

Abstract
We have carried out a mechanistic investigation of aerobic dehydrogenation of cyclohexanones and cyclohexenones to phenols with a Pd(TFA)2/2-dimethylaminopyridine (2-Me2Npy) catalyst system. Numerous experimental methods, including kinetic studies, filtration tests, Hg poisoning experiments, transmission electron microscopy (TEM), and dynamic light scattering (DLS) provide compelling evidence that the initial PdII catalyst mediates the first dehydrogenation of cyclohexanone to cyclohexenone, after which it evolves into soluble Pd nanoparticles that retain catalytic activity. This nanoparticle formation and stabilization is facilitated by each of the components in the catalytic reaction, including the ligand, TsOH, DMSO, substrate, and cyclohexenone intermediate.
Introduction
Phenols and phenol derivatives are common structural motifs in pharmaceuticals, bulk chemicals, and polymers.1 These molecules exhibit diverse substitution patterns, often containing multiple functional groups. Traditional synthetic methods to prepare substituted arenes include nucleophilic and electrophilic aromatic substitution and metal-catalyzed cross-coupling and C–H functionalization reactions. Catalytic dehydrogenation of saturated C–C bonds in carbocyclic structures represents a compelling alternative strategy for the preparation of substituted arenes.2–4 Until recently, most precedents for such reactions have been limited to reactions of simple unsubstituted precursors, such as cyclohexene and cyclohexanone,5 to afford benzene and phenol, respectively. In 2011, we reported two different catalyst systems for the dehydrogenation of cyclohexanones: (1) Pd(DMSO)2(TFA)2 (TFA = trifluoroacetate), which promotes a single dehydrogenation step to afford cyclohexenone products6 (Scheme 1A), and (2) Pd(TFA)2/2-dimethylaminopyridine (2-Me2Npy), which promotes a double dehydrogenation process to afford phenol products3a (Scheme 1B). In both cases, the reactions proceed under relatively mild conditions (60–80 °C) and exhibit broad substrate scope. These results raised a number of questions concerning the origin of the product selectivity with the two different catalyst systems.
Scheme 1.
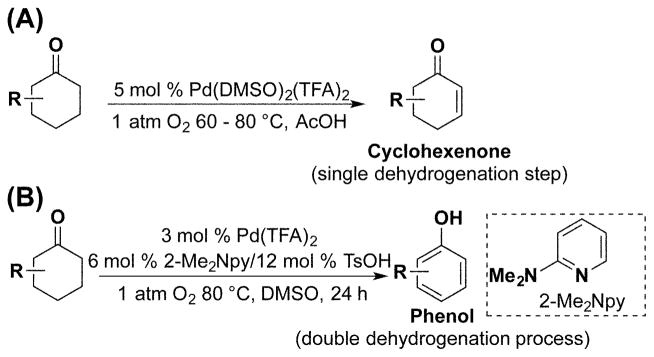
Pd-Catalyzed Methods for Chemoselective Dehydrogenation of Cyclohexanones.
We recently investigated the mechanism of the Pd(DMSO)2(TFA)2-catalyzed oxidative dehydrogenation of cyclohexanone,7 and the results support a catalytic cycle in which cyclohexanone undergoes reversible coordination to PdII, followed by turnover-limiting cleavage of the α-C–H bond (Scheme 2). The resulting PdII-enolate intermediate undergoes rapid β–H elimination to afford the cyclohexenone product and a PdII-hydride. Oxidation of a PdII-hydride is expected to proceed via reductive elimination of TFAH followed by aerobic oxidation of Pd0 to regenerate the PdII catalyst.8 The high chemoselectivity for cyclohexenone product over phenol was traced to the role of DMSO as a ligand for PdII. DMSO has minimal influence on the rate of the PdII-catalyzed dehydrogenation of cyclohexanone and stabilizes the homogeneous catalyst to prevent decomposition into Pd black. In contrast, DMSO strongly inhibits the conversion of cyclohexenone to phenol, thereby providing the basis for highly chemoselective formation of the enone product.
Scheme 2.

Proposed Mechanism for Pd(DMSO)2(TFA)2-Catalyzed Dehydrogenation of Cyclohexanone.
In our initial report describing Pd(TFA)2/2-Me2Npy-catalyzed dehydrogenation of cyclohexanone to phenol, cyclohexenone was observed as an intermediate in the reaction (Figure 1).3a,4a The reaction time course was fit to a sequential A → B → C kinetic model; however, an initial burst of cyclohexanone-to-cyclohexenone conversion was evident at early reaction time points, followed by more uniform steady-state kinetics. Here, we present a more thorough kinetic and mechanistic analysis of Pd(TFA)2/2-Me2Npy-catalyzed dehydrogenation of cyclohexanones and cyclohexenones, with an emphasis on establishing the identity of the active catalytic species. The results suggest that an initial, highly active molecular PdII species evolves into soluble Pd nanoparticles that serve as the active catalyst during steady-state dehydrogenation of the substrate. Factors that contribute to nanoparticle formation and the implications of these results for catalytic dehydrogenation are discussed.
Figure 1.

Kinetic time course of the dehydrogenation of cyclohexanone (
 ) to phenol (■), with cyclohexenone (
) to phenol (■), with cyclohexenone (
 ) observed as the intermediate. Reaction conditions: [cyclohexanone] = 1.0 M (0.5 mmol), [Pd(TFA)2] = 0.05 M (0.025 mmol), [2-Me2Npy] = 0.1 M (0.05 mmol), [TsOH] = 0.2 M (0.1 mmol), DMSO for total volume of 0.5 mL, 1 atm O2, 80 °C. Figure adapted from reference 3a.
