

Abstract
Primary open angle glaucoma (POAG) is a common late-onset neurodegenerative disease. Ocular hypertension represents a major risk factor, but POAG etiology remains poorly understood. Some cases of early-onset congenital glaucoma and adult POAG are linked to mutations in myocilin, a secreted protein of poorly defined function. Transgenic overexpression of myocilin in Drosophila and experiments in mice and human populations implicate the unfolded protein response (UPR) in the pathogenesis of glaucoma. We postulate that compromised ability of the UPR to eliminate misfolded mutant or damaged proteins, including myocilin, causes endoplasmic reticulum stress, resulting in functional impairment of trabecular meshwork cells that regulate intraocular pressure. This mechanism of POAG is reminiscent of other age-dependent neurodegenerative diseases that involve accumulation of protein aggregates.
Keywords: Primary open angle glaucoma, ocular hypertension, myocilin, neurodegenerative disease
Glaucoma and myocilin
Glaucoma refers to a collection of diseases which are characterized by progressive degeneration of the optic nerve. Symptoms become manifest as impairments in the peripheral visual field which, if left untreated, culminate in bilateral blindness. Primary open angle glaucoma (POAG) is an idiopathic late-onset disorder accompanied by elevated intraocular pressure that results from compromised outflow of fluid in the anterior chamber of the eye (Box 1). The mechanism that leads to optic nerve degeneration remains unclear. The incidence of POAG is high, affecting approximately 1 out of 100 people over the age of 40 with especially high incidence among individuals of African or Hispanic descent. In the USA alone, two million people are afflicted with this disease, and worldwide, the total is 60 million [1].
Congenital open angle glaucoma is a relatively rare inherited disorder that results in early childhood onset of glaucoma. In 1997, Stone et al. discovered that administration of cortisol to primary cell cultures of human trabecular meshwork cells, which regulate outflow through Schlemm's canal to control intraocular pressure (Box 1), induced expression of a protein, originally named “trabecular meshwork inducible glucocorticoid response (TIGR)” protein, but more commonly referred to as “myocilin”[2]. The myocilin gene, mapped to chromosome 1q21-31, contains promoter elements that are regulated by cortisol [3-5]. Myocilin is a member of the olfactomedin protein family with an N-terminal leucine zipper motif and a large C-terminal olfactomedin homology domain (Box 2). Association studies and analyses of pedigrees have shown that mutations in myocilin, especially its olfactomedin domain, are associated with a substantial fraction (>10 %) of juvenile onset glaucoma patients [2,6,7] and a smaller portion (~4%) of adult POAG cases in African-American [8], Canadian [9], Indian [10], Japanese [11], Greek [12], South African [13], Brazilian [14], Peruvian [15], Ghanaian [16], Swiss [17], and Dutch [18] populations. However, in some populations, including Moroccan [19], Middle Eastern [20], and Finnish populations [21], case-control studies failed to identify POAG-associated polymorphisms in myocilin. Mutations in several other proteins, notably CYP1B1 [22,23], an enzyme involved in drug metabolism and lipid synthesis, have also been implicated as risk factors for POAG, demonstrating that the underlying genetic susceptibility for POAG is complex.
Trabecular meshwork cells secrete myocilin into the aqueous humor, where it may play a role in extracellular matrix-mediated cell adhesion [24] or cell migration [25], similar to the roles of other olfactomedin proteins in early development (Box 2). Its still poorly understood function during development may be recapitulated as a protective mechanism for tissue maintenance at adult age when stress due to ocular hypertension or high circulating levels of cortisol activates the expression of myocilin. In the endoplasmic reticulum (ER), the calcium-dependent endoprotease calpain II cleaves myocilin at residues that link the leucine zipper motif and the C-terminal olfactomedin domain [26]. This proteolytic cleavage appears to represent a regulatory step for cellular processing of myocilin. It is of interest to note that the olfactomedin domain contains a high affinity calcium binding site [27]. Truncated myocilin that lacks the olfactomedin domain and mutant myocilins with mutations in the olfactomedin domain are retained in the ER [28,29].
Here, we propose that accumulation of overexpressed or misfolded proteins, whether myocilin or any other protein associated with age-dependent onset of glaucoma, leads to ER stress when the unfolded protein response (UPR) pathway falls short of removing these proteins through ubiquitination, thiol reduction and subsequent proteolysis via the proteasome (Figure 1) [30-32]. Thus, the UPR emerges as a focal mechanism for the pathogenesis of POAG. As with other complex diseases, individual disease susceptibility can be regarded as the cumulative sum of liabilities arising from polymorphisms in a range of cellular components that include not only myocilin or other proteins previously implicated in POAG, but also polymorphisms that contribute to variation in the efficacy of the UPR.
Figure 1.
