

Abstract
γ-Secretase is an aspartyl intramembranal protease composed of presenilin, Nicastrin, Aph1 and Pen2 with 19 transmembrane domains. γ-Secretase cleaves the amyloid precursor proteins (APP) to release Aβ peptides that likely play a causative role in the pathogenesis of Alzheimer disease (AD). In addition, γ-secretase cleaves Notch and other type I membrane proteins. γ-Secretase inhibitors (GSIs) have been developed and used for clinical studies. However, clinical trials have shown adverse effects of GSIs that are potentially linked with non-discriminatory inhibition of Notch signaling, overall APP processing and other substrate cleavages. Therefore, these findings call for the development of disease modifying agents that target γ-secretase activity to lower Aβ42 production without blocking the overall processing of γ-secretase substrates. γ-Secretase modulators (GSMs) originally derived from non-steroidal anti-inflammatory drugs (NSAIDs) display such characteristics and are the focus of this review. However, first generation GSMs have limited potential due to low potency and undesired neuropharmacokinetic properties. This generation of GSMs has been suggested to interact with the APP substrate, γ-secretase or both. To improve the potency and brain availability, second generation GSMs including NSAID-derived carboxylic acid and non-NSAID-derived heterocyclic chemotypes as well as natural product-derived GSMs have been developed. Animal studies of this generation of GSMs have shown encouraging preclinical profiles. Moreover, using potent GSM photoaffinity probes, multiple studies unambiguously have showed that both carboxylic acid and heterocyclic GSMs specifically target presenilin, the catalytic subunit of γ-secretase. In addition, two types of GSMs have distinct binding sites within the γ-secretase complex and exhibit different Aβ profiles. GSMs induce a conformational change of γ-secretase to achieve modulation. Various models are proposed and discussed. Despite the progress of GSM research, many outstanding issues remain to be investigated to achieve the ultimate goal of developing GSMs as effective AD therapies.
γ-Secretase and Aβ peptides
γ-Secretase modulators (GSMs) have emerged to the forefront of Alzheimer disease (AD) research due to their potential as disease modifying agents and despite an unclear mechanism of action. GSMs are a class of compounds that selectively reduce the formation of pathogenic Aβ42 species and yet do not affect the total amount of Aβ produced.(1) Moreover, they have little effect on γ-secretase-dependent Notch processing since the generation of Notch intracellular domain (NICD) is not inhibited.(1) Several reviews (2–7) have highlighted the progress made in developing the next generation of GSMs. This review focuses on recent progress in molecular probe development and studies toward elucidating the mechanism of action of GSMs.
Although the precise pathological mechanism of AD remains elusive, it is widely believed that Aβ peptides, the major constituents of amyloid plaques,(8, 9) play a central role in AD through a process named the “amyloid cascade hypothesis”.(10) In this hypothesis, Aβ peptides form a neurotoxic species that triggers a pathological cascade and ultimately leads to neurodegeneration and dementia. Aβ peptides are excised from the amyloid precursor protein (APP) through two proteases: β- and γ-secretases (Fig. 1A). This process also generates sAPPβ and APP intracellular C-terminal domain (AICD), which could have different biological roles.(11) Alternatively, APP can be processed by α- and γ-secretases to generate αCTF, sAPPα, P3 and AICD with varying biological activities.(11) Recent studies suggest that α-secretase cleavage can function as a negative feedback regulator to modulate γ-secretase for Aβ production,(12, 13) in addition to competing with β-secretase for APP substrates.(14–17)
Figure 1.

(A) Illustration of APP processing by α-, β-, and γ-secretases and the corresponding products. (B) Sequence of the membrane and nearby regions of the β-CTF substrate and relevant cleavages. Thick horizontal arrows represent the hypothesized processive cleavage by γ-secretase. Vertical red arrows show locations of γ, ζ, and ε cleavages.
γ-Secretase cleaves APP at multiple sites including γ-, ζ- and ε-cleavages(18, 19) (Fig. 1B) to generate Aβ species with heterogeneous C-termini, which are 37–46 amino acids long.(20, 21) Compelling evidence indicates that these Aβ peptides can be generated through a processive mechanism that travels from the ε-site to the γ-site and removes three to four amino acids at each step.(22) It has also been proposed that there are two γ-secretase product lines; one from Aβ49 to Aβ46, Aβ43, Aβ40 and Aβ37; and the other from Aβ48 to Aβ45, Aβ42 and Aβ38. However, recent studies showed that Aβ38 can be generated from Aβ42 and Aβ43 (23), suggesting that both product lines can be crossed with various combinations. Furthermore, multiple studies have shown that the γ- and ε-cleavages are not always correlated (12, 24–30). Mutations in APP and PS1 lead to different effects on γ- and ε-cleavages, and even within γ-sites (such as Aβ42 and Aβ38) (12, 24, 27–29). In addition, interaction of γ-secretase with other proteins and/or different assay conditions can dissociate these events.(25, 26, 30) Whether these findings reflect that γ- and ε-cleavages are differentially regulated during sequential processing or just indicate that they represent independent events merits further investigation.
Among the different forms of Aβ species, the role of Aβ40 and Aβ42 in AD has been intensively investigated. While both Aβ40 and Aβ42 have been implicated in AD,(10) Aβ42 is more prone to aggregation and is believed to play a critical role in the initiation of AD pathogenesis.(31, 32) However, recent studies suggest that the ratio of Aβ42/Aβ40, rather than the total amount of Aβ, exhibits a better correlation with the age of onset of FAD.(33) Moreover, in vitro and animal studies showed that Aβ40 can play a role in preventing Aβ42 aggregation and therefore reduction of Aβ40 that alters the ratio of Aβ42/Aβ40 may lead to enhanced amyloidogenesis.(34–39) Direct evidence demonstrating that Aβ40 inhibits amyloid deposition came from the studies of bitransgenic (BRL-Aβ40/Tg2576) mice in which the over-expression of Aβ40 peptide significantly reduced the amyloid deposition.(35)
Non-selective inhibition of γ-secretase drastically affects the processing and metabolism of APP proteins, which have been shown to regulate various neuronal and synaptic functions conferred by distinct APP domains.(11, 40) Furthermore, the accumulation of APP βCTF that results from γ-secretase inhibition has been implicated in neurotoxicity. (41) Also, it has been shown that γ-secretase inhibitors (GSIs) can cause Aβ elevation when administered at low concentrations and withdrawing of GSIs leads to a rebound increase in Aβ plasma levels. (42). In addition, it has been found that an increased concentration of βCTF can augment the Aβ42/Aβ40 ratio (43). Together these data suggest that total inhibition of APP processing could actually aggravate AD pathology.
Autosomal dominant inheritance of mutations in three genes—the amyloid precursor protein (APP), Presenilin-1 (PS1) and Presenilin-2 (PS2)–causes early-onset and familial AD (FAD).(44–46) Although how these FAD mutations cause the disease is controversial,(47) it appears that the overwhelming majority of mutations lead to an increase in the ratio of Aβ42/Aβ40,(48) further supporting the Aβ hypothesis. It is noteworthy to mention a recent discovery showing that an APP mutation, which reduces Aβ production, protects against AD and age-related cognitive decline,(49) providing another line of support for the amyloid cascade hypothesis.
Notch1 was the second γ-secretase substrate identified after APP, and functional γ-secretase knockouts result in a notch phenotype. (50–52) The Notch signaling pathway plays an essential role in cell fate decisions during development. (53) Notch signaling also plays an important role in the adult brain, which includes the maintenance and differentiation of neuronal stem cells, structure and synaptic plasticity as well as neuron survival.(54, 55) In addition, Notch can act as a proto-oncogene or tumor suppressor in some cancers.(56) Notch1 is processed at least three times (S1–S3 cleavages) for its signaling. First, Notch is cleaved by a furin-like protease (S1 site) in the Golgi that converts a single chain into a heterodimer.(57) Next, ligand binding to Notch triggers two sequential proteolytic events (S2 and S3): Notch is cut by ADAM metalloproteases at site 2 (S2) and then by γ-secretase at site 3 (S3), which is within the transmembrane domain(58) and analogous to the ε-site of APP (Fig. 2).(19) Following the S3 cleavage, the Notch intracellular domain (NICD) is released from the membrane tether and translocates to the nucleus, where it activates transcription of target genes. NICD binds the CSL (CBF-1/Su(H)/Lag-1) transcription factor, thereby dissociating co-repressors and recruiting co-activators such as mastermind (MamL), ultimately leading to the activation of effector genes.(53, 57) There are five Notch ligands (Dll-1,-3,-4, Jagged-1, -2) and four mammalian Notch receptors (N1-4). All four receptors have been shown to be cleaved by γ-secretase.(59)
Figure 2.

Illustration of the notch signaling cascade (A) depicting activation by a sending cell, which induces S2 cleavage by an ADAM protease, followed by S3 cleavage by γ-secretase within the membrane domain. Subsequently, notch intracellular domain (NICD) is released from the membrane and translocates to the nucleus where it can turn on target genes. (B) Sequence of the membrane domain and S3 site cleavage of the Notch-1 receptor.
