

Conspectus
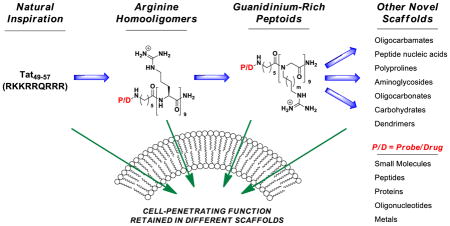
All living systems require biochemical barriers. As a consequence, all drugs, imaging agents, and probes have targets that are either on, in, or inside of these barriers. Fifteen years ago, we initiated research directed at more fully understanding these barriers and at developing tools and strategies for breaching them that could be of use in basic research, imaging, diagnostics and medicine. At the outset of this research and to a lesser extent now, the “rules” for drug design biased the selection of drug candidates to mainly those with an intermediate and narrow log P. At the same time, it was becoming increasingly apparent that Nature had long ago developed clever strategies to circumvent these “rules”. In 1988, for example, independent reports appeared documenting the otherwise uncommon passage of a protein (HIV-Tat) across a membrane. A subsequent study called attention to a highly basic domain in this protein (Tat49–57) being responsible for its cellular entry. This conspicuously contradictory behavior, i.e., a polar, highly charged peptide passing through a non-polar membrane, set the stage for learning how Nature had gotten around the current “rules” of transport. As elaborated in our studies and discussed herein, the key strategy used in Nature rests in part on the ability of a molecule to change its properties as a function of microenvironment, being a polarity chameleon – i.e., being polar in a polar milieu and relatively non-polar in a non-polar environment. Because this research originated in part with the protein Tat and its basic peptide domain, Tat49–57, the field focused heavily on peptides, even limiting its nomenclature to names such as ‘cell-penetrating peptides,’ ‘cell-permeating peptides,’ ‘protein transduction domains,’ and ‘membrane translocating peptides’ to note a few. Starting in 1997, through a systematic reverse engineering approach, we established that the ability of Tat49–57 to enter cells is not a function of its peptide backbone, but rather the number and spatial array of its guanidinium groups. These function-oriented studies allowed one to design more effective peptidic agents and to think beyond the confines of peptidic systems to new and even more effective non-peptidic agents. Because the function of passage across a cell membrane is not limited to or even best achieved with the peptide backbone, we referred to these agents by their shared function, i.e., ‘cell-penetrating molecular transporters’. The scope of this molecular approach to breaching biochemical barriers has expanded remarkably in the past 15 years, enabling or enhancing the delivery of a wide range of cargos into cells and across other biochemical barriers; creating new tools for research, imaging, and diagnostics; and introducing new therapies into clinical trials.
Introduction
Since the first designed cell-penetrating guanidinium-rich molecular transporters were reported in 2000, numerous new classes of guanidinium-rich molecular transporters have been described, including guanidinium-rich peptoids, carbamates, carbonates, carbohydrates, nucleic acids and dendrimers.1–3 These molecular transporters have been shown to deliver a variety of cargos, including small molecules, peptides, proteins, imaging agents, metals, siRNA, PNAs, plasmids, quantum dots, xenon cages, vesicles and vaults, across a variety of cellular and tissue barriers, including bacterial, algal and mammalian cell membranes, human skin and the blood brain barrier (BBB).2–6 Companies have been launched based on this technology, and the number of publications pertinent to this subject has increased every year, now averaging over 1 paper per day (Figure 1). According to PubMed, the seminal Wender and Futaki papers, which are only 12 years old, have been cited collectively almost 1500 times.1,7
Figure 1.
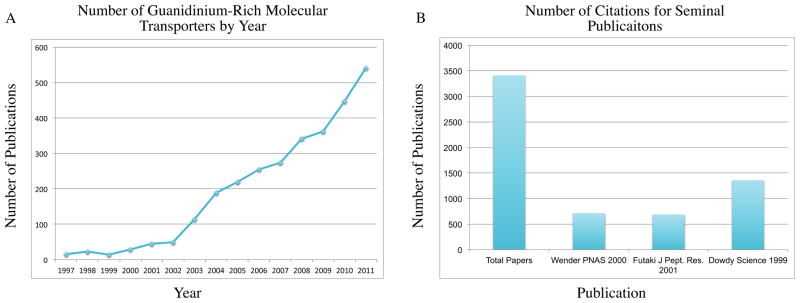
Molecular transporter publications. (A) Number of publications per year with a focus on guanidinium-rich molecular transporters. These numbers were determined by adding the number of publications per year, according to Web of Science, that contained either the keywords “cell penetrating peptide(s),” “guanidinium-rich molecular transporter(s),” “octaarginine (s),” “oligoarginine (s),” “nonaarginine(s),” or “tat peptide(s),” and subtracting references containing multiple keywords to remove duplicates. (B) Number of publications that cite three of the major seminal works in this area. “Total Papers” is the area under the curve in Figure 1A.
The striking growth in research on molecular transporters has been further fueled by the growing academic and clinical interest in biologics, including antibodies, peptides, proteins, and oligonucleotides, which are expected to dominate the top ten drugs by 2014.8 New delivery strategies would enhance the performance of such biologics against extracellular targets, and also open a vast range of opportunities associated with intracellular and tissue targets protected by other barriers (e.g., BBB, ocular, lung, skin, aural).
Here we present a brief overview of the development of designed cell-penetrating, guanidinium-rich molecular transporters inspired by Tat. We have divided this scientific journey into three post-1988 periods: “Initial Discoveries” (1997–2001), “Insights and Advances” (2002–2007), and “The Next Generation” from 2008 to present (and beyond). Major milestones are displayed in Figure 2 and impressively covered in recommended reviews.2,3,9,10
Figure 2.

Timeline for representative seminal publications on guanidinium-rich molecular transporters.