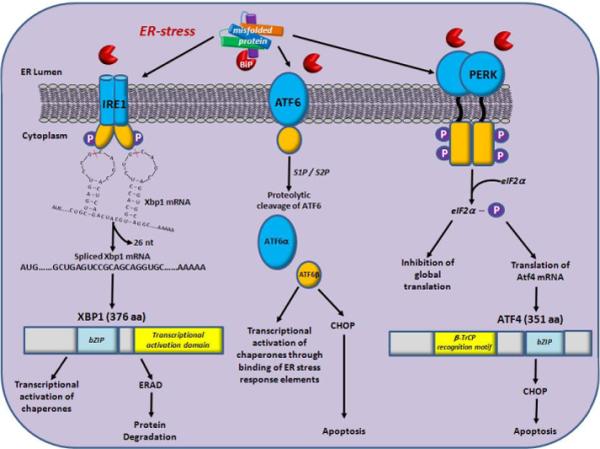
Signaling pathways of the UPR in response to ER-stress. The UPR is regulated through signaling cascades from three sensor proteins: IRE1, ATF6 and PERK. During non-stressed conditions, BiP (red pie) is bound to the three sensors to maintain their inactive state. When misfolded proteins accumulate in the lumen of the ER, BiP dissociates from the sensors and binds to unfolded and misfolded proteins. This results in activation of the IRE1, ATF6 and PERK signaling cascades. Dissociation of BiP from IRE1 results in its dimerization which activates its kinase and RNAse activities to promote splicing of Xbp1 mRNA resulting in a frame shift of the Xbp1 gene. Translation of spliced Xbp1 produces an active transcription factor which subsequently induces the translation of ER chaperones and genes involved in ER-associated protein degradation (ERAD). ATF6 is a basic leucine-zipper (bZIP) containing transcription factor. During ER-stress, BiP dissociates and ATF6 is translocated to the Golgi apparatus where it is cleaved by two proteases (serine protease site-1 protease (S1P) and serine protease site-2 protease (S2P). The cytosolic fragment of ATF6 migrates to the nucleus and activates transcription of UPR-responsive genes such as chaperones and CHOP, through promoter binding at their ER-stress response element binding sites. PERK is a serine/threonine protein kinase activated by ER-stress through dimerization and autophosphorylation upon dissociation of BiP. Activated PERK phosphorylates eIF2α, resulting in the inhibition of global translation. However, ATF4 is translated because it contains multiple 5’ open reading frames. ATF4 induces CHOP (a proapoptotic transcription factor) which activates the apoptosis pathway.
The UPR and ER stress: A common pathway for neurodegenerative diseases
The UPR is a conserved cellular pathway among all eukaryotes that protects cells against the accumulation and aggregation of misfolded proteins during protein synthesis (Figure 1) [33,34]. It does so by halting protein translation and up-regulating the expression of chaperones that facilitate protein folding. When the extent of aggregation of misfolded or damaged proteins exceeds the capacity of the UPR, it is no longer able to protect the ER from stress, and this results in activation of apoptosis. The pancreatic ER kinase-like ER kinase (PERK) attenuates protein synthesis by phosphorylating the α-subunit of elF2 that regulates initiation of mRNA translation [35]. The molecular chaperone Bip/Grp78 (glucose regulated protein 78) interacts with misfolded proteins in the lumen of the ER and facilitates their transport into the cytosol [36], where subsequent ubiquitination tags them for proteasome-mediated proteolytic degradation [37]. This process is accompanied by activation of the ER stress sensor, inositol requiring transmembrane kinase/endonuclease-1α (IRE-1α), which through splicing activates the transcription factor XBP1 (X-box binding protein 1) mRNA; this allows XBP1 to direct upregulation of the expression of target genes, associated with ER stress, including enhanced expression of UPR components [38,39]. Splice products of ATF6 (activating transcription factor 6) also facilitate this upregulation of gene expression [40]. This feed-forward mechanism aimed at upregulation of the UPR is designed to aid the removal of misfolded proteins to relieve ER stress and restore homeostasis of protein synthesis. Failure of the UPR to accomplish this mission results in the activation of apoptosis.
Given the prominent evolutionarily conserved protective function of the UPR, it is not surprising that compromised ability of the UPR to remove misfolded aggregated proteins is central to a wide range of neurodegenerative disorders. In neurodegenerative disorders that are classified as tauopathies, accumulation of the microtubule-associated tau protein causes activation of the UPR [41-43]. Neurodegeneration in the hippocampus during the development of Alzheimer's disease (AD) shows increased expression of UPR markers like PERK and IRE1α, as well as BiP/GRP78 [44].
A mouse model of Huntington's disease also implicates the UPR. In this model neurodegeneration occurs as a result of accumulation of a Huntingtin construct containing 95 polyglutamine repeats. Adenoviral delivery of XBP1 reduces accumulation of this protein [45].
Studies in mouse and rat models of Parkinson's disease (PD) also point at the UPR. Here, BiP/GRP78 may form a complex with α-synuclein, a protein of unknown function that forms aggregates associated with neurodegeneration in PD. Association of BiP/GRP78 with α-synuclein lowers its neurotoxicity [46]. Oxidative damage is a major contributor to the etiology of PD [47,48] and environmental toxins that produce reactive oxygen species contribute to PD risk. A direct link between oxidative stress and the UPR was demonstrated by a proteomics analysis of rotenone-induced oxidative stress in a human neuroblastoma cell line, which showed activation of the UPR in response to an increase in reactive oxygen species [49].
If accumulation of misfolded proteins is at the root of the development of at least the most common forms of glaucoma, it stands to reason that various contributing factors converge on the UPR as a central pathogenic mechanism. Thus, we can place POAG in the same context as other adult late-onset neurodegenerative diseases that result from failure of the UPR to remove excess protein aggregates from the ER. There is increasing evidence in support of this hypothesis.
Studies on flies, mice and people implicate the UPR
The Drosophila ocular hypertension model
Studies on glaucoma in people have been limited to studies using perfused post-mortem eyes, primary cell cultures of trabecular meshwork cells, or generally underpowered genetic case-control studies using pedigree designs or association analyses with a limited number of candidate genes. Drosophila melanogaster provides a powerful genetic model in which large numbers of genetically identical individuals can be grown rapidly under controlled environmental conditions without regulatory restrictions. Despite profound anatomical differences between the fly eye and the mammalian eye, we can expect that similar principles underlie the response to ocular hypertension and cellular injury if it is mediated by fundamental evolutionarily conserved mechanisms.
The binary GAL4-UAS expression system allows targeted expression of transgenes in Drosophila [50]. In this system a cell specific promoter drives expression of the yeast transcription factor GAL4. When we cross a homozygous line containing the promoter-GAL4 construct to a homozygous line that carries a transgene inserted behind a GAL4 promoter, known as UAS (upstream activator sequence), the heterozygous offspring produces the transgene in cells in which GAL4 is expressed. This system has been used to drive transgenic expression of human myocilin in the Drosophila eye under a gmr-GAL4 driver. A surprising phenotype emerges: Transgenic flies show intermittent extrusion of fluid through the lenses of the ommatidia leaving a crusty salt residue on the ocular surface. Histological examination reveals distended ommatidia (Figure 2) [30,51]. The most parsimonious explanation for this phenotype is an increase in intraocular pressure. The same phenotype is observed with wild-type myocilin and overexpression of myocilin mutants that have been previously associated with congenital open angle glaucoma in human subjects.