The wide spectrum of γ-secretase substrates has made it even more challenging to develop target-based therapy. More than 90 putative γ-secretase substrates have been reported,(60) reflecting the diverse functions of this protease. However, it is worth considering that many of the experimental studies have only demonstrated that γ-secretase can cleave these protein substrates. Deeper investigation is required in order to determine how many of these proteins are bone fide physiological substrates of γ-secretase, and which ones are most likely to cause detrimental side effects when γ-secretase is inhibited. The Phase III clinical trial of semagacestat, a non-selective γ-secretase inhibitor, was terminated due to slightly worse cognition scores and an increase in the risk of skin cancer compared to placebo.(61) Although the precise mechanism that caused these adverse effects is unknown, increased incidents in skin cancer are likely associated with γ-secretase dependent Notch1 signaling that functions as a tumor suppressor.(62, 63) In addition, semagacestat treatment also led to a lightening in hair color,(64) which could be associated with tyrosinase, a substrate of γ-secretase.(65) Therefore, it’s critical to know how many substrates are affected by in vivo inhibition of γ-secretase, and what the consequences of these events are.
γ-Secretase is an intramembranal complex which relies on the assembly of an active enzyme complex that is composed of a quartet of proteins: Nicastrin (NCT), Presenilin (PS), Pen-2, and Aph-1 with 19 putative transmembrane domains (Fig. 3).(66) All four proteins are obligatory for cellular γ-secretase activity.(67) PS is the catalytic subunit of γ-secretase,(68–70) and belongs to a unique family of GxGD type aspartyl proteases.(71, 72) Recently, the crystal structure of a PS/signal peptide peptidase (SPP) homologue (PSH) from the archaeon Methanoculleus marisnigri has offered insights into how the transmembrane domains and catalytic dyad are organized in PS1.(73) Both the PS1 and PS2 polypeptides undergo endoproteolysis, whereby the N- and C-terminal cleavage products (NTF and CTF) remain associated as heterodimeric integral membrane proteins.(74) There are two isoforms of presenilin: PS1 and PS2, and three isoforms of Aph-1: Aph-1aS, Aph-1aL and Aph-1b. At least six active γ-secretase complexes have been reported (2 presenilins × 3 Aph-1s).(76, 77) Remarkably, PS1 and PS2 are not engaged in the same complex albeit both of them co-exist in the same cells,(75) indicating a tight and precise control of the assembly of the γ-secretase complex. Aph-1 and NCT play critical roles in the assembly, trafficking, and stability of γ-secretase as well as substrate recognition.(66, 78, 79) Lastly, Pen-2 facilitates the endoproteolysis of PS into its N-terminal (NTF) and C-terminal (CTF) fragments thereby yielding a catalytically competent enzyme.(66, 78, 80–82) Although a γ-secretase complex of ~200 kDa, which contains only one of each subunit, is catalytically active,(83) the endogenous γ-secretase complex appears to possess a higher molecular weight ranging from 500–2,000 kDa.(83–87) Taken together, these studies suggest that the quaternary protein complex(83) may be the basic functional γ-secretase unit in cells, and additional cofactors and/or varying stoichiometry of subunits exist in the high molecular weight γ-secretase complexes for modulating γ-secretase activity and specificity. Nonessential factors, such as CD147, TMP21, γ-secretase activating protein (GSAP), β-arrestin-1 β-arrestin-2, Erin-2, syntexin-1, voltage-dependent anion channel 1 (VDAC1), contactin-associated protein 1 (CNTNAP1), TPPP and NDUFS7 have been found to be associated with the γ-secretase complex and modulate γ-secretase activity and specificity;(26, 88–94) however, the functional significance of some of these interactions has been contended.(95, 96) (97) Moreover, γ-secretase has been shown to interact with tetraspanin-enriched microdomains, or lipid rafts.(98) It has been suggested that different γ-secretase complexes can contribute to substrate specificity,(99, 100) which is exemplified by genetic knockout of Aph-1b in a mouse AD model that improved the disease-relevant phenotypic features without Notch-related side effects.(100)
Figure 3.

The four essential components of γ-secretase. Presenilin, the catalytic center, is depicted in zymogen form before endoproteolysis of Exon 9 and according to the predicted structure by Li et al (73). Stars represent the relative location of the two aspartic acid residues required for catalysis.
Another unique feature of γ-secretase is that only a small fraction of the four protein complex is catalytically active(85, 101) and the total amount of PS protein is not always correlated with γ-secretase activity.(75, 101, 102) Lai et al found that less than 14% of PS1 is engaged in active γ-secretase complexes.(75) Activity-based probes designed from transition state GSIs have been used broadly to study the active γ-secretase complex because they do not bind to the inactive complex. (68, 75, 99, 102–104)
Discovery and Development of GSMs
1) First Generation NSAID GSMs
The concept of γ-secretase modulation was discovered when a subset of NSAIDs, such as ibuprofen, indomethacin and sulindac sulfide, were found to selectively lower the formation of Aβ42 in favor of Aβ38 without inhibiting Notch1 cleavage.(1) Furthermore, the effect of these NSAIDs on Aβ modulation was dissociated from their COX activity.(1) GSMs have many unique characteristics, which include: 1) reducing Aβ42 production; 2) promoting shorter forms of Aβ species (Aβ38 or Aβ37); 3) having no significant effect on the total amount of Aβ produced nor accumulation of βCTF; and 4) lacking inhibitory effect on Notch cleavage and other substrates. Not surprisingly, these ideal properties have inspired the development of GSMs as potential disease modifying agents for AD treatment. Of note, although the role of Aβ37 or Aβ38 in AD is unknown, it is believed that the short forms are less pathogenic than Aβ42.
The NSAID GSMs selectively lower the formation of Aβ42 with a concomitant increase in the generation of Aβ38, without inhibiting the proteolysis of Notch1. The first generation GSMs include the NSAIDs: ibuprofen, indomethacin, sulindac sulfide, flurbiprofen and the close analog CHF5074 (Fig. 4). These compounds provided the first evidence that γ-secretase could be specifically modulated to reduce the more pathogenic Aβ42 species. However, their weak in vitro potencies (Aβ42 IC50 > 10 μM) and poor brain penetration has limited their development. Despite its weak potency (Aβ42 IC50 ~ 200–300 μM),(105) R-flurbiprofen (tarenflurbil) was advanced into clinical studies and a hint of efficacy was seen in a Phase II trial in a subgroup of patients with mild AD.(106) However, the Phase III clinical trial of R-flurbiprofen did not achieve statistically significant improvement compared to placebo.(107) R-flurbiprofen is a weak GSM and whether it crossed the blood brain barrier and significantly lowered Aβ42 levels in the clinical studies is unknown. Chiesi has prepared flurbiprofen analogs with improved Aβ42 inhibitory potency leading to CHF5074 (Aβ42 IC50 = 41 μM).(108) This compound has been advanced into clinical trials and was found to lower the levels of the soluble CD40 ligand, a marker of microglia activation, but not Aβ42 in both plasma and CSF so it is now being referred to as a microglial modulator.(109)
Figure 4.
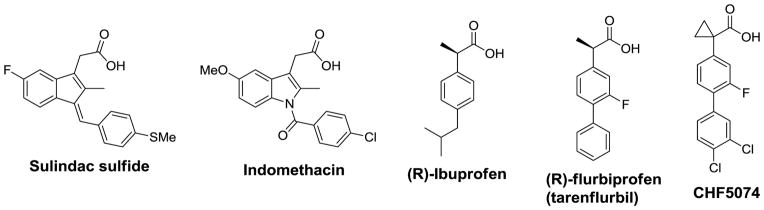
Structures of NSAID-based GSMs.
2) Second Generation GSMs
A key goal in the development of second generation GSMs has been to improve the potency and brain availability, and advances toward this end have resulted in GSMs with encouraging preclinical profiles in recent years.(2, 3) Structurally, second generation GSMs can be generally divided into three categories: NSAID-derived carboxylic acid GSMs, non-NSAID-derived heterocyclic GSMs and natural product-derived GSMs.
NSAID-derived carboxylic acid GSMs
Next-generation NSAID-derived GSMs, including GSM-1, GSM-2, GSM-10h, EVP-0015962, JNJ-40418677, and BIIB042, with improved in vitro potency and brain penetration have been reported (Fig. 5A). Merck and GSK have substituted the core aryl ring with a piperidine ring and optimized the substituent on the piperidine nitrogen to generate a potent series of piperidine acetic acid GSMs. This series is exemplified by GSM-1 and close analogs GSM-2 and GSM-10h, which have become the prototypical 2nd generation acid GSMs and have been extensively investigated from cellular to animal studies (see Table 1). Overall, this class of GSMs reduces the production of Aβ42 and promotes the generation of Aβ38 and has little effect on Aβ40 production, total Aβ levels, AICD and Notch1 processing.
Figure 5.

Structures of 2nd generation NSAID-derived GSMs with acetic acid chemotype.
Table 1.