Part I: 1997–2001: Initial Discoveries
Traditional “rules” for drug design logically posit that drugs with intracellular targets should exhibit a balance of hydrophilicity and lipophilicity, the former for dissolution in polar body fluids and the latter for passage through non-polar membranes. The findings of Green and Lowenstein11 and Frankel and Pabo12 that the HIV-1 Tat protein, unlike most proteins, readily enters cells thus flew in the face of conventional wisdom. The anomalous behavior of HIV-1 Tat was further addressed by Lebleu and coworkers, who discovered that the amphipathic alpha-helical region of this protein, thought to have been responsible for its cellular uptake, was in fact not required. Counter-intuitively, the cell-penetrating function of the protein was imparted by the adjacent cation-rich region (RKKRRQRRR), often referred to as Tat49–57 or the Tat 9-mer.13 Uptake studies with the peptide at 4°C indicated that endocytotic pathways were not solely involved for fluoresceinated peptides, providing evidence that the cell-penetrating function of the Tat peptide involves a novel mechanism.2,13
It was clear from the seemingly contradictory behavior of Tat49-57 – a highly water soluble molecule that readily crosses the non-polar cell membrane – that there was a potentially powerful lesson to be learned and potentially exploited in the design of new and more effective drug delivery strategies. We thus set out to identify the key features of Tat49-57 that enable its passage through cellular membranes.1 Through a series of systematic N- and C-termini truncations of Tat49-57, we found that the full-length nonamer provided optimal cell uptake. An alanine scan, in which each residue of Tat49-57 was individually replaced by an alanine, showed that all substitutions reduced uptake, except substitution of the only noncharged residue, glutamine, which produced a 9-mer with cell uptake similar to the native peptide.1 To address the then unknown relative contributions of lysines and arginines to the cell-penetrating function, homooligomers of lysine and arginine were analyzed for cell uptake.1,14 This breakthrough study showed that homooligomers of lysine were less effective than Tat49-57 in cell-penetrating function, but, significantly, homooligomers of arginine were dramatically better than Tat49-57. Thus the cationic charge of lysines alone is not sufficient for uptake while cationic guanidinium groups of arginines and their number are critical (5–20 arginines, with 7–9 being the best compromise of cost and performance).
We proposed that the difference between arginines and lysines could arise from the ability of the guanidinium group of arginine, unlike the ammonium group of a lysine, to form a bidentate hydrogen bond with anionic cell surface phosphates, carboxylates and/or sulfates, as the initiating event of cellular entry.15 We tested this hypothesis by replacing the hydrogens of the guanidinium groups with methyl groups incapable of hydrogen bonding. While still charged, the N-methylated guanidinium groups were profoundly less effective at entering cells.
Uptake of both the unnatural D-amino acid analog of Tat49-57, d-Tat49-57, and the retro-inverso analogs Tat57-49 and d-Tat57-49 by flow cytometry revealed an aspect of these molecular transporters that proved critical for design: cell-penetrating function is retained despite changes in backbone stereochemistry.1 These experiments also indicated that the mechanism of cell uptake of Tat49-57 and analogs was not receptor-mediated given that enantiomeric molecules exhibited similar function.
Contemporaneously, Futaki and coworkers examined a series of natural Tat-related, RNA- and DNA-binding peptides, many of which were arginine-rich and entered cells.7 They proposed that there was likely a common, but unspecified, mechanism for cell uptake of arginine-rich peptides. Our structure-function studies provided a more specific structural view, indicating that uptake was a function of the number and array of guanidinium groups.
Given that changes in stereochemistry of guanidinylated peptides did not dramatically affect cellular uptake, it was expected that the natural peptide backbone would not be required for cell-penetrating function. To test this hypothesis, a series of peptoid molecular transporters with guanidinium-containing sidechains was synthesized.1 Remarkably, these molecules exhibited uptake similar to that of the oligoarginines. Longer spacing between the peptoid backbone and guanidinium functionality also improved cellular uptake, suggesting that molecular flexibility is beneficial to function. Importantly, this study established for the first time that the function of Tat49-57 can be mimicked by a variety of guanidinium-rich oligomers, thereby providing a rationale for the design of new molecular transporters that could be tailored to specific needs of stability, biodegradability, toxicity, or intracellular targeting.
The collective advances of this period serve as a powerful exemplification of function- and synthesis-informed design, i.e. “function-oriented synthesis” (FOS).16 FOS is based in part on the view that while Nature has produced solutions to many problems, they are neither directed at, nor optimized for human use. Thus the ability of the polar Tat49-57 peptide to enter cells, while inspiring, can be mimicked, if not exceeded, through synthesis-informed design once the structural determinants of function are identified. With the importance of guanidinium number and array established, it became possible to design new guanidinium-rich transporters with improved performance and ease of synthesis.
The clinical potential of this technology was first addressed in a preclinical study of transporter-mediated drug delivery into human skin. The cyclic peptide, cyclosporin A (CsA), was chosen as the drug candidate because it is effective against several skin disorders, including psoriasis, but causes off-target toxicity when delivered systemically.17 By itself, CsA does not penetrate skin. Thus, in a first-in-class study, we sought to determine whether a CsA-oligoguanidinium conjugate would cross the stratum corneum and enter skin. Previous studies had shown that the Tat transporter crosses the blood-brain barrier, demonstrating the potential for breaching tissue barriers in addition to cellular membranes.18 To visualize penetration into skin, biotinylated hepta-D-arginine-CsA conjugates (biotin-r7-CsA) were applied with vehicle to a human skin graft on the back of a nude mouse, and after two hours skin biopsies were obtained and the tissue cryosections stained with either fluorescein- or peroxidase-labeled streptavidin.17 Visualization of the labeled tissue revealed that nearly all keratinocytes in the epidermis and a large number in the dermis contained biotin-r7-CsA, indicating that the transporter conjugates were indeed able to penetrate skin. The CsA-oligoarginine conjugates entered the clinic, passing phase I safety studies. Conjugate uptake was also observed but the pH-based release of free CsA was too slow, prompting the introduction of a more effective release strategy.19
While drug release is not required for all applications, it is for some, including CsA. We thus turned to bioactivatable release strategies, for which release would occur only upon cell entry. Our test cargo was the octapeptide, ψε RACK, which reduces ischemic damage by modulating e protein kinase C activity.20 Like many peptides, ψεRACK is not cell permeable. However, a conjugate of ψεRACK and heptaarginine attached through a redox-cleavable disulfide linker rapidly entered cardiomyocytes and released free ψεRACK peptide, thereby reducing ischemic damage in intact rat hearts.
