 (0.5 x CMC) | 144.3 |
| DDM | 0.0087 (1 x CMC) | 142.6 |
| DDM | 0.0174 (2 x CMC) | 142.6 |
| CHAPS | 0.245 (0.5 x CMC) | 85.6 |
| CHAPS | 0.49 (1 x CMC) | 136.8 |
| CHAPS | 0.98 (2 x CMC) | 123 |
BCMO1 activity was tested as described in Materials and Methods.
Isolation and determination of purified human BCMO1 enzymatic activity
BCMO1 activity was found in the soluble protein fraction and the addition of detergents failed to increase enzymatic activity. Therefore, we purified BCMO1 in detergent-free buffer. Frozen cell pellets of Sf9 cells infected with recombinant BCMO1 baculovirus were thawed and re-suspended as described in the Materials and Methods section. After lysis, the soluble fraction was separated from inclusion bodies by centrifugation and the supernatant was applied to a Co2+ column. Here it was observed that recombinant human BCMO1 could be purified almost free of contaminating proteins as contrasted to its purification with a Co2+-resin (data not shown). After the column was washed, BCMO1 was eluted first with 5 mM and then with 50 mM imidazole. All eluted fractions contained highly pure protein (Fig. 3A).
Figure 3. Purification of human BCMO1 and its enzymatic properties.

SDS-polyacrylamide gel electrophoresis of Co2+ metal affinity chromatography purified BCMO1 (A), further purification on size exclusion chromatography(B), and concentrated sample after size exclusion chromatography (C). Products formation from all-trans-β,β-carotene by BCMO1 after Co2+-column chromatography (E and F) compared with concentrated purified enzyme after size exclusion column chromatography (G). The enzyme was purified and assayed as described under “Materials and Methods”. 1, all-trans-β,β-carotene. 2, all-trans-retinal oxime (syn). 3, all-trans-retinal oxime (anti). (H) Talon purified recombinant human BCMO1 was incubated in the presence of increasing concentrations of β,β-carotene for 8 min at 28°C. Purified recombinant human BCMO1 displayed Michaelis-Menten kinetics.
Next, we determined if there was a difference in enzymatic activity between fractions eluted in buffer containing 5 mM or 50 mM imidazole. Equal volumes of fractions were used for enzymatic assays with β,β-carotene. Under the applied conditions, the enzyme reaction displayed a relatively short window of linearity with time in product formation of about 10 min, kinetics that could be explained by limited substrate availability in the enzymatic assay. We delivered β,β-carotene in 3% (w/v) OTG and have previously shown that the maximum loading capacity of OTG micelles is 20 μM [50]. Results clearly showed that both conditions dispayed comparable high enzymatic activity as assayed by quantification of the β,β-carotene cleavage product (18.5 pmoles × min−1 × μg−1 enzyme and 14.8 pmoles × min−1 × μg−1 enzyme; Fig. 3E and F, respectively). From this data, we calculated the turnover number to be about 8.5 and 7.5 molecules of β,β-carotene × sec−1. All fractions containing BCMO1 were then pooled, concentrated and loaded onto a Superdex™ 200 10/300 GL size exclusion column. The eluted BCMO1 fractions contained protein free of contaminants after this final purification (Fig. 2B). Fractions were pooled and concentrated (Fig. 3C) and 15 μg of the resulting purified protein were used for enzymatic assays. Concentrated recombinant human BCMO1 remained enzymatically active after the size exclusion purification step (Fig. 3D–G). Notably the type of reducing agent included in the purification buffers had a dramatic effect on enzymatic activity. When ascorbic acid and/or DTT were added to all buffers instead of TCEP, only low activity was detected after the Co2+-purification and this residual activity was abolished after the final size exclusion step (data not shown). This result agrees with published data indicating that the relative activity of purified recombinant human BCMO1 was only 32% when DTT was a component of the enzymatic assays instead of TCEP [15]. TCEP also was the reducing agent of choice in assays with another family member [51].
