

Abstract
Increasing evidence indicates that myeloid-derived suppressor cells (MDSCs) negatively regulate immune responses during tumor progression, inflammation and infection. However, the underlying molecular mechanisms of their development and mobilization remain to be fully delineated. Kruppel-like factor KLF4 is a transcription factor that has an oncogenic function in breast cancer development, but its function in tumor microenvironment, a critical component for tumorigenesis, has not been examined. By using a spontaneously metastatic 4T1 breast cancer mouse model and an immunodeficient NOD/SCID mouse model, we demonstrated that KLF4 knockdown delayed tumor development and inhibited pulmonary metastasis, which was accompanied by decreased accumulation of MDSCs in bone marrow, spleens and primary tumors. Mechanistically, we found that KLF4 knockdown resulted in a significant decrease of circulating GM-CSF, an important cytokine for MDSC biology. Consistently, recombinant GM-CSF restored the frequency of MDSCs in purified bone marrow cells incubated with conditioned medium from KLF4 deficient cells. In addition, we identified CXCL5 as a critical mediator to enhance the expression and function of GM-CSF. Reduced CXCL5 expression by KLF4 knockdown in primary tumors and breast cancer cells was correlated with a decreased GM-CSF expression in our mouse models. Finally, we found that CXCL5/CXCR2 axis facilitated MDSC migration and that anti-GM-CSF antibodies neutralized CXCL5-induced accumulation of MDSCs. Taken together, our data suggest that KLF4 modulates maintenance of MDSCs in bone marrow by inducing GM-CSF production via CXCL5 and regulates recruitment of MDSCs into the primary tumors through the CXCL5/CXCR2 axis, both of which contribute to KLF4-mediated mammary tumor development.
Keywords: KLF4, Myeloid-derived suppressor cells, Tumor development, GM-CSF, CXCL5
Introduction
Kruppel-like factor 4 (KLF4) is a transcription factor originally cloned by three groups 1–3. It regulates expression of many genes involved in cell cycle progression and epithelial differentiation 4, 5. KLF4 has been reported to function as a tumor suppressor in a variety of tumors, including colon adenomas, gastric cancer, and intestinal adenomas 6. However, KLF4 is overexpressed in up to 70% of primary breast ductal carcinoma 7, suggesting an oncogenic role of KLF4 in breast cancer development. Our recent studies indicate that KLF4 is required for the maintenance of breast cancer stem cells 8, thus revealing a novel mechanism by which KLF4 promotes breast tumorigenesis. However, other mechanisms including the role of KLF4 in the primary tumor microenvironment in KLF4-mediated tumorigenesis remain unknown.
Myeloid-derived suppressor cells (MDSCs), as mainly characterized by CD11b+ and Gr-1+ double positive myeloid cells in mice, have been identified as one of counter-regulatory mechanisms in inflammatory diseases and tumors 9, 10. MDSCs reside in bone marrow, peripheral blood, lymphoid tissues, and tumor tissues, and are closely correlated with tumor burden in a number of experimental models 11–15. A hallmark of MDSCs is the heterogeneity of their morphology, phenotypes and functions 9, 16. It has been reported that GM-CSF is one of the important cytokines for the maturation and homeostasis of MDSCs in human and mouse 17, 18. Recruitment of MDSCs into the tumor involves the CXCL5/CXCR2 and SDF-1/CXCR4 chemokine receptor axes in a transforming growth factor beta (TGF-β) deletion mouse model 19. More mechanistic studies on MDSC biology at a molecular level are clearly needed because of the importance of MDSCs in tumor biology and immunology and the complexity of MDSCs 9.
Inflammation as an inducer of MDSCs has been linked to carcinogenesis in a number of epithelial tissues 20. The relationship between KLF4 and inflammation has recently been established as shown by KLF4 acting on T cells, monocytes, macrophage, and Th17 cells 21–25. However, whether KLF4 regulates MDSCs remains unknown. We recently found that histidine decarboxylase (HDC) as a target of KLF4 26 is primarily expressed in CD11b+Ly6G+ immature myeloid cells, which also represent MDSCs, within the bone marrow 27. HDC knockout mice exhibited an inflammation-associated carcinogenesis phenotype through reduced myeloid maturation of CD11b+Ly6G+ cells. These observations suggest a possible regulation of MDSCs by KLF4 during tumorigenesis and led us to hypothesize that KLF4 regulation of MDSCs is one of the mechanisms whereby KLF4 promotes breast cancer development. Indeed, our current study using KLF4 knockdown breast cancer models suggest that KLF4-mediated regulation of MDSCs plays a critical role in mammary tumor growth and pulmonary metastasis.
Methods
Generation of KLF4 knockdown stable 4T1 cells
Plasmids containing KLF4 specific shRNA (psiLv-U6-KLF4, designated as siKLF4) or the control shRNA vector (psiLv-U6, designated as siCon) were obtained from GeneCopoeia. 1.6 μg plasmids were transfected into mouse 4T1 cells using Lipofectamine™ 2000 (Invitrogen). 24 h after transfection, cells were trypsinized and reseeded into 10-cm culture plates. 4T1 cells (4T1-Luc2) were purchased from Caliper LifeSciences. Cells were selected with puromycin at a final concentration of 2μg/ml for 3 weeks. KLF4 knockdown human breast cancer MDA-MB-231 stable cells were generated as previously described 8. The 4T1 cells and MDA-MB-231 cells were maintained in RPMI-1640 or Dulbecco’s modified Eagle’s medium respectively, supplemented with 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% heat-inactivated FBS (Invitrogen) at 37°C in a humidified atmosphere of 5% CO2. To collect supernatant, 1× 106 siCon and siKLF4 4T1 cells were cultured in 6-well plates in RPMI-1640 medium supplemented with 10% FBS. Supernatant was then collected after 24 h seeding.
4T1 metastatic mouse model
Five female BALB/c mice in each group were inoculated in the abdominal mammary glands with 1 × 105 4T1 cells. Tumor growth was monitored and tumor volume was calculated as described previously 8. All mouse procedures have been approved by IACUC at University of South Carolina. To test the role of MDSCs in tumor development, 5 × 105 4T1 cells, without or with MDSCs (1 × 105) derived from siCon cell- and siKLF4 cell-inoculated mice were injected into the mammary gland. The tumor weight and lung nodules were recorded. The same experiment was repeated four times.
Nonobese diabetic/severe combined immune deficiency (NOD/SCID) mouse tumor model
siKLF4 and siCon MDA-MB-231 stable cell lines were generated as previously described 8. Both cell lines were diluted to 5 × 105 cells per 20 μl in PBS. Five NOD/SCID female mice (National Cancer Institute, Fredrick, MD) in the age of 6–8 weeks were injected with siKLF4 or siCon MDA-MB-231 cells subcutaneously into the mammary fat pad number four. 28 days after tumor cell implantation, bone marrow, spleens and tumor cells were collected and stained with anti-CD11b-FITC and anti–Gr-1-PE antibodies. Representative results after flow cytometric analysis were shown from five independent experiments.
In vitro culture of bone marrow cells
Bone marrow cells from siCon cell- and siKLF4 cell-inoculated mice were extracted. 1 × 108 bone marrow cells were sequentially incubated and purified with 25 μl Biotin-conjugated Gr-1 Ab and 200 μl anti-Biotin microbeads (Miltenyi Biotech). MDSCs were cultured in 10-cm plates (5× 106 cells/plate) for a total of 6 days. Recombinant mouse GM-CSF (100 ng/ml), IL-4 (50 ng/ml), or IL-6 (50 ng/ml) (Sigma-Aldrich) were added directly to the culture medium on day 0. For MDSC maintenance, 1 × 107 bone marrow cells were cultured with CXCL5 (25 ng/ml) and mouse anti-GM-CSF monoclonal antibody (250 ng/ml, ab9471) or mouse IgG (250 ng/ml) (eBioscience). 6 days later, bone marrow cells were collected and MDSC population was detected by FACS. To examine GM-CSF expression in bone marrow, mammary tumor tissues (50 mg) from siCon and siKLF4 cell-inoculated mice were cut into 1 mm × 1 mm pieces and incubated with 1 × 106 bone marrow cells. 24 h later, bone marrow cells were collected and RT-PCR was performed.
T cell suppression assay
Splenocytes were isolated from wild type BALB/c mice and CD4+ T or CD8+ cells were sorted using Miltenyi Biotech magnetic beads as described in Materials and Methods of the main text. Different numbers of gamma-irradiated (9 Gy) MDSCs derived from siCon cell- and siKLF4 cell-inoculated mouse spleens or tumors, as indicated in the figures, were cocultured with purified CD4+ T cells (5 × 105) or CD8+ cells (1 × 106) stimulated with Con A (5 μg/mL) in 24-well plates. T-cell proliferation was determined after 72 h culture by pulsing with 1 μCi/well [3H] thymidine (PerkinElmer Life Sciences, Boston, MA) during the final 12 h of culture. Cultures were harvested and thymidine incorporation was measured by scintillation counting (Perkin Elmer). Data are expressed as cpm (mean ± SE) of triplicate cultures. Three independent experiments were performed.
Arginase activity
1 × 106 gamma-irradiated MDSCs derived from siCon cell- and siKLF4 cell-inoculated mouse spleens were cultured in 24-well plates for 24 h. Cells were collected and lysed with 200 μl of lysis buffer (0.1% Triton X-100 plus 1 tablet of protease inhibitor mixture). 10 μl of 10 mM MnCl2 was added. Arginase was activated by heating the solution for 10 min at 55°C. The lysate was incubated with 100 μl of 0.5 M L-arginine (pH 9.7) for 1 h at 37°C. The reaction was stopped with 800 μl stop solution (96% H2SO4:85% H3PO4:H2O ratio, 1:3:7). The urea concentration was measured at 540 nm after addition of 40 μl of a-isonitrosopropiophenone, followed by heating at 100°C for 30 min. A standard curve consisting of serial dilutions of urea was run in parallel. Data are presented as mean ± SE of triplicate cultures. Three independent experiments were performed.