The rapid clinical translation of the guanidinium-rich transporter technology with CsA and the ψεRACK peptide is a testament to the robustness of this technology and the value of new delivery strategies. Of special significance, this research established that peptides, previously thought to be poor drug candidates because of cell uptake problems, could be directly used as leads and drugs, thereby making the need for peptidomimetics unnecessary in many cases.
Part II: 2002–2007: Insights and Advances
The period from approximately 2002 to 2007 was marked by many advances enabled by the earlier structure-function studies. During this period, much effort was directed at mechanisms of cell entry, designing new transporters and exploring new cargos.
Our finding that the number and spatial array of guanidinium groups are the key determinants of transporter cellular uptake1 paved the way for the design of several new guanidinium-rich transporter scaffolds (Figure 4). We first showed that homooligomers of D-arginine readily enter cells, which inspired the initial study of guanidinium-rich peptoids. That these peptoids showed better uptake with longer side chains, led in turn to the synthesis of over 60 “spaced” transporters, each containing seven guanidinium groups interdigitated by one or more aminocaproic acid groups.21 The maximally spaced system (RXRXRXRXRXRXR) performed better than the unspaced heptamer (RRRRRRR). Impressive contemporaneous studies from other groups further expanded the types of amide scaffolds that exhibit cellular penetration, including β-peptides22,23 and polyproline scaffolds.24 Marking a departure from the peptide and amide theme, we reported the first non-amide linked transporters in 2002, showing that oligocarbamates decorated with guanidinium groups work as well as, and for selected cases better than, the corresponding arginine oligomers.25 Similarly, guanidinylated carbohydrates also exhibit cellular uptake.26,27 In 2002 the first branched guanidinium-rich molecular transporters were reported.28 Branched scaffolds including dendrimers have also been reported by our group and the groups of Goodman and Harth.29–31
Figure 4.

Timeline for the development of different scaffolds for guanidinium-rich transporters.
Direct guanidinylation of cargo also proved effective for enabling cell entry. For example, per-guanidinylated tobramycin and neomycin B showed enhanced cellular uptake.32,33 Guanidinylation of oligonucleotide scaffolds also proved effective, working with guanidinylated peptide nucleic acids,34 base guanidinylation,35 or guanidinylation along the backbone.36,37
The period of 2002–2007 witnessed an expansion in the types of cargos delivered using guanidinium-rich transporters. In 2003, we reported that cysteine-flanked guanidinium-rich transporters can non-covalently complex and deliver DNA plasmids into cells both in vitro and in vivo.38 Using DNA as a template, the oligomers were designed to form a disulfide-linked polymer that packages and delivers DNA. This noncovalent complexation strategy has also been used by us and others to deliver gene-silencing siRNA.39–41 In 2006, Kim and coworkers reported that noncovalent complexes of cholesterol-nonaarginine and vascular endothelial growth factor siRNA entered cells in vitro and in vivo.39 Subsequently, Kumar and coworkers reported that conjugates of R9 and rabies virus glycoprotein (RVG) complexed and delivered siRNA across the blood brain barrier.40 In both cases the guanidinium-rich subunit was used to complex the siRNA, while delivery was mediated by the conjugated cholesterol or RVG peptide putatively through receptor-mediated endocytosis. Our siRNA delivery studies used a similar strategy (vide infra).41 Guanidinium-rich transporters have delivered morpholinos, uncharged oligonucleotide mimics which modify gene expression.42 Guanidinium-rich transporters have also delivered a variety of nanocarriers, including cargo-loaded liposomes and nanoparticles,9 as well as metal complexes including indium, technetium, gadolinium, caged contrast agents, and even iron nanoparticles.6,43
A long-standing question with guanidinium-rich transporters is their mechanism(s) of cellular uptake. Over 30 mechanistic studies have addressed this question.2 Many articles start with a variation on the statement that the mechanism is “unclear.” It is, however, important to differentiate “unclear” from a function that depends on many variables, including transporter type, cargo type and size, cell type, and method of uptake analysis. Historically, chemists have unequivocally shown that many processes, even simple substitution reactions, can proceed through several pathways depending on many variables. Cell entry is not dissimilar. That noted, there are points of general agreement common to several mechanisms. As we showed in our early mechanistic studies on methylated guanidinium groups, positively-charged guanidinium groups are ideally suited to bind to negatively-charged cell surface carboxylates, sulfates and phosphates through electrostatic association and bidentate hydrogen bonding, initiating cell entry.15 We further showed that membrane potential influences uptake of small molecule oligoarginine conjugates. One mechanistic hypothesis, called ‘adaptive translocation,’ is that the poly-cationic guanidinium-rich transporter forms a complex with oppositely charged groups on the surface of the cell using bifurcated hydrogen bonds (Figure 5). This association diminishes the polarity of both participating groups by forming an ion pair, now attached to the cell membrane. The number of guanidinium groups influences the persistence of this association like the length of a Velcro strip. While this mechanism could operate for small cargos, as the cargo size increases, endocytotic mechanisms are expected to compete and/or dominate.44 Because of the structural variety of guanidinium-rich transporters, the variation in cell type and tissue being traversed, and the large variation in cargo size, structure, physical properties, and type (from DNA to small molecules), a universal mechanism for guanidinium-rich transporter cell entry is unlikely. Even the co-occurrence of competing pathways is suggested by our single molecule studies, in which two types of behavior of individual molecules on a living cell were directly observed.44
Figure 5.