) observed as the intermediate. Reaction conditions: [cyclohexanone] = 1.0 M (0.5 mmol), [Pd(TFA)2] = 0.05 M (0.025 mmol), [2-Me2Npy] = 0.1 M (0.05 mmol), [TsOH] = 0.2 M (0.1 mmol), DMSO for total volume of 0.5 mL, 1 atm O2, 80 °C. Figure adapted from reference 3a.
Results and Discussion
Qualitative Observations of Additive Effects on Dehydrogenation Reactions
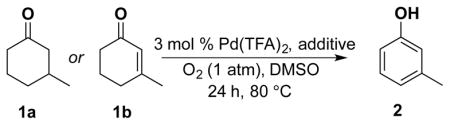
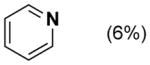
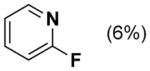
Dehydrogenation of cyclohexanone and cyclohexenone derivatives 1a and 1b with the Pd(TFA)2/2-Me2Npy catalyst is most effective when TsOH is included as a cocatalyst (Table 1, entry 5). In our original report, these observations were rationalized by comparison to results with other ligands, such as pyridine and 2-fluoropyridine. Use of pyridine as a ligand affords only 16 % yield in the conversion of 1a into 2 (entry 2), while the yield with 2-fluoropyridine considerably improved (44 % yield of 2, entry 3). We speculated that TsOH could protonate the dimethylamino group of 2-Me2Npy,9–11 thereby enhancing the electron deficiency of the pyridine ligand and improving the catalytic reactivity relative to 2-Me2Npy alone (compare entries 4 and 5).
Table 1.
Ligand and Additive Effects on Pd-Catalyzed Dehydrogenation of 3-Methylcyclohexanone, 1a, and 3-Methylcyclohexenone, 1b.
| ||||
|---|---|---|---|---|
| Entry | Additive | Substrate: 1a | 1b | |
| Yield of 2 (%)a | ||||
| 1 | none | 28 | 52 | |
| 2 |
|
16 | 39 | |
| 3 |
|
44 | 59 | |
| 4 |
|
/TsOH (0%) | 24 | 53 |
| 5 | /TsOH (12%) | 79 | 74 | |
| 6 |
|
/TsOH (0%) | 45 | 98 |
| 7 | /TsOH (6%) | 48 | 95 | |
| 8 | /TsOH (12%) | 50 | 81 | |
| 9 |
|
/TsOH (0%) | 45 | 78 |
| 10 | /TsOH (6%) | 50 | 90 | |
| 11 | /TsOH (12%) | 57 | 86 | |
| 12 | Pd/C instead of Pd(TFA)2 | 1 | < 2 | |
| 13 | Pd/Al2O3 instead of Pd(TFA)2 | 0 | < 1 | |
Entries 1–5, 12, and 13 obtained from reference 3a.
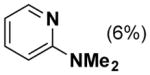
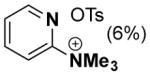
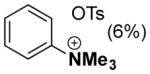
In order to test this hypothesis further, we prepared the 2-trimethylammonium-substituted pyridine, [2-Me3Npy][OTs], and tested its utility as a ligand in the absence of TsOH. Use of Pd(TFA)2/[2-Me3Npy][OTs] as a catalyst affords a lower yield of phenol (45 %) with cyclohexanone 1a as the substrate but an improved yield (98 %) with cyclohexenone 1b as the substrate (Table 1, entry 6). Addition of catalytic quantities of TsOH to these reactions has only marginal effect on the outcome of these reactions (entries 7 and 8). As control experiments, we performed dehydrogenation reactions with the trimethylphenylammonium salt [Me3NPh][OTs] in place of the pyridine ligand. Phenol yields from these reactions (entries 9–11) are quite similar to those obtained with the ammonium-substituted pyridine ligand, [2-Me3Npy]+. Overall, these results fail to provide a definitive conclusion about the origin of the observed ligand effects; however, they suggest that the catalyst may not consist of a simple pyridine-ligated PdII complex.
The beneficial effect of non-pyridine-containing ammonium salts is reminiscent of literature reports that ammonium salts promote formation and stabilization of Pd nanoparticles.12 Moreover, several qualitative observations are consistent with in situ conversion of the molecular PdII precursor into nanoparticulate or heterogeneous Pd species during the reactions. For example, opaque dark-red solutions are observed during the course of the reactions, and formation of Pd black and/or Pd mirror are evident in these reactions.
Dehydrogenation of Cyclohexanone to Cyclohexenone: Examination of Early Time Points
The qualitative observations outlined above prompted us to probe the reaction time courses in more detail. These studies required the use of freshly prepared stock solutions of the substrate and catalyst in order to obtain reliable kinetic data. Even then, quantitative results could only be compared among data acquired from a series of experiments performed in parallel (cf. Figure S1).
The kinetic order in [Pd] was evaluated to probe the nature of the catalyst. Pd(TFA)2 concentrations of 0, 1, 2.5, 5, 7.5, and 10 mM were used and the initial rate of the reaction was monitored through 10% conversion of the substrate. The time-course data show clear evidence of an initial kinetic burst followed by a slower steady-state reaction (Figure 2A; for additional data, see Figure S2). Linear fits of the burst and steady-state reaction periods intersect at a cyclohexenone concentration approximately equal to the concentration of Pd(TFA)2 used in the reaction (Figure 2A and 2B), implying that the burst corresponds to a single turnover of the PdII catalyst. The rate (d[cyclohexenone]/dt) during the burst period exhibits a first-order dependence on [Pd] (Figure 3A). During the post-burst period, the rate is proportional to [Pd]2 (Figure 3B). Further analysis of these data is complicated by consumption of cyclohexenone in the second dehydrogenation step to the phenol product. Nevertheless, these initial-rate data all suggest a change in the active catalytic species after the first turnover.
Figure 2.