Figure 2.

Consequences of overexpression of human myocilin in the Drosophila eye. (A) shows a normal Drosophila eye. (B) illustrates the fluid extrusion phenotype that results from expression of human myocilin. (C) shows a histological section through a normal Drosophila eye with a regular array of ommatidia. In transgenic flies that express myocilin this regular array of ommatidia is disrupted and they appear distended, as shown in (D). (E) shows a region where a dimple appears (between arrows) underneath which ommatidia appear to have regained their normal shape. This dimpled region has likely undergone recent fluid extrusion resulting in temporary relieve of hydrostatic pressure in the underlying tissue.
Western blots with antibodies against myocilin revealed aggregates in the transgenic flies, except for a truncated myocilin mutant that lacks the olfactomedin domain, which suggests that this domain functions as a protein-protein interaction motif that facilitates aggregation [30]. Transcriptional profiling of transgenic flies using expression microarrays showed extensive changes in transcript abundance levels, including transcripts corresponding to the UPR. Activation of the fluorescent UPR marker, xbp1-EGFP, can visualize induction of the UPR directly [30,52]. Based on the assumption of evolutionary conservation, these observations suggest that myocilin-associated ocular hypertension in the human eye results from protein aggregation and induction of the UPR in trabecular meshwork cells. This process occurs at a late age with wild-type myocilin, but might be accelerated by myocilin mutants to account for juvenile onset glaucoma.
Mice with glaucoma
The differences in eye structure between vertebrates and invertebrates impose limitations on the use of the Drosophila model. To fill the need for a vertebrate animal model to study POAG several laboratories pursued the generation of transgenic mice [53-55]. Introduction of a bacterial artificial chromosome construct that carries the full length mouse myocilin gene, modified to include the Y437H mutation that causes severe glaucoma in people, results in mice that develop symptoms that closely resemble POAG [54]. At about three months of age, these mice show an increase in intraocular pressure and axonal degeneration in the optic nerve. These mice express Y437H-myocilin in the sclera and irido-corneal angle, but the protein is not secreted. Rather it accumulates in the ER of trabecular meshwork cells and leads to apoptosis. Indeed, these mice show upregulation of the pro-apoptotic factor, CCAAT/enhancer-binding protein homologous protein (CHOP), which suppresses expression of the mitochondrial anti-apoptotic protein Bcl-2 and enables caspase 3 activation to initiate apoptosis. Topical application of the chemical chaperone 4-phenylbutyrate reduces intraocular pressure and ER stress in Y437H-myocilin mice as well as in wild-type mice in which the N-glycosylation inhibitor tunicamycin is used to elicit ER stress [56,57]. Results from these studies are consistent with conclusions drawn from the transgenic Drosophila ocular hypertension model; they point at failure of the UPR to alleviate ER stress with resulting apoptosis as a central molecular mechanism that mediates glaucoma.
Human cell lines and translational studies in human populations
Myocilin expressed in transiently transfected HEK293 cell lines localizes to the ER, where - as mentioned above - cleavage by calpain II separates its N-terminal and C-terminal domains [26]. This proteolytic cleavage prevents aggregation of myocilin. Mutant myocilins form aggregates that accumulate in the ER, and this may be due to inhibition of endoproteolytic processing. Among these mutant myocilins, the P370L mutation which is associated with a severe glaucoma phenotype is also the most potent inhibitor of proteolytic processing in the ER [58]. Expression of myocilin-green fluorescent protein fusion constructs in transfected CHO-K1, HEK293 and human trabecular meshwork cells resulted in accumulation of insoluble aggregates of misfolded myocilin and the induction of apoptosis. In these studies sodium 4-phenylbutyrate relieved ER stress and reduced the rate of apoptosis [59]. Oxidative stress, which has been proposed as a contributing factor to the pathogenesis of glaucoma, exacerbated ER stress that was induced by expression of wild-type or mutant myocilins in stably transfected HEK293 cells [60].
Direct evidence that identifies components of the UPR as risk factors for POAG in human populations comes from association studies that focused on candidate genes identified in the Drosophila ocular hypertension model [31]. Disease-control studies for POAG do not provide sufficient power for genome-wide association studies (GWAS) due to the generally small number of individuals in these study populations relative to the number of independently segregating polymorphisms and the consequent insurmountable multiple testing penalty. For this reason, association studies have been limited to candidate genes, mostly those previously implicated in POAG, such as myocilin [61], optineurin [62], WDR36 [63, 64], and CYP1B1 [65-67].
To escape from such “search for the lost key under the streetlight” approaches, unbiased GWAS can be pursued in Drosophila disease models, which can identify conserved orthologues as novel candidate genes for subsequent focused studies in human populations. This translational strategy resulted in the identification of 16 candidate genes, which are orthologous between Drosophila and humans, with 62 polymorphic markers, including myocilin and components of the UPR [30]. Tests of association between these polymorphic markers in two Caucasian study populations of patients with POAG and age-matched controls from replicate study populations in Salt Lake City (207 POAG cases, 270 controls) and San Diego (471 POAG cases, 151 controls) showed associations of single nucleotide polymorphisms (SNPs) with disease status for two components of the UPR, PDIA5 and BIRC6 [31]. PDIA5 encodes a protein disulfide isomerase that facilitates the formation of disulfide bonds during protein folding [68]. BIRC6 encodes a ubiquitin ligase that mediates removal of misfolded proteins by targeting them to the proteasome for degradation, thereby contributing protection against apoptosis (Figure 1) [69]. Thus, both of these gene products promote cellular homeostasis by providing protection against accumulation of misfolded proteins to reduce ER stress. This study did not detect associations with myocilin, but found a significant association between a polymorphism in optineurin, previously associated with POAG, in the Salt Lake City - but not in the San Diego - population. These discrepancies can be due to the limited sample size and the relatively rare incidence (~4%) of association of myocilin with adult onset POAG. We can expect replication when the assumption that common alleles underlie a common disease is valid. However, when the genetic basis of disease susceptibility is complex, as is the case for POAG, many rare alleles can predispose to disease risk and it is unlikely that the same rare alleles will segregate at similar frequencies in different populations. In light of these considerations, it is especially significant to find associations between variants in the same two UPR genes in both populations. This further supports the notion that polymorphisms in UPR-related genes play a prominent role in determining genetic susceptibility for POAG.