We also determined KM and VMAX values for the reaction by incubating enzyme extracts in the presence of increasing amounts of β,β-carotene (2 μM to 20 μM) for eight min. The reaction was stopped by the addition of 2 M hydroxylamine in 50 % (v/v) methanol. Then substrate and products were extracted and subjected to HPLC analysis for quantification. From assays performed in triplicate, we estimated the KM values to be 13.7 μM and VMAX to be 392.4 pmol of all-trans-retinal × min−1 × mg protein−1 (Fig. 3H).
Determination of the oligomeric state of recombinant human BCMO1
The oligomeric state of BCMO1 was determined from the results of gel-filtration assays. First, the Superdex™ 200 10/300 GL size exclusion column was equilibrated with a mixture of proteins with known molecular weights. Then elution times of these standards were compared with that of BCMO1 (Fig. 4A). A standard curve was generated by plotting the elution volumes of the standard proteins on the x-axis against the logarithm of their molecular weights on the y-axis. With the elution volume of BCMO1 (14 mL) used as the x-value for the formula y = − 0.2189x + 4.8392, the antilog (10) gave a result of 60 kDa for BCMO1 (Fig. 4B). As the theoretical molecular weight of recombinant human BCMO1 including its C-terminal TEV-, 6x His- and 1D4-tags is 65.2 kDa, we conclude that purified recombinant human BCMO1 exists as a monomer in solution.
Figure 4. BCMO1 is a monomeric protein.

(A) Size exclusion chromatogram of purified human BCMO1 along with size exclusion standards, and (B) molecular mass calibration curve for the Superdex™ 200 10/300 GL column. 1, protein aggregates and bovine thyroglobulin (670 kDa). 2, bovine γ-globulin (158 kDa). 3. C-terminal tagged human BCMO1 (65.2 kDa) 4, chicken ovalbumin (44 kDa). 5, horse myoglobin (17 kDa). 6, vitamin B12 (1,35 kDa). Elution volumes were plotted against absorption units at 280 nm. (B) Calibration curve generated with standards bovine thyroglobulin (670 kDa), bovine γ-globulin (158 kDa), chicken ovalbumin (44 kDa), horse myoglobin (17 kDa) and vitamin B12 (1,35 kDa). Elution volumes were plotted against logarithms of molecular weights of standards. The elution volume of 14 yielded a MW of 60 kDa for BCMO1.
BCMO1 exists as a soluble enzyme in cells and tissues
Recombinant human BCMO1 was soluble and its enzymatic activity was not dependent on a membrane-like environment. This finding was surprising because the enzyme, like its family members, must access its lipophilic substrate in the cellular environment of mammalian tissues. Moreover, the solved crystal structure of RPE65 revealed that this enzyme contains a cluster of hydrophobic residues that could be involved in membrane binding (28). Three of those regions are modeled in Fig. 5A. Sequence alignments of the three mammalian CCOs reveal regions with significant diversity that could underlie their differential membrane binding affinities (Fig. 5A). Mechanisms by which CCOs bind membranes, facilitating access to their lipophilic substrates, are still a matter of debate. One mechanism hypothesizes that hydrophobic interactions between amino acid side chains and the hydrophobic core of the lipid bilayer are responsible for membrane binding. Additional sequence comparisons and hydropathy plots of the fourth region, potentially involved in membrane anchoring, can be modeled as an amphipathic alpha helix that could favorably interact with membranes (Fig. 5B). Such modeling is supported by the structures of ACO and VP14 that exhibit analogous regions of their sequences. The calculated zero (Ho) and first (μH) hydrophobic moments for these structuresare similar in BCMO I and RPE65 with BCMO I displaying a slightly higher overall hydrophobicity and RPE65 evidencing a slightly higher dipole moment. For RPE65, posttranslational modifications such as palmitoylation of Cys112 have been described [52]. This Cys residue is conserved in BCMO1 and could help anchor the protein to membranes. Previous