

Abstract
Resolution of Chlamydia genital tract infection is delayed in the absence of MyD88. In these studies, we first used bone marrow chimeras to demonstrate a requirement for MyD88 expression by hematopoietic cells in the presence of a wild-type epithelium. Using mixed bone marrow chimeras we then determined that MyD88 expression was specifically required in the adaptive immune compartment. Furthermore, adoptive transfer experiments revealed that CD4+ T cell expression of MyD88 was necessary for normal resolution of genital tract infection. This requirement was associated with a reduced ability of MyD88−/− CD4+ T cells to accumulate in the draining lymph nodes and genital tract when exposed to the same inflammatory milieu as wild-type CD4+ T cells. We also demonstrated that the impaired infection control we observed in the absence of MyD88 could not be recapitulated by deficiencies in TLR or IL-1R signaling. In vitro, we detected an increased frequency of apoptotic MyD88−/− CD4+ T cells upon activation in the absence of exogenous ligands for receptors upstream of MyD88. These data reveal an intrinsic requirement for MyD88 in CD4+ T cells during Chlamydia infection and indicate that the importance of MyD88 extends beyond innate immune responses by directly influencing adaptive immunity.
INTRODUCTION
Chlamydia trachomatis infections of the female reproductive tract can result in serious pathophysiology including pelvic inflammatory disease (PID), chronic pelvic pain, ectopic pregnancy, and infertility (Reviewed in (1)). The immune response to Chlamydia is dually responsible for resolution of infection and the development of genital tract pathology. Due to its obligate intracellular lifecycle, Chlamydia is able to evade innate defense mechanisms that are effective against extracellular bacteria, and innate immune responses have been repeatedly correlated with the development of oviduct pathology (2-6). In contrast, studies in the mouse model have revealed that the adaptive immune response is crucial for eradication of both primary (7) and secondary infection (8). In addition, CD4+ Th1 cells are crucial for protection in both mice (8-13) and women (14-16). CD4+ T cells directly interact with infected epithelial cells and promote eradication of infection via IFNγ dependent and independent mechanisms (11, 12, 17, 18).
Recognition of pathogens by pattern recognition receptors (PRRs) expressed by innate immune cells is crucial for effective induction of an adaptive immune response (19), but overly robust innate immune activation results in tissue damage. Chlamydiae stimulate several PRRs including Toll-like receptor 2 (TLR2) (5, 20), TLR3 (21), TLR4 (22, 23), and nucleotide-binding oligomerization domain-containing protein 1 (NOD1) (24). Mice deficient in TLR2 develop reduced levels of oviduct pathology in response to Chlamydia muridarum infection, but resolution of infection is not impacted by the absence of this receptor (5). TLR4 and NOD1 do not appear to play a central role in either tissue damage or induction of a protective immune response in the mouse model (5, 24). These findings were corroborated by a study of women with Chlamydia trachomatis PID, which revealed that women with specific polymorphisms in TLR1, a receptor that signals by forming heterodimers with TLR2 (25), exhibited decreased rates of pregnancy, whereas no such association was found with polymorphisms in TLR4 (26). A Dutch study found a nonsignificant association of the TLR4 +896 G allele with tubal factor infertility (27).
MyD88 is an adaptor molecule that is central to signaling via all TLRs except for TLR3 and is required for signaling by the interleukin-1 (IL-1) family of cytokine receptors (28-32). Recognition of ligands by these receptors induces conformational changes that promote homotypic interactions between the Toll/interleukin-1 receptor (TIR) domain of these receptors and those of intracellular adaptor molecules including MyD88 (33-35). Stabilized oligomers of MyD88 then interact via death domains with IL-1 receptor associated kinase (IRAK)1, IRAK2, and IRAK4 to form a Myddosome complex (34, 36-39). This signal transduction cascade leads to NF-κB and AP-1 mediated transcription of pro-inflammatory genes. MyD88 is thus central to promoting innate immune activation and has been implicated in promoting resistance to a multitude of pathogens in the mouse model (Reviewed in (40)). In humans, loss-of-function mutations in MyD88 (41) and IRAK4 (42) have been associated with the development of severe and potentially fatal bacterial infections in children.
The importance of MyD88 in promoting adaptive immune responses to pathogens in murine models has been repeatedly attributed to its central role in innate immune activation. However, a requirement for MyD88 expression by adaptive immune cells has also been observed in models of infection and autoimmunity. In a murine model of Toxoplasma gondii infection, control of infection was impaired even when MyD88-deficient adaptive immune cells were activated in the presence of normal antigen presenting cells (43). These findings were recapitulated in two independent studies of murine lymphocytic choriomeningitis virus (LCMV) infection, which demonstrated that both CD4+ and CD8+ T cell survival was reduced in the absence of intrinsic expression of MyD88 (44, 45). A requirement for MyD88 expression by CD4+ T cells was also demonstrated in a model of colitis where MyD88-deficient CD4+ T cells exhibited reduced accumulation and cytokine production both in vitro and in vivo (46, 47). Finally, a recent publication demonstrated that CD4+ T cell expression of MyD88 was required for Th17 differentiation and the development of experimental autoimmune encephalitis (EAE) (48). Although the precise mechanism(s) behind this requirement for MyD88 in adaptive immune cells has not been determined, receptors upstream of MyD88 have been implicated in direct co-stimulation of T cells (49-54).
MyD88-mediated signals promote cytokine production by innate immune cells in response to Chlamydia infection (5, 20, 55, 56). In addition, MyD88−/− mice exhibit significantly impaired control of Chlamydia muridarum genital tract infection (55, 57, 58). Prolonged infection was associated with early reductions in natural killer (NK) cell IFNγ production in the cervix and a decreased frequency of CD4+ T cells in the upper genital tract. However, Chlamydia-specific CD4+ T cell proliferation and IFNγ production remained largely intact in the draining lymph nodes, although a small increase in IL-4 production was detected (58).
The development of a vaccine against Chlamydia requires delineation of immune mechanisms of protection from those that cause pathology. Activation of receptors upstream of MyD88, including TLR2 (5) and IL-1R (59), results in the development of oviduct damage in the mouse model. Although it is clear from these findings that MyD88-mediated signals promote tissue-damaging responses to chlamydial infection, detection of prolonged genital tract infection in MyD88−/− mice indicates that this molecule also participates in protective immunity. A rapid and robust CD4+ T cell response is key for control of chlamydial infection and protection from disease (10, 60, 61), and MyD88 expression by adaptive immune cells has been implicated in promoting optimum responses in other models (43, 45-47). However, these studies utilized models of systemic infection, and an intrinsic requirement for MyD88 in adaptive immune cells has not been previously explored in a model of mucosal infection. Using bone marrow chimeric mice and CD4+ T cell adoptive transfer experiments we have determined that intrinsic expression of MyD88 in CD4+ T cells is required for accumulation of CD4+ T cells in infected tissues and efficient resolution of Chlamydia genital tract infection. In vitro and in vivo experiments suggest that the CD4+ T cell specific effects of MyD88 are independent of TLR or IL-1R activation.
MATERIALS AND METHODS
Strains, cell lines, and culture conditions
C. muridarum Nigg was used for all experiments and was isolated as previously described (62, 63). All chlamydial strains were propagated in L929 cells (64). Bacteria were titrated by plaque assay (63) or as inclusion forming units (IFU) using fluorescently tagged anti-chlamydial lipopolysaccharide monoclonal antibody (Bio-Rad, Hercules, CA) (65).
Animals
C57BL/6 (CD45.2+), B6129SF2/J (C57BL/6;129S), B6.129P2(SJL)-Myd88tm1.1Defr/J (MyD88−/−), B6.129S7-Rag1tm1Mom/J (Rag1−/−), B6.129S7-Ifngtm1Ts/J (IFNγ−/−), B6.129S7-Ifngr1tm1Agt/J (IFNγR−/−), B6.SJL-Ptprca Pep3b/BoyJ (CD45.1+), B6.PL-Thy1a/CyJ (CD90.1+), B6;129S1-Tlr3tm1Flv/J (TLR3−/−) were obtained from The Jackson Laboratory (Bar Harbor, ME). TLR2−/−TLR4−/−, TLR7−/− (66), and TLR9−/− (67) mice were kindly provided by Dr. Shizuo Akira. Mice were given food and water ad libitum in an environmentally controlled room with a cycle of 12 hours of light and 12 hours of darkness. The University Institutional Animal Care and Use Committee approved all animal experiments.
Murine infection and monitoring
Female mice of at least 6 weeks of age were subcutaneously injected with 2.5 mg of medroxyprogesterone (Depo-Provera®; Upjohn, Kalamazoo, MI) 5 to 7 days prior to infection to induce a state of anestrous (68). Mice were intravaginally inoculated with 1×105 IFU of C. muridarum Nigg diluted in 30 μl of sucrose-sodium phosphate-glutamic acid (SPG) buffer unless otherwise indicated. Mice were monitored for cervicovaginal shedding via endocervical swabs (69), and IFU were calculated as previously described (65). Bacterial burden was measured in the oviducts, lungs, liver, and spleen by plaque assay (63). C57BL/6 mice were used as controls for all strains of knockout mice except TLR3−/− mice, which are on a mixed C57BL/6 and 129S background. Thus, F2 hybrids of C57BL/6 and 129S mice were used as controls for those mice.