Concluding remarks
Despite the prevalence of POAG and many years of research efforts, the molecular mechanisms that lead to progressive optic nerve degeneration remain poorly understood. A major challenge for the future is to elucidate the mechanism that links ocular hypertension to optic nerve degeneration and the molecular mechanisms that underlie forms of glaucoma that are not associated with ocular hypertension (normal tension glaucoma). The discovery of myocilin and its association with juvenile onset and adult onset glaucoma has provided a molecular handle on this disease. Most studies, however, have focused on identifying functions of myocilin, which are a priori of interest, but not necessarily relevant to the etiology of POAG. The functions of myocilin are likely associated primarily with embryonic development, in line with other members of the olfactomedin family (Box 2). Myocilin expression in adult eyes is low under normal conditions, but can be activated by stress, in particular high circulating levels of cortisol, which forms a link between the well-known risk for glaucoma as a consequence of diabetes.
The central theme that crystallizes from recent studies represents a paradigm shift in our views of the causal cellular mechanisms that lead to ocular hypertension as a preamble to the development of glaucoma. The new picture that emerges indicates that excessive overproduction of normal myocilin or production of misfolded mutant myocilin, or any other protein that harbors polymorphisms associated with POAG, forms protein aggregates that in susceptible individuals (depending on genetic composition) surpass the ability of the UPR to eliminate them via the proteasome. This results in ER stress and subsequent triggering of apoptosis of trabecular meshwork cells that regulate intraocular pressure. Thus, POAG may result from late adult onset protein aggregation followed by neurodegeneration, similar to other late-onset neurodegenerative diseases, such as Alzheimer's disease and Parkinson's disease.
This hypothesis suggests a molecular mechanism for this prevalent neurodegenerative disorder that is testable and can be validated in tractable model organisms. In addition, the UPR presents new targets for preventative and therapeutic applications, as already demonstrated by encouraging ameliorating effects of 4-phenylbutyrate in model systems [56,57,59]. Identifying the full complement of common and rare SNPs associated with disease risk and evaluating their relative contributions to propensity for disease development in different populations remains a major task for future studies aimed at applying genomic information to patient-tailored personal prevention and therapy. Finally, development of effective therapies for the treatment of POAG that target the UPR by preventing accumulation of protein aggregates also may prove to be beneficial for the treatment of a range of other late-onset neurodegenerative diseases.
Highlights.
➢ Accumulation of protein aggregates may overwhelm the UPR and result in ER stress.
➢ Impairment of the UPR which results in ER stress may be a common cause for POAG.
➢ Aspects of POAG resemble other neurodegenerative protein aggregation diseases.
Box 1. Circulation through the anterior chamber of the eye.
The aqueous humor provides nutrition to the lens and is produced by the epithelium of the ciliary body, located at the base of the ciliary muscles that anchor the lens. The aqueous humor flows through the pupil in the iris from the posterior chamber to the anterior chamber of the eye, where it drains in the iridocorneal angle via Schlemm's canal, named after the German anatomist, Friedrich Schlemm (Figure I). Fluid is returned to the bloodstream via the anterior ciliary veins. The outflow of fluid from the eye through Schlemm's canal is regulated by cells of the trabecular meshwork. Intraocular pressure in normal eyes is around 15 mm Hg. When outflow capacity through the canal of Schlemm is compromised, the pressure in the eye increases. Ocular hypertension is one of the principal diagnostics of POAG and lowering intraocular pressure, either through pharmacological or surgical means, is currently the only effective treatment to prevent progression of the disease.
Figure I. Circulation of aqueous humor through the anterior chamber of the eye. Red arrows indicate the flow of fluid from the posterior chamber through the iris into the anterior chamber of the eye, where it leaves through the canal of Schlemm.

Box 2. Olfactomedin proteins: Modulators of development.
Myocilin is a member of the family of olfactomedin-related proteins. The first olfactomedin was discovered in the olfactory neuroepithelium of the bullfrog where it is secreted into the extracellular mucus matrix in association with chemosensory dendritic cilia of olfactory sensory neurons [70]. The human and mouse olfactomedin families consist of 13 genes that contain conserved olfactomedin homology domains of about 250 amino acid residues. Several members of the olfactomedin family have been associated with early development of the nervous system. In zebrafish, olfactomedin 1 regulates elongation of retinal axons through interactions with the Wnt signaling pathway [71]. Morpholino antisense oligonucleotides targeting translation of olfactomedin 2 in this system, give rise to impaired development of branchiomotor neurons and disruption of branchiomotor axon guidance, and poorly developed caudal pharyngeal arches, olfactory pits, eyes and optic tectum [72]. These effects may involve Pax6 signaling [72]. In addition to myocilin, several other members of the olfactomedin family are expressed in the eye [73]. An olfactomedin-related protein is also associated with hematopoiesis [74]. It is reasonable to suspect that myocilin, like other closely related members of the olfactomedin protein family, may play a similar role in early development.