Immunohistochemistry (IHC) and Western blotting
Paraffin-embedded tumor sections were fixed in 4% paraformaldehyde and incubated with a Biotin-conjugated Gr-1 Ab (BD PharMingen). A streptavidin-conjugated HRP was applied and peroxidase activity was localized with diaminobenzidine (Vectastain ABC kit, Vector Laboratories). CXCL5 and GM-CSF expression in tumor and lung tissues of siCon and siKLF4 cell-inoculated mice was examined by Western blotting. The antibodies used were as follows: rabbit polyclonal anti-CXCL5 (ab18134, 1:1000, Abcam), rabbit polyclonal anti-GM-CSF (ab9471, 1:1000, Abcam) and rabbit polyclonal anti-β-actin (1:1000, Sigma). β-actin was used as an internal control.
In Vitro cell migration assay
For supernatant-mediated migration, 2 × 105 sorted MDSCs from spleen of BALB/c mice were seeded onto the top chamber of transwell insert with 8 μm pore size (Corning Life Sciences). The inserts were placed into 24-well plates that contained supernatants of siCon and siKLF4 4T1 cells with recombinant mouse GM-CSF (100 ng/ml), IL-4 (50 ng/ml), or IL-6 (50 ng/ml). Migrated MDSCs were counted (ten fields per well, triplicate for each experimental groups) 8 h after incubation.
Flow cytometric assay to measure MDSC population
Flow cytometric assay was performed as previously described8. Specifically, single live cells were gated as determined by forward and side scattering, and FITC-CD11b and PE-Gr-1 antibodies were used to measure the percentage of CD11b+/Gr-1+ MDSCs.
Immunofluorescence
Primary tumor tissues were fixed in 4% paraformaldehyde at 4°C for 12 h, dehydrated in 30% sucrose overnight, and embedded in OCT. Serial sections (8 μm thick) were cut throughout the entire tumor tissues. Frozen sections were fixed in acetone, incubated with 5% BSA/PBS, and incubated with FITC-conjugated rat anti-CD3 antibody (1:100, eBioseicence, San Diego, CA) overnight at 4°C. Slides were mounted in ProLong Gold Mounting Medium containing DAPI (Invitrogen). The tissue sections were visualized and recorded under a Nikon ECLIPSE E600 microscope (Nikon Inc. Melville, NY).
Cytokine analysis
Supernatants of 4T1 cells and sera from tumor-bearing mice were collected. Cytokine concentration was determined using Bio-plex chemiluminescence assay system according to the manufacturer’s instructions (Bio-Rad).
Quantitative real-time RT-PCR (qRT-PCR)
Total RNA was prepared using Trizol Reagent (Invitrogen). First-strand cDNA synthesis was performed using Oligo (dT) and Superscript III reverse transcriptase (Invitrogen). Quantitative real-time PCR was performed as described previously 8. Quantitative real-time PCR primer sequences were listed in Supplementary Table 1.
Statistical analysis
Data were expressed as mean ± SD from three independent experiments. Statistical analysis was performed using Student’s t test to assess differences between experimental groups. P < 0.05 was considered statistically significant.
Results
KLF4 knockdown suppresses mammary tumor growth and pulmonary metastasis
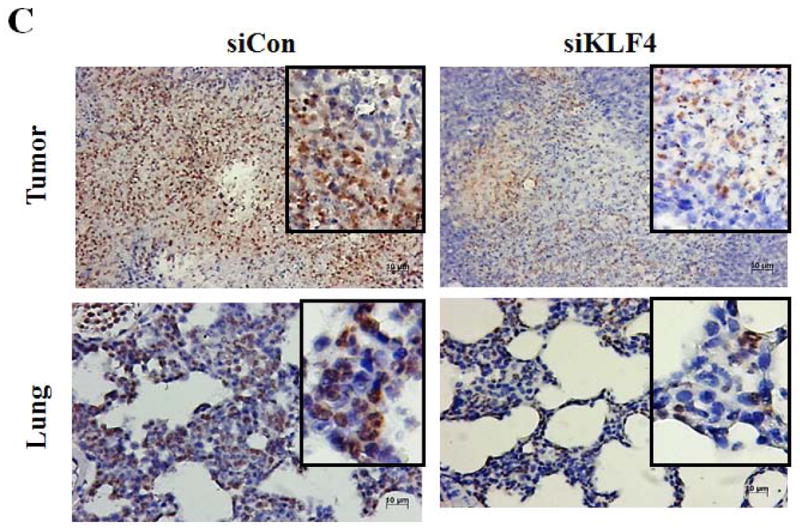
Recent studies have shown that MDSCs promote tumor growth and metastasis in several tumor models 28. To establish a functional link between KLF4 and MDSCs, we used a 4T1 orthotopic mammary tumor model, which shares many characteristics with human breast cancer, particularly its ability to spontaneously metastasize to lungs. KLF4 knockdown and control siRNA stable 4T1 cells (designated as siKLF4 and siCon respectively) were generated (Supplementary Fig 1). As shown in Figure 1A, tumors in siCon cell-inoculated congenic BALB/c mice were observed as early as day 9 and the tumor size reached to 18.2 ± 1.6 mm in diameter. However, the primary mammary tumors in siKLF4 cell-inoculated mice were detectable on day 14 and the tumor size was only 11.3 ± 1.4 mm in diameter (Figure 1A and B). We next examined whether KLF4 expression contributed to pulmonary metastasis. Haematoxylin and eosin (HE) staining showed that the tumors formed in siCon cell-inoculated mice gave rise to 13.2 ± 1.8 metastatic nodules per lung (Figure 1C). However, there were much fewer metastatic nodules per lung (2.3 ± 0.4) in mice implanted with siKLF4 cells (Figure 1C and D). These data were in agreement with our previous results showing that KLF4 knockdown delayed the onset of mammary tumor development and inhibited lung metastasis in immunodeficient NOD/SCID mice inoculated with MDA-MB-231 human breast cancer cells 8.
Figure 1. KLF4 knockdown delays growth of 4T1 primary tumors and suppresses lung metastasis.
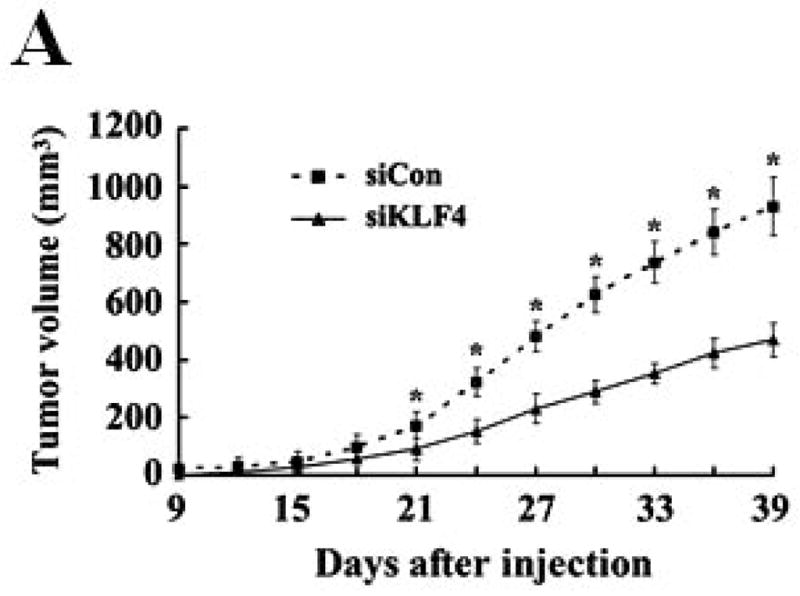
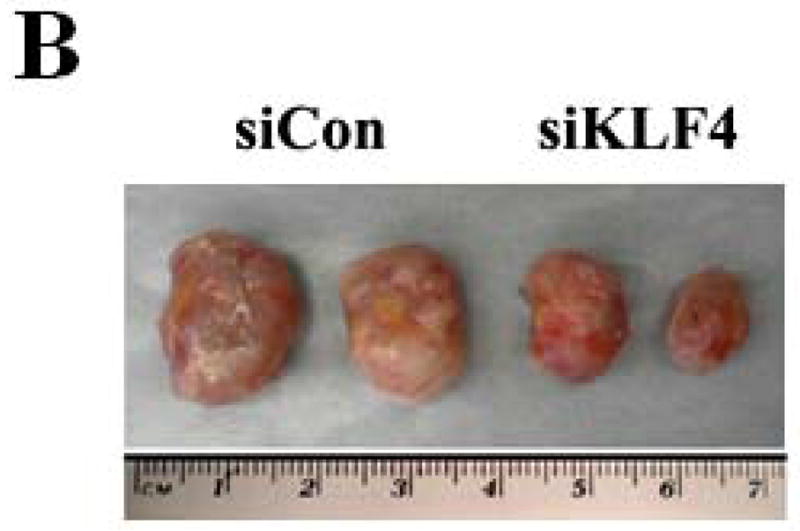

(A) Tumor growth was monitored in BALB/c mice implanted with control and KLF4 knockdown 4T1 cells (designated as siCon and siKLF4, respectively). Tumor size was measured every 3 days and tumor volume was calculated. Data were expressed as mean ± SD from three independent experiments. * indicates P < 0.05 versus siCon group; (B) Representative tumors in above mice were shown. (C) Numbers of lung nodules in above tumor-bearing mice were plotted. Data were expressed as mean ± SD from three independent experiments. * indicates P < 0.05 versus siCon group. (D) Representative lung metastases in above mice were shown. Scale bar, 20 μm. Arrows indicated the lung metastases.
Knockdown of KLF4 reduces MDSC accumulation and activities
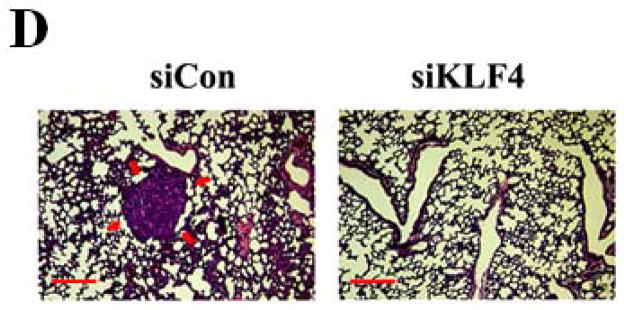
MDSCs are significantly expanded in bone marrow and spleens of tumor-bearing mice as well as in cancer patients of all stages 29. To examine whether MDSCs were involved in KLF4-mediated tumor progression, MDSC populations in bone marrow, spleen and tumor were examined by flow cytometric analysis. Nine days after implantation of 4T1 cells, KLF4 knockdown significantly decreased the percentage of MDSCs in bone marrow and spleen when compared to siCon counterparts (Figure 2A and B). This phenomenon disappeared on day 28 (data not shown). However, at this time point, KLF4 knockdown apparently inhibited MDSC accumulation in tumors (Figure 2B, right panel). To rule out the possibility that MDSC accumulation in 4T1 tumors is cell type-specific, MDA-MB-231 human breast cancer cells was also utilized. Consistently, KLF4 knockdown in MDA-MB-231 cells inhibited the number of MDSCs in bone marrow, spleen and tumor tissue compared to siCon counterparts in immunodeficient NOD/SCID mice (Supplementary Fig 2). We also performed IHC staining with an anti-Gr-1 antibody, which is a reliable approach to examine MDSCs 19. As expected, there were significantly reduced Gr-1 positive cells in primary tumors and lungs of the siKLF4 cell-inoculated mice compared to that of the siCon counterparts (Figure 2C).
