

Abstract
Homeostatic plasticity refers to mechanisms that the cell or network engage in order to homeostatically maintain a preset level of activity. These mechanisms include compensatory changes in cellular excitability, excitatory and inhibitory synaptic strength and are typically studied at a developmental stage when GABA or glycine are inhibitory. Here we focus on the expression of homeostatic plasticity in the chick embryo spinal cord at a stage when GABA is excitatory. When spinal activity is perturbed in the living embryo there are compensatory changes in postsynaptic AMPA receptors and in the driving force for GABAergic currents. These changes are triggered by reduced GABAA receptor signaling, which appears to be part of the sensing machinery for triggering homeostatic plasticity. We compare and contrast these findings to homeostatic plasticity expressed in spinal systems at different stages of development, and to the developing retina at a stage when GABA is depolarizing.
Keywords: Neuropharmacology - Special Issue - Homeostatic Synaptic Plasticity
Introduction
To understand how appropriately behaving neural circuits develop, we must understand the rules that underlie the maturation of network excitability. This maturation is first accomplished in a very dynamic period of embryonic development when there are several challenges to excitability, such as: connections being added and removed, cells becoming larger, channel conductances changing, and GABAergic postsynaptic currents converting from excitatory to inhibitory. Errors in this complicated process could have profound effects on circuit excitability. Recent work has identified an important process that is thought to ensure that networks achieve and maintain appropriate spiking activity levels. Homeostatic plasticity is thought to maintain network activity levels within a physiologically appropriate range by coordinately adjusting intrinsic cellular excitability, excitatory and inhibitory synaptic strength.
Almost 20 years ago, Eve Marder and Gina Turrigiano carried out experiments that launched the field of homeostatic plasticity (Turrigiano et al., 1994; Turrigiano et al., 1995). These experiments demonstrated that a rhythmically bursting cell could homeostatically recover it’s bursting activity after being stripped of all it’s synaptic inputs and grown in culture for several days. In this case the cell adjusted its repertoire of voltage-gated conductances to recover its rhythmic activity. The findings suggested the possibility that cells had a preset level and pattern of spiking activity, had mechanisms for detecting they were not experiencing such activity, and surprisingly could execute a program to recover this level and pattern of activity. Gina Turrigiano then extended these experiments by demonstrating that spiking activity levels in cultured synaptically connected networks of visual cortical neurons were homeostatically maintained (Turrigiano et al., 1998). Following perturbations to spiking activity levels in these cultured networks, compensatory changes were observed in intrinsic cellular excitability, and in excitatory and inhibitory synaptic strength ((Turrigiano et al., 1998; Desai et al., 1999; Kilman et al., 2002). For instance, following 2-day activity blockade intrinsic cellular excitability and excitatory synaptic strength increased, while inhibitory synaptic strength decreased. One form of homeostatic synaptic plasticity observed in these and other studies has received considerable attention, and has been called synaptic scaling since the entire distribution of miniature postsynaptic current (mPSC) amplitudes change by a particular scaling factor. Since these early experiments synaptic scaling has now been identified in many different systems, in culture and to a lesser extent in vivo as well (Turrigiano, 2012).
Experiments describing synaptic scaling are typically carried out on developing neurons. Cultures are derived from neurons harvested from the later embryo or first postnatal week and allowed to grow for 1 – 2 weeks, at which point synaptic networks have formed. In vivo work is typically done in the first few weeks of postnatal life. What has become clear is that homeostatic plasticity can be expressed differently depending on the stage of development (Burrone et al., 2002; Wierenga et al., 2006). Virtually all of these studies are carried out at a stage when GABAergic currents are inhibitory, and thus are weakened following activity blockade (Kilman et al., 2002). On the other hand less work has focussed on early stages of development, when GABA is still depolarizing and excitatory. In order to determine if synaptic scaling happens at this earlier developmental stage, we have carried out several studies, taking advantage of the accessibility of the in vivo embryonic chick spinal network. This review will focus on homeostatic synaptic plasticity in the embryonic spinal network, when GABA is excitatory, and compare these findings to other systems at different stages of development.
Spontaneous network activity in chick embryo spinal circuitry
Spontaneous network activity (SNA) is experienced in many different developing systems, at stages where GABA is depolarizing and excitatory (Blankenship and Feller, 2010). SNA is expressed in the spinal cord, retina, hippocampus, brain stem, superior colliculus, and others, shortly after synaptic connections first form, (Gummer and Mark, 1994; Fortin et al., 1995; Itaya et al., 1995; Lippe, 1995; Feller, 1999; Ho and Waite, 1999; O’Donovan, 1999; Wong, 1999; Ben-Ari, 2001). SNA exists as episodic bursts of spiking activity that last for seconds and are followed by longer lasting quiescent periods, as shown in Figure 1 (muscle nerve recordings in chick embryo spinal preparation). These episodes are a consequence of the highly excitable nature of a recurrently connected circuit, in which both glutamatergic and GABAergic neurotransmission is excitatory in early development, thus virtually all cells act to excite their synaptic partners (Ben-Ari et al., 1989; O’Donovan et al., 1998b; O’Donovan, 1999; Rivera et al., 1999; Blankenship and Feller, 2010). The chloride-mediated GABAA current is depolarizing because chloride is pumped into the cell and is maintained at a high intracellular concentration such that the reversal potential for GABA is significantly more depolarized than the resting membrane potential (Ben-Ari et al., 2007). During an episode of SNA a significant portion of neurons are recruited and experience increases in cytoplasmic calcium, important in several aspects of development (Katz and Shatz, 1996; Zhang and Poo, 2001; Spitzer, 2002). In the developing spinal cord embryonic limb movements are generated by spontaneous network activity (Bekoff et al., 1975; Hamburger, 1977; O’Donovan et al., 1998a), which are known to be important in motoneuron axonal pathfinding (Hanson and Landmesser, 2004) and for proper muscle and joint development (Ruano-Gil et al., 1978; Toutant et al., 1979; Roufa and Martonosi, 1981; Persson, 1983; Hall and Herring, 1990; Jarvis et al., 1996).
Figure 1.

Schematic of the isolated spinal cord and extracellular suction electrode recording showing 2 episodes of SNA. These recordings represent population motoneuron recordings.
Homeostatic plasticity in developing spinal neurons has been recognized for years. Synaptic scaling of excitatory mPSCs has been shown in dissociated spinal neurons in culture (O’Brien et al., 1998). The homeostatic nature of the embryonic spinal cord preparation at stages when GABA is excitatory and SNA is expressed has long been recognized. When either glutamatergic or GABAergic transmission was blocked in the isolated in vitro spinal cord preparation, SNA was only temporarily prevented, and recovered within an hour, now generated by the remaining transmitter system (Chub and O’Donovan, 1998). Below we focus on homeostatic mechanisms that appear to maintain the levels of SNA in the developing spinal cord.