Glossary
- Allele
a sequence variant of a gene at a specific locus
- Alzheimer's disease (AD)
a progressive neurodegenerative disease of advanced age that results in loss of memory, dementia, and behavioral impairments, and is characterized by neuritic plaques and tangles in the brain
- Analyses of pedigrees
a family based design to assess the evidence for a genetic association with a disease or particular phenotype
- Apoptosis
a process, also known as programmed cell death, mediated through specific transcription factors in which a cell triggers its own systematic destruction
- Association studies
tests for a correlation between a phenotype (e.g. disease status) and genetic variation to identify candidate genes or genomic regions which contribute to that phenotype
- Aqueous humor
a transparent, gelatinous fluid secreted from the ciliary epithelium and located in the anterior and posterior chambers of the eye
- Axon
a projection of a neuron that transmits electrical impulses away from the neuron's cell body to different neurons, muscles and glands
- Bacterial artificial chromosome (BAC)
a class of vectors used to clone genomic DNA sequences in bacterial cells
- Case-control studies
a genetic association study that compares the frequency of alleles or genotypes at genetic marker loci between cases (having the disease or trait of interest) and controls (unaffected individuals), in order to determine whether a statistical association exists between the disease trait and the genetic marker
- Chaperones
proteins that assist in the non-covalent folding and unfolding as well as the assembly and disassembly of proteins or RNA
- Cortisol
a steroid hormone released from the adrenal gland in response to stress
- Endoplasmic reticulum
the primary subcellular organelle, consisting of a system of membrane bound cavities, responsible for protein synthesis, posttranslational modification, folding and trafficking
- Endoproteolysis
the breaking of peptide bonds of nonterminal amino acids in proteins
- Etiology
the factors or mechanisms that result in a disease
- Fusion construct
a construct in which two or more genes that code for separate proteins are joined to produce a single polypeptide chain
- Heterozygous
the term used to describe the genotype of a diploid organism with two different alleles at a given DNA locus
- Hippocampus
a part of the cerebral cortex, located in the medial temporal lobe, underneath the cortical surface, which functions in the acquisition of memory
- Homeostasis
regulation of the internal environment to maintain a stable, constant condition
- Homozygous
the term used to describe the genotype of a diploid organism with two identical alleles at a given DNA locus
- Huntington's disease (HD)
a late age-onset lethal human disease of nerve degeneration characterized by involuntary movement and inherited as an autosomal dominant phenotype
- Iridocorneal angle
the acute angle between the iris and the cornea located in the anterior chamber of the eye, responsible for the flow of aqueous humor
- Leucine zipper
a protein-protein interaction domain with leucine residues every seven amino acids in the dimerization domain
- Mitochondria
eukaryotic intracellular organelles that procure energy as chemical bonds of adenosine triphosphate (ATP) by mediating aerobic respiration
- Multiple testing penalty
a statistical term that refers to the need to adjust p-values for the number of tests performed to control for false positives
- Neurodegenerative disease
diseases, such as Alzheimer's, Parkinson's and Huntington's disease, which involve the progressive loss of structure or function of neurons
- N-glycosylation
a post-translational modification by which oligosaccharides are attached to specific asparagine (N) residues of proteins targeting them for secretion or transport to the cell surface
- Ocular hypertension
intraocular pressure above the normal range between 10 and 21 mm Hg
- Ommatidia
Structural iterative units of the compound eyes of insects (750-800 in Drosophila), each of which is composed of a cluster of photoreceptor cells surrounded by cone cells and pigment cells
- Orthologs
genes in different species that are similar in their nucleotide sequences and may have evolved from a common ancestral gene
- Oxidative stress
imbalance in the reduction-oxidation state of a cell, either by overproduction of reactive oxygen species, or by dysfunction of the antioxidant systems
- Parkinson's disease (PD)
a neurodegenerative disease in which dopaminergic neurons of the nigrostriatal pathway are compromised, characterized by difficulty in initiating motion, stooped posture, and resting tremor
- Phenotype
an organism's observable trait such as morphology, behavior or development as a result of the organism's genotype
- Phosphorylation
a post-translational modification in which a phosphate group is added to a protein thereby altering its function or activity
- Polymorphisms
DNA sequence variants in the genome
- Promoter elements
regions of DNA located upstream of a gene that initiate transcription
- Proteasome
a protein complex that degrades damaged or superfluous proteins by proteolysis
- Protein translation
the process that occurs on ribosomes by which a polypeptide is synthesized through the sequential addition of specific amino acids encoded by a messenger RNA template
- Proteolysis
the enzymatic degradation of proteins
- Reactive oxygen species (ROS)
Oxygen radicals and non-radicals that are oxidizing agents and/or are easily converted into free radicals
- Sclera
the opaque, fibrous, protective, outer layer of the eye containing collagen and elastic fiber, commonly referred to as the “white of the eye”
- Splicing
the process by which non-coding introns are removed from pre-mRNA and exons are joined to produce the mature protein coding mRNA