Generation of Bone Marrow Chimeras
Mice were injected subcutaneously with 2.5 mg of depot medroxyprogesterone acetate (Depo-Provera®; Upjohn, Kalamazoo, MI) 5 days before irradiation. Recipient mice were prepared for the immunocompromised state that results from irradiation by replacing their normal diet with antibiotic food (1.2% sulfamethoxazole and 0.2% trimethoprim; Lab Diet, St. Louis, MO) and sterile acidified water (PH 2.5-3) for 10 days prior to the procedure. Mice were irradiated with 2 doses of 500 rads of X-ray irradiation separated by 6 hours. Immediately following the final dose of irradiation, mice were reconstituted by intravenous (i.v.) injection of 7×106 bone marrow cells from MyD88−/− (CD45.2+) or WT (CD45.1+) mice. MyD88−/− cells were injected into WT (CD45.1+) recipients and WT (CD45.1+) cells into WT (CD45.2+) recipients. Mice were maintained on acidified water and antibiotic food for 4 weeks following irradiation. Chimerism was verified after 6 weeks using flow cytometry. Mice were re-injected with Depo-Provera® after 6 weeks and infected with 1×106 IFU of C. muridarum Nigg 1 week later. Data are from one representative experiment of two with 5-6 mice per group.
Generation of Mixed Bone Marrow Chimeras
Recipient mice were fed antibiotic food (1.2% sulfamethoxazole and 0.2% trimethoprim; Lab Diet, St. Louis, MO) and sterile acidified water (PH 2.5-3) for 10 days prior to irradiation. Recipient mice were treated with two doses of 450 rads (900 rads total) of X-ray irradiation separated by 6 hours. Immediately after irradiation, Rag1−/− mice were reconstituted with 2.5×106 cells from a Rag1−/− donor and 2.5×106 cells from either a MyD88−/−, WT (CD45.1+), or IFNγ−/− donor. IFNγR−/− mice were injected with 2.5×106 cells from a Rag1−/− donor and 2.5×106 cells from a WT (CD45.1+) donor. Six weeks after injection, mice were bled to determine the level of engraftment and were injected with Depo-Provera®. Chimeras were infected with 1×106 IFU of C. muridarum Nigg. Groups consisted of 4-7 mice per donor: recipient combination.
CD4+ T cell Transfer into Rag1−/− mice
CD4+ T cells were isolated from the spleens of naïve C57BL/6 or MyD88−/− mice by negative magnetic selection (CD4+ T cell isolation kit II; Miltenyi Biotech, Auburn, CA). The purity of CD4+ T cells was determined to be > 93% for both strains by flow cytometry prior to transfer. Rag1−/− mice were injected i.v. with 4×106 CD4+ T cells from either strain. The frequency of CD4+ T cells in the peripheral blood was determined using flow cytometry at 3 weeks after transfer. Mice were injected with Depo-Provera® 4 weeks after transfer and were infected with C. muridarum 5 weeks after transfer. Data are presented from one representative experiment of three with 5-6 mice per group. Rag1−/− mice that did not receive a CD4+ T cell transfer were infected in 3 independent experiments with a total of 15 mice.
CD4+ T cell co-transfer experiment
Ten days prior to receiving the CD4+ T cell transfer, WT recipient mice (CD45.2+ CD90.1+ CD90.2−) were injected with Depo-Provera®. On the day of transfer, CD4+ T cells were isolated from the spleens of naïve WT (CD45.1+CD90.2+) or MyD88−/− (CD45.2+CD90.2+) mice by negative magnetic selection (CD4+ T cell isolation kit II; Miltenyi Biotech, Auburn, CA). Recipient mice were injected i.v. with 4×106 cells from both WT and MyD88−/− mice (8×106 cells total) and infected intravaginally with 1×106 IFU of C. muridarum Nigg. Four pools of cells per strain were processed independently from the beginning of the experiment and transferred into groups of 3 mice. The average purity of CD4+ T cells in these preparations was 88% for both strains of mice. Ten days post infection, mice were euthanized, and single cell suspensions were generated from their genital tracts and iliac nodes. The cervix and uterine horns were treated with collagenase I (1 mg/ml; Sigma-Aldrich, St. Louis, MO) while the oviducts and iliac lymph nodes were mechanically disrupted using the previously described protocol (6, 70). Cells from the cervix, uterine horns, and oviducts were pooled for analysis. Donor-derived cells were enriched using a CD90.2 positive selection kit (Miltenyi Biotech, Auburn, CA) prior to surface staining. The frequency of CD4+ T cells from each strain of mice was determined by flow cytometry. Cells were stained for the following cell surface markers: anti-CD4 PE (Clone: RM4-5), anti-CD3 V450 (Clone: 500A2), anti-CD90.1 FITC (Clone: OX-7), anti-CD90.2 PE-Cy7 (Clone: 53-2.1), anti-CD45.1 PerCP-Cy5.5 (Clone: A20), and anti-CD45.2 Allophycocyanin (Clone: 104). Accumulation of T cells from each strain was determined by gating on CD3+CD4+ T cells that were CD90.2+CD90.1−. These donor-derived cells were then divided into populations that were MyD88−/− (CD45.2+CD45.1−) or WT (CD45.1+CD45.2−). All antibodies were from BD Biosciences (San Jose, CA). Data shown represent the frequencies of donor cells in either the genital tract or iliac lymph nodes from 4 groups of three recipient mice.
In vitro analysis of CD4+ T cell apoptosis
Naïve CD4+ T cells were isolated from the spleens of MyD88−/− (CD45.2) and WT (CD45.1) mice via negative magnetic selection (Mouse Naïve CD4+ T cell isolation kit; STEMCELL Technologies, Vancouver, BC). Isolated T cells were combined at a 1:1 ratio (1×105 cells per strain) in a 96 well plate in complete medium. Complete medium consisted of DMEM (Thermo Fisher Scientific; Pittsburgh, PA), 10% fetal bovine serum (Thermo Scientific Hyclone, Pittsburgh, PA), vancomycin (100 μg/ml; Sigma-Aldrich, St. Louis, MO), gentamicin (50 μg/ml; Life Technologies Gibco, Grand Island, NY), Glutamax (1 mg/ml; Life Technologies Gibco, Grand Island, NY), 2-mercaptoethanol (50 μM; Sigma-Aldrich, St. Louis, MO), and non-essential amino acids (0.5 mg/ml; Life Technologies Gibco, Grand Island, NY). Cells were incubated with the indicated combination of the following reagents: plate-bound anti-CD3 (1 μg/ml, Clone 145-2C11; eBioscience, San Diego, CA), soluble anti-CD28 (1 μg/ml, Clone 37.51; Biolegend, San Diego, CA), IL-2 (5 ng/ml; Peprotech, Rocky Hill, NJ), IL-12p70 (10 ng/ml; Peprotech, Rocky Hill, NJ), and anti-IL-4 (1 μg/ml; Clone: 11B11; eBioscience, San Diego, CA). After three days in culture, cells were either directly stained for flow cytometry or were transferred to a new 96-well plate and incubated with complete medium without any stimulatory reagents for an additional 24 hours prior to antibody staining. Surface staining was conducted using the following antibody combination: anti-TCR β chain V450 (Clone: H57-597), anti-CD4 PE-Cy7 (Clone: RM4-5), anti-CD45.2 Allophycocyanin (Clone: 104), and anti-CD25 PE (Clone: PC61). After surface staining, apoptosis was measured by staining with Annexin V-FITC according to the manufacturer’s instructions (Annexin V-FITC Apoptosis Detection Kit I). Approximately 10 minutes prior to analysis, 5 μl of 7-Amino-Actinomycin D (7-AAD) was added to the samples. All antibodies and reagents used for staining were from BD Biosciences (San Jose, CA). Apoptosis was analyzed by gating on CD4+CD25high T cells that were CD45.2+ (MyD88−/−) or CD45.2− (WT). Data shown represent the mean ± SEM of triplicate wells for each condition from one representative experiment of two.
Statistics
Comparison of the course of infection was conducted via Two-way repeated measures (RM) ANOVA with Bonferroni post-test analysis. A Log-rank (Mantel-Cox) Test was used to compare the duration of infection. Significant differences in the frequency of cells accumulating in the genital tract and lymph nodes in the T cell co-transfer experiment was determined via Mann-Whitney U-test. Apoptosis was compared between strains under different stimulatory conditions in vitro by Two-way ANOVA with Bonferroni post-test analysis. Prism software (GraphPad Software, LaJolla, CA) was utilized for all statistical analysis. Values of P < 0.05 were considered significant.
RESULTS
MyD88 is required in hematopoietic cells for normal resolution of Chlamydia muridarum genital tract infection
Epithelial cells represent the primary niche for Chlamydia in the genital tract (71-73), and MyD88 participates in Chamydia-induced cytokine production by these cells (20, 56). We first sought to determine if the prolonged infection detected in the absence of MyD88 (55, 57, 58) could be observed for mice with a MyD88 deficient hematopoietic compartment and a wild-type epithelium. Bone marrow chimeras can be utilized for this purpose because hematopoietic cells are more sensitive to irradiation than epithelial or stromal cells. After irradiation, the hematopoietic compartment can be reconstituted with bone marrow from a donor strain of mice while the epithelium retains the genotype of the recipient strain. Wild-type (WT; CD45.2+ or CD45.1+) mice were irradiated, and their bone marrow was reconstituted with either WT (CD45.1+) or MyD88−/− (CD45.2+) cells. Recipient mice with a MyD88 deficient epithelium were not included in this analysis due the potentially confounding effects resulting from the enhanced sensitivity of MyD88 deficient epithelial cells to irradiation (74). The frequency of donor derived CD45+ cells was above 90% for all of the mice (data not shown). In addition, the frequency of CD45+ cells in the peripheral blood that were CD3+CD4+ T cells was similar between the strains (WT donor: 5.4 ± 1.0%; MyD88−/− donor: 6.5 ± 0.87%; P > 0.05 by Mann-Whitney U-test). Indeed, MyD88−/− bone marrow has been previously observed to have no deficiency in its ability to reconstitute irradiated recipient mice (75).
When mice with WT stromal/epithelial cells were reconstituted with MyD88−/− bone marrow (MyD88−/− Donor: WT Recipient), infection was significantly increased (P < 0.0001 by Two-Way RM ANOVA; Fig. 1A) and prolonged (P < 0.01 by Log-rank test; Fig. 1B) relative to that observed for recipients of wild-type bone marrow (WT Donor: WT Recipient mice). These data demonstrate that MyD88−/− hematopoietic cells fail to resolve Chlamydia muridarum infection normally, even in the presence of wild-type epithelial cells.