Mechanisms of uptake (adaptive translocation and endocytosis). 1. The guanidinium-group forms a bidentate bond with negative phosphates, sulfates, and carboxylates on the cell surface. 2. and 3. The charge-neutralized species moves through the membrane, in a process termed ‘adaptive translocation,’ driven into the cell by the membrane potential. 4. In the reverse of 1, the oligoguanidinium transporter dissociates from the membrane once inside the cell.
For many cargos, covalent conjugation to a transporter will not change the bioactivity of the cargo. For others, the transporter-cargo conjugate could serve as a prodrug that is then cleaved, releasing free drug, after cellular entry. Early attempts had used pH-based cargo release,17 but this approach increased release rates at the expense of shelf stability. To avoid this problem we focused on bioactivatable release systems that could be initiated with esterases, phosphatases, proteases, or intracellular redox events. We attached the cargo to the transporter through a disulfide bond, which would cleave upon encountering intracellular glutathione (Figure 6A).19 This release system takes advantage of the higher levels of intracellular over extracellular glutathione. Luciferin was used as a drug surrogate to simulate drug uptake, release, and interaction with its intracellular target, which in the case of luciferase produces a photon of light per molecule of luciferin, allowing one to quantify uptake and release in real time in both cells and animals.19,45 This bioactivatable release has since been used by our group for the delivery of taxol-releasing conjugates that overcome resistant ovarian cancer.46
Figure 6.

Activatable and targeted release strategies. (A) Redox-releasable linker. The disulfide is cleaved by intracellular glutathione, liberating a free thiol which cyclizes into the nearby carbonate, releasing free drug. (B) Activatable transporters. Oligoarginine is linked to a negatively-charged attenuating sequence through a protease-cleavable linker. In the presence of the specific protease, the linker is cleaved and the polyanion dissociates from oligoarginine. The free guanidinium groups can then interact with the surface of the cell and facilitate uptake.
The period from 2002–2007 witnessed advances of exceptional significance with respect to targeted delivery. For context, monoclonal antibodies, the current workhorse of targeted delivery, rely on the thermodynamics of cell surface antigen binding. Both the Tsien and our group investigated whether transporters could be used for “kinetic targeting” by connecting the oligoguanidinium sequence to an oligoanion sequence through a peptide that is cleaved by a cell surface protease (Figure 6B). The oligoanion sequence hydrogen bonds to the oligoguanidinium groups, preventing interaction with the surface of the cell membrane and thereby preventing cell uptake. However, once the connecting peptide sequence is cleaved, the oligoanion sequence dissociates from the transporter, allowing the transporter to enter proximate cells. The Tsien group reported on this strategy with a matrix metallo-protease-cleavable connecting sequence,47 while our group used a prostate-specific antigen-cleavable sequence.48 The Tsien group has advanced this technology for use in tumor imaging.49 While still new, this approach to targeting offers many advantages over monoclonal antibody targeting.
Part III: 2008-Today: The Next Generation
While the first period of transporter research established the structural basis for guanidinium-rich transporter uptake, and the second used that knowledge to expand the scope, the third generation is now harnessing the power of this technology in medicine and research (Figure 7).
Figure 7.

New technologies and advances in guanidinium-rich transporter research including (A) the ability of guanidinium-rich transporters to avoid Pgp-mediated efflux and overcome multidrug resistance, (B) an oligocarbonate oligomerization to more step-economically synthesize transporters, (C) the application of amphipathic oligocarbonate transporters for intracellular delivery of siRNA, and (D) delivery of guanidinium-rich transporter conjugates and complexes into algae.
In 2008 we explored whether transporters could address export-based drug resistance, a major cause of chemotherapy failure. Multidrug resistant cancer can be attributed to many factors, though it is often dominated by the overexpression of membrane-associated efflux pumps, most commonly P-glycoprotein (P-gp), which expel drugs from the cell membrane.50 Ironically, many drugs succumb to export as they are unwittingly designed to be membrane soluble. In contrast, transporter-drug conjugates have physical properties distinct from the drug alone and rapidly pass through membranes. Rather remarkably, conjugation of octaarginine through a bioactivatable disulfide linker to Taxol provides a conjugate that overcomes resistance to Taxol in Taxol-resistant ovarian carcinoma cells (Figure 7A).46 The Pgp-evading transporter-drug conjugate renders the drug water-soluble, eliminating the need for toxic excipients (e.g., CremophoreEL) and minimizing volume of formulation. This strategy to overcome resistance was demonstrated in animal models of ovarian cancer and in ex vivo human ovarian cancer patient samples.51 Remarkably, the Taxol-octaarginine conjugates significantly outperformed Taxol alone in all patient samples. The ability to overcome Pgp-based drug resistance with oligoarginine conjugates is a promising and general strategy that could improve the prognosis for cancer treatments and other diseases associated with drug efflux and resistance.
The power of FOS is that it enables one to design for both superior function and step-economical synthesis. Regarding the latter, while octaarginine transporters have been made on GMP scale, and a segment doubling strategy was developed as a step-saving method to access homooligomers,52 the number of synthetic steps in these approaches scales linearly with transporter length. To address this problem, an oligomerization strategy was introduced, allowing for the assembly of transporters of various lengths in a single operation, simply by varying the initiator-to-monomer ratio. Advantages of an oligomerization approach include fewer workups and purifications, and lower costs while allowing rapid access to new transporters as a result of the flexibility and speed of the synthesis (time economy).
In 2008, the Kiessling and Tew labs independently reported the ring-opening metathesis polymerization (ROMP) of functionalized norbornene- and oxanorbornene-derived monomers to access guanidinylated oligomers.53,54 Both groups subsequently showed that block copolymers containing guanidinium-rich sections also facilitated cell uptake.55,56 Recently Matile and coworkers developed a disulfide polymerization strategy to access guanidinylated oligomers.57
In 2009, in collaboration with Hedrick (IBM) and Waymouth (Stanford), we reported a metal-free, organocatalytic ring-opening oligomerization to produce guanidinium-rich oligocarbonates (Figure 7B).58 These oligocarbonate transporters enter cells, and can be used with our disulfide linker technology to release conjugated small molecule cargos. Importantly, the probe or linker entity can be easily incorporated into the transporter as the initiator for oligomerization. The organocatalytic process also avoids metal catalyst residues.