Figure 2. KLF4 knockdown is associated with reduced MDSC accumulation and attenuated MDSC function.


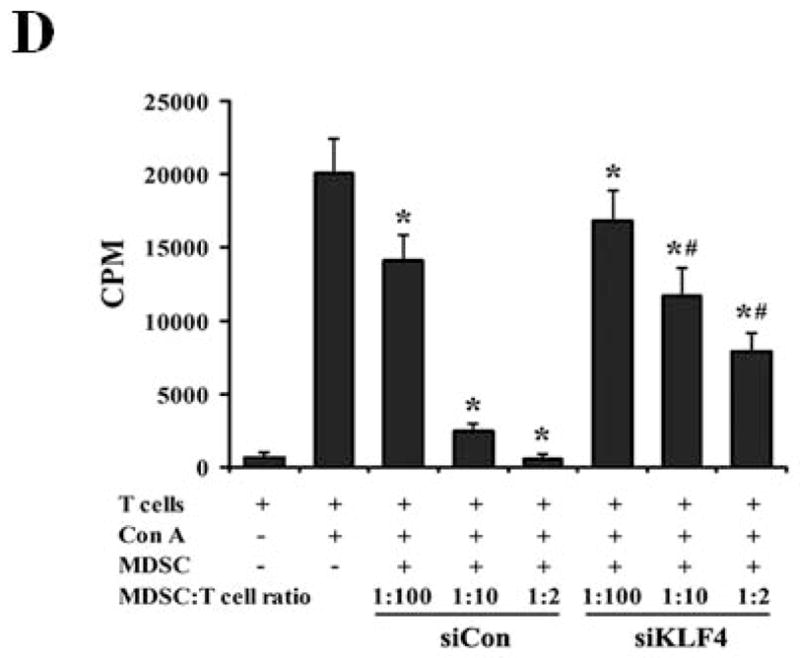
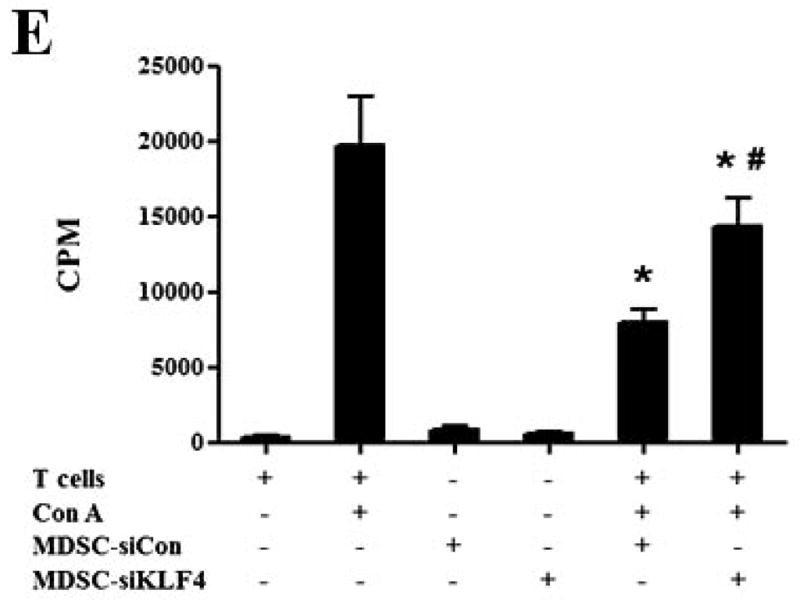
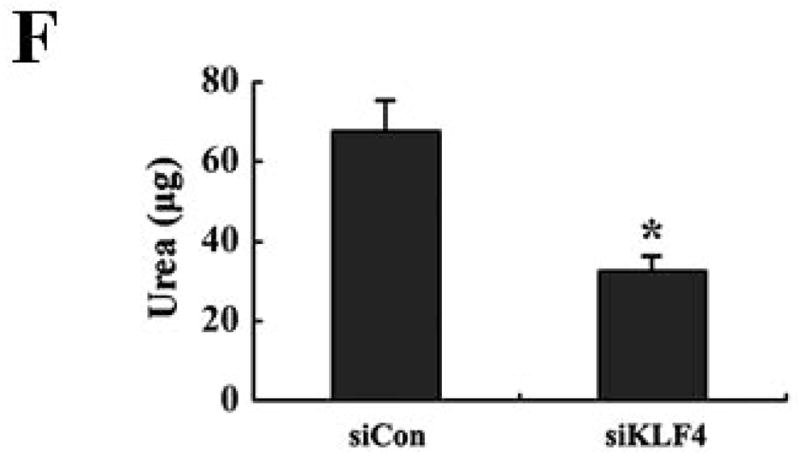
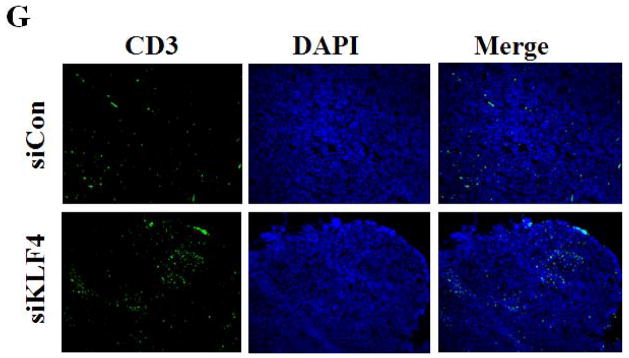
(A) 9 and 28 days after implantation of the BALB/c mice with siCon- and siKLF4 4T1 cells, bone marrow, spleen and tumor cells were collected from these mice and stained with anti-CD11b-FITC and anti–Gr-1-PE antibodies and MDSCs were analyzed by flow cytometry. A representative FACS analysis was shown from five independent experiments. The numbers in bold indicated the mean percentage of MDSCs. (B) The percentage of MDSCs in bone marrow and spleen 9 days after implantation of tumor cells (left) and the percentage of MDSCs in tumors 28 days after implantation of tumor cells (right) were plotted. Results were presented as the mean ± SD from five independent experiments (n=5). * indicates P < 0.05 versus siCon group; (C) Immunohistochemistry (IHC) staining of MDSCs in tumors and lung metastases. Scale bar, 20 μm. (D) [3H] incorporation assay was performed to detect the proliferation of CD4+ T cells in the presence of MDSCs as described in Materials and Methods. Data were expressed as cpm (mean ± SD) of triplicate cultures. Three independent experiments were performed. * indicates P < 0.05 versus Con A group; # indicates P < 0.05 versus siCon group at the same ratio. (E) Similar to (D) except CD8+ cells were used instead of CD4+ cells and the ratio of MDSCs to T cells was fixed to 1:10. (F) Arginase activities of MDSCs in spleens from above tumor-bearing mice were measured as explained in Materials and Methods. Data were presented as the mean ± SD of triplicate cultures. Three independent experiments were performed. * indicates P < 0.05 versus siCon group. (G) Increased T cell infiltration in tumor upon KLF4 knockdown. Primary tumor tissues were fixed. Frozen sections were made and stained with an FITC-conjugated anti-CD3 antibody. Representative fluorescence images of tumor tissues from siCon andsiKLF4 cell-inoculated mice were shown. DAPI was used to stain the nuclei of cells. Magnification, 10 x.
To rule out the possibility that the reduced MDSC population after KLF4 knockdown was merely the direct result of decreased tumor burden, we examined the functions of MDSCs from control cell- and KLF4 knockdown cell-inoculated mice by using the same amount of MDSCs. MDSCs have the ability to inhibit T cell activation in different tumor models 30, 31. We tested whether MDSCs derived from siCon- and siKLF4 cell-inoculated mice exhibited different immunosuppressive functions. As shown in Figure 2D, irradiated MDSCs from siKLF4 cell-inoculated mouse spleens inhibited T-cell proliferation significantly less than their siCon counterparts. The similar assay using MDSCs purified from mouse tumors confirmed this observation (Figure 2E). To further test the functional influence of MDSCs by KLF4, an arginase assay was performed. Consistent with repressed T cell proliferation upon KLF4 knockdown, the arginase activities in MDSCs from siKLF4 cell-inoculated mice were decreased by 2.11-fold when compared to siCon counterparts (Figure 2F). Furthermore, we examined the infiltration of T cells into tumor sites by CD3 immunofluorescence staining. We found that there were more T cells accumulated in siKLF4 cell-inoculated mice than those in siCon group (Figure 2G), suggesting that suppression of KLF4 expression in tumor cells greatly restores the infiltration of anti-tumor T cells.
MDSCs in tumor-bearing mice with KLF4 deficiency in tumor cells reduce tumorigenesis in vivo
The significantly decreased population and compromised function of MDSCs in siKLF4 cell-inoculated mice were accompanied by decreased tumor size and lung metastasis, suggesting that KLF4-modulated MDSCs may play a determinant role in mammary tumor development in the 4T1 mouse tumor model. To further test this possibility, 4T1 cells were mixed with the same amount of MDSCs purified from spleen of mice bearing siCon and siKLF4 4T1 tumors and injected into the mammary fat pads of BALB/c mice. Four weeks later, tumor weight and lung metastasis were examined in these mice. We observed a significant increase of tumor weight and lung nodules in mice received 4T1 coinjection with MDSCs derived from siCon tumor-bearing mice (V-MDSC) compared to 4T1 alone (Figure 3A and B), confirming a tumor promoting effect of MDSCs. Importantly, 4T1 coinjected with siKLF4 tumor-derived MDSCs (S-MDSC) resulted in delayed tumor growth and less lung metastasis compared to 4T1 coinjection with siCon tumor-derived MDSCs (Figure 3A and B). These results thus suggest that KLF4 deficiency-induced inhibition of tumor growth and lung metastasis is mediated by functionally compromised MDSCs.