Figure 1.

MyD88 is required in hematopoietic cells for normal resolution of C. muridarum genital tract infection. (A) Bone marrow chimeras were generated with the following strain combinations: WT Donor: WT recipient (black circles) and MyD88−/− Donor: WT Recipient (white triangles). Mice were intravaginally infected with C. muridarum, and the course of infection was monitored with lower genital tract swabs. Data points represent the mean ± SEM of 5-6 mice per group from one representative experiment of two. Significance determined via Two-way RM ANOVA with Bonferroni post-test. Comparison of strains on individual days: * P < 0.05, *** P < 0.001. Comparison of groups over the interval measured: P < 0.0001 for WT Donor: WT Recipient vs. MyD88−/− Donor: WT recipient. (B) Infection was significantly prolonged in mice reconstituted with MyD88−/− bone marrow (dashed line) relative to mice with WT bone marrow (solid line). Data points represent the first day of a negative titer in the lower genital tract for the mice described in (A). P < 0.01 by Log-rank (Mantel-Cox) Test.
MyD88 expression by adaptive immune cells is required for normal resolution of C. muridarum genital tract infection
MyD88-mediated signals promote activation of innate immune cells in response to Chlamydia muridarum (5, 55). In addition, MyD88 expression by adaptive immune cells has been shown to be important in murine models of infection and autoimmunity (43-45, 76). We sought to determine if there was a role for MyD88 in promoting resolution of chlamydial infection in mice with a WT antigen presenting cell (APC) compartment and MyD88 deficiency solely in the adaptive immune cells. To this end, we generated mixed bone marrow chimeras based on the experimental design used by LaRosa et al (43). WT, MyD88−/−, or IFNγ−/− bone marrow was combined at a 1:1 ratio with Rag1−/− bone marrow and transferred into irradiated Rag1−/− recipients (Fig. 2A, 2B). Rag1−/− mice have normal APCs but no adaptive immune cells. Thus, the bone marrow from WT, MyD88−/−, or IFNγ−/− mice served as the only source of adaptive cells, while Rag1−/− bone marrow acted as a source of functional APCs. Irradiation of the Rag1−/− recipients provided a niche for engraftment of the donor-derived bone marrow. Rag1−/− mice were used as recipients to ensure that all T cells were derived from the donor because irradiation cannot eliminate 100% of recipient cells. Chimeras were generated with IFNγ−/− adaptive immune cells as a positive control for defects in T cell mediated resolution of infection. In addition, irradiated IFNγR−/− mice were reconstituted with mixed WT + Rag1−/− bone marrow as another positive control (Fig. 2A, 2B) due to the central role for IFNγ signaling at the level of the genital tract epithelium (12, 13, 17, 70, 77). Verification of chimerism at six weeks after transfer was performed using antibodies for disparate markers present on the WT (CD45.1+) and Rag1−/− (CD45.2+) donor cells. Analysis of cells in the peripheral blood revealed, as expected, that 100% of CD3+CD4+ and CD3+CD8+ T cells in irradiated Rag1−/− recipients were derived from the WT (CD45.1+) donor while 53 ± 3% of Ly6G/Chigh innate cells were derived from the WT donor. In addition, the frequency of CD3+ CD4+ T cells in the peripheral blood did not significantly differ between the groups (data not shown). We were unable to verify the frequencies of MyD88−/− or IFNγ−/− adaptive immune cells since there is no disparate marker between these strains and Rag1−/− mice. However, an identical irradiation protocol, and the same pool of Rag1−/− bone marrow cells were used for all of the groups.
Figure 2.
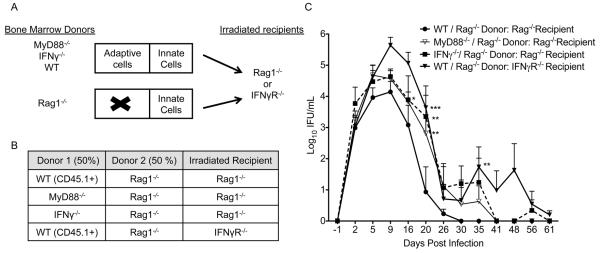
MyD88 expression and IFNγ production by adaptive immune cells as well as IFNγR expression by the stromal compartment is required for normal resolution of C. muridarum genital tract infection. (A, B) Bone marrow chimeras were generated with the following donor: recipient combinations. WT + Rag1−/− Donors: Rag1−/− Recipient (black circles); MyD88−/− + Rag1−/− Donors: Rag1−/− Recipient (white triangles); IFNγ−/− + Rag1−/− Donors: Rag1−/− Recipient (black square, dashed line); WT + Rag1−/− Donors: IFNγR−/− recipient (black triangle). (C) Data points represent the mean ± SEM of 4-7 mice per group. Significance determined via Two-way RM ANOVA with Bonferroni post-test. Comparison of individual days: * P < 0.05, ** P < 0.01, *** P < 0.001 chimeras vs. WT + Rag1−/− Donors: Rag1−/− Recipient group. Comparison of groups over the interval measured: P < 0.05 for WT + Rag1−/− Donors: Rag1−/− Recipient group vs. each of the three other groups. P > 0.05 for MyD88−/− + Rag1−/− Donors: Rag1−/− Recipient vs. IFNγ−/− + Rag1−/− Donors: Rag1−/− Recipient.
The chimeras were intravaginally infected with C. muridarum 7 weeks after bone marrow transfer. Mice with a MyD88−/− or IFNγ−/− adaptive compartment exhibited a significantly increased infection relative to mice with a WT adaptive immune compartment (Fig. 2C). The course of infection did not differ between mice lacking either MyD88 or IFNγ in their adaptive immune cells (Fig. 2C). Mice with WT adaptive immune cells but IFNγR−/− stromal/epithelial cells also exhibited a significantly increased infection compared to mice with a WT adaptive immune compartment and IFNγ responsive stromal/epithelial cells (Fig. 2B). In addition, comparison of the bacterial burden in the lower genital tract between days 5 and 16 revealed a significantly increased infection only for IFNγR−/− recipient mice (P < 0.01 Two-Way RM ANOVA) and not for the Rag1−/− recipients with MyD88−/− or IFNγ−/− adaptive cells (P > 0.05 Two-Way RM ANOVA). These data show that IFNγ production by innate immune cells is crucial during the early days of infection while MyD88 expression and IFNγ production by adaptive immune cells are required later.
MyD88 expression by CD4+ T cells is required for normal resolution of C. muridarum genital tract infection
The mixed bone marrow chimera experiment (Fig. 2) essentially permitted observation of the role of MyD88 in CD4+ T cells because neither a deficiency in antibody nor CD8+ T cells influences resolution of primary infection with C. muridarum (10, 78). In order to specifically analyze the role of MyD88 in CD4+ T cells, we compared the course of infection in Rag1−/− mice that received CD4+ T cells from either MyD88−/− mice or WT mice. Prior to infection, the frequency of CD3+CD4+ T cells in the peripheral blood did not significantly differ between the strains (WT: 4.96 ± 0.36%; MyD88−/−: 3.70 ± 0.61% of CD45+ cells; P > 0.05 by Student’s t-test). The course of infection in the lower genital tract was both significantly elevated (P < 0.01 by Two-way RM ANOVA) and prolonged (P < 0.001 by Log-rank Test) upon transfer of MyD88−/− CD4+ T cells relative to transfer of WT CD4+ T cells (Fig. 3A, 3B). The median day of resolution of infection for mice with WT CD4+ T cells was day 23 and with MyD88−/− CD4+ T cells, it was day 47 (Fig. 3B). However, Rag1−/− mice that did not receive a T cell transfer shed high levels of bacteria until they began to exhibit symptoms of systemic illness including tachypnea, hunching, lethargy, and death between days 14 and 25 post-infection (Fig. 3C). This was observed in a total of 15 mice from three independent experiments. A group of moribund Rag1−/− mice was sacrificed on day 25 post-infection, and Chlamydia was detected in the oviducts, lungs, liver and spleen (Fig. 3D). Thus, although clearance of infection was impaired in the absence of MyD88 in CD4+ T cells, infection eventually resolved, and dissemination of infection was prevented.
Figure 3.

MyD88 is intrinsically required in CD4+ T cells for efficient resolution of C. muridarum from the lower genital tract. (A) Rag1−/− mice were injected with 4×106 CD4+ T cells isolated from the spleens of naïve WT (black squares) or MyD88−/− mice (white circle, dashed line) and intravaginally inoculated with C. muridarum 5 weeks later. Data points represent the mean ± SEM of 5-6 mice per group from one representative experiment of three. P < 0.01 for WT CD4+ T cell recipients vs. MyD88−/− CD4+ T cell recipients over the interval measured via Two-way RM ANOVA with Bonferroni post-test. Comparison of individual days: * P < 0.05, ** P < 0.01, *** P < 0.001. (B) Infection was significantly prolonged when Rag1−/− mice were reconstituted with MyD88−/− CD4+ T cells (black line) compared to WT CD4+ T cells (dashed line). P < 0.001 by Log-rank (Mantel-Cox) Test. (C) Rag1−/− mice (white circle) infected with C. muridarum exhibited a significantly increased infection in the lower genital tract relative to C57BL/6 mice (black squares) starting on day 7. Data points represent the mean ± SEM of 4 mice per group from one representative experiment of three. *** P < 0.001 for individual days by Two-Way ANOVA with Bonferroni post-test. (D) Titration of organs from moribund Rag1−/− mice sacrificed on day 25 post infection. Bars represent the mean ± SEM of plaque assay titers from for three mice.