(A) Initial kinetic time courses for the formation of cyclohexenone from cyclohexanone with extrapolation towards the inflection point between the burst and post burst periods for 2.5 (
) and 5.0 mol % (
) Pd(TFA)2. All kinetic time course data is in Figure S2. (B) Plot of burst intercept with respect to [Pd] clearly demonstrates an initial burst during the first catalytic turnover. Reaction conditions: [cyclohexanone] = 100 mM (0.05 mmol), [2-Me2Npy] = 0.01 M (0.005 mmol), [TsOH] = 0.02 M (0.01 mmol), DMSO for VTotal of 0.5 mL, 1 atm O2, 80 °C. [Pd(TFA)2] = 0, 1.0, 2.5, 5.0, 7.5, 10.0 mM
Figure 3.

(A) Plot of d[Cyclohexenone]/dt during the initial burst with respect to [Pd] shows a first order dependence, while (B) plot of d[Cyclohexenone]/dt during the post burst shows a second order dependence on [Pd] (The curve is a second order curve fit of rate = kobs[Pd]2). Reaction conditions: [cyclohexanone] = 100 mM (0.05 mmol), [2-Me2Npy] = 0.01 M (0.005 mmol), [TsOH] = 0.02 M (0.01 mmol), DMSO for VTotal of 0.5 mL, 1 atm O2, 80 °C. [Pd(TFA)2] = 0, 1.0, 2.5, 5.0, 7.5, 10.0 mM
Dehydrogenation of Cyclohexenone to Phenol: Examination of Induction Period
Dehydrogenation of cyclohexenone to phenol was investigated independently. In contrast to the kinetic burst observed in the ketone-to-enone dehydrogenation step, dehydrogenation of cyclohexenone exhibits an induction period (Figure 4A). The length of the induction periods varies for different concentrations of cyclohexenone (25 – 400 mM), with shorter induction periods at higher [cyclohexenone] (Figure 4B and S3).
Figure 4.

(A) Selected initial kinetic time courses for the formation of phenol from cyclohexenone at varying [cyclohexenone]. All kinetic time course data is in Figure S3. (B) Plot of induction time with [cyclohexenone]. Reaction conditions: [Pd(TFA)2] = 0.005 M (0.0025 mmol), [2-Me2Npy] = 0.01 M (0.005 mmol), [TsOH] = 0.02 M (0.01 mmol), DMSO for VTotal of 0.5 mL, 1 atm O2, 80 °C. [cyclohexenone] = 25, 50, 100 (
), 200 (
), 300 (■), 400 mM
The induction period could arise from an autocatalytic phenomenon, in which one of the reaction products (e.g., phenol or water) accelerates the reaction. The possibility was tested by performing reactions with 12 mol % phenol or 10 mol % H2O13 in the initial reaction mixture. No rate increase was observed; the rates were identical to the experiments lacking inclusion of the products (Figure S4). Another rationale for the induction period, also consistent with the kinetic burst in Figure 2, is a cyclohexenone-induced transformation of the initial molecular PdII catalyst into a nanoparticulate or heterogeneous Pd catalyst. This proposal implies that the new catalyst exhibits greater reactivity for the cyclohexenone-to-phenol dehydrogenation step relative to the PdII precatalyst.
Distinguishing Between Homogeneous, Heterogeneous, or Soluble Nanoparticulate Active Catalyst
The Pd(TFA)2/2-Me2Npy dehydrogenation catalyst system is rare in its ability to achieve complete transformation of cyclohexanones to phenols. This reactivity bridges the gap between the homogeneous and heterogeneous catalytic dehydrogenation reactions. Pd(DMSO)2(TFA)2 mediates selective dehydrogenation of cyclohexanones to cyclohexenones,6 and mechanistic studies supported the involvement of a homogeneous catalyst.7 Heterogeneous Pd catalysts, such as Pd/C, promote dehydrogenation of cyclohexenones to phenols under forcing conditions (e.g., 200 °C),2a–d,3i but such catalysts are not active under the mild conditions used for the present reactions (cf. Table 1).
The mechanistic data presented above suggest that the Pd(TFA)2/2-Me2Npy catalyst may transform from a molecular species into a nanoparticle catalyst during the reaction. Support for Pd nanoparticles includes kinetic bursts and induction periods, difficulties in obtaining reliable kinetic data, and observation of opaque dark red solutions and the formation of Pd black/mirror. Additionally, ammonium salts,12 strong acids,14 solvents of high dielectric constants14a (e.g. εDMSO = 46.715), and Lewis bases, such as sulfoxides,16,17 are known to promote the formation and/or stabilization of noble-metal nanoparticles. In particular, Hiemstra and coworkers have highlighted the special ability of DMSO as a solvent to support Pd nanoparticles formed via in situ reduction of Pd(OAc)2 in Wacker-type aerobic oxidative cyclization of allylic N-hydroxymethyl carbamates.17,18 In the following series of additional tests, we provide further evidence that the initial molecular PdII(TFA)2/2-Me2Npy catalyst system evolves in situ into a soluble nanoparticle catalyst.14,19,20
A) Filtration Tests
Palladium black and/or mirror are often observed from the Pd(TFA)2/2-Me2Npy-catalyzed dehydrogenation reactions. Filtration tests were carried out to assess whether these heterogeneous Pd products catalyze the cyclohexanone-to-cyclohexenone or the cyclohexenone-to-phenol dehydrogenation reactions.
In the first series of filtration tests, dehydrogenations of cyclohexanone and cyclohexenone were carried out independently under standard reaction conditions for 24 h. The resulting reaction mixtures were cooled to facilitate precipitation of the heterogeneous material, and the homogeneous, dark-red supernatants were removed from the respective reaction mixtures. Both supernatants and precipitates were used again for dehydrogenation of the respective substrates, following addition of another equivalent of cyclohexanone or cyclohexenone. The supernatant retains good catalytic activity, albeit with some variability in product yields (Tables S1 and S2), while the heterogeneous material shows negligible activity (≤ 5% yield of dehydrogenation products).