Blocking activity in vivo triggers synaptic scaling in embryonic spinal motoneurons
The chick embryo allows one to monitor SNA by assessing the duration of embryonic movements observed through a window in the shell of the egg. In order to test whether blocking SNA in ovo would trigger homeostatic synaptic scaling we infused the voltage-gated Na+-channel blocker lidocaine into the egg from embryonic day 8 to 10 (E8-10) (Gonzalez-Islas and Wenner, 2006). We confirmed that this was sufficient to dramatically reduce embryonic movements in this 2-day period. Following activity blockade the spinal cord was isolated at E10 and ventral root recordings were monitored to assess SNA frequency in lidocaine-free saline solution. These activity-blocked cords were more excitable in that the frequency of episodes of SNA was twice that of controls (Figure 2A).
Figure 2.
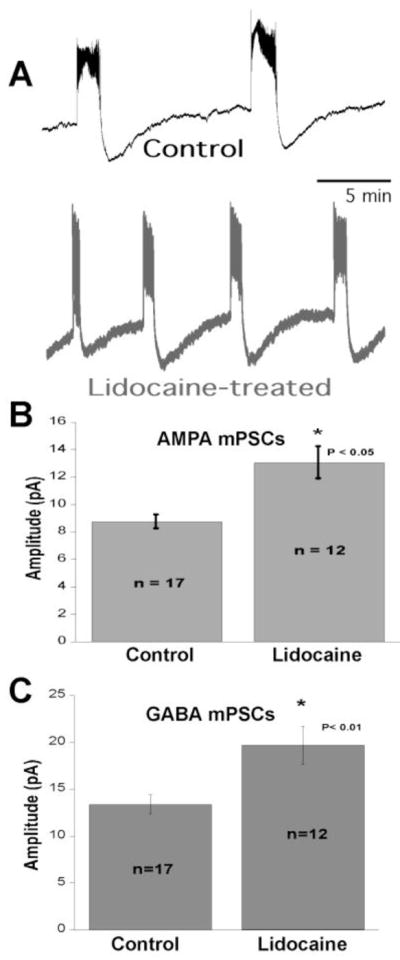
The effects of activity-blockade. A) Representative ventral root recordings showing frequency of spontaneous network activity from control and lidocaine-treated embryos. B–C) Average amplitude of AMPA mPSCs (B) and GABA mPSCs (C) from activity-reduced and control motoneurons.
To determine if synaptic strengthening contributed to this increased excitability, AMPAergic and GABAergic mPSCs in identified spinal MNs were recorded. Compensatory increases in both AMPAergic and GABAergic mPSC amplitude were observed following activity-blockade (Figure 2B–C). The increase in GABAergic synaptic strength was compensatory because of the depolarizing nature of GABA at this early embryonic stage. The frequency of SNA in activity-blocked isolated spinal preparations was reduced toward control levels by acute weakening of GABAergic or AMPAergic synaptic currents by bath application of sub-maximal concentrations of antagonists (Gonzalez-Islas and Wenner, 2006). This suggested that the synaptic strengthening contributed to the hyperexcitable nature of the lidocaine-treated cord. Further, the increases in mPSC amplitude expressed the scaling profiling in that the entire distribution of mPSCs increased by a scaling factor (1.3 to 1.4, (Gonzalez-Islas and Wenner, 2006; Garcia-Bereguiain et al., 2013)).
More recently, we have carried out experiments where SNA was increased in ovo rather than reduced (Gonzalez-Islas et al., 2012). This was accomplished by injecting an endocannabinoid receptor (CB1) antagonist into the egg at E8. This appears to increase embryonic kicking by increasing the frequency of mEPSCs which trigger spiking activity in spinal neurons and increase the likelihood of initiating an episode of SNA. In these activity-increased embryos GABAergic and glutamatergic mPSCs were reduced in a compensatory manner.
By regulating synaptic strength during embryonic development, the expression of SNA likely acts to play an important role in the maturation of AMPA and GABA synaptic strength in a coordinated manner, and be critical for setting the excitability of the early spinal network. Because SNA is observed in seemingly all developing networks it is reasonable to postulate that synaptic scaling may be regulated by SNA in other developing circuits. These findings raised 3 important questions: 1) How did the network sense the activity perturbations that triggered the scaling responses? 2) What was the mechanism(s) that mediated an increase in glutamatergic mPSC amplitude? 3) What was the mechanism(s) that mediated an increase in GABAergic mPSC amplitude?
GABAergic transmission is a critical component of the triggering of synaptic scaling in embryonic motoneurons
Understanding how cells or networks sense altered activity levels is one of the major current interests in the field, and certain parameters have been suggested, including spike rate and depolarization (Liu et al., 1998; Marder and Prinz, 2002; Davis, 2006; Rich and Wenner, 2007; Turrigiano, 2007). Many believe that reductions in neuronal spiking, and/or levels of depolarization lead to reductions in calcium entry, which then trigger synaptic scaling. However, several studies show that hyperpolarization, or reducing the spike rate of the postsynaptic cell did not trigger the expected compensatory changes in mPSC amplitude (Paradis et al., 2001; Burrone et al., 2002; Hartman et al., 2006; Pratt and Aizenman, 2007). In these studies, however, neurotransmission was not blocked in the postsynaptic neuron, so that if reduced neurotransmission triggers scaling then no change would be expected in these activity-reduced neurons. In fact, recent work suggests the possibility that reduced neurotransmission can trigger a local version of synaptic scaling (Stellwagen and Malenka, 2006; Sutton et al., 2006; Hou et al., 2008; Jakawich et al., 2010; Beique et al., 2011; Lee, 2012; Turrigiano, 2012; Wang et al., 2012).