Effect of 2nd generation acid GSMs on the production of Aβ peptides
| Acid GSM | Cell-based Aβ42 IC50 | In vivo studies | Aβ profile | ||
|---|---|---|---|---|---|
| 38 | 40 | 42 | |||
| GSM-1 | 348 nM(123) | guinea pig(124) | ↑ | - | ↓ |
| GSM-10h | 300 nM(114) | rat(115), mouse(125) | ↓ | ||
| GSM-2 | 65 nM(41) | mouse(41) (126) | ↓ | ||
| EVP-0015962 | 67 nM(117) | mouse(117) | ↑ | - | ↓ |
| JNJ-40418677 | 200 nM(118) | mouse(118) | ↑ | - | ↓ |
| BIIB042 | 170 nM(119) | mouse, rat, monkey(119) | ↓ | ||
The first in vitro characterization of GSM-1 appeared in 2008 where it was shown to significantly decrease Aβ42 and increase Aβ38 in cells expressing either WT PS1 or WT PS2.(28) In contrast, cells expressing PS1 L166P or PS2 N141I FAD mutants showed no change in Aβ42 with GSM-1 treatment, but a robust increase in Aβ38 was still observed. (28) This observation translated to an in vivo setting since administration of GSM-1 to Tg2576 mice resulted in a dose-dependent reduction in brain Aβ42 and an increase in Aβ38, whereas administration to APP-Swe/PS2N141I double transgenic mice showed no significant change in brain Aβ42 despite robust increases in Aβ38. Furthermore, the levels of Aβ40 and total Aβ were unchanged, which is consistent with the profile of the NSAID GSMs discussed above. Therefore, one must be careful when transgenic PS FAD models are chosen for GSM studies because certain mutants could give rise to false-negative results for the effect of GSMs on Aβ42. However, a study by Kretner et al that looked at over 20 different PS1 FAD mutations found that the majority of the mutations responded well to GSM-1, with the exception of L166P.(110) This finding suggests that GSMs could be considered as a candidate therapy for prevention trials in asymptomatic Alzheimer’s disease patients with PS1 FAD mutations.(111, 112) The Aβ profile in response to GSM-1 has also been characterized for a number of FAD-associated APP mutations in both cell-free and cell-based assays.(113) GSM-1 lowered Aβ42 robustly for each APP mutant, but the reciprocal increase in Aβ38 was attenuated in several cases (i.e. T43F, V44F, and I45F); and for certain mutants Aβ39 (i.e., V46I and V46F) and Aβ41 (i.e., V44F) were lowered by GSM-1 treatment. Therefore, it appears that the relationship between Aβ42 and Aβ38 is not always interdependent, and the effect of GSMs on each FAD mutant should be considered independently.
GSM-10h, a pyridyl analog of GSM-1 with lower lipophilicity, has also demonstrated excellent bioavailability and good CNS penetration.(114) Additionally, acute and sub-chronic administration of GSM-10h to rats decreased Aβ42 in plasma, CSF and brain.(115) Furthermore, GSM-10h did not cause Aβ-rebound in rat plasma nor accumulation of β-CTF.(116)
Recently, a study was conducted comparing the efficacy of two GSIs (LY450139 and BMS-708,163) and GSM-2, a piperidine acetic acid (Fig. 5A).(41) These compounds were administered to wild-type and 5.5 month-old Tg2576 mice for 8 days and Y-maze tests were conducted to evaluate spatial working memory. Only GSM-2 ameliorated the cognitive deficit in Tg2576 mice. While all three drugs reduced hippocampal Aβ42 levels, β-CTF levels increased with the two GSIs, but were unchanged with GSM-2. Subchronic treatment with LY450139 actually impaired normal cognitive function in WT mice, while treatment with GSM-2 had no effect. This data suggests that the cognitive impairment associated with GSI treatment could be due, at least in part, to β-CTF elevation.(41)
EnVivo and Janssen have returned to the phenyl acetic acid core of flurbiprofen and added additional substituents on the core aryl ring to generate potent compounds such as EVP-0015962(117) and JNJ-40418677.(118) Chronic treatment with EVP-0015962 in Tg2576 mice reduced soluble (Tris buffered saline extractable), insoluble (formic acid extractable), and aggregated Aβ42; amyloid plaque load in the hippocampus; and cognitive deficits in the contextual fear-conditioning test.(117) The major concern with this compound is its high lipophilicity with a clogP of 6.8 (measured logD = 3.88) and it remains to be seen if the promising preclinical profile can be matched by an acceptable safety profile. Acute treatment with JNJ-40418677 reduced brain Aβ42 levels in wild-type mice with a concomitant increase in Aβ38, while total Aβ levels in brain were not affected. In contrast, chronic administration in Tg2576 mice from 6 to 13 months of age resulted in dose-dependent reductions of all Aβ species in soluble and deposited fractions. As with the other 2nd generation acids, the increase in potency of JNJ-40418677 came at the expense of increased lipophilicity. No data was reported on the safety profile other than 7 months of dosing was tolerated with no weight loss.(118) Biogen has disclosed a phenyl acetic acid GSM (BIIB042) which appears to be a hybrid of flurbiprofen and GSM-1.(119) High drug concentrations in the brain appear to be necessary to achieve robust lowering of Aβ42 and this may be due in part to the high protein binding of BIIB042 (>99.9% protein-bound in all species tested).
The carboxylic acid moiety is critical for both first and second generation NSAID and NSAID-derived GSMs. Multiple studies have shown that if a carboxylic acid GSM is converted to the corresponding ester or amide (Fig. 5B), the compound behaves as an inverse GSM (iGSM) and actually increases Aβ42 production (Fig. 5C).(120–123)
non-NSAID-derived heterocyclic GSMs
The first examples of non-NSAID-derived heterocyclic GSMs were reported in the patent literature by Neurogenetics in 2004 and by Eisai in 2005 and are characterized by the presence of an arylimidazole moiety.(3) Since then several additional members from this class have been disclosed (Fig. 6).
Figure 6.

Structures of non-NSAID-derived heterocyclic GSMs containing the aryl imidazole chemotype.
In contrast to the NSAID-like GSMs, the imidazole GSMs alter the cleavage site preference of γ-secretase such that both Aβ42 and Aβ40 decrease, while Aβ37 and Aβ38 increase albeit to different degrees depending on the compound (Table 2).(127–129) The prototypical imidazole GSM is exemplified by E2012 and has been used as a standard by many labs. E2012 lowers Aβ42, Aβ40 and Aβ39 and raises Aβ37 and, to a lesser extent, Aβ38 (Table 2) (128–130). E2012 entered phase I trials in 2006, and it represents the first non-NSAID GSM to enter clinical development. Following the observation of lenticular opacity in a 13-week rat safety study, clinical development of E2012 was temporarily halted. Subsequent safety studies in rats and monkeys, however, did not show ocular toxicity, and the clinical trial was allowed to proceed in April of 2008. Dose-dependent reductions of Aβ40/42 were observed in plasma in the Phase I clinical trial (131). Eisai recently reported that E2012 was not developed further in favor of an improved compound, E2212. E2212 was reported to be more potent both in vitro and in vivo than E2012 and to have a wider safety margin. The first human study began in January 2010 (doses ranging from 10 mg to 250 mg, ClinicalTrials.gov identifier: NCT01221259), however the present status of development is not known.
Table 2.
Effect of 2nd generation non-acid GSMs on the production of Aβ peptides
| Non-Acid GSM | Cell-based Aβ42 IC50 | In vivo data | Aβ profile | ||||
|---|---|---|---|---|---|---|---|
| 37 | 38 | 39 | 40 | 42 | |||
| E2012 | 92 nM(130), 143 nM(137) | rat (130), dog(128), guinea pig(137) |
|
↑ | ↓ | ↓ | ↓ |
| GSM-A | 8–33 nM(124) | guinea pig(124) | ↑ | ↓ | ↓ | ||
| NGP-555 | 10–29 nM(127), | mouse(127) | ↑ | ↑ | ↓ | ↓ | |
| GSM-53 | 33 nM(133) | rat, dog, monkey(134) | ↑ | ↑ | ↓ | ↓ | ↓ |
| GSM-35 | 44 nM(135) | rat(135) | |||||
| RO-02 | ~14 nM(136) | ↑ |
|
↓ | ↓ | ||
| AZ4800 | 26 nM(129) | mouse(129) | ↑ |
|
↓ | ↓ | ↓ |
| AZ3303 | 74 nM(129) | mouse(129) |
|
↑ | ↓ | ↓ | ↓ |
| AZ1136 | 990 nM (129) |
|
↓ | ↑ | ↓ | ↓ | |
| JNJ-42601572 | 16 nM(138) | rat & mouse(138), dog(139) |
|
↑ | ↓ | ↓ | |
| JNJ-16 | 56 nM (140) | dog(140) | ↓ | ||||
| PF-118 | <10 nM | ||||||
| Merck-8 | 36–77 nM(141, 142) | ||||||
| RO-18 | 158 nM | ||||||
| BMS-3 | < 10 nM | ||||||
| Satori-1 | 100 nM(143) | ↑ | ↓ |
|
- | ↓ | |
| SPI-1810 | 100 nM(144) | mouse(144) | ↑ | ↓ |
|
- | ↓ |
Neurogenetics has recently disclosed a detailed in vitro and in vivo characterization of imidazole GSM compound 4,(127) which was identified as NGP-555 (132). This compound reduced Aβ42 and Aβ40 levels while concomitantly elevating levels of Aβ38 and Aβ37 without inhibiting NICD or AICD formation (Table 2). Administration of NGP-555 to 8-month old Tg2576 mice for 7 months showed significant reduction in plaque density and amyloid deposition. The compound appeared to be well tolerated with no change in body weights and intestinal goblet cell densities. However, in contrast to results from acute dosing and cell-based assays where an increase in Aβ38 levels was observed, all brain Aβ peptides (Aβ42, Aβ40 and Aβ38) were lowered in the soluble-DEA-extractable, denaturing-SDS-extractable and formic acid-extractable brain fractions from Tg2576 mice dosed chronically with NGP-555 from 8- to 15 months of age. The reasons for the Aβ38 lowering, most surprisingly in the soluble fraction from DEA brain extracts, are not known. Nonetheless, this is a significant study since it was the first demonstration that a non-NSAID GSM could lower plaque density and amyloid load in a transgenic mouse model of AD.