Accumulation of MyD88−/− CD4+ T cells is impaired relative to WT CD4+ T cells
After demonstrating a role for MyD88 in CD4+ T cells in resolution of C. muridarum infection (Fig. 3), we sought to define the mechanism responsible for this requirement. MyD88-mediated signals have been implicated in the survival of CD4+ and CD8+ T cells in other models of infection (43-45). To determine if MyD88−/− CD4+ T cells exhibited impaired accumulation in the genital tract and iliac nodes when exposed to the same inflammatory milieu as WT CD4+ T cells, we conducted a co-transfer experiment where a 1:1 ratio of MyD88−/− and WT CD4+ T cells was transferred into immunologically normal mice expressing a disparate allele of CD90 (CD90.1+) (Fig. 4A). Mice were intravaginally infected with C. muridarum at the time of T cell transfer. By day 10 post-infection, there was a significantly decreased frequency of MyD88−/− CD4+ T cells in both the iliac lymph nodes and genital tract (Fig. 4B). This difference was particularly striking in the genital tract where an average of 74% of donor cells were from the WT donor (Fig. 4B-4E). These data indicate that MyD88 expression by CD4+ T cells augments accumulation of these cells in the iliac lymph nodes and genital tract.
Fig. 4.

A significantly decreased frequency of MyD88−/− CD4+ T cells relative to WT CD4+ T cells was detected in the iliac lymph nodes and genital tract. (A) Schematic of CD4+ T cell co-transfer experiment. (B) Frequency of donor WT (CD45.1+CD45.2−CD90.2+) and MyD88−/− (CD45.1−CD45.2+CD90.2+) CD4+ T cells isolated from the iliac lymph nodes and genital tract of recipient mice (CD90.2−CD90.1+) on day 10 post infection. Bars represent the mean ± SEM of the frequency of CD3+CD4+ T cells recovered from 4 groups of 3 mice. * P < 0.05 by Mann Whitney U-test. (C,D,E) Representative flow plots of cells detected in the genital tract. (C) Gated off of forward and side scatter properties of intact cells and singlets. (D) Cells from gate created in (C). (E) Cells from gate created in (D).
Mice deficient in receptors upstream of MyD88 do not recapitulate the phenotype of MyD88−/− mice
Detection of decreased accumulation of MyD88−/− CD4+ T cells compared to WT CD4+ T cells (Fig. 4B) exposed to the same inflammatory milieu indicated that a MyD88-mediated signal might act to directly co-stimulate T cells during chlamydial infection. Signaling through several Toll-like receptors (TLRs) including TLR2 (49), TLR3 (50), TLR4 (51), TLR5 (52), TLR7 (52), and TLR9 (50) has been observed to directly co-stimulate T cells and promote their survival. We sought to determine if a deficiency in any of these receptors could recapitulate the significantly prolonged infection we observed in the absence of MyD88. We have previously observed that mice deficient in TLR2 or TLR4 resolve infection from the lower genital tract normally (5). We next infected mice deficient in both TLR2 and TLR4 in order to determine if these receptors served redundant roles; however, infection resolved with normal kinetics in the absence of both of these receptors (Fig. 5A). Although TLR3 signaling is not MyD88 dependent, we sought to determine if TLR3-mediated signals could promote resolution of infection since poly(I:C) can promote the survival of CD4+ T cells (50)(Fig. 5B), and Chlamydia has been demonstrated to stimulate TLR3 (79). Resolution of infection from mice deficient in TLR3 was normal. Bacteria in lysosomes have been shown to stimulate TLR7 (80), but we determined that TLR7 was not required for normal resolution of infection (Fig. 5C). Chlamydia possesses unmethylated deoxycytidyl-phosphate-deoxyguanosine (CpG) dinucleotides (81), which represent potential ligands for TLR9 (67). However, TLR9−/− mice also resolved infection normally (Fig. 5D). The role of TLR5 was not examined because Chlamydiae are non-motile bacteria that do not express flagellin.
Figure. 5.

TLR deficiencies do not impair resolution of C. muridarum. Mice deficient in (A) TLR2 and TLR4 (white triangles), (B) TLR3 (white diamonds, dotted line), (C) TLR7 (white circles), or (D) TLR9 (white squares) were intravaginally infected with C. muridarum, and the course of infection was compared to the appropriate control strain. C57BL/6 controls were used for all strains except for TLR3−/− mice, where the F2 generation of C57BL/6 and 129S mice was used. No differences were detected between the course of infection in the lower genital tract of any of the knockout strains and their matching control (P > 0.05 by Two-way RM ANOVA). Data points represent the mean ± SEM of 5 mice per strain and are from one representative experiment of three for the TLR2−/−TLR4−/− and TLR9−/− mice and from a single experiment with TLR7−/− and TLR3−/− mice.
MyD88 is also required for signaling via the IL-1 family of cytokine receptors, which includes receptors for IL-1, IL-18, and IL-33. Mice deficient in the IL-1 receptor exhibit an increased bacterial burden in the lower genital tract, but infection is not significantly prolonged relative to WT mice (59). We have observed that IL-18 deficient mice resolve infection from the genital tract normally (data not shown), which is in agreement with the normal resolution of infection observed in NLRP3−/− mice that have significantly impaired IL-18 production in response to Chlamydia (59). Finally, we did not pursue evaluation of IL-33 deficient mice since IL-33 induces Th2 responses and is not likely to promote resolution of chlamydial infection (32).
MyD88 deficient cells exhibit impaired survival upon activation in vitro
Our in vivo data indicate that deficiencies in receptors upstream of MyD88 do not recapitulate the phenotype of MyD88−/− mice (Fig. 5). These findings are similar to what has been described in other murine models (43, 45). We then sought to determine if impaired accumulation of MyD88−/− T cells would occur in vitro in the absence of TLR or IL-1R agonists, supporting the hypothesis that the accumulation defect observed in vivo (Fig. 4B) was independent of receptors upstream of MyD88. Naïve CD4+ T cells (CD25−CD44−) were isolated from the spleens of MyD88−/− (CD45.2) and WT (CD45.1+) mice. These cells were mixed at a 1:1 ratio and stimulated in vitro with different combinations of the following reagents: anti-CD3 (1 μg/ml), anti-CD28 (1 μg/ml), IL-2 (5 ng/ml), IL-12p70 (10 ng/ml), and anti-IL-4 (1μg/ml) (Fig. 6). A co-culture of MyD88−/− and WT T cells was performed in order to prevent confounding effects that could result from differences in the inflammatory milieu. After 3 days in culture, activated T cells (CD25+) were either examined for their viability based on Annexin V and 7-amino-actinomycin D (7-AAD) staining or were removed from culture and replated for an additional 24 hours with media alone prior to analysis of apoptosis. After 3 days and 3 days + 24 hrs rest, the level of CD25 expression by CD25+CD4+ T cells did not differ between the strains (Fig. 6A, 6B). However, the frequency of apoptotic (AnnexinV+7AAD−) CD4+CD25+ T cells was significantly increased in the absence of MyD88 under all stimulatory conditions tested (Fig. 6C, 6E, 6F). After an additional 24 hrs without stimulation, there was no longer a difference in the frequency of apoptotic cells in the group that had been stimulated with anti-CD3 alone, but the frequency of apoptotic cells was significantly increased in the absence of MyD88 under all other stimulatory conditions (Fig. 6D). The frequency of cells from either strain that up-regulated CD25 upon incubation with media alone for three days was negligible, so that group was not included in analysis of apoptosis. These data indicate that activated MyD88 deficient CD4+ T cells have an increased propensity towards apoptosis even in the absence of exogenous TLR/IL-1R ligands.
Figure 6.

MyD88−/− CD4+ T cells exhibit increased apoptosis when activated in vitro. Naïve CD4+ T cells from MyD88−/− and WT (CD45.1+) mice were co-cultured in vitro in the presence of the indicated stimulatory reagents for three days. (A, B) The level of CD25+ expression by activated CD4+ CD25+ T cells from each strain was determined (A) after 3 days or (B) after incubation with media alone for an additional 24 hrs. (C, D) The frequency of CD4+ CD25+ cells that were apoptotic (Annexin V+ 7AAD−) was determined after (C) 3 days or (D) 3 days + 24 hrs in media alone. *** P < 0.001 by Two-Way ANOVA with Bonferroni post-test. Bars represent the mean ± SEM of triplicate wells from one representative experiment of two. (E,F) Representative flow plots of T cells stimulated for 3 days with anti-CD3 and anti-CD28 antibodies. (E) Gate shown represents CD4+ cells that are CD25 high. (F) Plots were derived by first gating on CD4+CD25 high cells from (E) and then dividing that population into cells that were either CD45.2−(WT) or CD45.2+ (MyD88−/−).
DISCUSSION
In this manuscript, we present the results of our studies examining the role of the adaptor molecule MyD88 in the development of an effective adaptive immune response to Chlamydia genital tract infection. In doing so we attempt to provide a mechanism for the significantly impaired resolution of infection previously observed in MyD88 deficient mice (55, 57, 58). The increased bacterial burden observed in the absence of MyD88 was associated with the development of severe oviduct pathology (57, 58), which indicated that MyD88-dependent signaling was a double-edged component of the immune response to Chlamydia genital tract infection. Mice deficient in receptors upstream of MyD88, including TLR2 knockout mice (5) and IL-1 receptor knockout mice (59), exhibit significantly reduced oviduct pathology. However, signaling via MyD88 is necessary to effectively control infection and prevent the tissue damage that can result from a significantly increased bacterial burden. The goal of the studies outlined in this manuscript was to determine if the role of MyD88 in promoting the adaptive immune response could be separated from its involvement in promoting tissue damaging innate immune responses.