We tested the possibility that reduced neurotransmission triggers synaptic scaling in embryonic spinal networks by blocking either GABAA or glutamatergic receptor (GABAAR or AMPAR) activation from E8 to E10. When effective concentrations of GABAergic or glutamatergic (AMPA and/or NMDA) antagonists were injected at E8, embryonic movements (SNA) were blocked for the first 1–2 hours (Figure 3A)(Wilhelm and Wenner, 2008). This is because each antagonist removes a significant contribution of the excitatory drive for SNA. However, the embryonic movements homeostatically recovered to control levels 10 – 12 hours after the injection of either the glutamatergic or GABAA antagonists (Figure 3A), even though the antagonists remained effective throughout the E8-10 period (Wilhelm and Wenner, 2008). Treating embryos with glutamatergic antagonists did not increase GABAergic or glutamatergic mPSC amplitude suggesting that neither reduced glutamatergic receptor activation nor the transient reduction in SNA triggered scaling. On the other hand, following in ovo treatment of GABAA antagonists (gabazine or bicuculline) AMPAergic and GABAergic mPSC amplitude scaled to even greater values (70–100%; Figure 3B) than following activity-block (~40%); mPSC frequency was no different than control. The findings suggested the possibility that GABAA receptors are part of the sensing machinery that triggers scaling. Therefore, it is possible that when spiking activity was blocked with lidocaine infusions, GABA release and GABAA transmission were reduced, and this reduction in receptor activation triggered synaptic scaling. GABA may be capable of triggering synaptic scaling because it is depolarizing at this stage and could lead to opening of voltage-gated calcium channels, thereby triggering calcium dependent pathways. Because GABA is depolarizing at these stages, and is developmentally advanced compared to AMPA receptors, it is recognized as a developmental trophic factor (Owens and Kriegstein, 2002; Kandler and Gillespie, 2005; Akerman and Cline, 2007; Ben-Ari et al., 2007) (Chen et al., 1995; Ben-Ari et al., 2004). This suggested the possibility that GABAA receptors may be part of the machinery that senses GABA levels as a proxy for spiking activity, and when GABA levels are perturbed, synaptic scaling is triggered in a direction to bring spiking activity back to some control set point.
Figure 3.
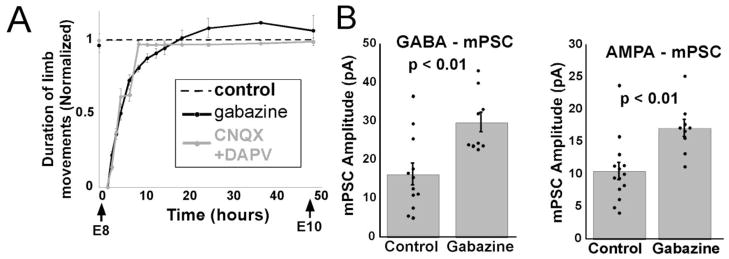
Blocking GABAA transmission triggers synaptic scaling. A) Embryonic movements recover to control levels (dotted line) 12 hours after injecting gabazine or APV/CNQX. B) GABAergic (left) and AMPAergic (right) mPSC amplitude increase following 2 days of GABAergic blockade. Dots indicate individual cells. Error, SE. Modified from Wilhelm and Wenner, 2008.
Cellular excitability is responsible for the initial recovery of SNA
Because we were able to follow the homeostatic recovery of the embryonic movements following the injection of antagonists we were able to recognize that the activity recovered to control levels by 12 hours (Figure 3A). This was somewhat surprising because quantal amplitude typically takes ~24 hours to clearly develop in other systems (Turrigiano et al., 1998; Stellwagen and Malenka, 2006). Therefore, we tested whether synaptic scaling was expressed by the time embryonic movements had first recovered (Wilhelm and Wenner, 2008). We found that AMPAergic and GABAergic mPSC amplitude had not increased in motoneurons treated with the GABAA antagonist for 12 hours (Figure 4A–B)
Figure 4.

Cellular excitability, but not mPSC amplitude, is increased by 12 hours of GABAergic block (gabazine). (A) The average amplitude of GABA (A) or AMPA (B) mPSCs is no different than control (dotted line) following 12 hours of gabazine. Gabazine treatment for 48 hours significantly increased the amplitude of mPSCs compared to control. Error bars, SE. Modified from Wilhelm and Wenner 2008. (C) Threshold current is significantly reduced after 12 and 48 hours of gabazine treatment. (D) responses to depolarizing pulse in control and gabazine-treated motoneurons. C–D modified from Wilhelm et al., 2009
Thus, embryonic movements had homeostatically recovered before any changes in quantal amplitude were observed, suggesting something else had mediated the recovery of embryonic SNA.
Homeostatic changes in intrinsic cellular excitability following reductions in spiking activity have been observed for many years and likely play a significant role in recovering or maintaining network activity levels following perturbations (Turrigiano et al., 1994; Thoby-Brisson and Simmers, 1998; Desai et al., 1999; Marder and Prinz, 2002; Karmarkar and Buonomano, 2006; Marder and Goaillard, 2006; Khorkova and Golowasch, 2007) including the chick embryo (Martin-Caraballo and Dryer, 2002; Casavant et al., 2004). Several different channels appear to contribute to the compensatory changes in cellular excitability including transient and calcium-activated potassium channels (IA and IK(Ca), respectively), the hyperpolarization activated cationic conductance (IH), and voltage gated sodium channels (INa). The findings have led to the suggestion that when activity is reduced, cells attempt to recover their activity levels by changing their constellation of channel conductances through homeostatic plasticity mechanisms. We therefore tested whether there were increases in cellular excitability in the first 12 hours that could contribute to the recovery of embryonic kicking (Wilhelm et al., 2009). We found that 12 hours of GABAergic blockade dramatically increased intrinsic cellular excitability in embryonic motoneurons. 12 hours after injecting gabazine, the threshold current was dramatically reduced and current injections produced higher firing frequencies compared to controls (Figure 4C–D). We found that these increases in excitability were mediated by reductions in IA, IK(Ca), and increases in INa. By 48 hours of GABAergic blockade cellular excitability was still increased compared to controls, but not to the extent observed at 12 hours. Together, the results suggested that reduced GABAergic transmission triggers changes in intrinsic cellular excitability that contribute to the recovery of SNA, and only after activity is recovered do we see the expression of synaptic scaling. It may be that while changes in different conductances can react quickly, more permanent changes in quantal amplitude take longer, but are more sustained.