Figure 3. MDSCs derived from KLF4 knockdown 4T1 cell-inoculated mice inhibit tumor growth and lung metastasis.
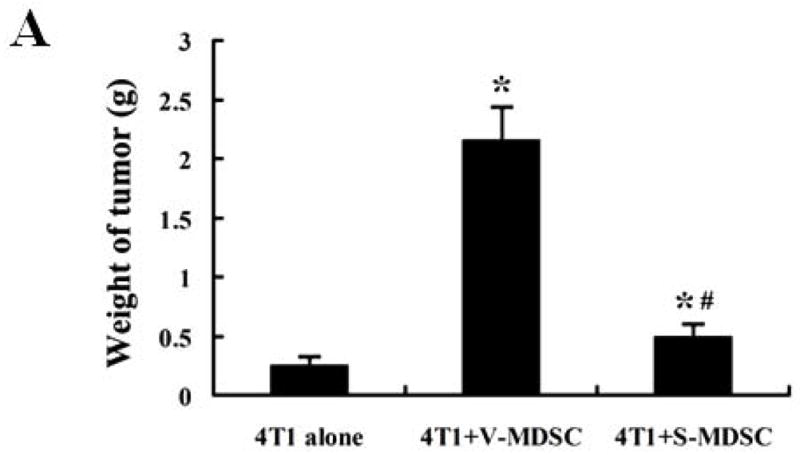
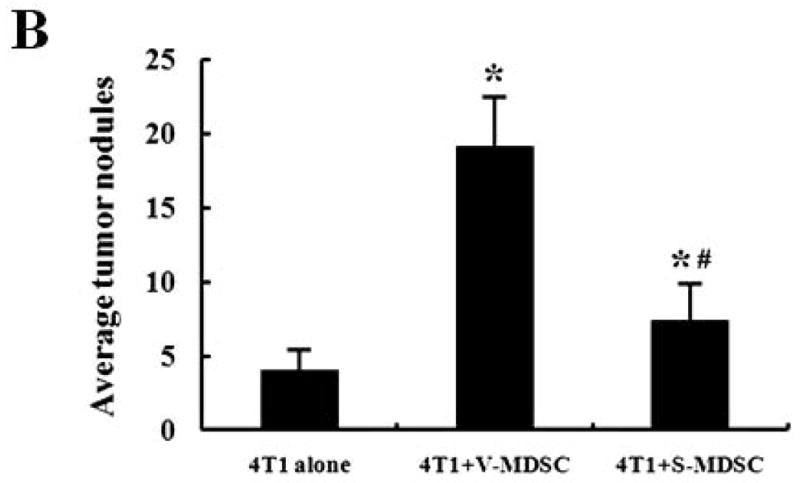
5 × 105 4T1 cells, without or with spleen MDSCs (1 × 105) derived from siCon cell- (V-MDSC) and siKLF4 cell-inoculated mice (S-MDSC) were injected into the mammary gland of BALB/c mice (n=5). The tumor weight (A) and lung nodules (B) were examined 21 days later. 4T1 alone was used as a control. Data were expressed as mean ± SD from three independent experiments. * indicates P < 0.05 versus 4T1 cells alone group; # indicates P < 0.05 versus 4T1+V-MDSC group.
GM-CSF is critical for KLF4-mediated migration and maintenance of MDSCs
A correlation between tumor burden and increased MDSCs suggests that tumor-derived factors may cause MDSC accumulation in cancer patients, such as granulocyte macrophage colony-stimulating factor (GM-CSF), interleukin-4 (IL-4), and interleukin-6 (IL-6) 17, 32. In an attempt to identify the possible chemokines in KLF4-mediated increase of MDSCs, a Bio-plex assay was performed. As shown in Figure 4A, siKLF4 cell-inoculated mice had decreased expression of GM-CSF, IL-6 and granulocyte colony-stimulating factor (G-CSF) and increased expression of interleukin-1β (IL-1β), IL-4, and tumor necrosis factor-alpha (TNF-α) in sera when compared to siCon cell-inoculated mice. Notably, GM-CSF expression in sera from siKLF4 cell-inoculated mice was dramatically decreased to an almost undetectable level. It has been demonstrated that 4T1 tumors activate and expand MDSCs through production of GM-CSF to impair antitumor T-cell response 33, 34. To test whether GM-CSF is involved in KLF4-mediated MDSC recruitment and to further rule out the direct effect of tumor burden in the mouse tumor model, an in vitro transwell migration assay was performed. Without GM-CSF treatment, the migratory capacity of MDSCs in the conditioned medium from siKLF4 cells (designated as siKLF4-CM) decreased by 2.48-fold relative to siCon countparts (Figure 4B). Notably, addition of recombinant GM-CSF in the medium not only increased the ability of cell migration in both groups but also dramatically eliminated differences between these two groups. Addition of IL-4 or IL-6 was unable to reverse cell migration suppressed by KLF4 knockdown (Figure 4B). Since GM-CSF is also important for maintenance of MDSCs 17, we postulated that GM-CSF was responsible for KLF4-mediated accumulation of MDSCs. We first isolated Gr-1+ cells from mouse bone marrow and this population was highly enriched with MDSCs (>95%) as measured by flow cytometry (Supplementary Fig 3). We then performed in vitro culture experiments with these purified cells. As shown in Figure 4C, the percentage of MDSCs incubated with siKLF4-CM was significantly reduced. In addition, exogenous GM-CSF restored the MDSC population in siKLF4-CM to the levels observed in siCon-CM (Figure 4C). However, neither IL-4 nor IL-6 showed any effect on MDSCs maintenance (Figure 4C, and data not shown). These data suggest that GM-CSF is critical for KLF4-mediated recruitment and maintenance of MDSCs.
Figure 4. GM-CSF is critical for KLF4-mediated migration and maintenance of MDSCs.


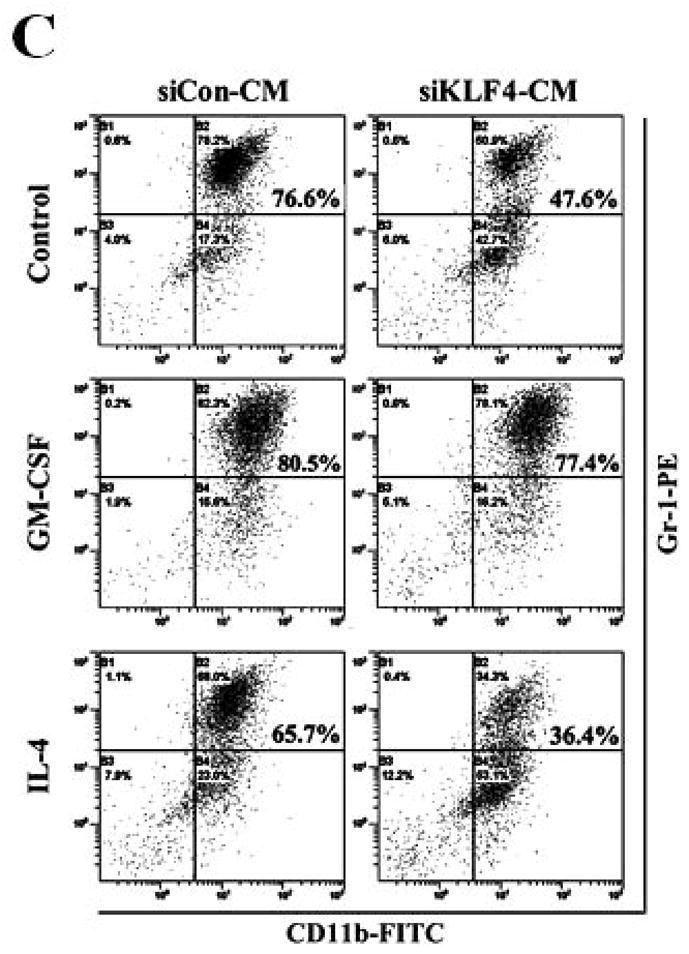
(A) Cytokine levels in sera from siCon and siKLF4 cell-inoculated mice were detected by Bio-plex assay. 28 days after tumor cell implantation, sera from tumor-bearing mice (n=3) were collected. Cytokine levels were determined using Bio-plex chemiluminescence assay system (Bio-Rad). * indicates P < 0.05, ** indicates P < 0.0001 versus siCon group. (B) Migratory ability of MDSCs to conditioned medium (CM) derived from siCon and siKLF4 4T1 cells was determined by a transwell migration assay. Recombinant mouse GM-CSF (100 ng/ml), IL-4 (50 ng/ml), and IL-6 (50 ng/ml) were added into the lower chamber. Migrated MDSCs were counted (ten fields per well, triplicate for each experimental groups) 8 h after incubation. Data were presented as mean ± SD from three independent experiments. * indicates P < 0.05 versus siCon group. (C) Maintenance of MDSCs in conditioned media derived from siCon and siKLF4 4T1 cells was evaluated by flow cytometry. Gr-1+ cells were purified from bone marrow of BALB/c mice and incubated with GM-CSF (100 ng/ml) or IL-4 (50 ng/ml) for 6 days. MDSCs were then analyzed by flow cytometry using anti-CD11b and anti-Gr-1 antibodies. A representative result of FACS analysis was shown from three independent experiments. The numbers in bold indicated the mean percentage of MDSCs.