studies have demonstrated that RPE65 interacts with membranes predominantly through hydrophobic interactions [47, 53, 54]. Here, we performed Triton X-114 phase separation experiments, as established by [48], on purified BSA, RPE microsomes with RPE65 as the single most abundant protein and on Talon-purified recombinant BCMO1 (Fig. 6A). As expected, hydrophilic BSA exclusively partitioned to the aqueous phase, lane b, and as reported previously, RPE65 partitioned to the detergent phase, lane c. In line with its observed solubility and activity in a membrane-free environment, recombinant BCMO1 was recovered entirely from the aqueous phase. Lane a contained the starting material independent of phase separation. Also to consider, recombinant BCMO1 was expressed as tagged protein which could result in aqueous solubility. To determine the localization of native BCMO1, we cloned its cDNA into the mammalian expression vector, pCDNA3.1 and the resulting BCMO1 plasmid construct was transfected into COS-7 cells. Then, these cells were seeded on coverslips, fixed and immunostained for BCMO1. Stained cells were viewed after confocal imaging to determine the localization of BCMO1. This analysis revealed a cytoplasmic localization of this enzyme (Fig. 6B) thereby corroborating our findings with the recombinant enzyme. To exclude that overexpression of BCMO1 could account for the cytoplasmic localization of BCMO1, we needed to analyze its localization in a mammalian tissue and used a freshly dissected liver of a 12-week- old mouse for this purpose. Mice express hepatic BCMO1 especially highly in Stellate cells [55]. We homogenized the liver tissue and subjected the cell homogenate to high-speed centrifugation to separate soluble from membrane fractions. We then re-solubilized the membrane pellet and subjected equal amounts of total protein of the membrane and soluble fractions to immunoblot analysis with antisera against murine BCMO1 and murine lecithin:retinol transferase (LRAT). The latter enzyme is an integral membrane protein that constitutes a marker for Stellate cells [56, 57]. BCMO1 was detected in the soluble hepatic protein fraction, whereas the lecithin:retinol transferase (LRAT) was exclusively present in the membrane fraction (Fig. 6C). Thus, BCMO1 is a soluble protein in its natural cellular environment.
Figure 5. Comparison of putative membrane-interacting regions between mammalian CCOs.
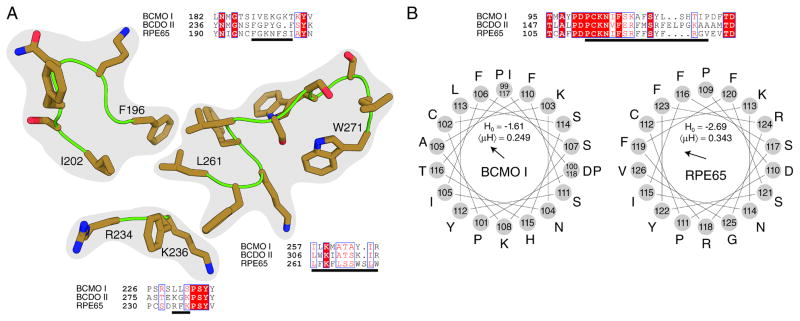
A, Sequence alignments of the three mammalian CCOs. Three regions identified as being involved in RPE65 membrane binding are shown as brown sticks (PDB accession code 4F2Z). Comparison of CCO sequences in these regions reveal a significant diversity that might underlie the differential membrane binding affinity of these enzymes. Strictly conserved residues are colored white on a boxed red background, whereas positions displaying sequence similarity across all three enzymes are colored red on a boxed white background. Residues underlined by black lines correspond to those shown as sticks. B, Sequence comparison and hydropathy plots of the fourth region potentially involved in membrane anchoring. This region of relatively high sequence conservation is disordered in all RPE65 crystal structures reported to date. However, the sequence can be modeled as an amphipathic alpha helix that could favorably interact with membranes. Such modeling is supported by the structures of ACO and VP14 which exhibit analogous regions of their sequences. The calculated zeroth (Ho) and first (μH) hydrophobic moments for these structures are similar between BCMO I and RPE65 with BCMO I displaying slightly higher overall hydrophobicity and RPE65 displaying a slightly higher dipole moment. The hydrophobic dipole vector is shown in the center of the helical wheel plot. Hydrophobic moments were calculated according to Eisenberg, et al. [63].
Figure 6. Partitioning of BCMO1 in Triton X-114 phase separations and BCMO1 solubility in tissues and cells.