A second series of tests employed hot filtration of the reaction mixture according to the protocol of Maitlis.21 Independent dehydrogenation reactions of cyclohexanone and cyclohexenone were carried out to low (≤ 20%) conversion, and the hot reaction mixtures were filtered through a layer of Celite 535 to remove particles greater than 3.3 nm22 or through a 200 nm PTFE filter. The filtrates were monitored for further conversion under the standard reaction conditions. Solvent and another equivalent of substrate were added to the Celite filtrand, and this mixture was resubjected to the reaction conditions. As shown in Figure 5 (see also Tables S3 and S4 and Figure S5), sustained catalytic activity was observed from the filtrate, often with somewhat lower yields of phenol after 24 h, while the filtrands showed no catalytic activity. Control experiments confirmed that the presence of Celite in the catalytic reactions (i.e., without filtration) does not inhibit product formation (Tables S3 and S4).
Figure 5.


Catalytic activity after filtration through Celite 535 using the following starting substrate: (A) cyclohexanone at t = 0.25 h and (B) cyclohexenone at t = 0.5 h. Reaction conditions: [substrate] = 1 M (0.5 mmol), [Pd(TFA)2] = 0.05 M (0.025 mmol), [2-Me2Npy] = 0.1 M (0.05 mmol), [TsOH] = 0.2 M (0.1 mmol), DMSO for VTotal of 0.5 mL, 1 atm O2, 80 °C. Similar results with 200 nm PTFE filters and the control reaction in the presence of Celite without filtration (See Tables S2 and S3 for details.).
Both filtration studies suggest that catalytic activity does not arise from the heterogeneous Pd material that forms in the reactions. The reduced activity of the filtrate can be attributed either to catalyst deactivation or loss of catalytically active material while performing the filtrations. The sustained catalytic activity observed with the filtrates, however, suggests that any non-molecular Pd catalysts formed under the reaction conditions retains solubility, a property consistent with low-nuclearity nanoparticles.
B) Hg and Poly(4-vinyl-pyridine) (PVPy) Poisoning Tests
Mercury forms amalgams with other metals, and it is commonly used to test for the role of heterogeneous catalysis.14,23 This assay can be problematic with homogeneous Pd-catalyzed reactions because Pd0 intermediates can react with Hg, thereby poisoning the homogeneous catalyst.14b,19b We employed a mercury test in the aerobic dehydrogenation of cyclohexanone to cyclohexenone with Pd(DMSO)2(TFA)2,7 a catalyst system that has been proposed to proceed via a homogeneous PdII/Pd0 cycle. For example, it exhibits clean, reproducible kinetics with no induction periods or kinetic bursts and no color changes during the reaction. Activity is retained for several hours with this catalyst following addition of Hg to the reaction mixture (Figure S6).
The Hg-poisoning test was similarly employed in the Pd(TFA)2/2-Me2Npy-catalyzed dehydrogenation of cyclohexanone. When 100 equiv of Hg was added at the start of the reaction, cyclohexenone formed rapidly in a kinetic burst, after which no additional formation of cyclohexenone nor conversion of cyclohexenone to phenol was observed (Figure 6A, red dashed line; for complete time course data, see Figure S7). Consistent with these observations, addition of 100 equiv of Hg to an ongoing reaction mixture (t = 4 h) led to immediate inhibition of the reaction (Figure 6A; blue solid line).
Figure 6.

Reaction inhibition of phenol formation with the addition of 100 equiv of Hg at t = 0 (
) and 4 h (
) using the following starting substrate: (A) cyclohexanone and (B) cyclohexenone. Lack of Hg (■) addition data is included. Dashed lines indicate cyclohexenone and solid lines indicate phenol formation. Additional time course data is in Figure S7. Reaction conditions: [substrate] = 1 M (0.5 mmol), [Pd(TFA)2] = 0.05 M (0.025 mmol), [2-Me2Npy] = 0.1 M (0.05 mmol), [TsOH] = 0.2 M (0.1 mmol), Hg = 500 mg (2.49 mmol), DMSO for VTotal of 0.5 mL, 1 atm O2, 80 °C.
Similar tests were performed for the dehydrogenation of cyclohexenone to phenol. Up to three catalytic turnovers were observed from a reaction in which Hg was added at the start of the reaction (Figure 6B; red line); however, addition of Hg to an ongoing reaction mixture (t = 4 h) led to immediate inhibition (Figure 6B; blue line).
Poly(4-vinylpyridine), PVPy, has been used as a selective poison for homogeneous Pd catalysts, without inhibiting the activity of heterogeneous Pd catalysts.14,20b This assay can have difficulty distinguishing between molecular Pd catalysts and soluble Pd nanoparticles because both can be poisoned by the polymer. Application of this assay to the dehydrogenation of cyclohexanone reveals that the catalyst is deactivated immediately upon addition of PVPy to the reaction mixture at t = 0 and 2 h (Figure 7A; for complete time course data, see Figure S8). Similar observations were made in the dehydrogenation of cyclohexenone to phenol (Figure 7B).
Figure 7.


Reaction inhibition of phenol formation with the addition of 25 equiv of poly(4-vinylpyridine) at t = 0 (
) and 2 h (
) using the following starting substrate: (A) cyclohexanone and (B) cyclohexenone. Lack of PVPy (■) addition data is included. Dashed lines indicate cyclohexenone and solid lines indicate phenol formation. Additional time course data is in Figure S8. Reaction conditions: [substrate] = 1 M (0.5 mmol), [Pd(TFA)2] = 0.05 M (0.025 mmol), [2-Me2Npy] = 0.1 M (0.05 mmol), [TsOH] = 0.2 M (0.1 mmol), PVPy = 65 mg, DMSO for VTotal of 0.5 mL, 1 atm O2, 80 °C.