The identification of the heterocyclic imidazole-containing GSM class by Neurogenetics and Eisai has spurred intense research activity throughout the industry as is evident by the large number of publications and patent applications related to this chemotype that have been published over the past several years (Fig. 6). For example, Merck/Schering Plough has reported analogs of E2012, exemplified by GSM-53, that incorporate a conformational constrained fused oxadiazine as an amide replacement.(133, 134) Merck, Hoffman LaRoche, AstraZeneca and Janssen have each disclosed variations of the arylimidazole series that incorporate an aminoheterocycle. For example, Merck replaced the methylenepiperidinone of E2012 with an aminopyridone to give GSM-35.(135) Hoffman LaRoche used a similar strategy, but replaced the pyridone with a pyrimidine to give the aminopyrimidine GSM RO-02. (136) AstraZeneca has also explored this chemical space and disclosed the Aβ profiles and binding characteristics of several aminopyrimidine GSMs, exemplified by AZ4800.(129) AZ4800 reduced Aβ42, Aβ40, and Aβ39 in HEK-APPs we cells and cell membranes, whereas Aβ38 and Aβ37 were increased by 750% and 300%, respectively. Interestingly, the close analog AZ3303 increased Aβ37 more than Aβ38, and another analog, AZ1136, actually decreased Aβ38 and increased Aβ39 (Table 2). Taken together with the Aβ profiles of E2012 and NGP-555, it is apparent that small structural changes can greatly influence the relative amounts of Aβ37 and Aβ38 that are generated, although the mechanistic basis for this is not clear.
Despite the improvement in potency for the 2nd generation GSMs, many are still very lipophilic which puts them at a higher risk of having off-target toxicity. As a result, it is clear from the recent patent literature that an important goal within industry is to lower the lipophilicity of candidate GSMs to improve the drug-like properties while maintaining the improved potency (improved lipophilic efficiency). For example, Janssen has removed the linker altogether and attached a triazolo-oxazine heterocycle directly to the arylimidazole to give JNJ-16.(140) This compound has a good pharmacokinetic profile in dog and lowered CSF Aβ42 by 30–40% (20 mg/kg). The reduced lipophilicity of JNJ-16 (clogP = 3.1) relative to earlier GSMs translated into an improved safety profile compared to JNJ-42601572. Another example can be seen in a patent application from Pfizer where the aryl core has been replaced with a bicyclic pyrido-pyrazinedione core as in PF-118. This compound is reported to lower Aβ42 in CHO-APP cells with an IC50 < 10 nM, while possessing improved lipophilicity (clogP = 3.1). (145)
Additional GSMs with distinct chemotypes
All of the non-NSAID derived heterocyclic GSMs discussed so far contain an aryl imidazole (or similar heterocycle such triazole or pyridine), but alternative cores are starting to emerge (Figure 7). For example, Merck has disclosed a series of GSMs where the aryl imidazole has been replaced with a 4-methoxyphenylpiperazine as in Merck-8.(141, 142) Furthermore, a recent patent from Hoffmann-La Roche highlighted a series of bridged amino-piperidines represented by RO-18 (146) in which a large portion of the exemplified compounds contain a thiadiazole left hand ring. Additionally, BMS has disclosed GSMs where the ubiquitous left-hand heterocycle (imidazole, triazole, pyridine) has been replaced with a nitrile.(147) For example, BMS-3 is reported to have an Aβ42 IC50 < 10 nM. Despite this structural diversity, the basic pharmacophore is maintained where two H-bond acceptors are separated by a conformationally constrained cyclic core with the presence of a lipophilic aryl group on the right hand side.
Figure 7.
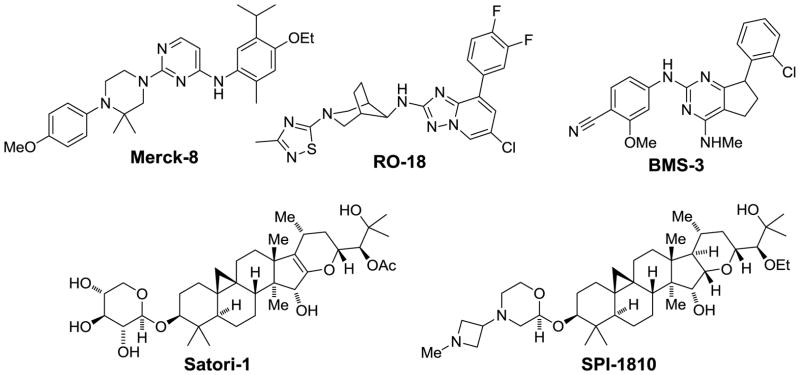
Additional GSMs with distinct chemotypes.
A truly structurally distinct chemotype has been introduced by Satori Pharmaceuticals (Figure 7). They have disclosed a new series of GSMs that were isolated from the black cohosh plant with the triterpene glycoside Satori-1 as the initial hit.(143) Subsequent optimization to improve metabolic stability and CNS disposition led to SPI-1810.(144) These GSMs have a distinct Aβ profile in that they lower both Aβ42 and Aβ38, but maintain total Aβ levels by raising Aβ39 and Aβ37.
Mechanism of action of GSMs
To determine the mechanism of action of GSMs, the following critical questions have to be addressed: 1) What are the targets of GSMs? 2) Do different classes occupy the same or overlapping binding sites of the target(s)? 3) What is the molecular basis for cleavage shifting and substrate specificity? 4) Do different classes of GSMs have similar mechanisms?
1) Notable chemical biology and biochemical techniques
Due to the lack of high resolution structural information and intrinsic complexity of the γ-secretase complex, investigators have had to use creative methods to study the mechanism of action of the diverse and myriad small molecules that target γ-secretase activity. Photoaffinity labeling (PAL) has widely been used for target identification of small molecules.(148) PAL has been instrumental in not only the identification of presenilin as the catalytic component of γ-secretase,(68) but also for determining the target of many γ-secretase inhibitors.(12, 149–151) Common cross-linking moieties include the photoreactive benzophenone, diazirine, and phenylazide motifs.(152) Many GSM photoprobes contain a biotin tag for affinity purification of the labeled enzyme (Fig. 8A). However, incorporation of a bulky biotin group could reduce the potency of parental compounds. Therefore, employing PAL with a smaller alkyne tag can be beneficial; moreover, the alkyne is more versatile because either a biotin or fluorescent tag can be “clicked” on using a copper catalyzed azide-alkyne cycloaddition (CuAAC) reaction (153, 154) (Fig. 8B).
Figure 8.

Structures of GSM derived photoaffinity probes containing (A) biotin or (B) clickable alkyne handle.
Another challenge is to detect small molecule induced conformational changes in γ-secretase within the lipid bilayer. Currently, three select approaches that have been used for such studies are Fluorescence Lifetime Imaging (FLIM), photophore walking, and the Surface Cysteine Accessibility Method (SCAM). By using FLIM, which measures the decay rate of a fluorophore rather than the intensity, one can measure the distance, or more importantly, changes in the distance between two Fluorescence Resonance Energy Transfer (FRET) pairs. Studies have looked at both the distance between the substrate and enzyme (APP-C-terminus and PS1-loop),(155) and distances from the CTF to NTF domains within PS1 itself.(155, 156) The conformational changes within presenilin have been studied by 2 methods: one used a pair of FITC and CY3 labeled antibodies which bind two different epitopes on presenilin-1 (155), while the other used a G-PS1-R fusion protein that has a GFP tag on the N-terminus and an RFP tag within the C-terminal loop of presenilin.(123, 156) Through these studies, it appears that γ-secretase adopts an “open” conformation when GSMs bind, resulting in an increased distance between the N and C termini of PS1 as measured by a longer fluorescence lifetime of the donor fluorophore.