A, Pure BSA, RPE65 microsomes, and Talon-purified recombinant BCMO1 were subjected to phase separation experiments as described under “Materials and Methods”. These experiments show that BCMO1 partitions into the aqueous phase similar to BSA and contrary to RPE65 which partitions into the detergent phase, as shown in previously reported studies [33, 47]. Lanes: a, Input, indicates the pure proteins and RPE65 microsomes before phase separation, b, the aqueous phase, and c, the detergent phase. Arrowheads indicate the positions of corresponding proteins as labeled on top of each Coomassie stained gel. B, Immunostaining of human BCMO1 (green) in Cos7 cells. The nucleus is stained with DAPI (blue). The merged image shows that BCMO1 is located in the cytoplasm. C, Immunoblot analyses for BCMO1 and LRAT in cytoplasmic and microsomal preparations of mouse liver. BCMO1 was detected in the cytoplasmic fraction whereas LRAT localized to microsomes.
Discussion
We established a protocol to express and purify human BCMO1 at high yield in an enzymatically active form and showed that this enzyme is a soluble monomeric protein. Moreover, the solubility of native BCMO1 was confirmed in both mice and mammalian cells. The KM value for β,β-carotene with the purified enzyme was within the range of previous estimates [15, 58]. However, the activity of this BCMO1 preparation was much higher than previously described. The purified enzyme had a turnover rate of about seven molecules of β,β-carotene per second, a number exceeding by more than 10-fold the turnover rates reported in previous studies of BCMO1 [15] and other family members such as RPE65 and NinaB [50, 59]. This disparity could be explained by the differing purification protocols and detergents employed. Additionally, the previous study of BCMO1 used substrate concentrations that exceeded the loading capacity of OTG micelles by more than 10-fold [15]. As we previously reported [50], the maximum loading capacity of a 3 % (w/v) OTG solution is 20 μM. Thus, most of the substrate was not dissolved in micelles and likely not available to BCMO1 in that study. The lower turnover rates reported for RPE65 and NinaB might be explained by the reactions catalyzed by these enzymes. The latter proteins catalyze either isomerase or combined carotenoid cleavage and isomerase reactions. The far more rapid conversion rate of β,β-carotene cleavage by BCMO1 implies that the carotenoid cleavage reaction displays faster kinetics than the rate-limiting isomerase reactions catalyzed by the two other enzymes. Moreover, it is noteworthy that purified BCMO1 catalyzed β,β-carotene cleavage without the addition of any cofactors. Yet it remains controversial as to whether these enzymes catalyze cleavage via a mono- or dioxygenase reaction mechanism. Thus, a monooygenase reaction mechanism was proposed for chicken BCMO1 [60]. In contrast, a plant family member catalyzes carotenoid cleavage via a dioxygenase reaction mechanism [61]. Hence our finding that BCMO1 did not require cofactors favors a dioxygenase reaction mechanism because an additional electron donor for the second oxygen atom would be required for a monooxygenase reaction. Alternatively, the electrons could also come from the substrate if this reaction follows an internal monooxygenase mechanism. Investigation of recombinant human BCMO1 could answer this question because its high turnover rate would allow short incubation times in labeling experiments to minimize oxygen exchange between the product and bulk water.
We provide evidence that BCMO1 is a soluble and monomeric protein. Interestingly, this interpretation does not agree with rapport previously published [15] in which authors found that purified recombinant human BCMO1 migrated at 230 kDa on a Sephadex S-300 size exclusion column indicating that this protein existed as a tetramer in its enzymatically active form. However, there was 1% OTG (3.5 x its CMC) in the gel filtration buffer indicating that BCMO1 was in complex with micelles and therefore migrated at a higher molecular weight on the size exclusion column [15]. Indeed, a later study considered this detergent effect and provided evidence that BCMO1 is indeed a monomer [18]. Data from another member of the family of non-heme iron oxygenases support the latter results. Thus most of our inclusion body purified and reconstituted ACO from Synechocystis sp. eluted as a monomer from the final size exclusion column with only a small amount detected as a dimer [41]. After addition of octylpolyoxyethylene (C8E4–8) the main peak shifted to a trimeric mass, probably because ACO forms a complex with detergent micelles. In both ACO crystal forms (two independent protein molecules in the asymmetric unit of iron-free ACO crystals and four in the asymmetric unit of Fe2+ soaked crystals), an asymmetric association of the crystallographically independent molecules was observed suggesting that this apocarotenoid oxygenase is a monomer as demonstrated by size exclusion chromatography [41]. The monotopic bovine membrane protein RPE65 also migrated as a monomer in complex with C8E4 detergent micelles on a size exclusion chromatogram [28]. However, several parallel-oriented RPE65 dimers were present in several different crystal forms which suggests that the retinoid isomerase functions as a dimer in vivo [33].