- α-Synuclein
a protein which forms fibrous aggregates associated with the pathology of Parkinson's disease
- Trabecular meshwork
a meshwork of connective tissue located at the angle of the anterior chamber of the eye which contains endothelium-lined spaces through which the aqueous humor drains from the eye
- Ubiquitination
the covalent attachment of a small 76 amino acid protein, ubiquitin, to a target protein to tag that protein for degradation by the proteasome
- Unfolded protein response (UPR)
a cellular stress response that is activated during accumulation of unfolded or misfolded proteins in the lumen of the endoplasmic reticulum
- Transcript
RNA that is synthesized from DNA with RNA polymerase
- Transcription factor
a protein that binds to specific DNA sequences to regulate the transcription of DNA to RNA
- Transfection
the introduction of foreign DNA into a eukaryotic cell using chemical or physical methods
- Transgene
DNA encoding a gene that has been isolated from one organism and inserted into a different organism through genetic engineering techniques
- Western blots
an analytical technique used to identify specific proteins in a given sample by staining with antibodies following gel electrophoresis and transfer to a membrane
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Quigley HA, Broman AT. The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol. 2006;90:262–267. doi: 10.1136/bjo.2005.081224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stone EM, et al. Identification of a gene that causes primary open angle glaucoma. Science. 1997;275:668–670. doi: 10.1126/science.275.5300.668. [DOI] [PubMed] [Google Scholar]
- 3.Sheffield VC, et al. Genetic linkage of familial open angle glaucoma to chromosome 1q21-q31. Nature Genetics. 1993;4:47–50. doi: 10.1038/ng0593-47. [DOI] [PubMed] [Google Scholar]
- 4.Michels-Rautenstrauss KG, et al. Juvenile open angle glaucoma: fine mapping of the TIGR gene to 1q24.3-q25.2 and mutation analysis. Hum. Genet. 1998;102:103–106. doi: 10.1007/s004390050661. [DOI] [PubMed] [Google Scholar]
- 5.Clark AF, et al. Glucocorticoid induction of the glaucoma gene MYOC in human and monkey trabecular meshwork cells and tissues. Invest. Ophthalmol. Vis. Sci. 2001;42:1769–1780. [PubMed] [Google Scholar]
- 6.Wiggs JL, et al. Prevalence of mutations in TIGR/Myocilin in patients with adult and juvenile primary open-angle glaucoma. American journal of human genetics. 1998;63:1549–1552. doi: 10.1086/302098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adam MF, et al. Recurrent mutations in a single exon encoding the evolutionarily conserved olfactomedin-homology domain of TIGR in familial open-angle glaucoma. Human molecular genetics. 1997;6:2091–2097. doi: 10.1093/hmg/6.12.2091. [DOI] [PubMed] [Google Scholar]
- 8.Liu W, et al. Low prevalence of myocilin mutations in an African American population with primary open-angle glaucoma. Mol. Vis. 2012;18:2241–2246. [PMC free article] [PubMed] [Google Scholar]
- 9.Faucher M, et al. Founder TIGR/myocilin mutations for glaucoma in the Quebec population. Human molecular genetics. 2002;11:2077–2090. doi: 10.1093/hmg/11.18.2077. [DOI] [PubMed] [Google Scholar]
- 10.Banerjee D, et al. Comprehensive analysis of myocilin variants in east Indian POAG patients. Mol. Vis. 2012;18:1548–1557. [PMC free article] [PubMed] [Google Scholar]
- 11.Mengkegale M, et al. Presence of myocilin sequence variants in Japanese patients with open-angle glaucoma. Mol. Vis. 2008;14:413–417. [PMC free article] [PubMed] [Google Scholar]
- 12.Wirtz MK, et al. Association of POAG risk factors and the Thr377Met MYOC mutation in an isolated Greek population. Invest. Ophthalmol. Vis. Sci. 2010;51:3055–3060. doi: 10.1167/iovs.09-4652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Whigham BT, et al. Myocilin mutations in black South Africans with POAG. Mol. Vis. 2011;17:1064–1069. [PMC free article] [PubMed] [Google Scholar]
- 14.Povoa CA, et al. Correlation between genotype and phenotype in primary open angle glaucoma of Brazilian families with mutations in exon 3 of the TIGR/MYOC gene. Arq. Bras. Oftalmol. 2006;69:289–297. doi: 10.1590/s0004-27492006000300002. [DOI] [PubMed] [Google Scholar]
- 15.Mendoza-Reinoso V, et al. Novel and known MYOC exon 3 mutations in an admixed Peruvian primary open-angle glaucoma population. Mol. Vis. 2012;18:2067–2075. [PMC free article] [PubMed] [Google Scholar]
- 16.Challa P, et al. Prevalence of myocilin mutations in adults with primary open-angle glaucoma in Ghana, West Africa. J. Glaucoma. 2002;11:416–420. doi: 10.1097/00061198-200210000-00008. [DOI] [PubMed] [Google Scholar]
- 17.Iliev ME, et al. Glaucoma phenotype in a large Swiss pedigree with the myocilin Gly367Arg mutation. Eye. 2008;22:880–888. doi: 10.1038/sj.eye.6702745. [DOI] [PubMed] [Google Scholar]
- 18.Hogewind BF, et al. Variable clinical spectrum of the myocilin Gln368X mutation in a Dutch family with primary open angle glaucoma. Curr. Eye Res. 2010;35:31–36. doi: 10.3109/02713680903374182. [DOI] [PubMed] [Google Scholar]
- 19.Melki R, et al. Mutational analysis of the Myocilin gene in patients with primary open-angle glaucoma in Morocco. Ophthalmic. Genet. 2003;24:153–160. doi: 10.1076/opge.24.3.153.15610. [DOI] [PubMed] [Google Scholar]
- 20.Ozgul RK, et al. Myocilin mt1 promoter polymorphism in Turkish patients with primary open angle glaucoma. Mol. Vis. 2005;11:916–921. [PubMed] [Google Scholar]
- 21.Lemmela S, et al. Exclusion of 14 candidate loci for primary open angle glaucoma in Finnish families. Mol. Vis. 2004;10:260–264. [PubMed] [Google Scholar]
- 22.Patel HY, et al. Screening glaucoma genes in adult glaucoma suggests a multiallelic contribution of CYP1B1 to open-angle glaucoma phenotypes. Clin. Experiment. Ophthalmol. 2012;40:e208–217. doi: 10.1111/j.1442-9071.2011.02714.x. [DOI] [PubMed] [Google Scholar]