GM-CSF is upregulated by KLF4 through CXCL5
We next determined if GM-CSF production in sera of 4T1 cell-inoculated mice was resulted from direct secretion by primary tumor cells. However, GM-CSF expression in KLF4 knockdown cells was not decreased compared to that in siCon cells as detected by both quantitative RT-PCR and Bio-plex assay (data not shown), raising the possibility that other factors secreted by primary tumor cells are responsible for GM-CSF expression. To test this, GM-CSF expression in bone marrow, the origin of MDSCs, incubated with siCon-CM and siKLF4-CM was determined. As expected, GM-CSF expression in bone marrow decreased by 56% in siKLF4-CM when compared to siCon-CM (Figure 5A). Consistently, we observed a 62.5% reduction of GM-CSF expression in bone marrow cells co-cultured with the siKLF4 tumor tissues compared to that with the siCon group (Figure 5B). These results indicate that decreased chemokine or cytokine secretion by siKLF4 cells resulted in lower GM-CSF expression in bone marrow. If this hypothesis is correct, the potential factors that regulate GM-CSF expression should be associated with the functions of MDSCs. Thus, we focused on those chemokines that had been reported to play critical roles in MDSC recruitment, such as chemokine (C-X-C motif) ligand 5 (CXCL5) and stromal derived factor 1 (SDF-1) 19. As shown in Figure 5C, in siKLF4 cell-inoculated mice, CXCL5 expression in primary breast tumors decreased by more than 90%. However, expression of other chemokines, including chemokine (C-X-C motif) ligand 1 (CXCL1), chemokine (C-X-C motif) ligand 10 (CXCL10), and SDF1, which are responsible for the recruitment of inflammatory cells into primary tumor sites 35, 36, showed only a moderate decrease or no change upon KLF4 knockdown (Figure 5C). These data suggest a possible up-regulation of CXCL5 by KLF4. Indeed, KLF4 knockdown reduced CXCL5 expression in 4T1 cells (Figure 5D) and in MDA-MB-231 human breast cancer cells (data not shown). In addition, an increased CXCL5 expression upon KLF4 overexpression in 4T1 and MDA-MB-231 cells was observed (Supplementary Fig 4). Importantly, in agreement with the in vivo data that decreased KLF4 was correlated with decreased CXCL5 and GM-CSF levels, recombinant CXCL5 upregulated GM-CSF expression in bone marrow by 2.6-fold (Figure 5E). Consistent with RNA expression, KLF4 knockdown in tumor cells also resulted in significantly decreased protein expression of CXCL5 and GM-CSF in tumor and lung tissues as examined by Western Blotting (Fig 5F).
Figure 5. GM-CSF is upregulated by KLF4 through CXCL5.

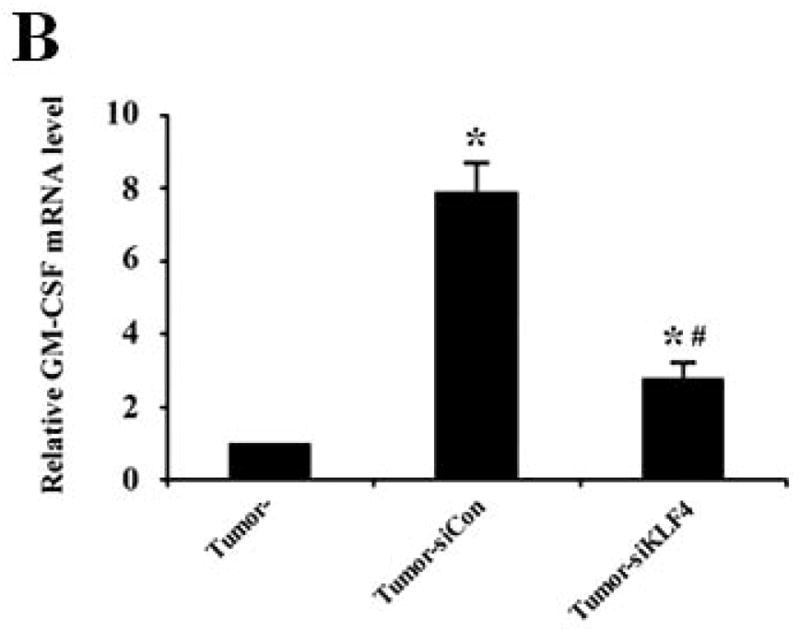
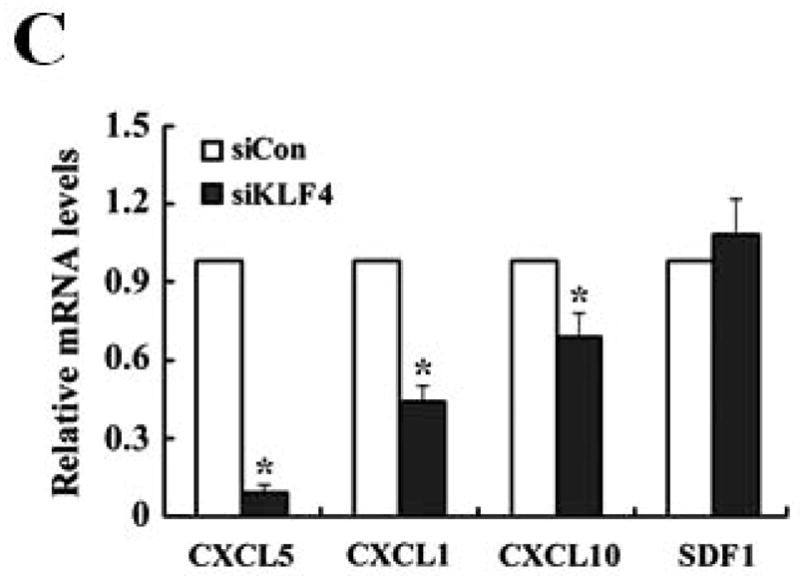

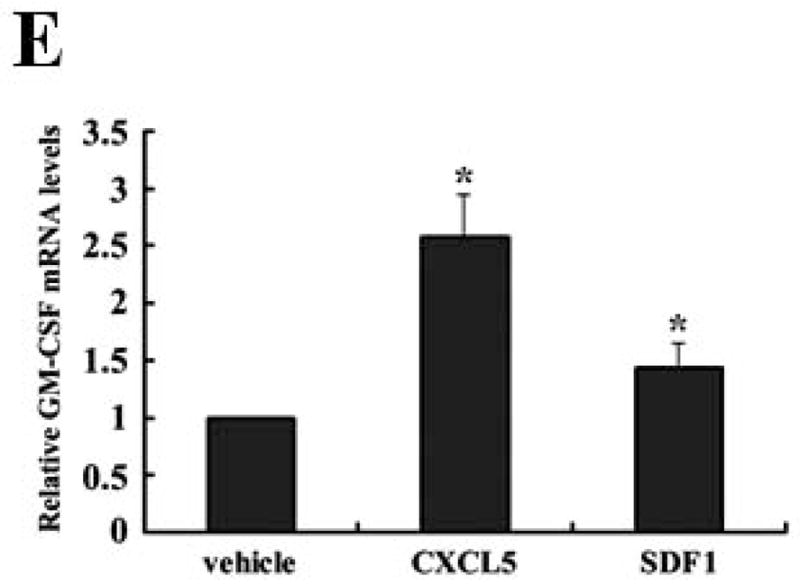
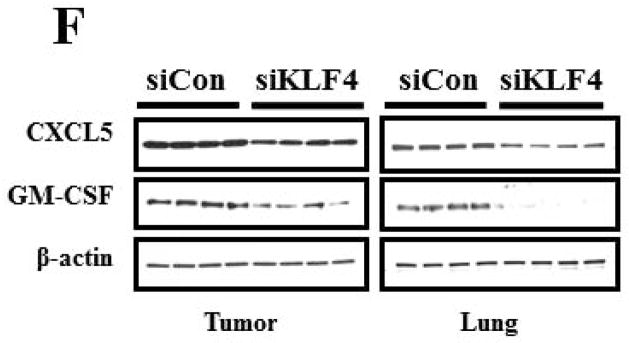
(A) Expression of VEGFR1, GM-CSF, CXCR2 and CXCR4 in MDSCs incubated with conditioned medium derived from siCon and siKLF4 cells (designated as siCon-CM and siKLF4-CM, respectively) was determined by quantitative RT-PCR. Values were expressed as mean ± SD of three independent experiments. * P < 0.05 versus siCon group. (B) GM-CSF expression in bone marrow cells was determined by quantitative RT-PCR upon incubation of 4T1 tumor tissues. 50 mg siCon and siKLF4 4T1 tumor tissues (Tumor-siCon and Tumor-siKLF4) were incubated with 1 × 106 bone marrow cells for 24 h followed by real time RT-PCR analysis. Bone marrow cells without tumor incubation (Tumor-) were used as a control. Values were expressed as means ± SD of three independent experiments. * P < 0.05 versus Tumor- group. # indicates P < 0.05 versus Tumor-siCon group. (C) Chemokine expression in mammary tumor tissues from control (siCon) and KLF4 knockdown (siKLF4) 4T1 cell-inoculated mice was determined by quantitative RT-PCR. Values were expressed as means ± SE of three independent experiments. * P < 0.05 versus siCon group. (D) Chemokine expression in 4T-1 control (siCon) and KLF4 knockdown (siKLF4) stable cells was determined by quantitative RT-PCR. Values were expressed as means ± SD of three independent experiments. * P < 0.05 vs siCon group. (E) GM-CSF expression was determined by quantitative RT-PCR after the bone marrow cells from BALB/c mice were treated with CXCL5 (25 ng/ml) and SDF1 (50 ng/ml) for 3 hrs. * P < 0.05 versus vehicle group. (F) Decreased expression of CXCL5 and GM-CSF in tumor and lung tissues of siCon and siKLF4 cell-inoculated mice by Western blotting. β-actin was used as an internal control. Results presented were from four different mice.
CXCL5 is important for the accumulation and migration of MDSCs
To investigate if regulation of CXCL5 by KLF4 was functionally relevant in our model, MDSC maintenance and migration were determined. As expected, CXCL5 induced a significant increase of MDSC accumulation in bone marrow (Figure 6A). To examine whether GM-CSF was involved in CXCL5-mediated accumulation of MDSCs, an anti-GM-CSF antibody was used to neutralize the effect of GM-CSF. As seen in Figure 6A, the effect of CXCL5 on MDSC accumulation was almost completely abrogated by anti-GM-CSF antibodies, indicating that upregulation of GM-CSF in bone marrow by CXCL5 contributes to accumulation of MDSCs. Next, we aimed to determine whether subsequent homing of these cells to the tumors is regulated by CXCL5. CXCL5 promoted the migration of MDSCs by 1.82-fold compared to vehicle control (Figure 6B). However, SDF1, another chemokine important for migration of MDSCs, had a smaller effect on MDSC recruitment (Figure 6B). In agreement of this observation, blockage of interaction between CXCL5 and its receptor chemokine (C-X-C motif) receptor 2 (CXCR2) with a specific CXCR2 antagonist, SB-265610, not only attenuated the basal migration ability, but also significantly inhibited the elevated migration of MDSCs in conditioned medium from wild type 4T1 cells (Figure 6C). Blocking the SDF1/CXCR4 signaling pathway by a chemokine (C-X-C motif) receptor 4 (CXCR4) antagonist, AMD3100, showed a much less effect than that by the CXCR2 antagonist (Figure 6C). These findings suggest that CXCL5/CXCR2 interaction plays a critical role in MDSC recruitment.