The results of the Hg poisoning experiments can be understood by recognizing that PdII exhibits dehydrogenation activity and is characterized by a kinetic burst in the conversion of cyclohexanone to cyclohexenone. Product formation observed when Hg is added to the initial reaction mixture can be attributed the PdII catalyst precursor. Once the molecular catalyst transforms into soluble nanoparticles, it succumbs to Hg poisoning. The PVPy results are consistent with the ability of PVPy to poison molecular or soluble-nanoparticle catalysts. Taken together, the Hg and PVPy poisoning experiments suggest that the active catalyst consists of soluble nanoparticles under steady-state turnover conditions.
Finke has described the use of phenanthroline as a quantitative poison to estimate the number of active sites with soluble nanoparticle catalysts.24 Quantitative poisoning studies of this type were attempted with the present reactions, but they proved to be unreliable because phenanthroline perturbs the Pd speciation under the reaction conditions (see Supporting Information for further details: Figures S9 – S11 and associated discussion). Other more commonly used quantitative poisons, such as CS2 and PPh3,25 are incompatible with the reaction temperature and oxidizing reaction conditions, respectively.
C) Transmission Electron Microscopy (TEM) and Dynamic Light Scattering (DLS)
Scanning and transmission electron microscopy (SEM, TEM) are commonly used to characterize Pd nanoparticle catalysts. 12,17b,26 These methods are typically performed ex situ and, therefore, are not ideally suited for characterization of catalysts that undergo changes during the reaction and/or are susceptible to changes in the process of sample preparation. Nevertheless, TEM images were obtained from samples of a cyclohexanone dehydrogenation reaction mixture. The images obtained from a reaction at t = 15 min revealed nanoparticles of approx 30 nm diameter (Figure 8). Larger particles, with broad size distribution, were observed following a 4 h reaction time (Figure S12).
Figure 8.

Transmission electron microscopy (TEM) image of palladium particles after 0.25 h of dehydrogenating 3-methylcyclohexanone. Reaction conditions: [ketone] = 2 M (1 mmol), [Pd(TFA)2] = 0.06 M (0.03 mmol), [2-Me2Npy] = 0.12 M (0.06 mmol), [TsOH] = 0.24 M (0.12 mmol), DMSO for VTotal of 0.5 mL, 1 atm O2, 80 °C.
Dynamic light scattering (DLS) is a non-invasive technique that can be used to analyze the catalyst in situ.27 Pd(TFA)2/2-Me2Npy in DMSO at 80 °C does not show evidence of particles in solution (Figure 9Ai; DLS detection limit ~ 1 nm). DLS analysis of the catalytic reaction mixture for dehydrogenation of cyclohexanone 1a revealed the presence of 3.2 nm diameter particles 5 min after initiating the reaction (15 % conversion) (Figure 9Aii). After 2 h, particles sizes of >300 nm were detected (Figure 9Aiii). Similar observations were made with the dehydrogenation of cyclohexenone to phenol (Figure 9B); however, larger particles were observed at earlier reaction times (e.g. ~350 nm particles at t = 5 min). The data cannot distinguish between the presence of large single particles versus aggregates of smaller particles, such as those evident in the TEM image in Figure 8. In a control experiment without substrate, DLS analysis of the catalyst system under the reaction conditions showed a slow background growth of Pd nanoparticles, with 18 nm particles observed at 24 h (Figure S13).
Figure 9.

Palladium particle sizes at different reaction times for the dehydrogenation of (A) 3-methylcyclohexanone and (B) 3-methylcyclohexenone at 5 min and 2 h. Reaction conditions: [ketone] = 2 M (1 mmol), [Pd(TFA)2] = 0.06 M (0.03 mmol), [2-Me2Npy] = 0.12 M (0.06 mmol), [TsOH] = 0.24 M (0.12 mmol), DMSO for VTotal of 0.5 mL, 1 atm O2, 80 °C.
These DLS data implicate continuous growth28 in the palladium particle size as the reaction proceeds,29 and the rate of Pd nanoparticle formation is faster when cyclohexenone is the substrate. The reduced rate of nanoparticle formation in cyclohexanone dehydrogenation reactions is attributed to the lower [cyclohexenone] at early time points. These results are consistent with our earlier observations that suggest cyclohexenone facilitates nanoparticle formation (cf. Figure 4). Previous results in the literature suggest that nanoparticle growth is often autocatalytic and results in sigmoidal kinetics,28 similar to the data in Figures 4A and S16. These experimental time courses can be fit to a simple kinetic model incorporating Pd nanoparticle nucleation and autocatalytic growth steps (see Supporting Information for details: Figures S16 and associated discussion). The autocatalytic mechanism for growth of Pd nanoparticles also provides a rationale for the second-order [Pd] dependence observed in the cyclohexanone dehydrogenation reaction (cf. Figure 3b).30
The influence of the 2-Me2Npy and TsOH additives were also investigated by DLS (Figures S14 and S15). In the absence of both additives, Pd nanoparticles take at least 2 h to become detectable by DLS, and this time period correlates with the initial detection of phenol in the reaction mixture. The presence of 2-Me2Npy in the reaction mixture (in the absence of TsOH) completely inhibits Pd nanoparticle formation over the first 4 h of the reaction, and no phenol formation is observed over a similar time period. In the presence of TsOH, both in the presence and in the absence of 2-Me2Npy, Pd-nanoparticle formation and phenol production are detected immediately after initiating the reactions. Higher overall product yields are observed in the presence of both TsOH and 2-Me2Npy ligand (i.e., after 24 h; cf. Table 1), but these DLS results suggest that the ligand effect is secondary to the TsOH effect in stimulating nanoparticle formation. Overall, the DLS data establish a correlation between the appearance of Pd nanoparticles and the onset of catalytic activity leading to phenol formation.