We also showed that this strategy could be used to produce amphipathic block co-oligomers that noncovalently complex, deliver and release siRNA in vitro with up to 90% knockdown of target protein (Figure 7C).41 Significantly, the carbonate scaffold is biodegradable, producing nontoxic components after cellular uptake, a unique feature that renders this technology particularly attractive for use in a range of imaging and therapeutic applications. Recently, the Tew group has also developed amphipathic siRNA complexation and delivery agents that were synthesized via the ROMP method, and showed effective siRNA delivery into primary human T cells.59
Recent work has also shown that guanidinium-rich transporters can be designed to target mitochondria27,60 and early endosomes,61 in addition to being directed to the nucleus.2,62 These studies represent an advancement of this technology to address subcellular organelle targeting. The increasing extent to which guanidinium-rich transporters are now employed in chemistry, biology, and medicine is also seen in the first report of induction of stem cell pluripotency using a recombinant fusion of oligoarginine and four separately identified transcription factors to reprogram murine and human somatic cells to induced pluripotent stem cells.63,64
We conclude with studies on yet another barrier problem, the cell wall, found in plants and other organisms. While chemical synthesis increasingly seeks greener and step- and time-economic strategies, synthetic biology using such organisms provides a powerful method to make many scaffolds. The synthesis of molecules by algae, for example, is a potentially inexpensive, scalable and solar-powered approach to a variety of chemical products – including biofuels, nanomaterials, recombinant proteins, medicinal leads and food additives – utilizing CO2 as a carbon source.65,66 Despite the interest in algal engineering, efforts to study or manipulate their metabolic processes are severely hindered by challenges encountered in the delivery of probes and genes across algal cell wall and membrane barriers.67 In collaboration with Lawrence Berkeley National Laboratory, the first molecular method for the delivery of small molecules and biomacromolecules into algae was developed using our transporter technology.5 In this seminal work, guanidinium-rich transporters were shown to mediate cell uptake of small molecule probes, as well as a 100 kDa protein complex containing a catalytically competent enzyme (Figure 7D), providing a versatile tool for studying algae and other non-mammalian systems with commercial promise.
Conclusion
Over the past 15 years, work in our and other laboratories has opened a broad range of new strategies for carrying drugs and probes across biochemical barriers. All variety of guanidinylated transporter scaffolds, including peptides, peptoids, oligocarbamates, oligocarbonates, and carbohydrates have been used to deliver cargos as varied as small molecules, DNA, siRNA, imaging agents, peptides, and metals. Biochemical barriers as diverse as the blood-brain barrier and the cell wall of algae have been breached with this technology. Given the increasing importance of barrier penetration and the incredible success of molecular transporters in breaching these barriers, molecular transporters can be expected to enable many advances in chemistry, biology, imaging, diagnostics, and medicine.
Acknowledgments
Support of this work through grants NIH-CA031841 and NIH-CA031845 from the National Institutes of Health are acknowledged. Support of this work through fellowships from the National Science Foundation (EGS, BMT, JRV) and the Stanford Graduate Fellowship (EGS, BMT) are acknowledged.
Biographies
Erika Geihe Stanzl received her B.A. degree (2008) from Harvard University. She is currently an NSF Graduate Fellow and Stanford Graduate Fellow in Paul Wender’s laboratory at Stanford University. During her Ph.D., her research has focused on technologies to deliver small molecules, proteins, and oligonucleotides across biological barriers.
Brian M. Trantow received a B.S. degree (2007) from Yale University and recently completed a Ph.D. at Stanford University as an NSF Graduate Fellow and Stanford Graduate Fellow in Paul Wender’s laboratory. His Ph.D. research focused on the development of new chemical tools and methods for probing biological systems, including transporter technologies for delivery across biological barriers.
Jessica R. Vargas received her B.S. degree in Biochemistry (2010) from Loyola Marymount University, and is currently an NSF Graduate Research Fellow at Stanford University. Her research in Paul Wender’s laboratory focuses on designing transporters for drug and probe delivery.
Paul A. Wender is a product of Wilkes, Yale and Columbia Universities, served on the Harvard faculty, is now Bergstrom Professor of Chemistry and Professor, by courtesy, of Chemical and Systems Biology at Stanford, and has the pleasure to be working with the co-authors and similarly gifted coworkers on major problems in synthesis, chemistry, biology and medicine.