Figure 6. CXCL5 is important for the accumulation and migration of MDSCs.
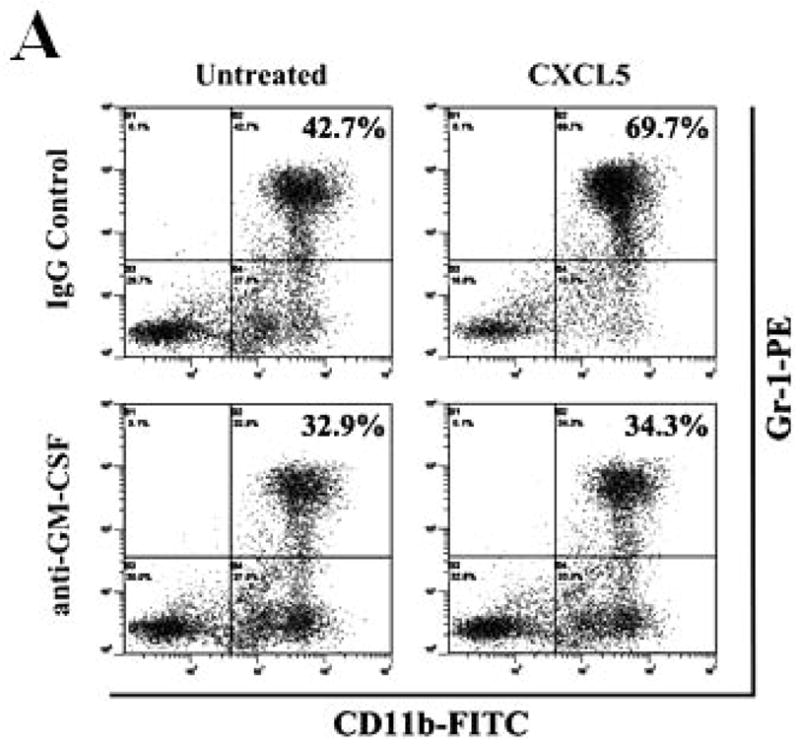
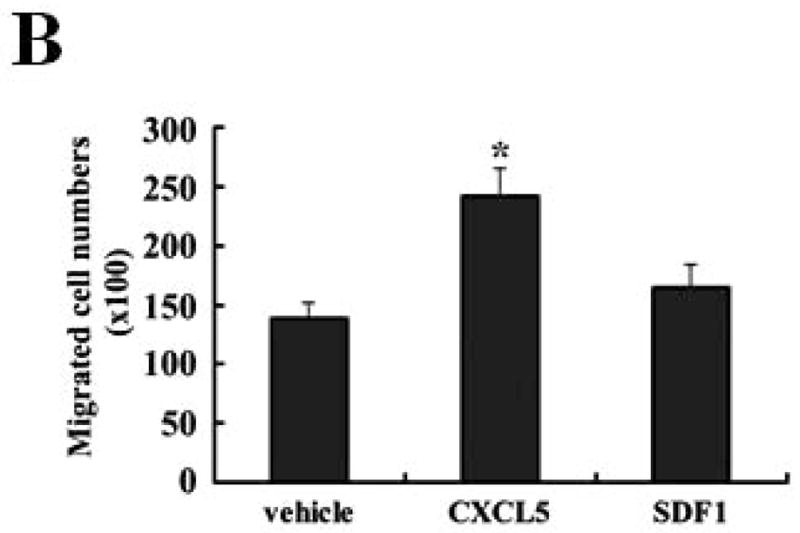
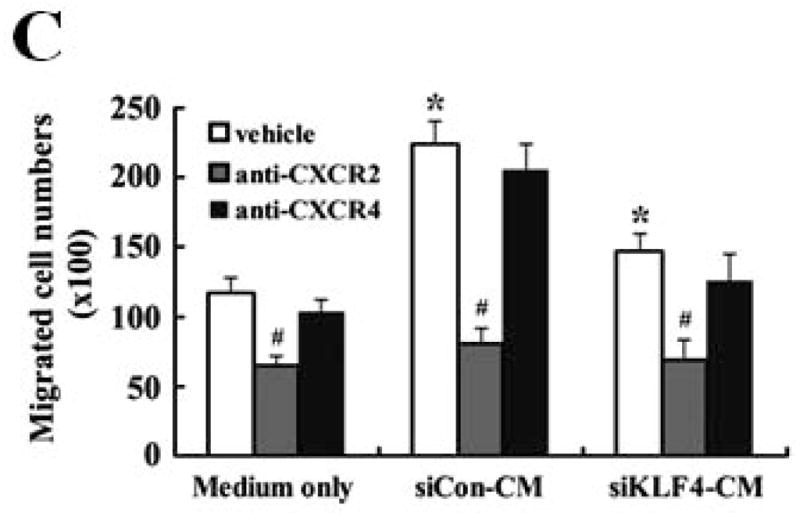
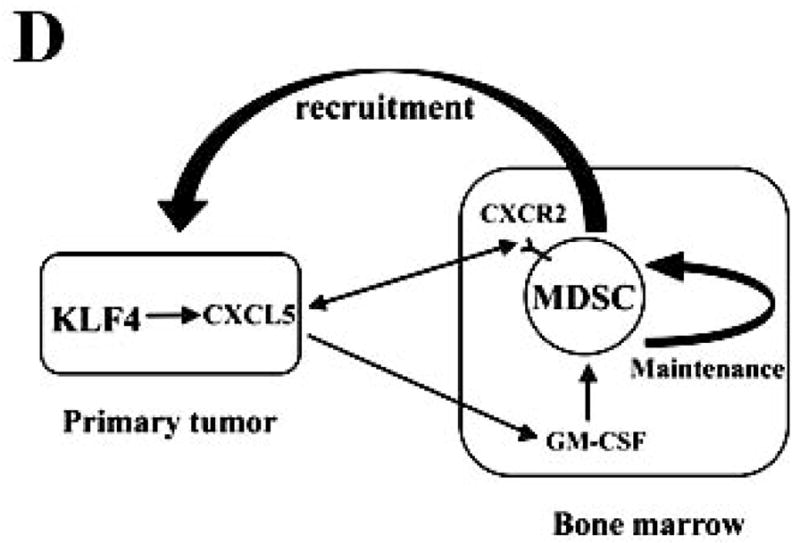
(A) Accumulation of MDSCs upon CXCL5 treatment was evaluated by flow cytometry. Bone marrow cells (1 × 107) were treated with CXCL5 (25 ng/ml) and anti-GM-CSF antibodies (250 ng/ml) or IgG Control (250 ng/ml) for a total of 6 days followed by flow cytometric analysis. A representative result of analysis was shown from three independent experiments. (B) Migratory ability of MDSCs upon CXCL5 stimulation was determined by a transwell migration assay. 2 × 105 sorted MDSCs from bone marrow of BALB/c type mice were seeded onto the top chamber of transwell inserts that were placed into the lower chamber containing medium supplemented with vehicle control, mouse recombinant CXCL5 (25 ng/ml) and SDF1 (50 ng/ml). Data were presented as mean ± SD from three independent experiments. * indicates P < 0.05 versus vehicle group. (C) Migratory ability of MDSCs was determined by a transwell assay upon blockage of CXCR2 and CXCR4 with specific antagonists. Before the assay was performed, 2 × 105 sorted MDSCs from bone marrow of wild type mice were incubated with SB-265610 (10 ng/ml) or AMD3100 (10 ng/ml), specific antagonists of CXCR2 and CXCR4 respectively, for 2 hrs. Data were presented as mean ± SD from three independent experiments. * indicates P < 0.05 versus vehicle group. # indicates P < 0.05 versus siCon group. (D) A proposed model showing that KLF4-mediated CXCL5 expression is critical not only for the recruitment of CXCR2-expressing MDSCs, but also for the activation of GM-CSF in bone marrow which is important for the maintenance of MDSCs.
Discussion
Our previous study demonstrated that KLF4 is required for maintenance of cancer stem cells, which contributes to mammary tumor development 8. In this study, by using a mouse 4T1 mammary tumor model and an immunodeficient NOD/SCID mouse model, we disclose that knockdown of KLF4 delays tumor growth and reduces pulmonary metastasis likely through decreased mobilization of MDSCs. MDSCs are potent suppressive agents of macrophage, T cells and NK cells, and polarize both innate and adaptive immunity toward a tumor-promoting type 2 phenotype 32, 37. Our studies therefore for the first time reveal that KLF4 may contribute to dictate the tumor microenvironment to anti-inflammatory/immune-suppressive responses during mammary tumor progression.
It should be noted that MDSC population is closely correlated with tumor burden 11–15. Because KLF4 knockdown significantly inhibited tumor growth and metastasis (Fig 1), it is concerned that decreased levels of MDSCs were the secondary effect of reduced tumor growth upon KLF4 knockdown. While it is difficult to examine if it is the case, several lines of evidence suggest that KLF4 directly modulates MDSCs. First, KLF4 knockdown changed the pattern of MDSCs in bone marrow at early time points (Fig 2A). Second, the same number of MDSCs from control cell- and KLF4 knockdown cell-inoculated mice had different effect on T cell function in vitro and tumor growth in vivo when mixed with 4T-1 tumor cells (Fig 3). Finally, although levels of GM-CSF, a critical cytokine for MDSC migration, was lower in KLF4 knockdown cell-inoculated mice compared to that in control cell-inoculated mice (Fig 4A), GM-CSF expression in KLF4 knockdown cells did not decrease compared to that in control cells (data not shown). Instead, the expression of CXCL5, a critical mediator that has been shown to increase the recruitment of MDSCs to the tumor19, significantly decreased in KLF4 knockdown 4T-1 cells and MDA-MB-231 cells when compared to the control cells (Fig 5C and 5D).