Conclusion and Implications for Pd-Catalyzed Aerobic Dehydrogenation of Cyclohexanone
This study has provided an in-depth analysis of the Pd(TFA)2/2-Me2Npy-catalyzed dehydrogenation of cyclohexanones to phenols, and the kinetic and mechanistic observations are summarized in Table 2. Observation of opaque dark-red solutions (Table 2, entry 1) provided the first indication that Pd nanoparticles might be forming under the catalytic conditions. Difficulty in obtaining reliable kinetic data, except under carefully controlled conditions with parallel experiments (entry 2), suggested that nanoparticles could contribute to the observed catalytic activity. The presence of a kinetic burst in the cyclohexanone-to-cyclohexenone dehydrogenation step and an induction period in the cyclohexenone-to-phenol step (entries 3 and 4) provided evidence that the PdII catalyst source transforms into a new catalytically active species during the reaction. Filtration experiments and the PVPy test results (entries 5 and 7) revealed that the catalytic activity is associated with a soluble Pd source, while the Hg test results (entry 6) supported either nanoparticulate or heterogeneous catalysis. Finally, DLS data (entry 8) revealed the continual growth of Pd nanoparticles during the catalytic reactions.
Table 2.
Summary of Observations to Support Formation of Catalytically Active Pd Nanoparticles from Pd(TFA)2/2-Me2Npy.
| Entry | Observation | Dehydrogenation Step
|
|
|---|---|---|---|
| Ketone to Enone | Enone to Phenol | ||
| 1 | Opaque, dark-red solutions | ✓ | ✓ |
| 2 | Reliable kinetics require carefully controlled conditions | ✓ | ✓ |
| 3 | Kinetic “burst” | ✓ | |
| 4 | Induction period | ✓ | |
| 5 | Filtrate remains catalytically active | ✓ | ✓ |
| 6 | Hg poisoning | ✓ | ✓ |
| 7 | PVPy poisoning | ✓ | ✓ |
| 8 | Pd particles detected by DLS and TEM | ✓ | ✓ |
Collectively, these results provide compelling evidence that the PdII catalyst precursor mediates initial dehydrogenation of cyclohexanone, after which it converts to soluble, catalytically active Pd nanoparticles. These observations may be contrasted with the catalytic inactivity of heterogeneous Pd, including those formed under the reaction conditions (Scheme 3) and traditional sources, such as Pd/C. The ability of Pd nanoparticles to mediate dehydrogenation of both cyclohexanone and cyclohexenone provides the basis for full conversion of cyclohexanone to phenol. This feature contrasts the activity of the homogeneous Pd(DMSO)2(TFA)2 catalyst, which is effective only for dehydrogenation of cyclohexanone, thereby enabling selective formation of cyclohexenone.7
Scheme 3.
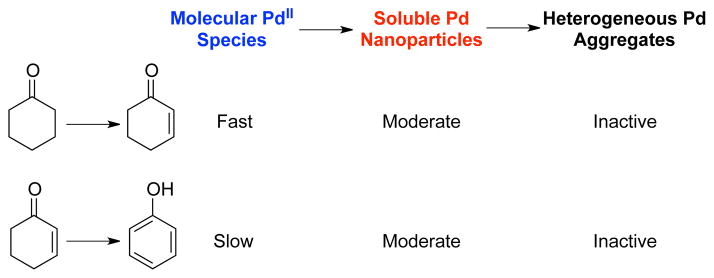
Proposed Conversion of Pd Species and Their Relative Catalytic Activities in the Dehydrogenation of Cyclohexanone and Cyclohexenone.
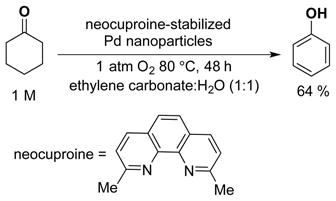
The insights gained from this study suggest that similar catalytic activity could be achieved from intentionally prepared Pd nanoparticles. In a preliminary test of this hypothesis, we synthesized well-defined, neocuproine-stabilized Pd nanoparticles in a mixture of ethylene carbonate and water, according to the method of Sheldon and coworkers.31 These nanoparticles were tested as catalysts in the dehydrogenation of cyclohexanone and led to a 64% yield of phenol at 80 °C (eq 1). This unoptimized result validates the proposed concept and offers the prospect that suitably stable, yet active, Pd-nanoparticle catalysts can be identified to enable broader application of aerobic dehydrogenation reactions in organic chemical synthesis.
 |
(1) |
Supplementary Material
Acknowledgments
Supporting Information. Experimental details for data acquisition and additional kinetic data are available free of charge via the Internet at http://pubs.acs.org.
The authors thank Dian Wang for assistance in modeling experimental time-course data and Prof. Nick Abbott and Dr. J. Muller (UW-Madison) for use of their dynamic light scattering apparatus and assistance with the DLS experiments. Financial support was provided by the NIH (R01-GM100143) and NSF (CHE-1041934; to DP).
References
- 1.Tyman JHP. Synthetic and Natural Phenols. Vol. 52 Elsevier; Amsterdam: 1996. [Google Scholar]
- 2.For early Pd/C precedents and reviews, see: Horning EC, Horning MG. J Am Chem Soc. 1947;69:1359.Horning EC, Horning MG, Walker GN. J Am Chem Soc. 1949;71:169.Fu PP, Harvey RG. Chem Rev. 1978;78:317.Bamfield P, Gordon PF. Chem Soc Rev. 1984;13:441.Neumann H, Jacobi von Wangelin A, Klaus S, Strübing D, Gördes D, Beller M. Angew Chem Int Ed. 2003;42:4503. doi: 10.1002/anie.200351484.