We first explored whether the prolonged infection observed in the absence of MyD88 could at least partially be attributed to a lack of MyD88 in circulating hematopoietic cells. We determined that bone marrow chimeras with a MyD88 deficient hematopoietic compartment and wild-type epithelial/stromal cells did indeed exhibit a significantly prolonged infection. Although the role of MyD88 in enhancing innate and adaptive responses to Chlamydia would seem intuitive, it was important to rule out the possibility that a deficiency in MyD88 expression by the epithelium of the genital tract was required to observe impaired resolution of infection. In vitro experiments have shown that genital tract epithelial cells produce significantly lower levels of cytokines in response to Chlamydia in the absence of MyD88 (56). In addition, MyD88 can directly interact with the IFNγ receptor (82), which could be important in IFNγ-mediated clearance of Chlamydia from epithelial cells. The significantly prolonged infection we observed for the bone marrow chimeras in the presence of a wild-type epithelium pointed to an important role for MyD88 expression by circulating immune cells, although it did not exclude the possibility that MyD88 signaling at the level of the epithelium participated in controlling infection.
An important technical point regarding the use of bone marrow chimeras in this model is that X-ray irradiation can influence the architecture of the genital tract. We observed that the bacterial burden in the lower genital tract of irradiated mice was 10 to 100 fold lower than that of non-irradiated mice at the peak of infection. In addition, at the time of sacrifice, the uterine horns and cervices of mice that had been irradiated where significantly thinner than those of non-irradiated mice. These effects may preclude the use of this type of experiment in describing immune mediators of genital tract pathology. It also prevented us from analyzing mice with a MyD88-deficient epithelium because these mice exhibit a further increase in susceptibility to radiation-induced damage due to a higher proliferative rate of epithelial cells (74). Despite these caveats, we determined that mice with MyD88 deficient hematopoietic cells exhibited a prolonged course of infection similar to that observed in MyD88−/− mice. These findings indicated that MyD88-mediated signals that directly promote epithelial damage, as has been described for IL-1 in vitro (83), could potentially be separated from MyD88-mediated responses essential for normal resolution of infection. In order to directly examine the role of MyD88 in the genital tract epithelium, a murine strain with a MyD88 deficiency solely in these epithelial cells, as has been described for epithelial cells of the gastrointestinal tract, will be required (84). We next used mixed bone marrow chimeras to more specifically explore the role of MyD88 in promoting clearance mediated by hematopoietic cells, with a focus on MyD88 expression by adaptive immune cells. The mixed bone marrow chimera experimental design was based on a previous manuscript, which showed that MyD88 expression by adaptive immune cells was required for control of Toxoplasma gondii infection (43). In these experiments, WT, MyD88−/−, or IFNγ−/− bone marrow was mixed with an equal ratio of Rag1−/− bone marrow and transferred into irradiated Rag1−/− recipients. The Rag1−/− bone marrow provided a large pool of normal innate immune cells for priming of the adaptive immune response. Mice with MyD88−/− or IFNγ−/− adaptive cells exhibited similar courses of infection, which were significantly increased compared to mice with WT adaptive cells. Although data regarding the course of infection that was obtained from independent experiments cannot be used to make definite comparisons between groups, the course of infection observed in the chimeras with a MyD88−/− adaptive immune compartment appeared to be less prolonged than that observed for the chimeras where the entire hematopoietic compartment did not express MyD88, indicating that MyD88 expression by APCs potentially plays a role in promoting resolution of Chlamydia infection. We also showed that mice with an IFNγR deficient epithelium but normal adaptive immune cells exhibited an early increase in infection that was not observed for the mice with MyD88−/− or IFNγ−/− adaptive cells but an IFNγ responsive epithelium. These findings indicate that IFNγ production by innate immune cells can contribute to early control of infection, but IFNγ provided by adaptive cells is critical for efficient resolution of infection. The mixed-bone marrow chimera experiment also revealed that the prolonged infection observed in the presence of MyD88−/− adaptive immune cells was similar to that observed when adaptive cells could not produce IFNγ. Thus, intrinsic expression of MyD88 may be required for an optimal T cell IFNγ response in the genital tract. However, this is not likely due to a decreased ability of MyD88 deficient CD4+ T cells to differentiate into Th1 cells because that has been repeatedly demonstrated to be unimpaired both in vitro and in the presence of normal APCs in vivo (45, 46, 48).
We used a CD4+ T cell transfer model to confirm our suspicions that MyD88 expression by CD4+ T cells was necessary for eradication of Chlamydia from the genital tract, since CD4+ T cells are the only adaptive immune cells required for clearance of primary infection in this model (10). Interestingly, the impaired resolution of infection observed for transfer of MyD88−/− CD4+ T cells to Rag1−/− mice was much more pronounced than what we observed in the mixed bone marrow chimera experiments. One potential explanation for this observation is that the length of infection was reduced in the bone marrow chimeras due to effects of irradiation on the architecture of the genital tract. It is also possible that since the mice were provided with a fixed number of CD4+ T cells in the T cell transfer model, they could not compensate for impairments in infection control with an increased release of adaptive immune cells from the bone marrow compartment. These findings also provided clues that impaired survival of T cells was responsible for the delayed resolution of infection, since release of cells from the bone marrow could replace failing adaptive immune cells in the bone-marrow chimera experiments, but this could not occur upon transfer of a finite number of CD4+ T cells into to the Rag−/− mice. Indeed, impaired survival of MyD88 deficient adaptive immune cells has been previously observed by others characterizing a requirement for MyD88 in CD4+ T cells (44, 46, 47) and CD8+ T cells (45).
Mechanistic experiments revealed that MyD88 deficient CD4+ T cells were impaired in their ability to accumulate in the genital tract and iliac lymph nodes when exposed to the same inflammatory milieu as WT CD4+ T cells. This is similar to what was observed in a CD4+ T cell transfer model of colitis, where naïve MyD88−/− CD4+ T cells exhibited impaired accumulation in a variety of organs when co-transferred with wild-type CD4+ T cells (47). This was also observed in mixed bone marrow chimeras infected with LCMV, where MyD88−/− CD8+ T cells exhibited significantly reduced accumulation in the spleen relative to WT T cells in the same mouse (45). Similar results were obtained upon transfer of LCMV specific MyD88 deficient and WT CD8+ T cells into WT recipients, and the detection of a significantly reduced number of MyD88 deficient CD8+ T cells responding to LCMV was associated with an increased rate of apoptosis and not a defect in proliferation (45). This would explain why we previously observed normal proliferation of Chlamydia-specific CD4+ T cells in from the iliac lymph nodes of MyD88−/− mice but a reduced frequency of these cells in the genital tract in the presence of a dramatically increased bacterial burden (58).
We attempted to find a receptor upstream of MyD88 signaling that would explain the deficiencies we observed in its absence. Our current and former studies show that mice with deficiencies in TLR2 (5), TLR2 and TLR4 (current work), TLR4 (5), TLR7 (current work), TLR9 (current work), IL-1R (59), and IL-18R (current work) do not exhibit delayed resolution of infection. These negative data could indicate that an untested TIR domain-containing receptor participates in MyD88 mediated control of infection or that a combination of deficiencies can explain this impaired resolution. However, it is also possible that MyD88 plays an unconventional role in promoting T cell survival. Our in vitro apoptosis assays were conducted without the addition of exogenous TLR/IL-1R ligands, and we still observed an increased rate of apoptosis in the absence of MyD88. Although we cannot rule out a role for autologous cytokine production or molecules released from dying cells in co-stimulating these cells, the fact that the level of CD25 expression by MyD88−/− and WT T cells was comparable under the stimulatory conditions tested, indicates that these cells were not exposed to different exogenous activating signals as would occur in the presence of signaling via mediators upstream of MyD88. Rather, these findings indicate that stimulatory signals are able to promote similar levels of activation in MyD88 deficient T cells, but defects arise after the divergence of activating and survival signals. This is similar to what was observed in a model of LCMV (45), where differentiation and activation of MyD88 deficient CD8+ T cells was normal, but accumulation was dramatically impaired. Interestingly, a number of MyD88-mediated signals that are TIR domain independent have been described such as a role for MyD88 in interacting with the IFNγR (82), Phosphatidylinositol 3-kinase (PI-3K) (85, 86), Fas-associated death domain protein (FADD)(87, 88), IFN regulatory factor (IRF)1 (89), IRF5 (89, 90), and IRF7 (91). FADD has been observed to prevent MyD88 mediated proinflammatory signals, so we could speculate that MyD88 could reciprocate by preventing FADD mediated proapoptotic signals (92). Our findings also indicate that the intrinsic requirement for MyD88 in CD4+ T cells for normal resolution of Chlamydia genital tract infection is not at specific to this model. That would explain why similar findings have been observed across several models without an upstream mechanism (43-45).
We were unable to find a receptor upstream of MyD88 that was required for resolution of Chlamydia from the genital tract. Direct stimulation of MyD88-dependent receptors on CD4+T cells does not significantly enhance protective immunity during chlamydial infection. In addition, activation of these pathways in CD4+ T cells has been shown to lead to detrimental responses in murine models, including induction of pathologic Th17 responses (48), EAE (48), and IBD (46, 47). These data indicate that activation of TLR and IL-1R receptors on CD4+ T cells should not be incorporated into a vaccination strategy. On the other hand, MyD88 augments the longevity of CD4+ T cells. In the absence of MyD88, small reductions in CD4+ T cell accumulation in the lymph nodes translate into dramatically decreased numbers of CD4+ T cells in the genital tract and impaired resolution of infection. Although the specific mechanisms whereby MyD88 promotes T cell longevity have not been determined, these studies show that MyD88 is clearly a necessary component of an effective adaptive immune response to Chlamydia. Determination of signaling pathways that promote CD4+ T cell survival would accelerate vaccine development.
ACKNOWLEDGEMENTS
We would like to thank Alison Logar for her expertise in flow cytometry and James Sikes for his technical assistance.
Footnotes
This work was supported by the NIH-NIAID via grants R01 A105624 and U19 A1084024 to T.D. and R01 A1067678 to U.N. Additional funding was provided by a grant from Children’s Hospital of Pittsburgh of the UPMC Health System to L.C.F.