- 23.Hilal L, et al. Screening of CYP1B1 and MYOC in Moroccan families with primary congenital glaucoma: three novel mutations in CYP1B1. Mol. Vis. 2010;16:1215–1226. [PMC free article] [PubMed] [Google Scholar]
- 24.Goldwich A, et al. Myocilin promotes substrate adhesion, spreading and formation of focal contacts in podocytes and mesangial cells. Histochem. Cell Biol. 2009;131:167–180. doi: 10.1007/s00418-008-0518-4. [DOI] [PubMed] [Google Scholar]
- 25.Kwon HS, Tomarev SI. Myocilin, a glaucoma-associated protein, promotes cell migration through activation of integrin-focal adhesion kinase-serine/threonine kinase signaling pathway. J. Cell. Physiol. 2011;226:3392–3402. doi: 10.1002/jcp.22701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanchez-Sanchez F, et al. Characterization of the intracellular proteolytic cleavage of myocilin and identification of calpain II as a myocilin-processing protease. J. Biol. Chem. 2007;282:27810–27824. doi: 10.1074/jbc.M609608200. [DOI] [PubMed] [Google Scholar]
- 27.Donegan RK, et al. The glaucoma-associated olfactomedin domain of myocilin is a novel calcium binding protein. J. Biol. Chem. 2012;287:43370–43377. doi: 10.1074/jbc.M112.408906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gobeil S, et al. Functional analysis of the glaucoma-causing TIGR/myocilin protein: integrity of amino-terminal coiled-coil regions and olfactomedin homology domain is essential for extracellular adhesion and secretion. Exp. Eye Res. 2006;82:1017–1029. doi: 10.1016/j.exer.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 29.Sohn S, et al. Expression of wild-type and truncated myocilins in trabecular meshwork cells: their subcellular localizations and cytotoxicities. Invest. Ophthalmol. Vis. Sci. 2002;43:3680–3685. [PubMed] [Google Scholar]
- 30.Carbone MA, et al. Overexpression of myocilin in the Drosophila eye activates the unfolded protein response: implications for glaucoma. PLoS One. 2009;4:e4216. doi: 10.1371/journal.pone.0004216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carbone MA, et al. Genes of the unfolded protein response pathway harbor risk alleles for primary open angle glaucoma. PLoS One. 2011;6:e20649. doi: 10.1371/journal.pone.0020649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Joe MK, et al. Accumulation of mutant myocilins in ER leads to ER stress and potential cytotoxicity in human trabecular meshwork cells. Biochem. Biophys. Res. Commun. 2003;312:592–600. doi: 10.1016/j.bbrc.2003.10.162. [DOI] [PubMed] [Google Scholar]
- 33.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 34.Gardner BM, et al. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb. Perspect. Biol. 2013;5 doi: 10.1101/cshperspect.a013169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rasheva VI, Domingos PM. Cellular responses to endoplasmic reticulum stress and apoptosis. Apoptosis. 2009;14:996–1007. doi: 10.1007/s10495-009-0341-y. [DOI] [PubMed] [Google Scholar]
- 36.Quinones QJ, et al. GRP78: a chaperone with diverse roles beyond the endoplasmic reticulum. Histol. Histopathol. 2008;23:1409–1416. doi: 10.14670/HH-23.1409. [DOI] [PubMed] [Google Scholar]
- 37.Goder V. Roles of ubiquitin in endoplasmic reticulum-associated protein degradation (ERAD). Curr. Protein Pept. Sci. 2012;13:425–435. doi: 10.2174/138920312802430572. [DOI] [PubMed] [Google Scholar]
- 38.Majumder M, et al. A novel feedback loop regulates the response to endoplasmic reticulum stress via the cooperation of cytoplasmic splicing and mRNA translation. Mol. Cell. Biol. 2012;32:992–1003. doi: 10.1128/MCB.06665-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takayanagi S, et al. Gene regulatory network of unfolded protein response genes in endoplasmic reticulum stress. Cell Stress Chaperones. 2013;18:11–23. doi: 10.1007/s12192-012-0351-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Logue SE, et al. New directions in ER stress-induced cell death. Apoptosis. 2013;18:537–546. doi: 10.1007/s10495-013-0818-6. [DOI] [PubMed] [Google Scholar]
- 41.Hoozemans JJ, Scheper W. Endoplasmic reticulum: the unfolded protein response is tangled in neurodegeneration. Int. J. Biochem. Cell Biol. 2012;44:1295–1298. doi: 10.1016/j.biocel.2012.04.023. [DOI] [PubMed] [Google Scholar]
- 42.Nijholt DA, et al. The unfolded protein response is associated with early tau pathology in the hippocampus of tauopathies. J. Pathol. 2012;226:693–702. doi: 10.1002/path.3969. [DOI] [PubMed] [Google Scholar]
- 43.Ferreiro E, Pereira CM. Endoplasmic reticulum stress: a new playER in tauopathies. J. Pathol. 2012;226:687–692. doi: 10.1002/path.3977. [DOI] [PubMed] [Google Scholar]
- 44.Viana RJ, et al. Endoplasmic reticulum enrollment in Alzheimer's disease. Mol. Neurobiol. 2012;46:522–534. doi: 10.1007/s12035-012-8301-x. [DOI] [PubMed] [Google Scholar]
- 45.Zuleta A, et al. AAV-mediated delivery of the transcription factor XBP1s into the striatum reduces mutant Huntingtin aggregation in a mouse model of Huntington's disease. Biochem. Biophys. Res. Commun. 2012;420:558–563. doi: 10.1016/j.bbrc.2012.03.033. [DOI] [PubMed] [Google Scholar]
- 46.Gorbatyuk MS, et al. Glucose regulated protein 78 diminishes alpha-synuclein neurotoxicity in a rat model of Parkinson disease. Mol. Ther. 2012;20:1327–1337. doi: 10.1038/mt.2012.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jenner P. Oxidative mechanisms in nigral cell death in Parkinson's disease. Mov. Disord. 1998;13(Suppl. 1):24–34. [PubMed] [Google Scholar]
- 48.Barnham KJ, et al. Neurodegenerative disease and oxidative stress. Nat. Rev. Drug Discov. 2004;3:205–214. doi: 10.1038/nrd1330. [DOI] [PubMed] [Google Scholar]