The monomeric state of BCMO1 has implications for rare and more common genetic polymorphisms in the human BCMO1 gene. Lindqvist and Anderson have already proposed that the enzyme exists in this form and showed that haplo-insufficiency of BCMO1 is associated with β,β-carotene accumulation and hypo-vitaminosis in a human subject [18]. Studies in mouse models show that a single copy of the other carotenoid oxygenase, BCDO2 also does not suffice to maintain carotenoid homeostasis [19]. In contrast, haplo-insufficiency of RPE65 does not affect visual chromophore regeneration in mice [62], even though the turnover rate of RPE65 is relatively low (see above). RPE65 extracts its substrate from membranes and this substrate formation is catalyzed by the membrane anchored LRAT protein with very rapid kinetics [47]. Yet little is known about how true carotenoid oxygenases, such as BCMO1, interact with their lipophilic substrates. Substrate availability could be a limiting factor for vitamin A production and could explain the consequences of haploid-insufficiency for this enzyme.
Finally, obtaining structural data for BCMO1, particularly in complex with its substrate, product and inhibitors such as fenretinide, should be a high priority. The overall structures of different family members are well conserved. However, no clear densities of a bound ligand have been collected in solved X-ray structures of ACO, RPE65, or VP14 [28, 41, 42]. Such data are critical for understanding the reaction mechanism and how the substrate is juxtaposed to the ferric iron in the active center of the enzyme. Establishment of a protocol for the purification of BCMO1 in a homogenous and enzymatically highly active form is an important step towards achieving such molecular insights. Comparisons between BCMO1 and RPE65 would also allow identification of amino acid residues that participate in the oxidative cleavage of the substrate versus the isomerization of the retinoid product. Only progress in understanding the molecular mechanisms that underlie the chemistry of vitamin A metabolism can guarantee that efforts to fight vitamin A deficiency and eye diseases will advance in parallel.
Highlights.
Purification of the human vitamin A forming enzyme BCMO1 in highly active monomeric form.
Characterization of the enzymatic properties of purified BCMO1.
Comparative analysis of membrane association properties of mammalian carotenoid cleavage oxygenase family members.
Characterization of BCMO1 as a soluble protein in cells and tissue.