While we are beginning to better understand the homeostatic responses that recover activity levels following GABAAR blockade, it is less clear how the activity recovers after glutamatergic blockade. Following in ovo injection of glutamatergic antagonists (CNQX & APV), kicking activity is blocked but then recovers to normal levels within 10 hours even though there are no compensatory changes in GABAergic or AMPAergic quantal amplitude (Wilhelm and Wenner, 2008). Additionally, glutamatergic blockade did not trigger any observed changes in cellular excitability (threshold current or voltage, passive membrane properties (Wilhelm et al., 2009)). So how does the spinal network activity recover following in ovo glutamatergic blockade? It is possible that there were homeostatic changes in the probability of GABAergic release, or alteration in expression or function of channels that were not assessed in our previous work, although we have no evidence to support this. Another possibility is suggested by 2 previous studies where SNA was temporarily blocked in the isolated in vitro chick cord by bath perfusion of glutamatergic antagonists (Chub and O’Donovan, 1998; Tabak et al., 2001). SNA recovered within 1–2 hours and eventually stabilized to a new slightly slower frequency of SNA. The recovery was at least partly mediated by a rapid strengthening of evoked GABAergic currents that occur in the absence of episodes of SNA (Tabak et al., 2001). This GABAergic strengthening was thought to occur because intracellular chloride accumulated to greater levels when glutamatergic antagonists initially blocked episodes of SNA, which normally reduce Cl−in. The chloride accumulation enhanced the driving force for these currents and allowed GABAergic currents to quickly compensate for the loss of the glutamatergic synaptic drive. Presumably, in embryos treated with glutamatergic antagonists in vivo, changes to intracellular chloride did not persist once the cords were isolated, drugs washed off, and SNA resumed, as no changes were seen in GABAergic mPSC amplitude (Wilhelm and Wenner, 2008).
AMPA scaling mechanism
Synaptic scaling of AMPAergic mPSCs has been shown to be mediated by changes in postsynaptic AMPA receptors in many different systems (O’Brien et al., 1998; Lee, 2012). However, it is less clear which receptor subunits are involved (Man, 2011; Lee, 2012; Shepherd, 2012; Turrigiano, 2012). Several studies in which network activity was blocked in cultured neurons suggest AMPAergic scaling was mediated by increases in GluA2-containing calcium impermeable AMPA receptors (CI-AMPARs), while other studies demonstrate the involvement of GluA2-lacking calcium permeable AMPA receptors (CP-AMPARs). AMPAergic scaling via either CI-AMPARs or CP-AMPARs in cortical pyramidal cells have also been identified following in vivo reductions of visual input (Goel et al., 2006; Gainey et al., 2009; Goel et al., 2011). Embryonic spinal motoneurons treated with lidocaine for 2 days showed increased mEPSC conductance, consistent with the idea that changes in postsynaptic receptors mediated AMPAergic scaling of mPSCs (Garcia-Bereguiain et al., 2013). Using multiple techniques we were able to establish that control mEPSCs were mediated by CI-AMPARs, while mEPSCs in activity-blocked motoneurons were mediated by CP-AMPARs. In fact, it appeared that AMPAergic scaling in chick embryo motoneurons occurred through the replacement of CI-AMPARs by the higher conductance CP-AMPARs. Similar results were obtained when scaling was triggered by GABAergic transmission blockade, suggesting that reductions in spiking activity or GABAergic transmission were triggering similar forms of scaling. While the mechanism of AMPAergic scaling was the same following the 2 treatments, the degree of CI-AMPAR replacement by CP-AMPARs was greater for embryos treated with the GABAergic antagonist. The findings of this study support a model for embryonic spinal networks at stages when GABA is depolarizing, where AMPAergic scaling is mediated by a proportional replacement of CI-AMPARs by CP-AMPARs at all glutamatergic synapses (Garcia-Bereguiain et al., 2013).
GABAergic scaling mechanisms
When network activity was blocked in cultured neural networks for days the amplitude of inhibitory GABAergic mPSCs were scaled downward (amplitude reduced)(Kilman et al., 2002; Wenner, 2011). The mechanisms that underlie GABAergic scaling following activity blockade of cultured networks included reductions in postsynaptic GABAA receptor number and the presynaptic vesicle concentration of GABA (Kilman et al., 2002; Wilson et al., 2005; Hartman et al., 2006; Swanwick et al., 2006; Peng et al., 2010; Wenner, 2011). Mechanisms underlying GABAergic synaptic scaling following activity perturbations in vivo are less well understood. If GABAergic scaling in embryonic spinal motoneurons was mediated by changes in postsynaptic receptors or neurotransmitter vesicle filling, then we would expect to see increases in mPSC conductance in activity-blocked embryos, as we saw for AMPAergic scaling. GABAergic mPSC conductance was no different in activity-blocked and control motoneurons, but rather the driving force for these currents was increased in treated embryos (Gonzalez-Islas et al., 2010). Control E10 spinal motoneurons maintain ~50mM intracellular Cl− (Cl−- was raised to significantly higher in), but following activity blockade Cl in levels (~100mM) as demonstrated with whole cell and perforated patch recordings. Similar increases in Cl−in were obtained for gabazine-treated embryos (unpublished observations). While this is quite distinct from what has been described in cultured systems, such changes have been observed in mature spinal neurons after injury (peripheral nerve injury and spinal cord injury)(Coull et al., 2003; De Koninck, 2007; Boulenguez et al., 2010). This raises the possibility that these injuries alter spiking activity or transmission in the network, which may trigger homeostatic mechanisms that lead to inappropriate levels of excitability commonly associated with these injuries.
Together these findings support the following model for synaptic scaling in the chick embryo spinal network (Figure 5): Reduced spiking activity leads to less GABA release and thus reduced GABAAR activation - supported by the observations that AMPAergic and GABAergic scaling mechanisms are the same for activity-blocked and GABAA-blocked embryos. Reductions in GABAA transmission, possibly through voltage-gated calcium channels, trigger AMPAergic and GABAergic upward scaling through CP-AMPAR insertion and chloride accumulation, respectively. Future studies in other systems will be necessary to establish if these findings translate to other developing circuits in the period when GABA is excitatory.
Figure 5.

Model summarizing our current thinking of homeostatic synaptic scaling in embryonic motoneurons following 2-day reductions in spiking activity. Schematic illustrates the concept that reduced spiking activity leads to reduced GABA release, and thereby reduced GABAA receptor activation. 2 days of reduced GABAA receptor activation lead to synaptic scaling through currently unknown pathways. AMPAergic scaling is mediated by the insertion of higher conductance calcium permeable AMPA receptors at the expense of calcium impermeable receptors. GABAergic scaling is mediated by the accumulation of chloride, thereby increasing driving force for GABAA-mediated currents.
Comparisons of homeostatic plasticity in spinal and other networks at different stages
Synaptic scaling has not been described in terms of a homeostatic recovery of spontaneous network activity in other networks, at stages when GABA is excitatory. However, evidence supporting a homeostatic recovery of a form of spontaneous network activity has been described in the developing retina at a stage when GABA is depolarizing (Hennig et al., 2011). Interestingly, later in development when GABA is no longer depolarizing, the homeostatic recovery does not occur. Like the embryonic spinal cord, homeostasis of network dynamics is triggered following a chronic inhibition of the GABAA receptor. Network features such as wave size (% of recruited neurons into a wave of SNA) are initially altered following application of a GABAA antagonist, but within hours homeostatically recover to levels observed before the application of the antagonist. Just days later, this homeostatic response is not observed. It is currently unknown what mechanisms underlie this form of network homeostasis, but synaptic scaling could certainly contribute, and the authors do propose the possibility that alterations in chloride accumulation could be involved (Hennig et al., 2011).