REFERENCES
- 1.Haggerty CL, Gottlieb SL, Taylor BD, Low N, Xu F, Ness RB. Risk of sequelae after Chlamydia trachomatis genital infection in women. J Infect Dis. 2010;201(Suppl 2):S134–155. doi: 10.1086/652395. [DOI] [PubMed] [Google Scholar]
- 2.Lee HY, Schripsema JH, Sigar IM, Lacy SR, Kasimos JN, Murray CM, Ramsey KH. A role for CXC chemokine receptor-2 in the pathogenesis of urogenital Chlamydia muridarum infection in mice. FEMS Immunology & Medical Microbiology. 2010;60:49–56. doi: 10.1111/j.1574-695X.2010.00715.x. [DOI] [PubMed] [Google Scholar]
- 3.Imtiaz MT, Distelhorst JT, Schripsema JH, Sigar IM, Kasimos JN, Lacy SR, Ramsey KH. A role for matrix metalloproteinase-9 in pathogenesis of urogenital Chlamydia muridarum infection in mice. Microbes.Infect. 2007;9:1561–1566. doi: 10.1016/j.micinf.2007.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lacy HM, Bowlin AK, Hennings L, Scurlock AM, Nagarajan UM, Rank RG. Essential role for neutrophils in pathogenesis and adaptive immunity in Chlamydia caviae ocular infections. Infect Immun. 2011;79:1889–1897. doi: 10.1128/IAI.01257-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Darville T, O’Neill JM, Andrews CW, Jr., Nagarajan UM, Stahl L, Ojcius DM. Toll-like receptor-2, but not toll-like receptor-4, is essential for development of oviduct pathology in chlamydial genital tract infection. J Immunol. 2003;171:6187–6197. doi: 10.4049/jimmunol.171.11.6187. [DOI] [PubMed] [Google Scholar]
- 6.Frazer LC, O’Connell CM, Andrews CW, Jr., Zurenski MA, Darville T. Enhanced neutrophil longevity and recruitment contribute to the severity of oviduct pathology during Chlamydia muridarum infection. Infect Immun. 2011;79:4029–4041. doi: 10.1128/IAI.05535-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rank RG, Soderberg LS, Barron AL. Chronic chlamydial genital infection in congenitally athymic nude mice. Infect Immun. 1985;48:847–849. doi: 10.1128/iai.48.3.847-849.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morrison SG, Su H, Caldwell HD, Morrison RP. Immunity to murine Chlamydia trachomatis genital tract reinfection involves B cells and CD4(+) T cells but not CD8(+) T cells. Infection and Immunity. 2000;68:6979–6987. doi: 10.1128/iai.68.12.6979-6987.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Igietseme JU, Ramsey KH, Magee DM, Williams DM, Kincy TJ, Rank RG. Resolution of murine chlamydial genital infection by the adoptive transfer of a biovar-specific TH1 lymphocyte clone. Regional Immunology. 1993;5:317–324. [PubMed] [Google Scholar]
- 10.Morrison RP, Feilzer K, Tumas DB. Gene knockout mice establish a primary protective role for major histocompatibility complex class II-restricted responses in Chlamydia trachomatis genital tract infection. Infection and Immunity. 1995;63:4661–4668. doi: 10.1128/iai.63.12.4661-4668.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cain TK, Rank RG. Local Th1-like responses are induced by intravaginal infection of mice with the mouse pneumonitis biovar of Chlamydia trachomatis. Infection and Immunity. 1995;63:1784–1789. doi: 10.1128/iai.63.5.1784-1789.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perry LL, Feilzer K, Caldwell HD. Immunity to Chlamydia trachomatis is mediated by T helper 1 cells through IFN-gamma-dependent and -independent pathways. J.Immunol. 1997;158:3344–3352. [PubMed] [Google Scholar]
- 13.Cotter TW, Ramsey KH, Miranpuri GS, Poulsen CE, Byrne GI. Dissemination of Chlamydia trachomatis chronic genital tract infection in gamma interferon gene knockout mice. Infection and Immunity. 1997;65:2145–2152. doi: 10.1128/iai.65.6.2145-2152.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cohen CR, Koochesfahani KM, Meier AS, Shen C, Karunakaran K, Ondondo B, Kinyari T, Mugo NR, Nguti R, Brunham RC. Immunoepidemiologic profile of Chlamydia trachomatis infection: importance of heat-shock protein 60 and interferon-gamma. J Infect.Dis. 2005;192:591–599. doi: 10.1086/432070. [DOI] [PubMed] [Google Scholar]
- 15.Debattista J, Timms P, Allan J. Reduced levels of gamma-interferon secretion in response to chlamydial 60 kDa heat shock protein amongst women with pelvic inflammatory disease and a history of repeated Chlamydia trachomatis infections. Immunol Lett. 2002;81:205–210. doi: 10.1016/s0165-2478(02)00036-6. [DOI] [PubMed] [Google Scholar]
- 16.Kimani J, Maclean IW, Bwayo JJ, MacDonald K, Oyugi J, Maitha GM, Peeling RW, Cheang M, Nagelkerke NJ, Plummer FA, Brunham RC. Risk factors for Chlamydia trachomatis pelvic inflammatory disease among sex workers in Nairobi, Kenya. J.Infect.Dis. 1996;173:1437–1444. doi: 10.1093/infdis/173.6.1437. [DOI] [PubMed] [Google Scholar]
- 17.Igietseme JU, Uriri IM, Hawkins R, Rank RG. Integrin-mediated epithelial-T cell interaction enhances nitric oxide production and increased intracellular inhibition of Chlamydia. J Leukoc Biol. 1996;59:656–662. doi: 10.1002/jlb.59.5.656. [DOI] [PubMed] [Google Scholar]
- 18.Jayarapu K, Kerr M, Ofner S, Johnson RM. Chlamydia-specific CD4 T cell clones control Chlamydia muridarum replication in epithelial cells by nitric oxide-dependent and -independent mechanisms. J Immunol. 2010;185:6911–6920. doi: 10.4049/jimmunol.1002596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pasare C, Medzhitov R. Toll-dependent control mechanisms of CD4 T cell activation. Immunity. 2004;21:733–741. doi: 10.1016/j.immuni.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 20.O’Connell CM, Ionova IA, Quayle AJ, Visintin A, Ingalls RR. Localization of TLR2 and MyD88 to Chlamydia trachomatis inclusions. Evidence for signaling by intracellular TLR2 during infection with an obligate intracellular pathogen. Journal of Biological Chemistry. 2006;281:1652–1659. doi: 10.1074/jbc.M510182200. [DOI] [PubMed] [Google Scholar]
- 21.Derbigny WA, Hong SC, Kerr MS, Temkit M, Johnson RM. Chlamydia muridarum infection elicits a beta interferon response in murine oviduct epithelial cells dependent on interferon regulatory factor 3 and TRIF. Infection and Immunity. 2007;75:1280–1290. doi: 10.1128/IAI.01525-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bulut Y, Faure E, Thomas L, Karahashi H, Michelsen KS, Equils O, Morrison SG, Morrison RP, Arditi M. Chlamydial heat shock protein 60 activates macrophages and endothelial cells through Toll-like receptor 4 and MD2 in a MyD88-dependent pathway. J Immunol. 2002;168:1435–1440. doi: 10.4049/jimmunol.168.3.1435. [DOI] [PubMed] [Google Scholar]
- 23.Heine H, Muller-Loennies S, Brade L, Lindner B, Brade H. Endotoxic activity and chemical structure of lipopolysaccharides from Chlamydia trachomatis serotypes E and L2 and Chlamydophila psittaci 6BC. Eur.J Biochem. 2003;270:440–450. doi: 10.1046/j.1432-1033.2003.03392.x. [DOI] [PubMed] [Google Scholar]
- 24.Welter-Stahl L, Ojcius DM, Viala J, Girardin S, Liu W, Delarbre C, Philpott D, Kelly KA, Darville T. Stimulation of the cytosolic receptor for peptidoglycan, Nod1, by infection with Chlamydia trachomatis or Chlamydia muridarum. Cell Microbiol. 2006;8:1047–1057. doi: 10.1111/j.1462-5822.2006.00686.x. [DOI] [PubMed] [Google Scholar]
- 25.Ozinsky A, Underhill DM, Fontenot JD, Hajjar AM, Smith KD, Wilson CB, Schroeder L, Aderem A. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc.Natl.Acad.Sci.U.S.A. 2000;97:13766–13771. doi: 10.1073/pnas.250476497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taylor BD, Darville T, Ferrell RE, Kammerer CM, Ness RB, Haggerty CL. Variants in toll-like receptor 1 and 4 genes are associated with Chlamydia trachomatis among women with pelvic inflammatory disease. J Infect Dis. 2012;205:603–609. doi: 10.1093/infdis/jir822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.den Hartog JE, Lyons JM, Ouburg S, Fennema JS, de Vries HJ, Bruggeman CA, Ito JI, Pena AS, Land JA, Morre SA. TLR4 in Chlamydia trachomatis infections: knockout mice, STD patients and women with tubal factor subfertility. Drugs Today (Barc) 2009;45(Suppl B):75–82. [PubMed] [Google Scholar]
- 28.Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9:143–150. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- 29.Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, Janeway CA., Jr. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol Cell. 1998;2:253–258. doi: 10.1016/s1097-2765(00)80136-7. [DOI] [PubMed] [Google Scholar]