The “photophore walking” approach (157) has been developed to detect conformational changes in the γ-secretase active site. Requirements of photophore walking probes include: 1) that they directly interact with the active site, and 2) that photoactivatable groups are incorporated into different side chains along the probe, and therefore crosslink to different subpockets within the active site. Since the efficiency of photolabeling depends on the contact region and proximity to residues within the active site, conformational changes induced by GSMs that alter the orientation or distance between a subpocket and the photophore can lead to different cross-linking efficiencies. By exploiting the complementarity of more than one probe, one can examine changes within the active site of γ-secretase by comparing differences in labeling efficiency of each probe in the presence or absence of a modifier. This approach has been used to characterize different inhibitors and GSMs,(12, 154, 158, 159) as well as to investigate the conformational changes caused by PS1 FAD mutants.(157) Importantly, the structure activity relationship (SAR) of the active-site directed photoprobes (68, 160–162) indicates that the subpockets within γ-secretase have enough plasticity for interacting with different sized side chains since substitution of Phe with BPA (benzoylphenyl alanine) at different positions did not alter the potency of these probes.(68, 160–162) This method allows investigation of endogenous γ-secretase in any cell type or tissue. The Surface Cysteine Accessibility Method (SCAM) is another interesting and unique methodology that has been used to characterize γ-secretase modulators. This method allows for identification of which amino acid residues are membrane embedded and what environmental changes, such as GSM binding, can alter the water accessibility of certain amino acids on presenilin.(123)
2) Mechanism of action
First generation NSAID GSMs
Initially it was reported that NSAIDs bound to the γ-secretase complex at some undefined allosteric site due to their non-competitive inhibition of γ-secretase(163) and non-competitive displacement of radiolabeled GSIs.(164, 165) Furthermore, it was shown that sulindac sulfide could also non-competitively displace [3H]L-685,458 from SPP, an aspartyl intramembrane protease, thus suggesting that NSAID GSMs also had a binding site for SPP. (166) Using FLIM-based FRET imaging, Lleó et al showed that the presence of NSAID GSMs resulted in an increase in the distance between APP-C-terminus and the loop region of PS-1 as determined by a measured increase in the lifetime of the donor fluorophore, the FITC labeled C-terminus of APP (155). Through similar methods they were also able to show an increase in the distance between PS1-NTF and PS1-CTF fragments upon GSM binding, suggesting a conformational change to PS1 upon NSAID binding (155) (Table 3).
Table 3.
Summary of Evidence for Target of NSAID GSMs
| GSM used in study | Method | Target | Ref. |
|---|---|---|---|
| Fenofibrate R-flurbiprofen |
Biotinylated probes label recombinant C100 and APP-CTF83 from cells | APP | (120) |
| Ibuprofen Flurbiprofen Indomethacin Fenofibrate |
Conformational changes induced by GSMs and/or C99, NotchΔEC, or helical peptide substrates were monitored using a FRET based FLIM assay. | APP/PS-1 border, PS-1 with docking |
(155) (167) |
| Sulindac Sulfide | Circular dichroism and electron spin resonance with SDS solubilized C100 and C100 mutant substrates | C100 and C100 dimer | (168) |
| Sulindac Sulfide Indomethacin |
Surface Plasmon Resonance (SPR) and solution state NMR | Aβ42 | (169) |
| Sulindac sulfide Sulindac sulfone R-flurbiprofen |
Noncompetitive displacement of [3H] GSIs | γ-secretase |
(164) (165) |
| Sulindac Sulfide | In vitro assay shows non-competitive inhibition of γ-secretase | γ-secretase Not APP |
(163) |
| Sulindac sulfide | Noncompetitive displacement of [3H]L-685,458 | SPP | (166) |
| R-flurbiprofen Indomethacin Fenofibrate Sulindac sulfide |
Monitored binding with TROSY-NMR of GSMs with [U-15N]C99; or titration of C99 while measuring 19F GSMs | Not APP | (170) |
| Sulindac Sulfide Flurbiprofen Sulindac Sulfone |
Measure binding with 15N HSQC and Surface Plasmon Resonance (SPR). | Not Aβ | (171) |
However, in 2008 Kukar et al published a paradigm-shifting paper suggesting that GSMs bound to the substrate APP rather than to the γ-secretase complex.(120) Using benzophenone and biotin containing molecular probes derived from fenofibrate and flurbiprofen (Flurbi-BpB, Fig. 8), they found probe incorporation in APP-CTF83 (αCTF), but not APP-CTF99 (βCTF), from CHAPSO solubilized H4-APP-alkaline phosphatase cells. Moreover, they were unable to find any labeling of γ-secretase complex subunits purified from CHO cells.(120) Both probes showed a dose-dependent (10–150 μM) increase in binding to a recombinant APP-C100-Flag substrate, which is essentially the βCTF substrate of γ-secretase required for Aβ production. Furthermore, they show that labeling of C100-Flag by the fenofibrate probe can be partially competed with 100 μM of multiple NSAID GSMs, and fenofibrate prefers binding to APP(C100)-Flag compared to Notch(C100)-Flag substrate. Using a series of truncated Aβ peptides, they mapped the binding site of the GSMs to Aβ28–36 (see Fig. 1), which includes the beginning of the transmembrane domain of APP.(120) This finding not only offers a straightforward explanation of substrate selectivity, but also provides an interesting mechanism for modulation of γ-secretase through targeting substrate, rather than the enzyme. Similarly, Espeseth et al had previously reported on a series of APP binding compounds that also inhibited Aβ42 production.(172)
Munter et al demonstrated that the GxxxG motif that corresponds to residues 29–33 within Aβ, was not only important for dimerization of the APP transmembrane domain, but that an increase in dimerization strength within the TM region is correlated with an increase in Aβ42 production relative to other Aβ species.(173) Conversely, if the GxxxG motif is mutated and/or disrupted so that dimerization is lost, then γ-secretase cleavage is altered so that there is an increase in Aβ38 production but a decrease in Aβ42.(173) This led to the hypothesis that GSMs may bind to the GxxxG motif in βCTF and alter the transmembrane dimerization of APP, resulting in modulation of cleavage from Aβ42 to Aβ38 production. Support for this hypothesis emerged from Richter et al’s work which suggests that sulindac sulfide, and to a lesser degree indomethacin, could inhibit dimerization of the APP TM domain in a β-galactose based dimerization assay using a ToxR fusion protein with residues 29–42 of the APP membrane.(169) They also showed that sulindac sulfide could directly bind to immobilized Aβ42 as measured by Surface Plasmon Resonance (SPR) and that incubation of 100 μM Aβ42 with 300 μM sulindac sulfide yielded NMR chemical shifts at several residues including a few within the purported binding domain of GSMs.(169) Further studies using SPR showed that sulindac sulfide prefers binding a C100 mutant that has an increased propensity for dimerization compared to wild type C100.(168) Similarly, sulindac sulfide prefers binding wild type with respect to a G33I mutant that disrupts the GxxxG motif and does not readily form dimers.(168) However, this work has not been repeated with more potent 2nd generation GSMs so the functional significance awaits further studies.
Beel et al studied the biochemical nature of the interaction between βCTF and GSMs by using recombinant purified [U-15N]C99 in LMPG micelles monitored by 1H-15N TROSY protein NMR; but they found no specific binding between C99 and R-flurbiprofen, fenofibrate, indomethacin or sulindac sulfide.(170) Instead, they only found a few chemical shifts that were non-specific in nature and did not correspond to the purported Aβ28–33 binding region of GSMs.(170) Interestingly, they found that GSMs did in fact bind to aggregated C99, and that the aggregated protein seems to promote the formation of GSM aggregates.(170) Similarly, in response to findings that GSMs bind directly to Aβ,(168, 169) Barrett et al performed additional SPR and protein NMR experiments with the Aβ42 peptide.(171) They again found that GSMs only non-specifically bind to Aβ and this binding can be eliminated with micelle formation.(171) Importantly, they also show using dynamic light scattering (DLS) that sulindac sulfide forms aggregates at concentrations above 50 μM.(171) Furthermore, Page et al examined the effect of GSMs on multiple APP FAD mutations together with systemic phenylalanine scanning mutagenesis near the γ-secretase cleavage site (including the GxxxG domain) and found that the overwhelming majority of mutants responded well to the second generation NSAID-derived GSM-1, and that the iGSM fenofibrate was also responsive to G33I and K28E mutations, thus further creating uncertainty about the binding of GSMs to the GxxxG domain.(113) Recently, NMR structural studies revealed that βCTF exists as a monomer and the GxxxG motif plays an important role in cholesterol binding.(174)
If the first hypothesis is that GSMs bind γ-secretase, and the second is that GSMs bind directly to the APP substrate, then the third is that the compounds bind both. The latter theory is supported by several FRET based FLIM assays with fluorescently tagged PS1 in APP/APLP2 knock out cells.(167) Uemura et al., show that the conformational changes induced by NSAID GSMs in PS1 first require substrate docking by either C99, NotchΔEC or a helical peptide.(167) It is interesting to note however that neither NotchΔEC nor the helical peptide contain the GxxxG motif, suggesting that substrate dimerization is not necessary for NSAID induced PS1 conformation changes.
If NSAIDs do indeed bind solely to APP, and this is how selectivity is achieved, then one would expect NSAIDs to be selective for APP and not bind other γ-secretase substrates. There is controversy over whether NSAID GSMs affect any substrates other than APP. Several groups have claimed that NSAIDs have an effect on Notch, by either reducing Nβ (175) or by binding to N100-Flag (120)—but in both of these studies higher concentrations of NSAIDs were required for Notch than for APP. However, other groups have found that NSAIDs have no effect on Nβ (176, 177) nor CD44-β or an APP-Notch TMD chimera.(177) Furthermore, NSAIDs have been shown to non-competitively compete for binding of SPP (166) and to also alter the cleavage site of the SPP substrate Prl.(178) These data suggest that NSAID GSM binding is unlikely to be entirely on the substrate, but could be on the interface between substrate and enzyme.