- 3.For recent examples: Izawa Y, Pun D, Stahl SS. Science. 2011;333:209. doi: 10.1126/science.1204183.Imahori T, Tokuda T, Taguchi T, Takahata H. Org Lett. 2012;14:1172. doi: 10.1021/ol300145g.Xie Y, Liu S, Liu Y, Wen Y, Deng GJ. Org Lett. 2012;14:1692. doi: 10.1021/ol3002442.Kandukuri SR, Oestreich M. J Org Chem. 2012;77:8750. doi: 10.1021/jo301088f.Simon MO, Girard SA, Li CJ. Angew Chem Int Ed. 2012;51:7537. doi: 10.1002/anie.201200698.Hajra A, Wei Y, Yoshikai N. Org Lett. 2012;14:5488. doi: 10.1021/ol302568b.Girard SA, Hu X, Knauber T, Zhou F, Simon MO, Deng GJ, Li CJ. Org Lett. 2012;14:5606. doi: 10.1021/ol3027279.Izawa Y, Zheng C, Stahl SS. Angew Chem Int Ed. 2013;52:3672. doi: 10.1002/anie.201209457.Sutter M, Sotto N, Raoul Y, Métay E, Lemaire M. Green Chem. 2013;15:347.Kim D, Min M, Hong S. Chem Commun. 2013;49:4021. doi: 10.1039/c3cc41296b.
- 4.For recent examples of related dehydrogenation reactions and reviews: Diao T, Wadzinski TJ, Stahl SS. Chem Sci. 2012;3:887. doi: 10.1039/C1SC00724F.Gao W, He Z, Qian Y, Zhao J, Huang Y. Chem Sci. 2012;3:883.Wei Y, Deb I, Yoshikai N. J Am Chem Soc. 2012;134:9098. doi: 10.1021/ja3030824.Muzart J. Eur J Org Chem. 2010;20:3779.Dobereiner GE, Crabtree RH. Chem Rev. 2010;110:681. doi: 10.1021/cr900202j.Choi J, MacArthur AHR, Brookhart M, Goldman AS. Chem Rev. 2011;111:1761. doi: 10.1021/cr1003503.
- 5.See, for example: Trost BM, Metzner PJ. J Am Chem Soc. 1980;102:3572.Wenzel TT. J Chem Soc Chem Commun. 1989:932.Sheldon RA, Sobczak JM. J Mol Catal. 1991;68:1.Kambourakis S, Frost JW. J Org Chem. 2000;65:6904. doi: 10.1021/jo000335z.Bercaw JE, Hazari N, Labinger JA. J Org Chem. 2008;73:8654. doi: 10.1021/jo8016296.Williams TJ, Caffyn AJM, Hazari N, Oblad PF, Labinger JA, Bercaw JE. J Am Chem Soc. 2008;130:2418. doi: 10.1021/ja076740q.Moriuchi T, Kikushima K, Kajikawa T, Hirao T. Tetrahedron Lett. 2009;50:7385.Yi CS, Lee DW. Organometallics. 2009;28:947. doi: 10.1021/om8010883.Zhang X, Wang DY, Emge TJ, Goldman AS. Inorg Chim Acta. 2011;369:253.
- 6.Diao T, Stahl SS. J Am Chem Soc. 2011;133:14566. doi: 10.1021/ja206575j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diao T, Pun D, Stahl SS. submitted for publication. [Google Scholar]
- 8.Recent studies suggest that aerobic oxidation of PdII–hydrides proceed via Pd0, as shown in Scheme 2. See: Popp BV, Stahl SS. J Am Chem Soc. 2007;129:4410. doi: 10.1021/ja069037v.Konnick MM, Stahl SS. J Am Chem Soc. 2008;130:5753. doi: 10.1021/ja7112504.Popp BV, Stahl SS. Chem Eur J. 2009;15:2915. doi: 10.1002/chem.200802311.Decharin N, Popp BV, Stahl SS. J Am Chem Soc. 2011;133:13268. doi: 10.1021/ja204989p.Konnick MM, Decharin N, Popp BV, Stahl SS. Chem Sci. 2011;2:326.
- 9.Burton AG, Frampton RD, Johnson CD, Katritzky AR. J Chem Soc, Perkin Trans 2. 1972:1940. [Google Scholar]
- 10.Dega-Szafran Z, Kania A, Nowak-Wydra B, Szafran M. J Mol Struct. 1994;322:223. [Google Scholar]
- 11.Under our catalytic conditions, only the monoprotonated pyridinium ligand is observable.
- 12.(a) Boutonnet M, Kizling J, Stenius P. Colloids Surf. 1982;5:209. [Google Scholar]; (b) Bönnemann H, Brijoux W, Brinkmann R, Dinjus E, Joussen T, Korall B. Angew Chem Int Ed. 1991;30:1312. [Google Scholar]; (c) Toshima N, Takahashi T. B Chem Soc Jpn. 1992;65:400. [Google Scholar]; (d) Reetz MT, Helbig W. J Am Chem Soc. 1994;116:7401. [Google Scholar]; (e) Reetz MT, Westermann E. Angew Chem Int Ed. 2000;39:165. doi: 10.1002/(sici)1521-3773(20000103)39:1<165::aid-anie165>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]; (f) Reetz MT, de Vries JG. Chem Commun. 2004:1559. doi: 10.1039/b406719n. [DOI] [PubMed] [Google Scholar]
- 13.As an addition experiment, 10 % dimethylsulfone was also added. Dimethylsulfone is formed from the oxidation of the DMSO solvent, which is common in the presence of H2O2: Tashlick I. US Patent 3069471. 1962Morimoto A, Nanbu N, Nanbu T. Japanese Patent JP54044611. 1979Patai S, Rappoport ZZ, Stirling C, editors. The Chemistry of Sulphones and Sulphoxides. John Wiley; Chichester: 1988.