References
- 1.Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: Peptoid molecular transporters. Proc Natl Acad Sci US A. 2000;97:13003–13008. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wender PA, Galliher WC, Goun EA, Jones LR, Pillow TH. The design of guanidinium-rich transporters and their internalization mechanisms. Adv Drug Deliv Rev. 2008;60:452–472. doi: 10.1016/j.addr.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wender PA, Cooley CB, Geihe EI. Beyond cell penetrating peptides: Designed molecular transporters. Drug Discovery Today: Technologies. 2012;9:e49–e55. doi: 10.1016/j.ddtec.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang J, Srinivasan A, Sun Y, Mrazek J, Shu Z, Kickhoefer VA, Rome LH. Vault nanoparticles engineered with the protein transduction domain, TAT48, enhances cellular uptake. Integr Biol. 2013;5:151–158. doi: 10.1039/c2ib20119d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hyman JM, Geihe EI, Trantow BM, Parvin B, Wender PA. A molecular method for the delivery of small molecules and proteins across the cell wall of algae using molecular transporters. Proc Natl Acad Sci US A. 2012;109:13225–13230. doi: 10.1073/pnas.1202509109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seward GK, Wei Q, Dmochowski IJ. Peptide-mediated cellular uptake of cryptophane. Bioconj Chem. 2008;19:2129–2135. doi: 10.1021/bc8002265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Futaki S, Suzuki T, Ohashi W, Yagami T, Tanaka S, Ueda K, Sugiura Y. Arginine-rich peptides: An abundant source membrane-permeable peptides having potential as carriers for intracellular protein delivery. J Biol Chem. 2001;276:5836–5840. doi: 10.1074/jbc.M007540200. [DOI] [PubMed] [Google Scholar]
- 8.Ho RJY, Chien JY. Drug delivery trends in clinical trials and translational medicine: Growth in biologic molecule development and impact on rheumatoid arthritis, Crohn’s disease, and colitis. J Pharm Sci. 2012;101:2668–2674. doi: 10.1002/jps.23154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Torchilin VP. Tat peptide-mediated intracellular delivery of pharmaceutical nanocarriers. Adv Drug Deliv Rev. 2008;60:548–558. doi: 10.1016/j.addr.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 10.Langel U, editor. Methods in Molecular Biology Series 683. Humana Press Inc; Totowa, NJ: 2011. Cell-Penetrating Peptides: Methods and Protocols. [Google Scholar]
- 11.Green MM, Loewenstein PMP. Autonomous functional domains of chemically synthesized human immunodeficiency virus Tat trans-activator protein. Cell. 1988;55:1179–1188. doi: 10.1016/0092-8674(88)90262-0. [DOI] [PubMed] [Google Scholar]
- 12.Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55:1189–1193. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- 13.Vivés L, Brodin P, Lebleu B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem. 1997;272:16010–16017. doi: 10.1074/jbc.272.25.16010. [DOI] [PubMed] [Google Scholar]
- 14.Mitchell DJ, Kim DT, Steinman L, Fathman CG, Rothbard JB. Polyarginine enters cells more efficiently than other polycationic homopolymers. J Peptide Res. 2000;56:318–325. doi: 10.1034/j.1399-3011.2000.00723.x. [DOI] [PubMed] [Google Scholar]
- 15.Rothbard JB, Jessop TC, Lewis RS, Murray BA, Wender PA. Role of membrane potential and hydrogen bonding in the mechanism of translocation of guanidinium-rich peptides into cells. J Am Chem Soc. 2004;126:9506–9507. doi: 10.1021/ja0482536. [DOI] [PubMed] [Google Scholar]
- 16.Wender PA, Verma VA, Paxton TJ, Pillow TH. Function-oriented synthesis, step economy, and drug design. Acc Chem Res. 2008;41:40–49. doi: 10.1021/ar700155p. [DOI] [PubMed] [Google Scholar]
- 17.Rothbard JB, Garlington S, Lin Q, Kirschberg T, Kreider E, McGrane PL, Wender PA, Khavari PA. Conjugation of arginine oligomers to cyclosporin A facilitates topical delivery and inhibition of inflammation. Nat Med. 2000;6:1253–1257. doi: 10.1038/81359. [DOI] [PubMed] [Google Scholar]
- 18.Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: Delivery of a biologically active protein into the mouse. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- 19.Jones LR, Goun EA, Shinde R, Rothbard JB, Contag CH, Wender PA. Releasable luciferin-transporter conjugates: Tools for the real-time analysis of cellular uptake and release. J Am Chem Soc. 2006;128:6526–6527. doi: 10.1021/ja0586283. [DOI] [PubMed] [Google Scholar]
- 20.Chen L, Wright LR, Chen CH, Oliver SF, Wender PA, Mochly-Rosen D. Molecular transporters for peptides: delivery of a cardioprotective εPKC agonist peptide into cells and intact ischemic heart using a transport system, R7. Chem Biol. 2001;8:1123–1129. doi: 10.1016/s1074-5521(01)00076-x. [DOI] [PubMed] [Google Scholar]
- 21.Rothbard JB, Kreider E, VanDeusen CL, Wright L, Wylie BL, Wender PA. Arginine-rich Molecular transporters for drug delivery: Role of backbone spacing in cellular uptake. J Med Chem. 2002;45:3612–3618. doi: 10.1021/jm0105676. [DOI] [PubMed] [Google Scholar]
- 22.Rueping M, Mahajan Y, Sauer M, Seebach D. Cellular uptake studies with Beta-peptides. ChemBioChem. 2002;3:257–259. doi: 10.1002/1439-7633(20020301)3:2/3<257::AID-CBIC257>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 23.Umezawa N, Gelman MA, Haigis MC, Raines RT, Gellman SH. Translocation of a β-peptide across cell membranes. J Am Chem Soc. 2002;124:368–369. doi: 10.1021/ja017283v. [DOI] [PubMed] [Google Scholar]
- 24.Fillon YA, Anderson JP, Chmielewski J. Cell-penetrating agents based on a polyproline helix scaffold. J Am Chem Soc. 2005;127:11798–11803. doi: 10.1021/ja052377g. [DOI] [PubMed] [Google Scholar]