Other studies carried out in the spinal cord have provided insights to the developmental progression of homeostatic plasticity in the cord. At very early stages of Xenopus laevis spinal development, before these networks are synaptically connected, perturbations of spiking activity trigger homeostatic adjustments of the relative number of excitatory versus inhibitory neurons within the spinal network (Borodinsky et al., 2004). There is no evidence that there are changes in neurotransmitter phenotypes once the synaptic circuitry is better established as in the chick embryo spinal cord.
Two separate studies carried out in Zebrafish larvae at similar stages, when chloride-mediated conductances were hyperpolarizing, demonstrate homeostatic plasticity, but arrive at different conclusions. Synaptic scaling can be triggered in vivo in zebrafish larvae when chloride-mediated conductances are hyperpolarizing. In the first study, spinal activity homeostatically recovered within hours after glutamatergic currents were blocked in vivo (Knogler et al., 2010). The authors show that mEPSCs scaled upward 48 hours after glutamatergic blockade or when spiking activity was blocked. However, scaling was not observed in the first 24 hours after glutamatergic blockade, well after swimming had recovered (6 hours). Further, the authors convincingly showed that the scaling of glutamatergic currents were very unlikely to have influenced the swimming behavior. These results suggested scaling did not play a role in the recovery or maintenance of swimming. In addition, no compensatory changes in mIPSCs or cellular excitability were observed. In contrast to the above study, an apparent homeostatic control of swimming activity through compensatory changes in synaptic strength has been described in the zebrafish at comparable stages (Mongeon et al., 2008). A mutant zebrafish lacking a functional glycine transporter experiences swimming abnormalities early in development but appears to recover normal swimming within days. In this study the recovery of the normal swimming behavior appeared to be accomplished through compensatory reductions in glycinergic synaptic strength mediated by a reduction in glycine receptors.
There are both similarities and differences in the expression of homeostatic plasticity in the embryonic spinal cord when GABA is depolarizing and zebrafish spinal studies where glycine is inhibitory. In each of the studies, synaptic perturbations in the living system led to altered spinal behavior that was then homeostatically recovered within hours, and synaptic changes were observed. The 2 zebrafish studies demonstrate distinct results in that one study suggests scaling occurs but does not contribute to the homeostasis of the swimming behavior, while the other study suggests compensatory changes in synaptic current do contribute to behavioral homeostasis. Interestingly, the results of the embryonic chick spinal cord support both of these seemingly distinct findings. In support of synaptic compensations contributing to behavioral homeostasis we found that after chronic in ovo activity blockade, the isolated spinal preparation (in absence of lidocaine) had increased SNA frequency, and that sub-maximal concentrations of glutamatergic or GABAergic antagonists acutely reduced SNA frequency back toward control levels (Gonzalez-Islas and Wenner, 2006). On the other hand, following 2 day in ovo GABAergic blockade, scaling was not observed until after the behavior had been recovered. Further, following chronic treatment with GABAA antagonists, GABAergic and AMPAergic mPSC amplitude was increased twice as much as after activity blockade, yet isolated preparations expressed control levels of SNA frequency (Wilhelm and Wenner, 2008). Therefore, while synaptic strengthening can contribute to behavioral recovery, it is too simplistic to assume that synaptic strengthening will always impact rhythmic behaviors in a straight-forward way. Clear differences were also observed in the chick and zebrafish studies. In the chick, compensatory changes in GABAergic and AMPAergic scaling and cellular excitability were triggered by reduced GABAAR activation. In the zebrafish, blockade of glutamatergic transmission triggered AMPAergic scaling only. It is possible that scaling is triggered by reduced GABAAR activation when GABA is depolarizing and could trigger calcium signaling cascades; later in development when GABA is no longer depolarizing, scaling could then be triggered by reduced AMPAR activation. These differences in homeostatic plasticity between embryonic and more mature networks may be a consequence of the developmental restriction of the expression of this plasticity, as described in other systems (Wierenga et al., 2006; Echegoyen et al., 2007; Hennig et al., 2011; Lee et al., 2013).
At even later stages of spinal development, the expression of homeostatic plasticity is different still. Synaptically connected organotypic rodent spinal slices exhibit homeostatic alterations in mPSC frequency following exposure to blockers of glutamatergic transmission, GABAergic/glycinergic transmission, or spiking activity in the second week in culture (Galante et al., 2000; Galante et al., 2001). In contrast to the chick embryo spinal cord (Gonzalez-Islas and Wenner, 2006; Wilhelm and Wenner, 2008) and dissociated spinal cultures (O’Brien et al., 1998) no changes were observed in mPSC amplitude (scaling) in these organotypic studies. This difference could be due to the duration of blocker application, different stages, or the specific sets of spinal neurons recorded.
Together, the results suggest that synaptic scaling, by itself, is not sufficient to explain the homeostatic recovery of network behavior. For instance it is clear that cellular excitability plays an important role as well. In order to understand the complete homeostatic process future studies will need to recognize the importance of the constellation of homeostatic mechanisms that are recruited, and the time course of each mechanism. Further, it is likely that different mechanisms and time courses will be distinct at different stages of development.
Highlights.
We discuss homeostatic plasticity in developing networks at a stage when GABA is excitatory.
We discuss the mechanisms of homeostatic plasticity in embryonic spinal networks.
We discuss the sensor for triggering homeostatic plasticity in embryonic spinal networks.