Acknowledgments
We are grateful to Dr. Leslie T. Webster, Jr. for critical comments on the manuscript. This research was supported, in whole or in part, by EY009339 (K.P.), and EY 020551 (J. v L.) grants from National Institutes of Health. K.P. is John H. Hord Professor of Pharmacology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rhinn M, Dolle P. Development. 2012;139:843–858. doi: 10.1242/dev.065938. [DOI] [PubMed] [Google Scholar]
- 2.Travis GH, Golczak M, Moise AR, Palczewski K. Annu Rev Pharmacol Toxicol. 2007;47:469–512. doi: 10.1146/annurev.pharmtox.47.120505.105225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hall JA, Grainger JR, Spencer SP, Belkaid Y. Immunity. 2011;35:13–22. doi: 10.1016/j.immuni.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noy N. Annu Rev Nutr. 2010;30:201–217. doi: 10.1146/annurev.nutr.28.061807.155509. [DOI] [PubMed] [Google Scholar]
- 5.Underwood BA. J Nutr. 2004;134:231S–236S. doi: 10.1093/jn/134.1.231S. [DOI] [PubMed] [Google Scholar]
- 6.Sommer A, Vyas KS. Am J Clin Nutr. 2012;96:1204S–1206S. doi: 10.3945/ajcn.112.034868. [DOI] [PubMed] [Google Scholar]
- 7.Sommer A. J Nutr. 2008;138:1835–1839. doi: 10.1093/jn/138.10.1835. [DOI] [PubMed] [Google Scholar]
- 8.Lietz G, Lange J, Rimbach G. Arch Biochem Biophys. 2010;502:8–16. doi: 10.1016/j.abb.2010.06.032. [DOI] [PubMed] [Google Scholar]
- 9.von Lintig J. Annu Rev Nutr. 2010;30:35–56. doi: 10.1146/annurev-nutr-080508-141027. [DOI] [PubMed] [Google Scholar]
- 10.Goodman DS, Huang HS. Science. 1965;149:879–880. doi: 10.1126/science.149.3686.879. [DOI] [PubMed] [Google Scholar]
- 11.Olson JA, Hayaishi O. Proc Natl Acad Sci U S A. 1965;54:1364–1370. doi: 10.1073/pnas.54.5.1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wyss A, Wirtz G, Woggon W, Brugger R, Wyss M, Friedlein A, Bachmann H, Hunziker W. Biochem Biophys Res Commun. 2000;271:334–336. doi: 10.1006/bbrc.2000.2619. [DOI] [PubMed] [Google Scholar]
- 13.Redmond TM, Gentleman S, Duncan T, Yu S, Wiggert B, Gantt E, Cunningham FX., Jr J Biol Chem. 2001;276:6560–6565. doi: 10.1074/jbc.M009030200. [DOI] [PubMed] [Google Scholar]
- 14.Paik J, During A, Harrison EH, Mendelsohn CL, Lai K, Blaner WS. J Biol Chem. 2001;276:32160–32168. doi: 10.1074/jbc.M010086200. [DOI] [PubMed] [Google Scholar]
- 15.Lindqvist A, Andersson S. J Biol Chem. 2002;277:23942–23948. doi: 10.1074/jbc.M202756200. [DOI] [PubMed] [Google Scholar]
- 16.Lampert JM, Holzschuh J, Hessel S, Driever W, Vogt K, Von Lintig J. Development. 2003;130:2173–2186. doi: 10.1242/dev.00437. [DOI] [PubMed] [Google Scholar]
- 17.Hessel S, Eichinger A, Isken A, Amengual J, Hunzelmann S, Hoeller U, Elste V, Hunziker W, Goralczyk R, Oberhauser V, von Lintig J, Wyss A. J Biol Chem. 2007;282:33553–33561. doi: 10.1074/jbc.M706763200. [DOI] [PubMed] [Google Scholar]
- 18.Lindqvist A, Sharvill J, Sharvill DE, Andersson S. J Nutr. 2007;137:2346–2350. doi: 10.1093/jn/137.11.2346. [DOI] [PubMed] [Google Scholar]
- 19.Amengual J, Lobo GP, Golczak M, Li HN, Klimova T, Hoppel CL, Wyss A, Palczewski K, von Lintig J. FASEB J. 2011;25:948–959. doi: 10.1096/fj.10-173906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.von Lintig J, Vogt K. J Nutr. 2004;134:251S–256S. doi: 10.1093/jn/134.1.251S. [DOI] [PubMed] [Google Scholar]
- 21.Lakshmanan MR, Chansang H, Olson JA. J Lipid Res. 1972;13:477–482. [PubMed] [Google Scholar]
- 22.Singh H, Cama HR. Biochim Biophys Acta. 1974;370:49–61. doi: 10.1016/0005-2744(74)90030-8. [DOI] [PubMed] [Google Scholar]