- 49.Bauereis B, et al. Proteomic insights into the protective mechanisms of an in vitro oxidative stress model of early stage Parkinson's disease. Neurosci. Lett. 2011;488:11–16. doi: 10.1016/j.neulet.2010.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 51.Borrás T, et al. Transcription profiling in Drosophila eyes that overexpress the human glaucoma-associated trabecular meshwork-inducible glucocorticoid response protein/myocilin (TIGR/MYOC). Genetics. 2003;163:637–645. doi: 10.1093/genetics/163.2.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ryoo HD, et al. Unfolded protein response in a Drosophila model for retinal degeneration. EMBO J. 2007;26:242–252. doi: 10.1038/sj.emboj.7601477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McDowell CM, et al. Mutant human myocilin induces strain specific differences in ocular hypertension and optic nerve damage in mice. Exp. Eye Res. 2012;100:65–72. doi: 10.1016/j.exer.2012.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zillig M, et al. Overexpression and properties of wild-type and Tyr437His mutated myocilin in the eyes of transgenic mice. Invest. Ophthalmol. Vis. Sci. 2005;46:223–234. doi: 10.1167/iovs.04-0988. [DOI] [PubMed] [Google Scholar]
- 55.Nguyen C, et al. Studies of scleral biomechanical behavior related to susceptibility for retinal ganglion cell loss in experimental mouse glaucoma. Invest. Ophthalmol. Vis. Sci. 2013;54:1767–1780. doi: 10.1167/iovs.12-10952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zode GS, et al. Topical ocular sodium 4-phenylbutyrate rescues glaucoma in a myocilin mouse model of primary open-angle glaucoma. Invest. Ophthalmol. Vis. Sci. 2012;53:1557–1565. doi: 10.1167/iovs.11-8837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zode GS, et al. Reduction of ER stress via a chemical chaperone prevents disease phenotypes in a mouse model of primary open angle glaucoma. J. Clin. Invest. 2011;121:3542–3553. doi: 10.1172/JCI58183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aroca-Aguilar JD, et al. Myocilin mutations causing glaucoma inhibit the intracellular endoproteolytic cleavage of myocilin between amino acids Arg226 and Ile227. J Biol. Chem. 2005;280:21043–21051. doi: 10.1074/jbc.M501340200. [DOI] [PubMed] [Google Scholar]
- 59.Yam GH, et al. Sodium 4-phenylbutyrate acts as a chemical chaperone on misfolded myocilin to rescue cells from endoplasmic reticulum stress and apoptosis. Invest. Ophthalmol. Vis. Sci. 2007;48:1683–1690. doi: 10.1167/iovs.06-0943. [DOI] [PubMed] [Google Scholar]
- 60.Joe MK, Tomarev SI. Expression of myocilin mutants sensitizes cells to oxidative stress-induced apoptosis: implication for glaucoma pathogenesis. The American journal of pathology. 2010;176:2880–2890. doi: 10.2353/ajpath.2010.090853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cheng JW, et al. Myocilin polymorphisms and primary open-angle glaucoma: a systematic review and meta-analysis. PLoS One. 2012;7:e46632. doi: 10.1371/journal.pone.0046632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cheng JW, et al. Meta-analysis of association between optineurin gene and primary open-angle glaucoma. Med. Sci. Monit. 2010;16:CR369–377. [PubMed] [Google Scholar]
- 63.Hewitt AW, et al. A Glaucoma Case-control Study of the WDR36 Gene D658G sequence variant. Am. J. Ophthalmol. 2006;142:324–325. doi: 10.1016/j.ajo.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 64.Hauser MA, et al. Distribution of WDR36 DNA sequence variants in patients with primary open-angle glaucoma. Invest. Ophthalmol. Vis. Sci. 2006;47:2542–2546. doi: 10.1167/iovs.05-1476. [DOI] [PubMed] [Google Scholar]
- 65.Lopez-Garrido MP, et al. Functional analysis of CYP1B1 mutations and association of heterozygous hypomorphic alleles with primary open-angle glaucoma. Clin. Genet. 2010;77:70–78. doi: 10.1111/j.1399-0004.2009.01284.x. [DOI] [PubMed] [Google Scholar]
- 66.Lopez-Garrido MP, et al. Heterozygous CYP1B1 gene mutations in Spanish patients with primary open-angle glaucoma. Mol. Vis. 2006;12:748–755. [PubMed] [Google Scholar]
- 67.Melki R, et al. CYP1B1 mutations in French patients with early-onset primary open-angle glaucoma. J. Med. Genet. 2004;41:647–651. doi: 10.1136/jmg.2004.020024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kozlov G, et al. A structural overview of the PDI family of proteins. FEBS J. 2010;277:3924–3936. doi: 10.1111/j.1742-4658.2010.07793.x. [DOI] [PubMed] [Google Scholar]
- 69.Bartke T, et al. Dual role of BRUCE as an antiapoptotic IAP and a chimeric E2/E3 ubiquitin ligase. Mol. Cell. 2004;14:801–811. doi: 10.1016/j.molcel.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 70.Yokoe H, Anholt RRH. Molecular cloning of olfactomedin, an extracellular matrix protein specific to olfactory neuroepithelium. Proc. Natl. Acad. Sci. U.S.A. 1993;90:4655–4659. doi: 10.1073/pnas.90.10.4655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nakaya N, et al. Zebrafish olfactomedin 1 regulates retinal axon elongation in vivo and is a modulator of Wnt signaling pathway. J. Neurosci. 2008;28:7900–7910. doi: 10.1523/JNEUROSCI.0617-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee JA, et al. Olfactomedin-2 mediates development of the anterior central nervous system and head structures in zebrafish. Mech. Dev. 2008;125:167–181. doi: 10.1016/j.mod.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mukhopadhyay A, et al. Bioinformatic approaches for identification and characterization of olfactomedin related genes with a potential role in pathogenesis of ocular disorders. Mol. Vis. 2004;10:304–314. [PubMed] [Google Scholar]
- 74.Zhang J, et al. Identification and characterization of a novel member of olfactomedin-related protein family, hGC-1, expressed during myeloid lineage development. Gene. 2002;283:83–93. doi: 10.1016/s0378-1119(01)00763-6. [DOI] [PubMed] [Google Scholar]