- 14.For reviews describing experimental methods to distinguish between homogeneous and heterogeneous catalytic reactions, see: Widegren JA, Finke RG. J Mol Catal A: Chem. 2003;198:317.Phan NTS, Van Der Sluys M, Jones CW. Adv Synth Catal. 2006;348:609.Crabtree RH. Chem Rev. 2011;112:1536. doi: 10.1021/cr2002905.
- 15.Hovermale RA, Sears PG. J Phys Chem. 1956;60:1579. [Google Scholar]
- 16.Man RWY, Brown ARC, Wolf MO. Angew Chem Int Ed. 2012;51:11350. doi: 10.1002/anie.201205057. [DOI] [PubMed] [Google Scholar]
- 17.(a) van Bentham RATM, Hiemstra H, Michels JJ, Speckamp WN. J Chem Soc, Chem Comm. 1994:357. [Google Scholar]; (b) van Benthem RATM, Hiemstra H, van Leeuwen PWNM, Geus JW, Speckamp WN. Angew Chem Int Ed. 1995;34:457. [Google Scholar]
- 18.Hiemstra has proposed that the Pd nanoparticles consist of Pd-oxide particles. We have no evidence for or against the presence of a (PdO)x component in the nanoparticles present in our reactions.
- 19.For precedents of PdII catalyst-precursors converting into nanoparticulate or heterogeneous catalysts, see ref. 17 and the following: Tibbitt JM, Gong WH, Schammel WP, Hepfer RP, Adamian V, Brugge SP, Metelski PD, Zhou C. US Patent 0118536. 2009Horrillo-Martínez P, Virolleaud MA, Jaekel C. ChemCatChem. 2010;2:175.
- 20.For precedents of heterogeneous Pd catalysts that serve as precursors to homogeneous catalyst, see ref. 14b and the following: Davies IW, Matty L, Hughes DL, Reider PJ. J Am Chem Soc. 2001;123:10139. doi: 10.1021/ja016877v.Sommer WJ, Yu K, Sears JS, Ji Y, Zheng X, Davis RJ, Sherrill CD, Jones CW, Weck M. Organometallics. 2005;24:4351.
- 21.Hamlin JE, Hirai K, Millan A, Maitlis PM. J Mol Catal. 1980;7:543. [Google Scholar]
- 22.Sulpizio TE. American Filtration & Separations Society. Boston: 1999. [Google Scholar]
- 23.Foley P, DiCosimo R, Whitesides GM. J Am Chem Soc. 1980;102:6713. [Google Scholar]
- 24.(a) Bayram E, Linehan JC, Fulton JL, Roberts JAS, Szymczak NK, Smurthwaite TD, Özkar S, Balasubramanian M, Finke RG. J Am Chem Soc. 2011;133:18889. doi: 10.1021/ja2073438. [DOI] [PubMed] [Google Scholar]; (b) Bayram E, Finke RG. ACS Catal. 2012;2:1967. [Google Scholar]
- 25.Gonzalez-Tejuca L, Aika K, Namba S, Turkevich J. J Phys Chem. 1977;81:1399. [Google Scholar]
- 26.(a) Schmid G. Polyhedron. 1988;7:2321. [Google Scholar]; (b) Vargaftik MN, Zagorodnikov VP, Stolarov IP, Moiseev II, Kochubey DI, Likholobov VA, Chuvilin AL, Zamaraev KI. J Mol Catal. 1989;53:315. [Google Scholar]; (c) Choi KM, Akita T, Mizugaki T, Ebitani K, Kaneda K. New J Chem. 2003;27:324. [Google Scholar]; (d) Diéguez M, Pàmies O, Mata Y, Teuma E, Gómez M, Ribaudo F, van Leeuwen PWNM. Adv Synth Catal. 2008;350:2583. [Google Scholar]; (e) Zalesskiy SS, Ananikov VP. Organometallics. 2012;31:2302. [Google Scholar]
- 27.(a) Pecora R. J Nanoparticle Res. 2000;2:123. [Google Scholar]; (b) Hintermair U, Hashmi SM, Elimelech M, Crabtree RH. J Am Chem Soc. 2012;134:9785. doi: 10.1021/ja3033026. [DOI] [PubMed] [Google Scholar]
- 28.For previous observation and analysis of nanoparticle nucleation and autocatalytic growth kinetics, see the following leading references: Turkevich J, Stevenson PC, Hillier J. Discuss Faraday Soc. 1951;11:55.Watzky MA, Finke RG. J Am Chem Soc. 1997;119:10382.Hornstein BJ, Finke RG. Chem Mater. 2004;16:139.Besson C, Finney EE, Finke RG. Chem Mater. 2005;17:4925. doi: 10.1021/ja0504439.Finney EE, Finke RG. J Colloid Interface Sci. 2008;317:351. doi: 10.1016/j.jcis.2007.05.092.Finney EE, Finke RG. Chem Mater. 2009;21:4468.
- 29.For an example of Pd nanoparticle growth during catalysis: Heckenroth M, Khlebnikov V, Neels A, Schurtenberger P, Albrecht M. ChemCatChem. 2011;3:167.
- 30.For leading references to second-order [catalyst] dependence arising from catalyst aggregation phenomena, see ref. 28c and the following: Steinhoff BA, Fix SR, Stahl SS. J Am Chem Soc. 2002;124:766. doi: 10.1021/ja016806w.Steinhoff BA, Stahl SS. J Am Chem Soc. 2006;128:4348. doi: 10.1021/ja057914b.
- 31.Mifsud M, Parkhomenko KV, Arends IWCE, Sheldon RA. Tetrahedron. 2010;66:1040. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.