- 23.Inesi G, Goodman JJ, Watanabe S. J Biol Chem. 1967;242:4637–4643. [PubMed] [Google Scholar]
- 24.Fidge NH, Smith FR, Goodman DS. Biochem J. 1969;114:689–694. doi: 10.1042/bj1140689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oberhauser V, Voolstra O, Bangert A, von Lintig J, Vogt K. Proc Natl Acad Sci U S A. 2008;105:19000–19005. doi: 10.1073/pnas.0807805105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moise AR, von Lintig J, Palczewski K. Trends Plant Sci. 2005;10:178–186. doi: 10.1016/j.tplants.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 27.Kiefer C, Hessel S, Lampert JM, Vogt K, Lederer MO, Breithaupt DE, von Lintig J. J Biol Chem. 2001;276:14110–14116. doi: 10.1074/jbc.M011510200. [DOI] [PubMed] [Google Scholar]
- 28.Kiser PD, Golczak M, Lodowski DT, Chance MR, Palczewski K. Proc Natl Acad Sci U S A. 2009;106:17325–17330. doi: 10.1073/pnas.0906600106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lobo GP, Isken A, Hoff S, Babino D, von Lintig J. Development. 2012;139:2966–2977. doi: 10.1242/dev.079632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mein JR, Dolnikowski GG, Ernst H, Russell RM, Wang XD. Arch Biochem Biophys. 2010;506:109–121. doi: 10.1016/j.abb.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu KQ, Liu C, Ernst H, Krinsky NI, Russell RM, Wang XD. J Biol Chem. 2006;281:19327–19338. doi: 10.1074/jbc.M512095200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lobo GP, Amengual J, Palczewski G, Babino D, von Lintig J. Biochim Biophys Acta. 2012;1821:78–87. doi: 10.1016/j.bbalip.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kiser PD, Farquhar ER, Shi W, Sui X, Chance MR, Palczewski K. Proc Natl Acad Sci U S A. 2012;109:E2747–2756. doi: 10.1073/pnas.1212025109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marlhens F, Bareil C, Griffoin JM, Zrenner E, Amalric P, Eliaou C, Liu SY, Harris E, Redmond TM, Arnaud B, Claustres M, Hamel CP. Nat Genet. 1997;17:139–141. doi: 10.1038/ng1097-139. [DOI] [PubMed] [Google Scholar]
- 35.Redmond TM, Yu S, Lee E, Bok D, Hamasaki D, Chen N, Goletz P, Ma JX, Crouch RK, Pfeifer K. Nat Genet. 1998;20:344–351. doi: 10.1038/3813. [DOI] [PubMed] [Google Scholar]
- 36.Jin M, Li S, Moghrabi WN, Sun H, Travis GH. Cell. 2005;122:449–459. doi: 10.1016/j.cell.2005.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Redmond TM, Poliakov E, Yu S, Tsai JY, Lu Z, Gentleman S. Proc Natl Acad Sci U S A. 2005;102:13658–13663. doi: 10.1073/pnas.0504167102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moiseyev G, Chen Y, Takahashi Y, Wu BX, Ma JX. Proc Natl Acad Sci U S A. 2005;102:12413–12418. doi: 10.1073/pnas.0503460102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.von Lintig J, Vogt K. J Biol Chem. 2000;275:11915–11920. doi: 10.1074/jbc.275.16.11915. [DOI] [PubMed] [Google Scholar]
- 40.Nikolaeva O, Takahashi Y, Moiseyev G, Ma JX. FEBS J. 2009;276:3020–3030. doi: 10.1111/j.1742-4658.2009.07021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kloer DP, Ruch S, Al-Babili S, Beyer P, Schulz GE. Science. 2005;308:267–269. doi: 10.1126/science.1108965. [DOI] [PubMed] [Google Scholar]
- 42.Messing SA, Gabelli SB, Echeverria I, Vogel JT, Guan JC, Tan BC, Klee HJ, McCarty DR, Amzel LM. Plant Cell. 2010;22:2970–2980. doi: 10.1105/tpc.110.074815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bertoni G. Plant Cell. 2010;22:2925. doi: 10.1105/tpc.110.220912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.MacKenzie D, Arendt A, Hargrave P, McDowell JH, Molday RS. Biochemistry. 1984;23:6544–6549. doi: 10.1021/bi00321a041. [DOI] [PubMed] [Google Scholar]