There is likely a complicated binding mechanism for GSMs, perhaps on the interface between the enzyme and the substrate or perhaps multiple binding sites are present, and unfortunately the high concentrations required due to low efficacy of first generation compounds complicates the interpretation of findings.
Second Generation GSMs
Despite the large structural variation among second generation GSMs, all work done to date on the more potent (IC50 < 300 nM) GSMs shows invariably that γ-secretase is indeed the target of these molecules. By immobilizing an imidazole based 2nd generation GSM, Kounnas et al first showed that this GSM could pull down components of the γ-secretase complex such as Pen-2, PS1-NTF and PS1-CTF, but not APP.(127) Soon to follow, several independent labs simultaneously designed photo-crosslinking probes based on multiple 2nd generation GSMs and indisputably show specific labeling of PS1-NTF but not APP (123, 136, 154, 179, 180) (Table 4).
Table 4.
Summary of Evidence for Target of 2nd Generation GSMs
| GSM used in study | Method | Target | Ref |
|---|---|---|---|
| GSM-1 (acid GSM) | Biotinylated or clickable photoprobes label PS1-NTF in cell membranes or with recombinant PS proteins. | PS1-NTF PS1-FL PS1-ΔE9 without APP |
(123) (154) |
| BB25 AR80 (acid GSM) |
Biotinylated photoprobes label PS1-NTF in cell membranes and live cells | PS1-NTF | (179) |
| GSM-97555 (imidazole GSM) | Pull-down with GSM immobilized Affigel matrix | Pen-2≫ PS1-NTF > PS1-CTF |
(127) |
| RO-57 (imidazole GSM) | Biotinylated photoprobes label PS-NTF in cell membranes | PS1-NTF PS2-NTF |
(136) |
| E2012 (imidazole GSM) | Clickable photoprobes label PS1-NTF in cell membranes, recombinant PS proteins, and live cells and neurons | PS1-NTF PS1-ΔE9 without APP |
(180) |
The NSAID-derived piperidine acetic acid GSM-1 directly binds to PS1-NTF using photoaffinity probes GSM-1-BpB, GSM-1-BPyne and GSM-5 (see Fig. 8).(123, 154) Furthermore, GSM-1-BpB was suggested to bind to residues 78–100 of TMD1 of PS1-NTF.(123) It appears that upon binding to this region of PS1, GSM-1 is able to induce an overall conformational change in γ-secretase as visualized by a FLIM study (123) as well as a conformational change within the active site of γ-secretase.(154) Interestingly, GSM-1-BpB was found to also bind full-length PS1,(123) the zymogen of γ-secretase.(68, 70) Importantly, GSM-1-BPyne and GSM-5 bind to a reconstituted PS1 mutant, PS1ΔE9, in liposomes without any substrates present.(154) Together, these data paint a very different picture than what has previously been hypothesized for NSAID GSMs: GSM-1 can bind PS1 independent of any substrates and can presumably bind an inactive enzyme. Interestingly, the GSM-1 probes were also able to specifically label SPP, (123, 154) a structurally related intramembrane aspartyl protease, which was also reported for the NSAID sulindac sulfide.(166)
The Roche imidazole based GSMs were also found to directly label PS1-NTF and PS2-NTF.(136) Competition studies with labeling of RO-57-BpB probe showed good competition with E2012, but not NGP-555 like GSMs—all of which belong to the imidazole class of 2nd generation GSMs. Moreover, sulindac sulfide (100 μM) could compete for RO-57-BpB binding but neither GSM-1 nor fenofibrate had any effect on RO-57-BpB labeling of PS1, although GSM-1 was found to partially block RO-57-BpB labeling of PS2, suggesting there could be partial overlap in binding sites.(136) This also raises a critical issue for cross-talk studies regarding concentrations and solubility of competing compounds. It can be addressed by conducting the competition in a dose responsive fashion in which compounds maintain solubility under assay conditions, further elucidating the nature of the competition.
Recently, using a series of reciprocal labeling experiments with GSM-1- and E2012-based photoaffinity probes, our groups have shown that the two compounds have distinct binding sites on PS1-NTF.(180) Moreover, unlike GSM-1-BPyne, E2012-BPyne labeling to PS1-NTF is significantly potentiated in the presence of L458, showing direct cross-talk between the E2012 binding site and the active site of the enzyme.(180) Surprisingly, binding of L458 has no effect on the RO-57-BPyne labeling,(180) suggesting that E2012 and RO-57 could have distinct effects even though both are from the same imidazole class of GSMs. In contrast to GSM-1-BpB, E2012-BPyne specifically labels PS1-NTF (active γ-secretase) but not full-length PS1 (inactive γ-secretase). Furthermore, the GSI BMS-708,163 binds to PS1-NTF, and the binding site does not overlap with the sites that interact with GSM-1 or E2012 (Fig. 9).(159, 180)
Figure 9.

Model for different binding sites of GSMs and GSIs. The active site of γ-secretase is represented by a pair of scissors. GSMs alter the “handle” of the scissors, thereby manipulating the way the enzyme cuts and/or the location of the cleavage sites. In contrast, an allosteric GSI will shut the blades, whereas a transition state analog (TSA) will block the blades of the scissors, preventing substrate binding and cleavage of the substrate.
Taken together, although both piperidine acetic acid GSMs and E2012-like GSMs target PS1, it appears they occupy different sites within the γ-secretase complex (Fig. 9). Consequently, they lead to varying pharmacological effects on Aβ species (Tables 1 and 2), such that acid GSMs reduce Aβ42 production and enhance Aβ38, whereas imidazole GSMs differentially decrease Aβ42 and Aβ40, and concurrently increase Aβ38 and/or Aβ37 levels. It is noteworthy to point out there is great diversity within the imidazole GSMs, which are exemplified by NGP-555, E2012 and RO-57 (Fig. 6), (136, 180). Therefore, the interplay between different subtypes of imidazole GSMs and other classes of GSMs should be carefully examined.
The next key question is how the binding of second generation GSMs to γ-secretase induces a conformational change that has been detected by FLIM (156), SCAM (123) or photophore walking (154) (Fig. 10A). We propose two alternative models of how GSM-induced conformation, such as the S1 subpocket alteration(154), leads to γ-secretase modulation (Fig. 10). (1) Acid GSMs mainly affect the sequential processing cycle of Aβ42 to Aβ38 (181), which has been suggested to be due to slower dissociation of the Aβ42 substrate from the γ-secretase complex allowing further processing to Aβ38 (Fig. 10B).(23) The overall result is Aβ42 reduction and Aβ38 elevation. However, E2012 and many imidazole GSMs are known to preferentially increase Aβ37 (Table 2), which presumably represents a fifth γ-secretase cleavage from the Aβ49 product line. Therefore, imidazole GSMs could bind to γ-secretase in a way that alters the sequential cycles of Aβ42 to Aβ38 and Aβ40 to Aβ37 to achieve γ-secretase modulation. (2) Alternatively, the GSM-induced conformational change could specifically block Aβ42 or Aβ40 production and potentiate the Aβ38 or Aβ37 generation based on the independent cleavage model in which all sites of cleavage are parallel (Fig. 10C). However, the newly discovered natural product GSMs (Fig. 7) inhibit both Aβ38 and Aβ42 while increasing Aβ37 and Aβ39, suggesting a different mechanism than imidazole and acid GSMs. These Satori compounds could be operating by a mechanism similar to model (1) where both the Aβ40 and Aβ42 peptide substrates have a slower dissociation rate, resulting in further processing to Aβ37 and Aβ39 peptides, or alternatively by model (2) in which Aβ38 and Aβ42 cleavage is specifically and independently blocked while Aβ37 and Aβ39 cleavage is enhanced. However, more information is clearly needed to determine how these natural product compounds compare with imidazole and acid GSMs.
Figure 10.

Proposed models for the mechanism of GSMs. A) GSM binding leads to a conformational change in the active site, such as the S1 subpocket. B) Sequential cleavage model: GSM binding has little effect on processivity of γ-secretase at 48 and 45 sites; however, a tighter association of γ-secretase with Aβ42 results in reduced release of Aβ42 and an increase in the generation of Aβ38.
C) Independent cleavage model: all cleavage sites are parallel; GSM binding inhibits Aβ42 cleavage site, but enhances Aβ38 cleavage and has little effect on other cleavages including AICD production.