- 25.Wender PA, Rothbard JB, Jessop TC, Kreider EL, Wylie BL. Oligocarbamate molecular transporters: Design, synthesis, and biological evaluation of a new class of transporters for drug delivery. J Am Chem Soc. 2002;124:13382–13383. doi: 10.1021/ja0275109. [DOI] [PubMed] [Google Scholar]
- 26.Maiti KK, Jeon OY, Lee WS, Kim DC, Kim KT, Takeuchi T, Futaki S, Chung SK. Design, synthesis, and membrane-translocation studies of inositol-based transporters. Angew Chem Int Ed. 2006;45:2907–2912. doi: 10.1002/anie.200600312. [DOI] [PubMed] [Google Scholar]
- 27.Maiti KK, Lee WS, Takeuchi T, Watkins C, Fretz M, Kim DC, Futaki S, Jones A, Kim KT, Chung SK. Guanidine-containing molecular transporters: Sorbitol-based transporters show high intracellular selectivity toward mitochondria. Angew Chem Int Ed. 2007;46:5880–5884. doi: 10.1002/anie.200701346. [DOI] [PubMed] [Google Scholar]
- 28.Futaki S, Nakase I, Suzuki T, Zhang, Sugiura Y. Translocation of branched-chain arginine peptides through cell membranes: Flexibility in the spatial disposition of positive charges in membrane-permeable peptides. Biochemistry. 2002;41:7925–7930. doi: 10.1021/bi0256173. [DOI] [PubMed] [Google Scholar]
- 29.Chung HH, Harms G, Min Seong C, Choi BH, Min C, Taulane JP, Goodman M. Dendritic oligoguanidines as intracellular translocators. Biopolym Pept Sci. 2004;76:83–96. doi: 10.1002/bip.10597. [DOI] [PubMed] [Google Scholar]
- 30.Wender PA, Kreider E, Pelkey ET, Rothbard J, VanDeusen CL. Dendritic molecular transporters: Synthesis and evaluation of tunable polyguanidino dendrimers that facilitate cellular uptake. Org Lett. 2005;7:4815–4818. doi: 10.1021/ol051496y. [DOI] [PubMed] [Google Scholar]
- 31.Huang K, Voss B, Kumar D, Hamm HE, Harth E. Dendritic molecular transporters provide control of delivery to intracellular compartments. Bioconj Chem. 2007;18:403–409. doi: 10.1021/bc060287a. [DOI] [PubMed] [Google Scholar]
- 32.Luedtke NW, Carmichael P, Tor Y. Cellular uptake of aminoglycosides, guanidinoglycosides, and poly-arginine. J Am Chem Soc. 2003;125:12374–12375. doi: 10.1021/ja0360135. [DOI] [PubMed] [Google Scholar]
- 33.Dix AV, Fischer L, Sarrazin S, Redgate CP, Esko JD, Tor Y. Cooperative, heparin sulfate-dependent cellular uptake of dimeric guanidinoglycosides. ChemBioChem. 2010;11:2302–2310. doi: 10.1002/cbic.201000399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou P, Wang M, Du L, Fisher GW, Waggoner A, Ly DH. Novel binding and efficient cellular uptake of guanidine-based peptide nucleic acids (GPNA) J Am Chem Soc. 2003;125:6878–6879. doi: 10.1021/ja029665m. [DOI] [PubMed] [Google Scholar]
- 35.Ohmichi T, Kuwahara M, Sasaki N, Hasegawa M, Nishikata T, Sawai H, Sugimoto N. Nucleic acid with guanidinium modification exhibits efficient cellular uptake. Angew Chem Int Ed. 2005;44:6682–6685. doi: 10.1002/anie.200500904. [DOI] [PubMed] [Google Scholar]
- 36.Deglane G, Kuwahara M, Sasaki N, Hasegawa M, Nishikata T, Sawai H, Sugimoto N. Impact of the guanidinium group on hybridization and cellular uptake of cationic oligonucleotides. ChemBioChem. 2006;7:684–692. doi: 10.1002/cbic.200500433. [DOI] [PubMed] [Google Scholar]
- 37.Dempcy OR, Almarsson O, Bruice TC. Design and synthesis of deoxynucleic guanidine: a polycation analogue of DNA. Proc Natl Acad Sci US A. 1994;91:7864–7868. doi: 10.1073/pnas.91.17.7864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Siprashvili Z, Scholl FA, Oliver SF, Adams A, Contag CH, Wender PA, Khavari PA. Gene transfer via reversible plasmid condensation with cysteine-flanked internally spaced arginine-rich peptides. Hum Gene Ther. 2003;14:1225–1233. doi: 10.1089/104303403767740768. [DOI] [PubMed] [Google Scholar]
- 39.Kim WJ, Christensen LV, Jo S, Yockman JW, Jeong JH, Kim YH, Kim SW. Cholesteryl oligoarginine delivering vascular endothelial growth factor siRNA effectively inhibits tumor growth in colon adenocarcinoma. Mol Ther. 2006;14:343–350. doi: 10.1016/j.ymthe.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 40.Kumar P, Wu H, McBride JL, Jung KE, Kim MH, Davidson BL, Lee SK, Shankar P, Manjunath N. Transvascular delivery of small interfering RNA to the central nervous system. Nature. 2007;448:39–43. doi: 10.1038/nature05901. [DOI] [PubMed] [Google Scholar]
- 41.Geihe EI, Cooley CB, Simon JR, Kiesewetter MK, Edward JA, Hickerson RP, Kaspar RL, Hedrick JL, Waymouth RM, Wender PA. Designed guanidinium-rich amphipathic oligocarbonate molecular transporters complex, deliver and release siRNA in cells. Proc Natl Acad Sci US A. 2012;109:13171–13176. doi: 10.1073/pnas.1211361109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li YF, Morcos PA. Design and synthesis of dendritic molecular transporter that achieves efficient in vivo delivery of morpholino antisense oligo. Bioconj Chem. 2008;19:1464–1470. doi: 10.1021/bc8001437. [DOI] [PubMed] [Google Scholar]
- 43.Bullok KE, Gammon ST, Violini S, Prantner AM, Villalobos VM, Piwnica-Worms D. Permeation peptide conjugates for in vivo molecular imaging applications. Mol Imaging. 2006;5:1–15. [PubMed] [Google Scholar]