- 45.Maeda T, Perusek L, Amengual J, Babino D, Palczewski K, von Lintig J. Mol Pharmacol. 2011;80:943–952. doi: 10.1124/mol.111.074732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Amengual J, Golczak M, Palczewski K, von Lintig J. J Biol Chem. 2012;287:24216–24227. doi: 10.1074/jbc.M112.353979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Golczak M, Kiser PD, Lodowski DT, Maeda A, Palczewski K. J Biol Chem. 2010;285:9667–9682. doi: 10.1074/jbc.M109.063941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bordier C. J Biol Chem. 1981;256:1604–1607. [PubMed] [Google Scholar]
- 49.Lobo GP, Hessel S, Eichinger A, Noy N, Moise AR, Wyss A, Palczewski K, von Lintig J. FASEB J. 2010;24:1656–1666. doi: 10.1096/fj.09-150995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oberhauser V, Voolstra O, Bangert A, von Lintig J, Vogt K. Proc Natl Acad Sci USA. 2008;105:19000–19005. doi: 10.1073/pnas.0807805105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marasco EK, Vay K, Schmidt-Dannert C. J Biol Chem. 2006;281:31583–31593. doi: 10.1074/jbc.M606299200. [DOI] [PubMed] [Google Scholar]
- 52.Kiser PD, Golczak M, Lodowski DT, Chance MR, Palczewski K. Proc Natl Acad Sci USA. 2009;106:17325–17330. doi: 10.1073/pnas.0906600106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hamel CP, Tsilou E, Harris E, Pfeffer BA, Hooks JJ, Detrick B, Redmond TM. J Neurosci Res. 1993;34:414–425. doi: 10.1002/jnr.490340406. [DOI] [PubMed] [Google Scholar]
- 54.Bavik CO, Busch C, Eriksson U. J Biol Chem. 1992;267:23035–23042. [PubMed] [Google Scholar]
- 55.Shmarakov I, Fleshman MK, D’Ambrosio DN, Piantedosi R, Riedl KM, Schwartz SJ, Curley RW, Jr, von Lintig J, Rubin LP, Harrison EH, Blaner WS. Arch Biochem Biophys. 2010;504:3–10. doi: 10.1016/j.abb.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Golczak M, Kiser PD, Sears AE, Lodowski DT, Blaner WS, Palczewski K. J Biol Chem. 2012;287:23790–23807. doi: 10.1074/jbc.M112.361550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moise AR, Dominguez M, Alvarez S, Alvarez R, Schupp M, Cristancho AG, Kiser PD, de Lera AR, Lazar MA, Palczewski K. J Am Chem Soc. 2008;130:1154–1155. doi: 10.1021/ja710487q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Leung WC, Hessel S, Meplan C, Flint J, Oberhauser V, Tourniaire F, Hesketh JE, von Lintig J, Lietz G. Faseb J. 2008 doi: 10.1096/fj.08-121962. [DOI] [PubMed] [Google Scholar]
- 59.Lyubarsky AL, Savchenko AB, Morocco SB, Daniele LL, Redmond TM, Pugh EN., Jr Biochemistry. 2005;44:9880–9888. doi: 10.1021/bi0505363. [DOI] [PubMed] [Google Scholar]
- 60.Leuenberger MG, Engeloch-Jarret C, Woggon WD. Angew Chem Int Ed Engl. 2001;40:2613–2617. doi: 10.1002/1521-3773(20010716)40:14<2613::AID-ANIE2613>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 61.Schmidt H, Kurtzer R, Eisenreich W, Schwab W. J Biol Chem. 2006;281:9845–9851. doi: 10.1074/jbc.M511668200. [DOI] [PubMed] [Google Scholar]
- 62.Wenzel A, Oberhauser V, Pugh EN, Jr, Lamb TD, Grimm C, Samardzija M, Fahl E, Seeliger MW, Reme CE, von Lintig J. J Biol Chem. 2005;280:29874–29884. doi: 10.1074/jbc.M503603200. [DOI] [PubMed] [Google Scholar]
- 63.Eisenberg D, Weiss RM, Terwilliger TC, Wilcox W. Faraday Symposia of the Chemical Society. 1982:109–120. [Google Scholar]