Summary and future perspective
APP is processed into three major species, sAPP, Aβ and AICD. GSMs cause a shift from Aβ42 to shorter less toxic Aβ species and have little effect on the generation of AICD and NICD, thus allowing their signaling roles to remain intact. As a result, GSI-mediated adverse effects should not be a concern, offering the hope that GSMs will become promising disease modifying agents. Indeed, comparative studies of GSIs and GSMs in mice have supported such a notion.(41) Moreover, γ-secretase contains distinct sites that interact with different GSMs, which highlights that γ-secretase can be modulated in multiple ways.(180) Although modulation of γ-secretase holds much promise, significant questions remain to be answered. First, although it is clear that GSMs differentiate from GSIs, it is not known if other safety issues will emerge with chronic treatment of GSMs. Second, while much progress has been made in understanding the target of GSMs, more work is needed to determine their precise binding sites and the molecular basis for their mechanism of action. The mechanism that regulates when γ-secretase cleavage of APP ends and Aβ is released is not well understood and it will be important to understand the influence GSMs have on that process. In addition, since different GSMs have distinct Aβ profiles, it may be difficult to determine the mechanism without considering the full Aβ profile including shorter Aβ peptides such as Aβ37. It will be important to find efficient ways to quantitatively measure all Aβ species. Furthermore, it has been shown recently that mutation of Lysine 624 of APP (K28A of Aβ) shifts the final γ-secretase cleavage site to favor shorter Aβ species such as Aβ1–33 and 1–34, suggesting a pivotal role for this charged residue in preventing the continuation of APP cleavage by γ-secretase.(182) Further studies that help elucidate the precise mechanism of action of GSMs are highly anticipated. Third, it would be interesting to consider if different classes of GSMs could be used as a combination therapy or in combination with a BACE inhibitor. Finally, it is unknown whether successful GSM clinical trials could be conducted without the availability of effective biomarkers for early diagnosis of AD. It has been suggested that the pathological process of AD starts more than 10 years before clinical symptoms manifest.(183) Since some 2nd generation GSMs have been found to be responsive to several PS1 and APP mutations,(110)(113) perhaps a prevention trial in asymptomatic patients with FAD mutations could be considered similar to the DIAN trial that is being planned.(111, 112) Clearly, development of GSMs for the treatment AD not only relies on discovery of effective drug candidates, but also is dependent on the progress of AD research in molecular pathogenesis, biomarkers, diagnosis and other therapeutic developments. Undoubtedly, with the recognition that AD is the fastest growing threat to human health, an interdisciplinary approach and significant effort are required to drive these critical issues toward resolution for the development of effective AD therapies.
Acknowledgments
Funding
This work was supported by NIH grants 1R01NS076117, 2R01AG026660 (YML) and training grant in the pharmacological sciences, NIH T32 GM073546 (CJC), Alzheimer Association IIRG-08-90824, the American Health Assistance Foundation (YML), Pfizer (YML), the Geoffrey Beene Cancer Research Center of MSKCC, Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research, the Experimental Therapeutics Center of MSKCC, and the William Randolph Hearst Fund in Experimental Therapeutics.
LIST OF ABBREVIATIONS
- αCTF
α-secretase cleaved C terminal fragment of APP
- βCTF
β-secretase cleaved C terminal fragment of APP
- AD
Alzheimer’s disease
- ADAM
A disintegrin and metalloproteinase domain-containing protein
- AICD
APP intracellular domain
- APH-1
Anterior pharynx defective-1
- APLP2
Amyloid beta precursor-like protein 2
- APP
Amyloid precursor protein
- Aβ
β-Amyloid peptide
- CHAPSO
3-[(3-Cholamidopropyl)dimethylammonio]-2-hydroxy-1-propanesulfonate
- COX
cyclooxygenase
- CSF
Cerebral spinal fluid
- CSL
CBF1/Su(H)/Lag-1, also known as RBP-Jκ family
- CuAAC
Copper(I)-catalyzed Azide-Alkyne Cycloaddition
- FAD
Familial Alzheimer’s disease
- FITC
Fluorescein isothiocyanate
- FLIM
Fluorescence-lifetime imaging microscopy
- FRET
Förster (Fluorescence) resonance energy transfer
- GSAP
γ-secretase activating protein
- GSI
γ-secretase inhibitor
- GSK
GlaxoSmithKline
- GSM
γ-secretase modulator
- HES1
Hairy and enhancer of split-1
- HEY
Hairy/enhancer-of-split related with YRPW motif protein
- iGSM
inverse γ-secretase modulator
- LMPG
lyso-myristoylphosphatidylglycerol
- MamL
Mastermind-like
- NCT
Nicastrin
- NICD
Notch intracellular domain
- NMR
Nuclear magnetic resonance
- NotchΔEC
Notch with extracellular domain removed
- NSAIDs
Non-steroidal anti-inflammatory drugs
- PAL
Photoaffinity labeling
- PEN2
Presenilin enhancer 2
- PS
Presenilin
- PS1-CTF
Presenilin1 C-terminal fragment
- PS1-NTF
Presenilin1 N-terminal fragment
- PS1ΔE9
Presenilin1 Exon9 removed
- sAPPα
Soluble APP, α-secretase cleaved
- sAPPβ
Soluble APP, β-secretase cleaved
- SPP
Signal peptide peptidase
- TM
Transmembrane
- TMD
Transmembrane domain
- TROSY
Transverse relaxation optimized spectroscopy
- TSA
Transition state analog
References
- 1.Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, Kang DE, Marquez-Sterling N, Golde TE, Koo EH. A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature. 2001;414:212–216. doi: 10.1038/35102591. [DOI] [PubMed] [Google Scholar]
- 2.Oehlrich D, Berthelot DJ, Gijsen HJ. gamma-Secretase Modulators as Potential Disease Modifying Anti-Alzheimer’s Drugs. J Med Chem. 2011;54:669–698. doi: 10.1021/jm101168r. [DOI] [PubMed] [Google Scholar]
- 3.Pettersson M, Kauffman GW, am Ende CW, Patel NC, Stiff C, Tran TP, Johnson DS. Novel gamma-secretase modulators: a review of patents from 2008 to 2010. Expert Opin Ther Pat. 2011;21:205–226. doi: 10.1517/13543776.2011.547479. [DOI] [PubMed] [Google Scholar]
- 4.Bulic B, Ness J, Hahn S, Rennhack A, Jumpertz T, Weggen S. Chemical Biology, Molecular Mechanism and Clinical Perspective of gamma-Secretase Modulators in Alzheimer’s Disease. Current neuropharmacology. 2011;9:598–622. doi: 10.2174/157015911798376352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wolfe MS. gamma-Secretase inhibitors and modulators for Alzheimer’s disease. J Neurochem. 2012;120(Suppl 1):89–98. doi: 10.1111/j.1471-4159.2011.07501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wagner SL, Tanzi RE, Mobley WC, Galasko D. Potential Use of gamma-Secretase Modulators in the Treatment of Alzheimer Disease. Arch Neurol. 2012:1–4. doi: 10.1001/archneurol.2012.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xia W, Wong ST, Hanlon E, Morin P. gamma-secretase modulator in Alzheimer’s disease: shifting the end. J Alzheimers Dis. 2012;31:685–696. doi: 10.3233/JAD-2012-120751. [DOI] [PubMed] [Google Scholar]
- 8.Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochemical and biophysical research communications. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 9.Glenner GG, Wong CW. Alzheimer’s disease and Down’s syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochemical and biophysical research communications. 1984;122:1131–1135. doi: 10.1016/0006-291x(84)91209-9. [DOI] [PubMed] [Google Scholar]
- 10.Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci. 1991;12:383–388. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- 11.Zheng H, Koo EH. Biology and pathophysiology of the amyloid precursor protein. Mol Neurodegener. 2011;6:27. doi: 10.1186/1750-1326-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tian Y, Bassit B, Chau D, Li YM. An APP inhibitory domain containing the Flemish mutation residue modulates gamma-secretase activity for Abeta production. Nat Struct Mol Biol. 2010;17:151–158. doi: 10.1038/nsmb.1743. [DOI] [PubMed] [Google Scholar]
- 13.Tian Y, Crump CJ, Li YM. Dual role of {alpha}-Secretase Cleavage in the regulation of {gamma}-secretase activity for amyloid production. J Biol Chem. 2010 doi: 10.1074/jbc.M110.128439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nitsch RM, Slack BE, Wurtman RJ, Growdon JH. Release of Alzheimer amyloid precursor derivatives stimulated by activation of muscarinic acetylcholine receptors. Science. 1992;258:304–307. doi: 10.1126/science.1411529. [DOI] [PubMed] [Google Scholar]