Chemokines secreted by primary tumor cells play critical roles in the recruitment of MDSCs into tumor microenvironment, which facilitates tumor development and progression 19, 35, 36, 38. Our data demonstrate that CXCL5/CXCR2 axis regulates the recruitment of MDSCs into tumor tissues. CXCL5 expression is dramatically reduced upon KLF4 knockdown both in 4T1 stable cells and in primary breast tumor tissues, indicating that CXCL5 may be a major contributing factor for the recruitment of MDSCs mediated by KLF4. It is likely that secretion of CXCL5 by primary tumors creates a chemokine gradient, which permits the attraction and incorporation of MDSCs into the tumor sites, thereby developing a conducive environment for tumor implantation and growth. Yang et al. disclose that elevated CXCL5 production but not the corresponding CXCR2 receptor regulates the recruitment of MDSCs in the PyVmT/Tgfbr2MGKO tumor microenvironment 19. In agreement with this observation, our studies showed that there was no difference of CXCR2 expression in bone marrow or Gr-1+ cells incubated with conditioned media from siCon and siKLF4 cells (Figure 5A and data not shown). Together, our data suggest that the tumor microenvironment created by KLF4-regulated CXCL5 is responsible for the recruitment of MDSCs. Although CXCL5 plays a critical role in primary tumor sites for the recruitment of MDSCs 19, its role in bone marrow microenvironment has not been reported. Our studies reveal that in addition to creating a local immunosuppressive microenvironment that is favorable for tumor growth, the function of CXCL5 on tumor development is also aided by its influence on distant organs including bone marrow. This has been supported by the following findings. First, CXCL5 activates expression of GM-CSF, one of the most important factors responsible for maintaining MDSC population 39 in bone marrow. Second, decreased CXCL5 secretion in primary tumor sites upon KLF4 knockdown was accompanied by a remarkably reduced GM-CSF production in sera, indicating the requirement of CXCL5 for the stimulation of GM-CSF. Based on these findings, we propose that KLF4-mediated CXCL5 secretion in primary tumors activates GM-CSF expression in bone marrow, which promotes maturation, maintenance, and migration of MDSCs (Figure 6D). In addition, local CXCL5 recruits CXCR2-expressing MDSCs, resulting in a favorable microenvironment for tumor development (Figure 6D).
MDSCs are significantly accumulated in bone marrow, spleen and primary tumor of tumor-bearing mice as well as in peripheral blood of cancer patients. In addition, MDSCs have also been implicated in tumor refractoriness to anti-VEGF treatment 40 and radiotherapy 41. Mounting evidence indicates that infiltrated MDSCs into tumor tissues create an environment that favors tumor progression. MDSCs downregulate immune surveillance and antitumor immunity by inhibiting T cell and B cell responses 42, and promote tumor angiogenesis and tumor progression by producing angiogenic factors (such as VEGF-a and PIGF) and matrix-degrading enzymes (such as MMP2 and MMP9) 20, 43. In addition, MDSCs infiltrated into the invasive front of tumor tissues facilitates tumor cell invasion and metastasis and results in increased survive of tumor cells. Our current study demonstrated that suppression of KLF4 expression in tumor cells inhibited tumor metastasis and decreased MDSC accumulation in tumor and lung tissues. Our further study will examine if and how the infiltrated MDSCs affect the survival, migration and invasion of tumor cells. Moreover, based on the decreased inhibition of T cell proliferation by MDSCs in siKLF4-inoculated mice, we are interested to know if the function of MDSCs is dependent on the interaction between MDSCs and siCon or siKLF4 tumor cells. If this is the case, we will investigate the underlying molecular mechanism. Another aspect of KLF4 function is the regulation of MDSC maintenance. It is known that MDSCs are sensitive to in vitro culture, as evidenced by loss of Gr-1 and acquisition of CD11 and F4/80 markers in some cells31, 44. As shown in Fig. 4C, after 6-day in vitro culture under siKLF4-CM, the proportion of Gr-1+/CD11b+ MDSCs was decreased, whereas the proportion of Gr-1-/CD11b+ (42.7%) was elevated (Fig 4C). We postulate that KLF4 controls the differentiation of MDSCs into F4/80+ macrophages (Gr-1-/CD11b+ cells in the figure).
MDSCs are now considered as one of the major factors responsible for tumor associated immune defects and are an attractive target for therapeutic intervention. Therefore, a comprehensive understanding of the molecular mechanisms for their recruitment and of their roles in tumor progression will provide valuable information for future drug design.
It is very interesting to know that KLF4 knockdown significantly represses MDSC population in bone marrow and spleen at early time points (Figure 2A and B), but not later (data not shown). These observations suggest that primary tumors establish a tumor-promoting systemic environment at a relatively early stage during their growth and possibly maintain this environment during all stages of tumor development. Alternatively, during the early stage of cancer development, primary tumor cells promote the establishment of a pre-metastatic niche that hosts the future tumor cells by mobilizing bone marrow-derived cells, such as MDSCs into peripheral sites including the lung 45. Consistent with this hypothesis, within several hours of incubation of bone marrow cells with conditioned medium from 4T1 cells, KLF4 knockdown significantly repressed the expression of vascular endothelial growth factor receptor 1 (VEGFR1) and CXCR4 (Figure 5A), which are highly expressed on hematopoietic stem and progenitor cells and implicated in the establishment of the pre-metastatic niche 38, 46. Future studies will be needed to examine the possible involvement of KLF4 in the establishment of a pre-metastatic niche and the underlying cellular and molecular mechanisms.
Although our current findings are consistent with those of others indicating that KLF4 may function as a tumor promoter in mouse mammary tumor models in vivo, a very recent report showed that enforced KLF4 expression in a 4T1 tumor model prevented mammary tumor development through inhibiting epithelial-to-mesenchymal transition (EMT) 47. In that study, the authors used a gain-of-function approach to study KLF4 function. However, we utilized a loss-of-function approach in both 4T1 and MDA-MB-231 cells. Among all different possibilities, we postulate that different KLF4 isoforms or the ratio between isoforms may be responsible for this discrepancy. It has been shown that different KLF4 isoforms exist in mouse and human although full length KLF4 is the predominant one 48, 49. Wei D et al. disclosed that full length KLF4 suppresses pancreatic tumorigenesis, whereas KLF4α has a completely opposite function 49. Based on these observations, we hypothesize that knockdown of KLF4 in our model may silence several KLF4 isoforms, including the one that suppresses tumor growth, resulting in alterations of the ratio between different KLF4 isoforms. This ratio may control the cellular localization of different of KLF4 isoforms and the overall function of KLF4. Therefore, identification of different KLF4 isoforms and their specific functions in breast cancer cells will be very helpful to elucidate the role of KLF4 in mammary tumor development.
Supplementary Material
Acknowledgments
This work was supported by an NIH/NIDDK grant to WA (1K01DK069489) and by the Center for Colon Cancer Research at University of South Carolina (5P20RR017698).
Abbreviations
- KLF4
Kruppel-like factor 4
- MDSCs
myeloid-derived suppressor cells
- GM-CSF
granulocyte macrophage colony-stimulating factor
- CM
conditioned medium
- CXCL5
chemokine (C-X-C motif) ligand 5
- SDF-1
stromal derived factor 1
- qRT-PCR
Quantitative real-time RT-PCR
- NOD/SCID
Nonobese diabetic/severe combined immune deficiency
References
- 1.Yet SF, McA’Nulty MM, Folta SC, Yen HW, Yoshizumi M, Hsieh CM, Layne MD, Chin MT, Wang H, Perrella MA, Jain MK, Lee ME. Human EZF, a Kruppel-like zinc finger protein, is expressed in vascular endothelial cells and contains transcriptional activation and repression domains. J Biol Chem. 1998;273:1026–31. doi: 10.1074/jbc.273.2.1026. [DOI] [PubMed] [Google Scholar]
- 2.Shields JM, Christy RJ, Yang VW. Identification and characterization of a gene encoding a gut-enriched Kruppel-like factor expressed during growth arrest. J Biol Chem. 1996;271:20009–17. doi: 10.1074/jbc.271.33.20009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garrett-Sinha LA, Eberspaecher H, Seldin MF, de Crombrugghe B. A gene for a novel zinc-finger protein expressed in differentiated epithelial cells and transiently in certain mesenchymal cells. J Biol Chem. 1996;271:31384–90. doi: 10.1074/jbc.271.49.31384. [DOI] [PubMed] [Google Scholar]
- 4.Chen X, Whitney EM, Gao SY, Yang VW. Transcriptional profiling of Kruppel-like factor 4 reveals a function in cell cycle regulation and epithelial differentiation. J Mol Biol. 2003;326:665–77. doi: 10.1016/S0022-2836(02)01449-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ghaleb AM, Nandan MO, Chanchevalap S, Dalton WB, Hisamuddin IM, Yang VW. Kruppel-like factors 4 and 5: the yin and yang regulators of cellular proliferation. Cell Res. 2005;15:92–6. doi: 10.1038/sj.cr.7290271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McConnell BB, Ghaleb AM, Nandan MO, Yang VW. The diverse functions of Kruppel-like factors 4 and 5 in epithelial biology and pathobiology. Bioessays. 2007;29:549–57. doi: 10.1002/bies.20581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Foster KW, Frost AR, McKie-Bell P, Lin CY, Engler JA, Grizzle WE, Ruppert JM. Increase of GKLF messenger RNA and protein expression during progression of breast cancer. Cancer Res. 2000;60:6488–95. [PubMed] [Google Scholar]