We compare and contrast homeostatic plasticity in spinal and other networks at different developmental stages.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- Akerman CJ, Cline HT. Refining the roles of GABAergic signaling during neural circuit formation. Trends Neurosci. 2007;30:382–389. doi: 10.1016/j.tins.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Beique JC, Na Y, Kuhl D, Worley PF, Huganir RL. Arc-dependent synapse-specific homeostatic plasticity. Proc Natl Acad Sci U S A. 2011;108:816–821. doi: 10.1073/pnas.1017914108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekoff A, Stein PS, Hamburger V. Coordinated motor output in the hindlimb of the 7-day chick embryo. Proc Natl Acad Sci U S A. 1975;72:1245–1248. doi: 10.1073/pnas.72.4.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y. Developing networks play a similar melody. Trends Neurosci. 2001;24:353–360. doi: 10.1016/s0166-2236(00)01813-0. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Cherubini E, Corradetti R, Gaiarsa JL. Giant synaptic potentials in immature rat CA3 hippocampal neurones. J Physiol (Lond) 1989;416:303–325. doi: 10.1113/jphysiol.1989.sp017762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Khalilov I, Represa A, Gozlan H. Interneurons set the tune of developing networks. Trends Neurosci. 2004;27:422–427. doi: 10.1016/j.tins.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Gaiarsa JL, Tyzio R, Khazipov R. GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev. 2007;87:1215–1284. doi: 10.1152/physrev.00017.2006. [DOI] [PubMed] [Google Scholar]
- Blankenship AG, Feller MB. Mechanisms underlying spontaneous patterned activity in developing neural circuits. Nat Rev Neurosci. 2010;11:18–29. doi: 10.1038/nrn2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borodinsky LN, Root CM, Cronin JA, Sann SB, Gu X, Spitzer NC. Activity-dependent homeostatic specification of transmitter expression in embryonic neurons. Nature. 2004;429:523–530. doi: 10.1038/nature02518. [DOI] [PubMed] [Google Scholar]
- Boulenguez P, Liabeuf S, Bos R, Bras H, Jean-Xavier C, Brocard C, Stil A, Darbon P, Cattaert D, Delpire E, Marsala M, Vinay L. Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nat Med. 2010;16:302–307. doi: 10.1038/nm.2107. [DOI] [PubMed] [Google Scholar]
- Burrone J, O’Byrne M, Murthy VN. Multiple forms of synaptic plasticity triggered by selective suppression of activity in individual neurons. Nature. 2002;420:414–418. doi: 10.1038/nature01242. [DOI] [PubMed] [Google Scholar]
- Casavant RH, Colbert CM, Dryer SE. A-current expression is regulated by activity but not by target tissues in developing lumbar motoneurons of the chick embryo. J Neurophysiol. 2004;92:2644–2651. doi: 10.1152/jn.00307.2004. [DOI] [PubMed] [Google Scholar]
- Chen G, Trombley PQ, van den Pol AN. GABA receptors precede glutamate receptors in hypothalamic development; differential regulation by astrocytes. J Neurophysiol. 1995;74:1473–1484. doi: 10.1152/jn.1995.74.4.1473. [DOI] [PubMed] [Google Scholar]
- Chub N, O’Donovan MJ. Blockade and recovery of spontaneous rhythmic activity after application of neurotransmitter antagonists to spinal networks of the chick embryo. J Neurosci. 1998;18:294–306. doi: 10.1523/JNEUROSCI.18-01-00294.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coull JA, Boudreau D, Bachand K, Prescott SA, Nault F, Sik A, De Koninck P, De Koninck Y. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature. 2003;424:938–942. doi: 10.1038/nature01868. [DOI] [PubMed] [Google Scholar]
- Davis GW. Homeostatic control of neural activity: from phenomenology to molecular design. Annu Rev Neurosci. 2006;29:307–323. doi: 10.1146/annurev.neuro.28.061604.135751. [DOI] [PubMed] [Google Scholar]
- De Koninck Y. Altered chloride homeostasis in neurological disorders: a new target. Curr Opin Pharmacol. 2007;7:93–99. doi: 10.1016/j.coph.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Desai NS, Rutherford LC, Turrigiano GG. Plasticity in the intrinsic excitability of cortical pyramidal neurons. Nat Neurosci. 1999;2:515–520. doi: 10.1038/9165. [DOI] [PubMed] [Google Scholar]
- Echegoyen J, Neu A, Graber KD, Soltesz I. Homeostatic plasticity studied using in vivo hippocampal activity-blockade: synaptic scaling, intrinsic plasticity and age-dependence. PLoS One. 2007;2:e700. doi: 10.1371/journal.pone.0000700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller MB. Spontaneous correlated activity in developing neural circuits. Neuron. 1999;22:653–656. doi: 10.1016/s0896-6273(00)80724-2. [DOI] [PubMed] [Google Scholar]
- Fortin G, Kato F, Lumsden A, Champagnat J. Rhythm generation in the segmented hindbrain of chick embryos. J Physiol (Lond) 1995;486:735–744. doi: 10.1113/jphysiol.1995.sp020849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galante M, Nistri A, Ballerini L. Opposite changes in synaptic activity of organotypic rat spinal cord cultures after chronic block of AMPA/kainate or glycine and GABAA receptors. J Physiol. 2000;523(Pt 3):639–651. doi: 10.1111/j.1469-7793.2000.t01-1-00639.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galante M, Avossa D, Rosato-Siri M, Ballerini L. Homeostatic plasticity induced by chronic block of AMPA/kainate receptors modulates the generation of rhythmic bursting in rat spinal cord organotypic cultures. Eur J Neurosci. 2001;14:903–917. doi: 10.1046/j.0953-816x.2001.01710.x. [DOI] [PubMed] [Google Scholar]
- Garcia-Bereguiain MA, Gonzalez-Islas C, Lindsly C, Butler E, Hill AW, Wenner P. In Vivo Synaptic Scaling Is Mediated by GluA2-Lacking AMPA Receptors in the Embryonic Spinal Cord. J Neurosci. 2013;33:6791–6799. doi: 10.1523/JNEUROSCI.4025-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Islas C, Wenner P. Spontaneous Network Activity in the Embryonic Spinal Cord Regulates AMPAergic and GABAergic Synaptic Strength. Neuron. 2006;49:563–575. doi: 10.1016/j.neuron.2006.01.017. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Islas C, Chub N, Garcia-Bereguiain MA, Wenner P. GABAergic synaptic scaling in embryonic motoneurons is mediated by a shift in the chloride reversal potential. J Neurosci. 2010;30:13016–13020. doi: 10.1523/JNEUROSCI.1659-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Islas CA, Garcia-Bereguiain MA, Wenner P. Tonic and transient endocannabinoid regulation of AMPAergic mPSCs and homeostatic plasticity in embryonic motor networks. Journal of Neuroscience. 2012 doi: 10.1523/JNEUROSCI.1229-12.2012. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gummer AW, Mark RF. Patterned neural activity in brain stem auditory areas of a prehearing mammal, the tammar wallaby (Macropus eugenii) Neuroreport. 1994;5:685–688. doi: 10.1097/00001756-199402000-00006. [DOI] [PubMed] [Google Scholar]