- 30.Jiang Z, Mak TW, Sen G, Li X. Toll-like receptor 3-mediated activation of NF-kappaB and IRF3 diverges at Toll-IL-1 receptor domain-containing adapter inducing IFN-beta. Proc Natl Acad Sci U S A. 2004;101:3533–3538. doi: 10.1073/pnas.0308496101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115–122. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- 32.Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, Zurawski G, Moshrefi M, Qin J, Li X, Gorman DM, Bazan JF, Kastelein RA. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 33.Latz E, Verma A, Visintin A, Gong M, Sirois CM, Klein DC, Monks BG, McKnight CJ, Lamphier MS, Duprex WP, Espevik T, Golenbock DT. Ligand-induced conformational changes allosterically activate Toll-like receptor 9. Nat Immunol. 2007;8:772–779. doi: 10.1038/ni1479. [DOI] [PubMed] [Google Scholar]
- 34.Motshwene PG, Moncrieffe MC, Grossmann JG, Kao C, Ayaluru M, Sandercock AM, Robinson CV, Latz E, Gay NJ. An oligomeric signaling platform formed by the Toll-like receptor signal transducers MyD88 and IRAK-4. J Biol Chem. 2009;284:25404–25411. doi: 10.1074/jbc.M109.022392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsukamoto H, Fukudome K, Takao S, Tsuneyoshi N, Kimoto M. Lipopolysaccharide-binding protein-mediated Toll-like receptor 4 dimerization enables rapid signal transduction against lipopolysaccharide stimulation on membrane-associated CD14-expressing cells. Int Immunol. 2010;22:271–280. doi: 10.1093/intimm/dxq005. [DOI] [PubMed] [Google Scholar]
- 36.Burns K, Martinon F, Esslinger C, Pahl H, Schneider P, Bodmer JL, Di Marco F, French L, Tschopp J. MyD88, an adapter protein involved in interleukin-1 signaling. J Biol Chem. 1998;273:12203–12209. doi: 10.1074/jbc.273.20.12203. [DOI] [PubMed] [Google Scholar]
- 37.Suzuki N, Suzuki S, Duncan GS, Millar DG, Wada T, Mirtsos C, Takada H, Wakeham A, Itie A, Li S, Penninger JM, Wesche H, Ohashi PS, Mak TW, Yeh WC. Severe impairment of interleukin-1 and Toll-like receptor signalling in mice lacking IRAK-4. Nature. 2002;416:750–756. doi: 10.1038/nature736. [DOI] [PubMed] [Google Scholar]
- 38.Muzio M, Ni J, Feng P, Dixit VM. IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signaling. Science. 1997;278:1612–1615. doi: 10.1126/science.278.5343.1612. [DOI] [PubMed] [Google Scholar]
- 39.Lin SC, Lo YC, Wu H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature. 2010;465:885–890. doi: 10.1038/nature09121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.von Bernuth H, Picard C, Puel A, Casanova JL. Experimental and natural infections in MyD88- and IRAK-4-deficient mice and humans. Eur J Immunol. 2012;42:3126–3135. doi: 10.1002/eji.201242683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.von Bernuth H, Picard C, Jin Z, Pankla R, Xiao H, Ku CL, Chrabieh M, Mustapha IB, Ghandil P, Camcioglu Y, Vasconcelos J, Sirvent N, Guedes M, Vitor AB, Herrero-Mata MJ, Arostegui JI, Rodrigo C, Alsina L, Ruiz-Ortiz E, Juan M, Fortuny C, Yague J, Anton J, Pascal M, Chang HH, Janniere L, Rose Y, Garty BZ, Chapel H, Issekutz A, Marodi L, Rodriguez-Gallego C, Banchereau J, Abel L, Li X, Chaussabel D, Puel A, Casanova JL. Pyogenic bacterial infections in humans with MyD88 deficiency. Science. 2008;321:691–696. doi: 10.1126/science.1158298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Picard C, Puel A, Bonnet M, Ku CL, Bustamante J, Yang K, Soudais C, Dupuis S, Feinberg J, Fieschi C, Elbim C, Hitchcock R, Lammas D, Davies G, Al-Ghonaium A, Al-Rayes H, Al-Jumaah S, Al-Hajjar S, Al-Mohsen IZ, Frayha HH, Rucker R, Hawn TR, Aderem A, Tufenkeji H, Haraguchi S, Day NK, Good RA, Gougerot-Pocidalo MA, Ozinsky A, Casanova JL. Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science. 2003;299:2076–2079. doi: 10.1126/science.1081902. [DOI] [PubMed] [Google Scholar]
- 43.LaRosa DF, Stumhofer JS, Gelman AE, Rahman AH, Taylor DK, Hunter CA, Turka LA. T cell expression of MyD88 is required for resistance to Toxoplasma gondii. Proc Natl Acad Sci U S A. 2008;105:3855–3860. doi: 10.1073/pnas.0706663105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou S, Kurt-Jones EA, Cerny AM, Chan M, Bronson RT, Finberg RW. MyD88 intrinsically regulates CD4 T-cell responses. J Virol. 2009;83:1625–1634. doi: 10.1128/JVI.01770-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rahman AH, Cui W, Larosa DF, Taylor DK, Zhang J, Goldstein DR, Wherry EJ, Kaech SM, Turka LA. MyD88 plays a critical T cell-intrinsic role in supporting CD8 T cell expansion during acute lymphocytic choriomeningitis virus infection. J Immunol. 2008;181:3804–3810. doi: 10.4049/jimmunol.181.6.3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fukata M, Breglio K, Chen A, Vamadevan AS, Goo T, Hsu D, Conduah D, Xu R, Abreu MT. The myeloid differentiation factor 88 (MyD88) is required for CD4+ T cell effector function in a murine model of inflammatory bowel disease. J Immunol. 2008;180:1886–1894. doi: 10.4049/jimmunol.180.3.1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tomita T, Kanai T, Fujii T, Nemoto Y, Okamoto R, Tsuchiya K, Totsuka T, Sakamoto N, Akira S, Watanabe M. MyD88-dependent pathway in T cells directly modulates the expansion of colitogenic CD4+ T cells in chronic colitis. J Immunol. 2008;180:5291–5299. doi: 10.4049/jimmunol.180.8.5291. [DOI] [PubMed] [Google Scholar]
- 48.Chang J, Burkett PR, Borges CM, Kuchroo VK, Turka LA, Chang CH. MyD88 is essential to sustain mTOR activation necessary to promote T helper 17 cell proliferation by linking IL-1 and IL-23 signaling. Proc Natl Acad Sci U S A. 2013;110:2270–2275. doi: 10.1073/pnas.1206048110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Imanishi T, Hara H, Suzuki S, Suzuki N, Akira S, Saito T. Cutting Edge: TLR2 directly triggers Th1 effector functions. J.Immunol. 2007;178:6715–6719. doi: 10.4049/jimmunol.178.11.6715. [DOI] [PubMed] [Google Scholar]
- 50.Gelman AE, Zhang J, Choi Y, Turka LA. Toll-like receptor ligands directly promote activated CD4+ T cell survival. J.Immunol. 2004;172:6065–6073. doi: 10.4049/jimmunol.172.10.6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reynolds JM, Martinez GJ, Chung Y, Dong C. Toll-like receptor 4 signaling in T cells promotes autoimmune inflammation. Proc Natl Acad Sci U S A. 2012;109:13064–13069. doi: 10.1073/pnas.1120585109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Caron G, Duluc D, Fremaux I, Jeannin P, David C, Gascan H, Delneste Y. Direct stimulation of human T cells via TLR5 and TLR7/8: flagellin and R-848 up-regulate proliferation and IFN-gamma production by memory CD4+ T cells. J Immunol. 2005;175:1551–1557. doi: 10.4049/jimmunol.175.3.1551. [DOI] [PubMed] [Google Scholar]
- 53.Takeda K, Tsutsui H, Yoshimoto T, Adachi O, Yoshida N, Kishimoto T, Okamura H, Nakanishi K, Akira S. Defective NK cell activity and Th1 response in IL-18-deficient mice. Immunity. 1998;8:383–390. doi: 10.1016/s1074-7613(00)80543-9. [DOI] [PubMed] [Google Scholar]
- 54.Tominaga K, Yoshimoto T, Torigoe K, Kurimoto M, Matsui K, Hada T, Okamura H, Nakanishi K. IL-12 synergizes with IL-18 or IL-1beta for IFN-gamma production from human T cells. Int Immunol. 2000;12:151–160. doi: 10.1093/intimm/12.2.151. [DOI] [PubMed] [Google Scholar]
- 55.Nagarajan UM, Ojcius DM, Stahl L, Rank RG, Darville T. Chlamydia trachomatis induces expression of IFN-gamma-inducible protein 10 and IFN-beta independent of TLR2 and TLR4, but largely dependent on MyD88. J Immunol. 2005;175:450–460. doi: 10.4049/jimmunol.175.1.450. [DOI] [PubMed] [Google Scholar]
- 56.Derbigny WA, Kerr MS, Johnson RM. Pattern recognition molecules activated by Chlamydia muridarum infection of cloned murine oviduct epithelial cell lines. The Journal of Immunology. 2005;175:6065–6075. doi: 10.4049/jimmunol.175.9.6065. [DOI] [PubMed] [Google Scholar]
- 57.Chen L, Lei L, Chang X, Li Z, Lu C, Zhang X, Wu Y, Yeh IT, Zhong G. Mice deficient in MyD88 Develop a Th2-dominant response and severe pathology in the upper genital tract following Chlamydia muridarum infection. J Immunol. 2010;184:2602–2610. doi: 10.4049/jimmunol.0901593. [DOI] [PubMed] [Google Scholar]
- 58.Nagarajan UM, Sikes J, Prantner D, Andrews CW, Jr., Frazer L, Goodwin A, Snowden JN, Darville T. MyD88 deficiency leads to decreased NK cell gamma interferon production and T cell recruitment during Chlamydia muridarum genital tract infection, but a predominant Th1 response and enhanced monocytic inflammation are associated with infection resolution. Infect Immun. 2011;79:486–498. doi: 10.1128/IAI.00843-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nagarajan UM, Sikes JD, Yeruva L, Prantner D. Significant Role of IL-1 Signaling, but Limited Role of Inflammasome Activation, in Oviduct Pathology during Chlamydia muridarum Genital Infection. J Immunol. 2012;188:2866–2875. doi: 10.4049/jimmunol.1103461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Riley MM, Zurenski MA, Frazer LC, O’Connell CM, Andrews CW, Jr., Mintus M, Darville T. The recall response induced by genital challenge with Chlamydia muridarum protects the oviduct from pathology but not from reinfection. Infect Immun. 2012;80:2194–2203. doi: 10.1128/IAI.00169-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gondek DC, Olive AJ, Stary G, Starnbach MN. CD4+ T cells are necessary and sufficient to confer protection against Chlamydia trachomatis infection in the murine upper genital tract. J Immunol. 2012;189:2441–2449. doi: 10.4049/jimmunol.1103032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.O’Connell CM, Ingalls RR, Andrews CW, Jr., Skurlock AM, Darville T. Plasmid-deficient Chlamydia muridarum fail to induce immune pathology and protect against oviduct disease. Journal of Immunology. 2007;179:4027–4034. doi: 10.4049/jimmunol.179.6.4027. [DOI] [PubMed] [Google Scholar]