- 44.Lee HL, Dubikovskaya EA, Hwang H, Semyonov AN, Wang H, Jones LR, Twieg RJ, Moerner WE, Wender PA. Single-molecule motions of oligoarginine transporter conjugates on the plasma membrane of Chinese hamster ovary cells. J Am Chem Soc. 2008;130:9364–9370. doi: 10.1021/ja710798b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wender PA, Goun EA, Jones LR, Pillow TH, Rothbard JB, Shinde R, Contag CH. Real-time analysis of uptake and bioactivatable cleavage of luciferin-transporter conjugates in transgenic reporter mice. Proc Natl Acad Sci US A. 2007;104:10340–10345. doi: 10.1073/pnas.0703919104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dubikovskaya EA, Thorne SH, Pillow TH, Contag CH, Wender PA. Overcoming multidrug resistance of small-molecule therapeutics through conjugation with releasable octaarginine transporters. Proc Natl Acad Sci US A. 2008;105:12128–12133. doi: 10.1073/pnas.0805374105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiang T, Olson ES, Nguyen QT, Roy M, Jennings PA, Tsien RY. Tumor imaging by means of proteolytic activation of cell-penetrating peptides. Proc Natl Acad Sci US A. 2004;101:17867–17872. doi: 10.1073/pnas.0408191101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goun EA, Shinde R, Dehnert KW, Adams-Bond A, Wender PA, Contag CH, Franc BA. Intracellular cargo delivery by an octaarginine transporter adapted to target prostate cancer cells through cell surface protease activation. Bioconj Chem. 2006;17:787–796. doi: 10.1021/bc0503216. [DOI] [PubMed] [Google Scholar]
- 49.Nguyen QT, Olson ES, Aguilera TA, Jiang T, Scadeng M, Ellies LG, Tsien RY. Surgery with molecular fluorescence imaging using activatable cell-penetrating peptides decreases residual cancer and improves survival. Proc Natl Acad Sci US A. 2010;107:4317–4322. doi: 10.1073/pnas.0910261107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Persidis A. Cancer multidrug resistance. Nat Biotechnol. 1999;17:94–95. doi: 10.1038/5289. [DOI] [PubMed] [Google Scholar]
- 51.Wender PA, Galliher WC, Bhat NM, Pillow TH, Bieber MM, Teng NH. Taxol-oligoarginine conjugates overcome drug resistance in-vitro in human ovarian carcinoma. Gynecol Oncol. 2012;126:118–123. doi: 10.1016/j.ygyno.2012.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wender PA, Jessop TC, Pattabiraman K, Pelkey ET, VanDeusen CL. An efficient, scalable synthesis of the molecular transporter octaarginine via a segment doubling strategy. Org Lett. 2001;3:3229–3232. doi: 10.1021/ol0161108. [DOI] [PubMed] [Google Scholar]
- 53.Kolonko EM, Kiessling LL. A polymeric domain that promotes cellular internalization. J Am Chem Soc. 2008;130:5626–5627. doi: 10.1021/ja8001716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gabriel GJ, Madkour AE, Dabkowski JM, Nelson CF, Nüsslein K, Tew GN. Synthetic mimic of antimicrobial peptide with nonmembrane-disrupting antibacterial properties. Biomacromolecules. 2008;9:2980–2983. doi: 10.1021/bm800855t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kolonko EM, Pontrello JK, Mangold SL, Kiessling LL. General synthetic route to cell-permeable block copolymers via ROMP. J Am Chem Soc. 2009;131:7327–7333. doi: 10.1021/ja809284s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Som A, Reuter A, Tew GN. Protein transduction domain mimics: The role of aromatic functionality. Angew Chem Int Ed. 2012;51:980–983. doi: 10.1002/anie.201104624. [DOI] [PubMed] [Google Scholar]
- 57.Bang EK, Gasparini G, Molinard G, Roux A, Sakai N, Matile S. Substrate-initated synthesis of cell-penetrating poly(disulfide)s. J Am Chem Soc. 2013;135:2088–2091. doi: 10.1021/ja311961k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cooley CB, Trantow BM, Nederberg F, Kiesewetter MK, Hedrick JL, Waymouth RM, Wender PA. Oligocarbonate molecular transporters: oligomerization-based syntheses and cell-penetrating studies. J Am Chem Soc. 2009;131:16401–16403. doi: 10.1021/ja907363k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tezgel AÖ, Gonzalez-Perez G, Telfer JC, Osborne BA, Minter LM, Tew GN. Novel protein transduction domain mimics as nonviral delivery vectors for siRNA targeting NOTCH1 in primary human T cells. Mol Ther. 2013;21:201–209. doi: 10.1038/mt.2012.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Horton KL, Stewart KM, Fonseca SB, Guo Q, Kelley SO. Mitochondria-penetrating peptides. Chem Biol. 2008;15:375–382. doi: 10.1016/j.chembiol.2008.03.015. [DOI] [PubMed] [Google Scholar]
- 61.Appelbaum JS, LaRochelle JR, Smith BA, Balkin DM, Holub JM, Schepartz A. Arginine topology controls escape of minimally cationic proteins from early endosomes to the cytoplasm. Chem Biol. 2012;19:819–830. doi: 10.1016/j.chembiol.2012.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Puckett CA, Barton JK. Fluorescein Redirects a Ruthenium-Octaarginine Conjugate to the Nucleus. J Am Chem Soc. 2009;131:8738–8739. doi: 10.1021/ja9025165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhou H, Wu S, Joo JY, Zhu S, Han DW, Lin T, Trauger S, Bien G, Yao S, Zhu Y, Siuzdak G, Schöler HR, Duan L, Ding S. Generation of Induced Pluripotent Stem Cells Using Recombinant Proteins. Cell Stem Cell. 2009;4:381–384. doi: 10.1016/j.stem.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim D, Kim CH, Moon JI, Chung YG, Chang MY, Han BS, Ko S, Yang E, Cha KY, Lanza R, Kim KS. Generation of Human Induced Pluripotent Stem Cells by Direct Delivery of Reprogramming Proteins. Cell Stem Cell. 2009;4:472–476. doi: 10.1016/j.stem.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Specht E, Miyake-Stoner S, Mayfield S. Micro-algae come of age as a platform for recombinant protein production. Biotechnol Lett. 2010;32:1373–1383. doi: 10.1007/s10529-010-0326-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Radakovits R, Jinkerson RE, Darzins A, Posewitz MC. Genetic Engineering of Algae for Enhanced Biofuel Production. Eukar Cell. 2010;9:486–501. doi: 10.1128/EC.00364-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Harris EH. The Chlamydomonas. Sourcebook. (2) 2009;1:293–302. [Google Scholar]