- Hall BK, Herring SW. Paralysis and growth of the musculoskeletal system in the embryonic chick. J Morphol. 1990;206:45–56. doi: 10.1002/jmor.1052060105. [DOI] [PubMed] [Google Scholar]
- Hamburger V. The developmental history of the motor neuron. Neurosci Res Program Bull. 1977;15(Suppl):iii-37. doi: 10.1007/978-1-4899-6743-5_5. [DOI] [PubMed] [Google Scholar]
- Hanson MG, Landmesser LT. Normal patterns of spontaneous activity are required for correct motor axon guidance and the expression of specific guidance molecules. Neuron. 2004;43:687–701. doi: 10.1016/j.neuron.2004.08.018. [DOI] [PubMed] [Google Scholar]
- Hartman KN, Pal SK, Burrone J, Murthy VN. Activity-dependent regulation of inhibitory synaptic transmission in hippocampal neurons. Nat Neurosci. 2006;9:642–649. doi: 10.1038/nn1677. [DOI] [PubMed] [Google Scholar]
- Hennig MH, Grady J, van Coppenhagen J, Sernagor E. Age-dependent homeostatic plasticity of GABAergic signaling in developing retinal networks. J Neurosci. 2011;31:12159–12164. doi: 10.1523/JNEUROSCI.3112-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho SM, Waite PM. Spontaneous activity in the perinatal trigeminal nucleus of the rat. Neuroreport. 1999;10:659–664. doi: 10.1097/00001756-199902250-00039. [DOI] [PubMed] [Google Scholar]
- Hou Q, Zhang D, Jarzylo L, Huganir RL, Man HY. Homeostatic regulation of AMPA receptor expression at single hippocampal synapses. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:775–780. doi: 10.1073/pnas.0706447105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itaya SK, Fortin S, Molotchnikoff S. Evolution of spontaneous activity in the developing rat superior colliculus. Can J Physiol Pharmacol. 1995;73:1372–1377. doi: 10.1139/y95-192. [DOI] [PubMed] [Google Scholar]
- Jakawich SK, Nasser HB, Strong MJ, McCartney AJ, Perez AS, Rakesh N, Carruthers CJ, Sutton MA. Local presynaptic activity gates homeostatic changes in presynaptic function driven by dendritic BDNF synthesis. Neuron. 2010;68:1143–1158. doi: 10.1016/j.neuron.2010.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis JC, Sutherland H, Mayne CN, Gilroy SJ, Salmons S. Induction of a fast-oxidative phenotype by chronic muscle stimulation: mechanical and biochemical studies. Am J Physiol. 1996;270:C306–312. doi: 10.1152/ajpcell.1996.270.1.C306. [DOI] [PubMed] [Google Scholar]
- Kandler K, Gillespie DC. Developmental refinement of inhibitory sound-localization circuits. Trends Neurosci. 2005;28:290–296. doi: 10.1016/j.tins.2005.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmarkar UR, Buonomano DV. Different forms of homeostatic plasticity are engaged with distinct temporal profiles. Eur J Neurosci. 2006;23:1575–1584. doi: 10.1111/j.1460-9568.2006.04692.x. [DOI] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- Khorkova O, Golowasch J. Neuromodulators, not activity, control coordinated expression of ionic currents. J Neurosci. 2007;27:8709–8718. doi: 10.1523/JNEUROSCI.1274-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilman V, van Rossum MC, Turrigiano GG. Activity deprivation reduces miniature IPSC amplitude by decreasing the number of postsynaptic GABA(A) receptors clustered at neocortical synapses. J Neurosci. 2002;22:1328–1337. doi: 10.1523/JNEUROSCI.22-04-01328.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knogler LD, Liao M, Drapeau P. Synaptic scaling and the development of a motor network. J Neurosci. 2010;30:8871–8881. doi: 10.1523/JNEUROSCI.0880-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK. Ca-permeable AMPA receptors in homeostatic synaptic plasticity. Front Mol Neurosci. 2012;5:17. doi: 10.3389/fnmol.2012.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KJ, Queenan BN, Rozeboom AM, Bellmore R, Lim ST, Vicini S, Pak DT. Mossy fiber-CA3 synapses mediate homeostatic plasticity in mature hippocampal neurons. Neuron. 2013;77:99–114. doi: 10.1016/j.neuron.2012.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippe WR. Relationship between frequency of spontaneous bursting and tonotopic position in the developing avian auditory system. Brain Res. 1995;703:205–213. doi: 10.1016/0006-8993(95)01096-3. [DOI] [PubMed] [Google Scholar]
- Liu Z, Golowasch J, Marder E, Abbott LF. A model neuron with activity-dependent conductances regulated by multiple calcium sensors. J Neurosci. 1998;18:2309–2320. doi: 10.1523/JNEUROSCI.18-07-02309.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man HY. GluA2-lacking, calcium-permeable AMPA receptors--inducers of plasticity? Curr Opin Neurobiol. 2011;21:291–298. doi: 10.1016/j.conb.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marder E, Prinz AA. Modeling stability in neuron and network function: the role of activity in homeostasis. Bioessays. 2002;24:1145–1154. doi: 10.1002/bies.10185. [DOI] [PubMed] [Google Scholar]
- Marder E, Goaillard JM. Variability, compensation and homeostasis in neuron and network function. Nat Rev Neurosci. 2006;7:563–574. doi: 10.1038/nrn1949. [DOI] [PubMed] [Google Scholar]
- Martin-Caraballo M, Dryer SE. Activity- and target-dependent regulation of large-conductance Ca2+-activated K+ channels in developing chick lumbar motoneurons. J Neurosci. 2002;22:73–81. doi: 10.1523/JNEUROSCI.22-01-00073.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mongeon R, Gleason MR, Masino MA, Fetcho JR, Mandel G, Brehm P, Dallman JE. Synaptic homeostasis in a zebrafish glial glycine transporter mutant. J Neurophysiol. 2008;100:1716–1723. doi: 10.1152/jn.90596.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien RJ, Kamboj S, Ehlers MD, Rosen KR, Fischbach GD, Huganir RL. Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron. 1998;21:1067–1078. doi: 10.1016/s0896-6273(00)80624-8. [DOI] [PubMed] [Google Scholar]
- O’Donovan MJ. The origin of spontaneous activity in developing networks of the vertebrate nervous system. Curr Opin Neurobiol. 1999;9:94–104. doi: 10.1016/s0959-4388(99)80012-9. [DOI] [PubMed] [Google Scholar]