- 63.O’Connell CM, Nicks KM. A plasmid-cured Chlamydia muridarum strain displays altered plaque morphology and reduced infectivity in cell culture. Microbiology. 2006;152:1601–1607. doi: 10.1099/mic.0.28658-0. [DOI] [PubMed] [Google Scholar]
- 64.O’Connell CM, Abdelrahman YM, Green E, Darville HK, Saira K, Smith B, Darville T, Scurlock AM, Meyer CR, Belland RJ. TLR2 activation by Chlamydia trachomatis is plasmid-dependent and plasmid-responsive chromosomal loci are coordinately regulated in response to glucose limitation by C. trachomatis but not by C. muridarum. Infect Immun. 2011;79:1044–1056. doi: 10.1128/IAI.01118-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Darville T, Andrews CW, Jr., Laffoon KK, Shymasani W, Kishen LR, Rank RG. Mouse strain-dependent variation in the course and outcome of chlamydial genital tract infection is associated with differences in host response. Infect Immun. 1997;65:3065–3073. doi: 10.1128/iai.65.8.3065-3073.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo H, Hoshino K, Horiuchi T, Tomizawa H, Takeda K, Akira S. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol. 2002;3:196–200. doi: 10.1038/ni758. [DOI] [PubMed] [Google Scholar]
- 67.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 68.Tuffrey M, Taylor-Robinson D. Progesterone as a key factor in the development of a mouse model for genital-tract infection with Chlamydia trachomatis. FEMS Microbiol.Let. 1981;12:111–115. [Google Scholar]
- 69.Kelly KA, Robinson EA, Rank RG. Initial route of antigen administration alters the T-cell cytokine profile produced in response to the mouse pneumonitis biovar of Chlamydia trachomatis following genital infection. Infection and Immunity. 1996;64:4976–4983. doi: 10.1128/iai.64.12.4976-4983.1996. published erratum appears in Infect Immun 1997 Jun;65(6):2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Scurlock AM, Frazer LC, Andrews CW, Jr., O’Connell CM, Foote IP, Bailey SL, Chandra-Kuntal K, Kolls JK, Darville T. IL-17 contributes to generation of Th1 immunity and neutrophil recruitment during Chlamydia muridarum genital tract infection but is not required for macrophage influx or normal resolution of infection. Infect Immun. 2011;79:1349–1362. doi: 10.1128/IAI.00984-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Swanson J, Eschenbach DA, Alexander ER, Holmes KK. Light and electron microscopic study of Chlamydia trachomatis infection of the uterine cervix. J.Infect.Dis. 1975;131:678–687. doi: 10.1093/infdis/131.6.678. [DOI] [PubMed] [Google Scholar]
- 72.Kiviat NB, Wolner-Hanssen P, Peterson M, Wasserheit J, Stamm WE, Eschenbach DA, Paavonen J, Lingenfelter J, Bell T, Zabriskie V. Localization of Chlamydia trachomatis infection by direct immunofluorescence and culture in pelvic inflammatory disease. Am J Obstet.Gynecol. 1986;154:865–873. doi: 10.1016/0002-9378(86)90473-4. [DOI] [PubMed] [Google Scholar]
- 73.Morrison RP, Caldwell HD. Immunity to Murine Chlamydial Genital Infection. Infection and Immunity. 2002;70:2741–2751. doi: 10.1128/IAI.70.6.2741-2751.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 75.Hao W, Liu Y, Liu S, Walter S, Grimm MO, Kiliaan AJ, Penke B, Hartmann T, Rube CE, Menger MD, Fassbender K. Myeloid differentiation factor 88-deficient bone marrow cells improve Alzheimer’s disease-related symptoms and pathology. Brain. 2011;134:278–292. doi: 10.1093/brain/awq325. [DOI] [PubMed] [Google Scholar]
- 76.Zhang Y, Jones M, McCabe A, Winslow GM, Avram D, Macnamara KC. MyD88 Signaling in CD4 T Cells Promotes IFN-gamma Production and Hematopoietic Progenitor Cell Expansion in Response to Intracellular Bacterial Infection. J Immunol. 2013;190:4725–4735. doi: 10.4049/jimmunol.1203024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rank RG, Ramsey KH, Pack EA, Williams DM. Effect of gamma interferon on resolution of murine chlamydial genital infection. Infect Immun. 1992;60:4427–4429. doi: 10.1128/iai.60.10.4427-4429.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Su H, Feilzer K, Caldwell HD, Morrison RP. Chlamydia trachomatis genital tract infection of antibody-deficient gene knockout mice. Infection and Immunity. 1997;65:1993–1999. doi: 10.1128/iai.65.6.1993-1999.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Derbigny WA, Shobe LR, Kamran JC, Toomey KS, Ofner S. Identifying a role for Toll-like receptor 3 in the innate immune response to Chlamydia muridarum infection in murine oviduct epithelial cells. Infect Immun. 2012;80:254–265. doi: 10.1128/IAI.05549-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mancuso G, Gambuzza M, Midiri A, Biondo C, Papasergi S, Akira S, Teti G, Beninati C. Bacterial recognition by TLR7 in the lysosomes of conventional dendritic cells. Nat Immunol. 2009;10:587–594. doi: 10.1038/ni.1733. [DOI] [PubMed] [Google Scholar]
- 81.Ouburg S, Lyons JM, Land JA, den Hartog JE, Fennema JS, de Vries HJ, Bruggeman CA, Ito JI, Pena AS, Lundberg PS, Morre SA. TLR9 KO mice, haplotypes and CPG indices in Chlamydia trachomatis infection. Drugs Today (Barc) 2009;45(Suppl B):83–93. [PubMed] [Google Scholar]
- 82.Sun D, Ding A. MyD88-mediated stabilization of interferon-gamma-induced cytokine and chemokine mRNA. Nat Immunol. 2006;7:375–381. doi: 10.1038/ni1308. [DOI] [PubMed] [Google Scholar]
- 83.Hvid M, Baczynska A, Deleuran B, Fedder J, Knudsen HJ, Christiansen G, Birkelund S. Interleukin-1 is the initiator of fallopian tube destruction during Chlamydia trachomatis infection. Cell Microbiol. 2007;9:2795–2803. doi: 10.1111/j.1462-5822.2007.00996.x. [DOI] [PubMed] [Google Scholar]
- 84.Kirkland D, Benson A, Mirpuri J, Pifer R, Hou B, DeFranco AL, Yarovinsky F. B cell-intrinsic MyD88 signaling prevents the lethal dissemination of commensal bacteria during colonic damage. Immunity. 2012;36:228–238. doi: 10.1016/j.immuni.2011.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ojaniemi M, Glumoff V, Harju K, Liljeroos M, Vuori K, Hallman M. Phosphatidylinositol 3-kinase is involved in Toll-like receptor 4-mediated cytokine expression in mouse macrophages. Eur J Immunol. 2003;33:597–605. doi: 10.1002/eji.200323376. [DOI] [PubMed] [Google Scholar]
- 86.Gelman AE, LaRosa DF, Zhang J, Walsh PT, Choi Y, Sunyer JO, Turka LA. The adaptor molecule MyD88 activates PI-3 kinase signaling in CD4+ T cells and enables CpG oligodeoxynucleotide-mediated costimulation. Immunity. 2006;25:783–793. doi: 10.1016/j.immuni.2006.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Aliprantis AO, Yang RB, Weiss DS, Godowski P, Zychlinsky A. The apoptotic signaling pathway activated by Toll-like receptor-2. EMBO J. 2000;19:3325–3336. doi: 10.1093/emboj/19.13.3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Horng T, Medzhitov R. Drosophila MyD88 is an adapter in the Toll signaling pathway. Proc Natl Acad Sci U S A. 2001;98:12654–12658. doi: 10.1073/pnas.231471798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Negishi H, Fujita Y, Yanai H, Sakaguchi S, Ouyang X, Shinohara M, Takayanagi H, Ohba Y, Taniguchi T, Honda K. Evidence for licensing of IFN-gamma-induced IFN regulatory factor 1 transcription factor by MyD88 in Toll-like receptor-dependent gene induction program. Proc Natl Acad Sci U S A. 2006;103:15136–15141. doi: 10.1073/pnas.0607181103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Takaoka A, Yanai H, Kondo S, Duncan G, Negishi H, Mizutani T, Kano S, Honda K, Ohba Y, Mak TW, Taniguchi T. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature. 2005;434:243–249. doi: 10.1038/nature03308. [DOI] [PubMed] [Google Scholar]
- 91.Honda K, Yanai H, Mizutani T, Negishi H, Shimada N, Suzuki N, Ohba Y, Takaoka A, Yeh WC, Taniguchi T. Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in Toll-like receptor signaling. Proc Natl Acad Sci U S A. 2004;101:15416–15421. doi: 10.1073/pnas.0406933101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bannerman DD, Tupper JC, Kelly JD, Winn RK, Harlan JM. The Fas-associated death domain protein suppresses activation of NF-kappa B by LPS and IL-1 beta. J Clin Invest. 2002;109:419–425. doi: 10.1172/JCI14774. [DOI] [PMC free article] [PubMed] [Google Scholar]