- 8.Yu F, Li J, Chen H, Fu J, Ray S, Huang S, Zheng H, Ai W. Kruppel-like factor 4 (KLF4) is required for maintenance of breast cancer stem cells and for cell migration and invasion. Oncogene. 2011;30:2161–72. doi: 10.1038/onc.2010.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Youn JI, Gabrilovich DI. The biology of myeloid-derived suppressor cells: the blessing and the curse of morphological and functional heterogeneity. Eur J Immunol. 2010;40:2969–75. doi: 10.1002/eji.201040895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182:4499–506. doi: 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–74. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, Padhya T, McCaffrey TV, McCaffrey JC, Gabrilovich DI. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol. 2009;182:5693–701. doi: 10.4049/jimmunol.0900092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bronte V, Apolloni E, Cabrelle A, Ronca R, Serafini P, Zamboni P, Restifo NP, Zanovello P. Identification of a CD11b(+)/Gr-1(+)/CD31(+) myeloid progenitor capable of activating or suppressing CD8(+) T cells. Blood. 2000;96:3838–46. [PMC free article] [PubMed] [Google Scholar]
- 14.Watanabe S, Deguchi K, Zheng R, Tamai H, Wang LX, Cohen PA, Shu S. Tumor-induced CD11b+Gr-1+ myeloid cells suppress T cell sensitization in tumor-draining lymph nodes. J Immunol. 2008;181:3291–300. doi: 10.4049/jimmunol.181.5.3291. [DOI] [PubMed] [Google Scholar]
- 15.Mundy-Bosse BL, Lesinski GB, Jaime-Ramirez AC, Benninger K, Khan M, Kuppusamy P, Guenterberg K, Kondadasula SV, Chaudhury AR, La Perle KM, Kreiner M, Young G, et al. Myeloid-Derived Suppressor Cell Inhibition of the IFN Response in Tumor-Bearing Mice. Cancer Res. 2011;71:5101–10. doi: 10.1158/0008-5472.CAN-10-2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dumitru CA, Moses K, Trellakis S, Lang S, Brandau S. Neutrophils and granulocytic myeloid-derived suppressor cells: immunophenotyping, cell biology and clinical relevance in human oncology. Cancer Immunol Immunother. 2012;61:1155–67. doi: 10.1007/s00262-012-1294-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morales JK, Kmieciak M, Knutson KL, Bear HD, Manjili MH. GM-CSF is one of the main breast tumor-derived soluble factors involved in the differentiation of CD11b-Gr1- bone marrow progenitor cells into myeloid-derived suppressor cells. Breast Cancer Res Treat. 2010;123:39–49. doi: 10.1007/s10549-009-0622-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lechner MG, Liebertz DJ, Epstein AL. Characterization of cytokine-induced myeloid–derived suppressor cells from normal human peripheral blood mononuclear cells. J Immunol. 2010;185:2273–84. doi: 10.4049/jimmunol.1000901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, Carbone DP, Matrisian LM, Richmond A, Lin PC, Moses HL. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008;13:23–35. doi: 10.1016/j.ccr.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tetreault MP, Wang ML, Yang Y, Travis J, Yu QC, Klein-Szanto AJ, Katz JP. Klf4 overexpression activates epithelial cytokines and inflammation-mediated esophageal squamous cell cancer in mice. Gastroenterology. 2010;139:2124–34. e9. doi: 10.1053/j.gastro.2010.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lebson L, Gocke A, Rosenzweig J, Alder J, Civin C, Calabresi PA, Whartenby KA. Cutting edge: The transcription factor Kruppel-like factor 4 regulates the differentiation of Th17 cells independently of RORgammat. J Immunol. 2010;185:7161–4. doi: 10.4049/jimmunol.1002750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alder JK, Georgantas RW, 3rd, Hildreth RL, Kaplan IM, Morisot S, Yu X, McDevitt M, Civin CI. Kruppel-like factor 4 is essential for inflammatory monocyte differentiation in vivo. J Immunol. 2008;180:5645–52. doi: 10.4049/jimmunol.180.8.5645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feinberg MW, Wara AK, Cao Z, Lebedeva MA, Rosenbauer F, Iwasaki H, Hirai H, Katz JP, Haspel RL, Gray S, Akashi K, Segre J, et al. The Kruppel-like factor KLF4 is a critical regulator of monocyte differentiation. Embo J. 2007;26:4138–48. doi: 10.1038/sj.emboj.7601824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liao X, Sharma N, Kapadia F, Zhou G, Lu Y, Hong H, Paruchuri K, Mahabeleshwar GH, Dalmas E, Venteclef N, Flask CA, Kim J, et al. Kruppel-like factor 4 regulates macrophage polarization. J Clin Invest. 2011;121:2736–49. doi: 10.1172/JCI45444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ai W, Liu Y, Langlois M, Wang TC. Kruppel-like factor 4 (KLF4) represses histidine decarboxylase gene expression through an upstream Sp1 site and downstream gastrin responsive elements. J Biol Chem. 2004;279:8684–93. doi: 10.1074/jbc.M308278200. [DOI] [PubMed] [Google Scholar]
- 27.Yang XD, Ai W, Asfaha S, Bhagat G, Friedman RA, Jin G, Park H, Shykind B, Diacovo TG, Falus A, Wang TC. Histamine deficiency promotes inflammation-associated carcinogenesis through reduced myeloid maturation and accumulation of CD11b+Ly6G+ immature myeloid cells. Nat Med. 2011;17:87–95. doi: 10.1038/nm.2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ko JS, Bukowski RM, Fincke JH. Myeloid-derived suppressor cells: a novel therapeutic target. Curr Oncol Rep. 2009;11:87–93. doi: 10.1007/s11912-009-0014-6. [DOI] [PubMed] [Google Scholar]
- 29.Nagaraj S, Gabrilovich DI. Myeloid-derived suppressor cells in human cancer. Cancer J. 2010;16:348–53. doi: 10.1097/PPO.0b013e3181eb3358. [DOI] [PubMed] [Google Scholar]
- 30.Bronte V, Serafini P, De Santo C, Marigo I, Tosello V, Mazzoni A, Segal DM, Staib C, Lowel M, Sutter G, Colombo MP, Zanovello P. IL-4-induced arginase 1 suppresses alloreactive T cells in tumor-bearing mice. J Immunol. 2003;170:270–8. doi: 10.4049/jimmunol.170.1.270. [DOI] [PubMed] [Google Scholar]
- 31.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181:5791–802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sinha P, Clements VK, Ostrand-Rosenberg S. Reduction of myeloid-derived suppressor cells and induction of M1 macrophages facilitate the rejection of established metastatic disease. J Immunol. 2005;174:636–45. doi: 10.4049/jimmunol.174.2.636. [DOI] [PubMed] [Google Scholar]
- 33.Serafini P, De Santo C, Marigo I, Cingarlini S, Dolcetti L, Gallina G, Zanovello P, Bronte V. Derangement of immune responses by myeloid suppressor cells. Cancer Immunol Immunother. 2004;53:64–72. doi: 10.1007/s00262-003-0443-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DuPre SA, Hunter KW., Jr Murine mammary carcinoma 4T1 induces a leukemoid reaction with splenomegaly: association with tumor-derived growth factors. Exp Mol Pathol. 2007;82:12–24. doi: 10.1016/j.yexmp.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 35.Sander LE, Sackett SD, Dierssen U, Beraza N, Linke RP, Muller M, Blander JM, Tacke F, Trautwein C. Hepatic acute-phase proteins control innate immune responses during infection by promoting myeloid-derived suppressor cell fuction. J Exp Med. 2010;207:1453–64. doi: 10.1084/jem.20091474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bose A, Taylor JL, Alber S, Watkins SC, Garcia JA, Rini BI, Ko JS, Cohen PA, Finke JH, Storkus WJ. Sunitinib facilitates the activation and recruitment of therapeutic anti-tumor immunity in concert with specific vaccination. Int J Cancer. 2010 doi: 10.1002/ijc.25863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol. 2007;179:977–83. doi: 10.4049/jimmunol.179.2.977. [DOI] [PubMed] [Google Scholar]
- 38.Mohle R, Bautz F, Rafii S, Moore MA, Brugger W, Kanz L. The chemokine receptor CXCR-4 is expressed on CD34+ hematopoietic progenitors and leukemic cells and mediates transendothelial migration induced by stromal cell-derived factor-1. Blood. 1998;91:4523–30. [PubMed] [Google Scholar]
- 39.Dolcetti L, Peranzoni E, Ugel S, Marigo I, Fernandez Gomez A, Mesa C, Geilich M, Winkels G, Traggiai E, Casati A, Grassi F, Bronte V. Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by GM-CSF. Eur J Immunol. 2010;40:22–35. doi: 10.1002/eji.200939903. [DOI] [PubMed] [Google Scholar]
- 40.Shojaei F, Wu X, Malik AK, Zhong C, Baldwin ME, Schanz S, Fuh G, Gerber HP, Ferrara N. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol. 2007;25:911–20. doi: 10.1038/nbt1323. [DOI] [PubMed] [Google Scholar]
- 41.Kodumudi KN, Weber A, Sarnaik AA, Pilon-Thomas S. Blockade of myeloid-derived suppressor cells after induction of lymphopenia improves adoptive T cell therapy in a murine model of melanoma. J Immunol. 2012;189:5147–54. doi: 10.4049/jimmunol.1200274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182:4499–506. doi: 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Balkwill F, Coussens LM. Cancer: an inflammatory link. Nature. 2004;431:405–6. doi: 10.1038/431405a. [DOI] [PubMed] [Google Scholar]
- 44.Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, Cho HI, Celis E, Quiceno DG, Padhya T, McCaffrey TV, McCaffrey JC, et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. 207:2439–53. doi: 10.1084/jem.20100587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peinado H, Lavotshkin S, Lyden D. The secreted factors responsible for pre-metastatic niche formation: old sayings and new thoughts. Semin Cancer Biol. 2011;21:139–46. doi: 10.1016/j.semcancer.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 46.Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, Zhu Z, Hicklin D, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820–7. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yori JL, Seachrist DD, Johnson E, Lozada KL, Abdul-Karim FW, Chodosh LA, Schiemann WP, Keri RA. Kruppel-like Factor 4 Inhibits Tumorigenic Progression and Metastasis in a Mouse Model of Breast Cancer. Neoplasia. 2011;13:601–10. doi: 10.1593/neo.11260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shields JM, Yang VW. Two potent nuclear localization signals in the gut-enriched Kruppel-like factor define a subfamily of closely related Kruppel proteins. J Biol Chem. 1997;272:18504–7. doi: 10.1074/jbc.272.29.18504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wei D, Wang L, Kanai M, Jia Z, Le X, Li Q, Wang H, Xie K. KLF4alpha up-regulation promotes cell cycle progression and reduces survival time of patients with pancreatic cancer. Gastroenterology. 2010;139:2135–45. doi: 10.1053/j.gastro.2010.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.