- O’Donovan MJ, Chub N, Wenner P. Mechanisms of spontaneous activity in developing spinal networks. J Neurobiol. 1998a;37:131–145. doi: 10.1002/(sici)1097-4695(199810)37:1<131::aid-neu10>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- O’Donovan MJ, Wenner P, Chub N, Tabak J, Rinzel J. Mechanisms of spontaneous activity in the developing spinal cord and their relevance to locomotion. Ann N Y Acad Sci. 1998b;860:130–141. doi: 10.1111/j.1749-6632.1998.tb09044.x. [DOI] [PubMed] [Google Scholar]
- Owens DF, Kriegstein AR. Is there more to GABA than synaptic inhibition? Nat Rev Neurosci. 2002;3:715–727. doi: 10.1038/nrn919. [DOI] [PubMed] [Google Scholar]
- Paradis S, Sweeney ST, Davis GW. Homeostatic control of presynaptic release is triggered by postsynaptic membrane depolarization. Neuron. 2001;30:737–749. doi: 10.1016/s0896-6273(01)00326-9. [DOI] [PubMed] [Google Scholar]
- Peng YR, Zeng SY, Song HL, Li MY, Yamada MK, Yu X. Postsynaptic spiking homeostatically induces cell-autonomous regulation of inhibitory inputs via retrograde signaling. J Neurosci. 2010;30:16220–16231. doi: 10.1523/JNEUROSCI.3085-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson M. The role of movements in the development of sutural and diarthrodial joints tested by long-term paralysis of chick embryos. J Anat. 1983;137:591–599. [PMC free article] [PubMed] [Google Scholar]
- Pratt KG, Aizenman CD. Homeostatic regulation of intrinsic excitability and synaptic transmission in a developing visual circuit. J Neurosci. 2007;27:8268–8277. doi: 10.1523/JNEUROSCI.1738-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich MM, Wenner P. Sensing and expressing homeostatic synaptic plasticity. Trends Neurosci. 2007;30:119–125. doi: 10.1016/j.tins.2007.01.004. [DOI] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- Roufa D, Martonosi AN. Effect of curare on the development of chicken embryo skeletal muscle in ovo. Biochem Pharmacol. 1981;30:1501–1505. doi: 10.1016/0006-2952(81)90373-7. [DOI] [PubMed] [Google Scholar]
- Ruano-Gil D, Nardi-Vilardaga J, Tejedo-Mateu A. Influence of extrinsic factors on the development of the articular system. Acta Anat (Basel) 1978;101:36–44. doi: 10.1159/000144947. [DOI] [PubMed] [Google Scholar]
- Shepherd JD. Memory, plasticity and sleep - A role for calcium permeable AMPA receptors? Front Mol Neurosci. 2012;5:49. doi: 10.3389/fnmol.2012.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer NC. Activity-dependent neuronal differentiation prior to synapse formation: the functions of calcium transients. J Physiol Paris. 2002;96:73–80. doi: 10.1016/s0928-4257(01)00082-1. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Ito HT, Cressy P, Kempf C, Woo JC, Schuman EM. Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell. 2006;125:785–799. doi: 10.1016/j.cell.2006.03.040. [DOI] [PubMed] [Google Scholar]
- Swanwick CC, Murthy NR, Kapur J. Activity-dependent scaling of GABAergic synapse strength is regulated by brain-derived neurotrophic factor. Mol Cell Neurosci. 2006 doi: 10.1016/j.mcn.2005.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabak J, Rinzel J, O’Donovan MJ. The role of activity-dependent network depression in the expression and self-regulation of spontaneous activity in the developing spinal cord. J Neurosci. 2001;21:8966–8978. doi: 10.1523/JNEUROSCI.21-22-08966.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoby-Brisson M, Simmers J. Neuromodulatory inputs maintain expression of a lobster motor pattern-generating network in a modulation-dependent state: evidence from long-term decentralization in vitro. J Neurosci. 1998;18:2212–2225. doi: 10.1523/JNEUROSCI.18-06-02212.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toutant JP, Toutant MN, Renaud D, Le Douarin GH. Enzymatic differentiation of muscle fibre types in embryonic latissimus dorsii of the chick: effects of spinal cord stimulation. Cell Differ. 1979;8:375–382. doi: 10.1016/0045-6039(79)90022-8. [DOI] [PubMed] [Google Scholar]
- Turrigiano G. Homeostatic signaling: the positive side of negative feedback. Curr Opin Neurobiol. 2007;17:318–324. doi: 10.1016/j.conb.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Turrigiano G. Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb Perspect Biol. 2012;4 doi: 10.1101/cshperspect.a005736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G, Abbott LF, Marder E. Activity-dependent changes in the intrinsic properties of cultured neurons. Science. 1994;264:974–977. doi: 10.1126/science.8178157. [DOI] [PubMed] [Google Scholar]
- Turrigiano G, LeMasson G, Marder E. Selective regulation of current densities underlies spontaneous changes in the activity of cultured neurons. J Neurosci. 1995;15:3640–3652. doi: 10.1523/JNEUROSCI.15-05-03640.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- Wang G, Gilbert J, Man HY. AMPA receptor trafficking in homeostatic synaptic plasticity: functional molecules and signaling cascades. Neural Plast. 2012;2012:825364. doi: 10.1155/2012/825364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenner P. Mechanisms of GABAergic Homeostatic Plasticity. Neural Plasticity. 2011 doi: 10.1155/2011/489470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierenga CJ, Walsh MF, Turrigiano GG. Temporal regulation of the expression locus of homeostatic plasticity. J Neurophysiol. 2006;96:2127–2133. doi: 10.1152/jn.00107.2006. [DOI] [PubMed] [Google Scholar]
- Wilhelm JC, Wenner P. GABAA transmission is a critical step in the process of triggering homeostatic increases in quantal amplitude. Proc Natl Acad Sci U S A. 2008;105:11412–11417. doi: 10.1073/pnas.0806037105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm JC, Rich MM, Wenner P. Compensatory changes in cellular excitability, not synaptic scaling, contribute to homeostatic recovery of embryonic network activity. Proc Natl Acad Sci U S A. 2009;106:6760–6765. doi: 10.1073/pnas.0813058106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson NR, Kang J, Hueske EV, Leung T, Varoqui H, Murnick JG, Erickson JD, Liu G. Presynaptic regulation of quantal size by the vesicular glutamate transporter VGLUT1. J Neurosci. 2005;25:6221–6234. doi: 10.1523/JNEUROSCI.3003-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RO. Retinal waves and visual system development. Annu Rev Neurosci. 1999;22:29–47. doi: 10.1146/annurev.neuro.22.1.29. [DOI] [PubMed] [Google Scholar]
- Zhang LI, Poo MM. Electrical activity and development of neural circuits. Nat Neurosci. 2001;4(Suppl):1207–1214. doi: 10.1038/nn753. [DOI] [PubMed] [Google Scholar]