- 15.Skovronsky DM, Moore DB, Milla ME, Doms RW, Lee VM. Protein kinase C-dependent alpha-secretase competes with beta-secretase for cleavage of amyloid-beta precursor protein in the trans-golgi network. J Biol Chem. 2000;275:2568–2575. doi: 10.1074/jbc.275.4.2568. [DOI] [PubMed] [Google Scholar]
- 16.Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M, Haass C, Fahrenholz F. Constitutive and regulated alpha-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci U S A. 1999;96:3922–3927. doi: 10.1073/pnas.96.7.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Postina R, Schroeder A, Dewachter I, Bohl J, Schmitt U, Kojro E, Prinzen C, Endres K, Hiemke C, Blessing M, Flamez P, Dequenne A, Godaux E, van Leuven F, Fahrenholz F. A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J Clin Invest. 2004;113:1456–1464. doi: 10.1172/JCI20864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao G, Mao G, Tan J, Dong Y, Cui MZ, Kim SH, Xu X. Identification of a new presenilin-dependent zeta-cleavage site within the transmembrane domain of amyloid precursor protein. J Biol Chem. 2004;279:50647–50650. doi: 10.1074/jbc.C400473200. [DOI] [PubMed] [Google Scholar]
- 19.Sastre M, Steiner H, Fuchs K, Capell A, Multhaup G, Condron MM, Teplow DB, Haass C. Presenilin-dependent gamma-secretase processing of beta-amyloid precursor protein at a site corresponding to the S3 cleavage of Notch. EMBO Rep. 2001;2:835–841. doi: 10.1093/embo-reports/kve180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qi-Takahara Y, Morishima-Kawashima M, Tanimura Y, Dolios G, Hirotani N, Horikoshi Y, Kametani F, Maeda M, Saido TC, Wang R, Ihara Y. Longer forms of amyloid beta protein: implications for the mechanism of intramembrane cleavage by gamma-secretase. J Neurosci. 2005;25:436–445. doi: 10.1523/JNEUROSCI.1575-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kakuda N, Funamoto S, Yagishita S, Takami M, Osawa S, Dohmae N, Ihara Y. Equimolar production of amyloid beta-protein and amyloid precursor protein intracellular domain from beta-carboxyl-terminal fragment by gamma-secretase. J Biol Chem. 2006;281:14776–14786. doi: 10.1074/jbc.M513453200. [DOI] [PubMed] [Google Scholar]
- 22.Takami M, Nagashima Y, Sano Y, Ishihara S, Morishima-Kawashima M, Funamoto S, Ihara Y. gamma-Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of beta-carboxyl terminal fragment. J Neurosci. 2009;29:13042–13052. doi: 10.1523/JNEUROSCI.2362-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Okochi M, Tagami S, Yanagida K, Takami M, Kodama TS, Mori K, Nakayama T, Ihara Y, Takeda M. gamma-Secretase Modulators and Presenilin 1 Mutants Act Differently on Presenilin/gamma-Secretase Function to Cleave Abeta42 and Abeta43. Cell reports. 2013;3:42–51. doi: 10.1016/j.celrep.2012.11.028. [DOI] [PubMed] [Google Scholar]
- 24.Hecimovic S, Wang J, Dolios G, Martinez M, Wang R, Goate AM. Mutations in APP have independent effects on Abeta and CTFgamma generation. Neurobiol Dis. 2004;17:205–218. doi: 10.1016/j.nbd.2004.04.018. [DOI] [PubMed] [Google Scholar]
- 25.Chen F, Hasegawa H, Schmitt-Ulms G, Kawarai T, Bohm C, Katayama T, Gu Y, Sanjo N, Glista M, Rogaeva E, Wakutani Y, Pardossi-Piquard R, Ruan X, Tandon A, Checler F, Marambaud P, Hansen K, Westaway D, George-Hyslop PS, Fraser P. TMP21 is a presenilin complex component that modulates γ-secretase but not ε-secretase activity. Nature. 2006;440:1208–1212. doi: 10.1038/nature04667. [DOI] [PubMed] [Google Scholar]
- 26.He G, Luo W, Li P, Remmers C, Netzer WJ, Hendrick J, Bettayeb K, Flajolet M, Gorelick F, Wennogle LP, Greengard P. Gamma-secretase activating protein is a therapeutic target for Alzheimer’s disease. Nature. 2010;467:95–98. doi: 10.1038/nature09325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moehlmann T, Winkler E, Xia X, Edbauer D, Murrell J, Capell A, Kaether C, Zheng H, Ghetti B, Haass C, Steiner H. Presenilin-1 mutations of leucine 166 equally affect the generation of the Notch and APP intracellular domains independent of their effect on Abeta 42 production. Proc Natl Acad Sci U S A. 2002;99:8025–8030. doi: 10.1073/pnas.112686799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Page RM, Baumann K, Tomioka M, Perez-Revuelta BI, Fukumori A, Jacobsen H, Flohr A, Luebbers T, Ozmen L, Steiner H, Haass C. Generation of Abeta38 and Abeta42 is independently and differentially affected by familial Alzheimer disease-associated presenilin mutations and gamma-secretase modulation. J Biol Chem. 2008;283:677–683. doi: 10.1074/jbc.M708754200. [DOI] [PubMed] [Google Scholar]
- 29.Czirr E, Cottrell BA, Leuchtenberger S, Kukar T, Ladd TB, Esselmann H, Paul S, Schubenel R, Torpey JW, Pietrzik CU, Golde TE, Wiltfang J, Baumann K, Koo EH, Weggen S. Independent generation of Abeta42 and Abeta38 peptide species by gamma-secretase. J Biol Chem. 2008;283:17049–17054. doi: 10.1074/jbc.M802912200. [DOI] [PubMed] [Google Scholar]
- 30.Quintero-Monzon O, Martin MM, Fernandez MA, Cappello CA, Krzysiak AJ, Osenkowski P, Wolfe MS. Dissociation between the processivity and total activity of gamma-secretase: implications for the mechanism of Alzheimer’s disease-causing presenilin mutations. Biochemistry. 2011;50:9023–9035. doi: 10.1021/bi2007146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y. Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43) Neuron. 1994;13:45–53. doi: 10.1016/0896-6273(94)90458-8. [DOI] [PubMed] [Google Scholar]
- 32.Jarrett JT, Berger EP, Lansbury PT., Jr The C-terminus of the beta protein is critical in amyloidogenesis. Ann N Y Acad Sci. 1993;695:144–148. doi: 10.1111/j.1749-6632.1993.tb23043.x. [DOI] [PubMed] [Google Scholar]
- 33.Kumar-Singh S, Theuns J, Van Broeck B, Pirici D, Vennekens K, Corsmit E, Cruts M, Dermaut B, Wang R, Van Broeckhoven C. Mean age-of-onset of familial alzheimer disease caused by presenilin mutations correlates with both increased Abeta42 and decreased Abeta40. Hum Mutat. 2006;27:686–695. doi: 10.1002/humu.20336. [DOI] [PubMed] [Google Scholar]
- 34.Deng Y, Tarassishin L, Kallhoff V, Peethumnongsin E, Wu L, Li YM, Zheng H. Deletion of presenilin 1 hydrophilic loop sequence leads to impaired gamma-secretase activity and exacerbated amyloid pathology. J Neurosci. 2006;26:3845–3854. doi: 10.1523/JNEUROSCI.5384-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim J, Onstead L, Randle S, Price R, Smithson L, Zwizinski C, Dickson DW, Golde T, McGowan E. Abeta40 inhibits amyloid deposition in vivo. J Neurosci. 2007;27:627–633. doi: 10.1523/JNEUROSCI.4849-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang R, Wang B, He W, Zheng H. Wild-type presenilin 1 protects against Alzheimer disease mutation-induced amyloid pathology. J Biol Chem. 2006;281:15330–15336. doi: 10.1074/jbc.M512574200. [DOI] [PubMed] [Google Scholar]
- 37.Murray MM, Bernstein SL, Nyugen V, Condron MM, Teplow DB, Bowers MT. Amyloid beta protein: Abeta40 inhibits Abeta42 oligomerization. J Am Chem Soc. 2009;131:6316–6317. doi: 10.1021/ja8092604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yan Y, Wang C. Abeta40 protects non-toxic Abeta42 monomer from aggregation. J Mol Biol. 2007;369:909–916. doi: 10.1016/j.jmb.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 39.Jan A, Gokce O, Luthi-Carter R, Lashuel HA. The ratio of monomeric to aggregated forms of Abeta40 and Abeta42 is an important determinant of amyloid-beta aggregation, fibrillogenesis, and toxicity. J Biol Chem. 2008;283:28176–28189. doi: 10.1074/jbc.M803159200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zheng H, Koo EH. The amyloid precursor protein: beyond amyloid. Mol Neurodegener. 2006;1:5. doi: 10.1186/1750-1326-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mitani Y, Yarimizu J, Saita K, Uchino H, Akashiba H, Shitaka Y, Ni K, Matsuoka N. Differential effects between gamma-secretase inhibitors and modulators on cognitive function in amyloid precursor protein-transgenic and nontransgenic mice. J Neurosci. 2012;32:2037–2050. doi: 10.1523/JNEUROSCI.4264-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lanz TA, Karmilowicz MJ, Wood KM, Pozdnyakov N, Du P, Piotrowski MA, Brown TM, Nolan CE, Richter KE, Finley JE, Fei Q, Ebbinghaus CF, Chen YL, Spracklin DK, Tate B, Geoghegan KF, Lau LF, Auperin DD, Schachter JB. Concentration-dependent modulation of amyloid-beta in vivo and in vitro using the gamma-secretase inhibitor, LY-450139. J Pharmacol Exp Ther. 2006;319:924–933. doi: 10.1124/jpet.106.110700. [DOI] [PubMed] [Google Scholar]
- 43.Yin YI, Bassit B, Zhu L, Yang X, Wang C, Li YM. {gamma}-Secretase Substrate Concentration Modulates the Abeta42/Abeta40 Ratio: IMPLICATIONS FOR ALZHEIMER DISEASE. J Biol Chem. 2007;282:23639–23644. doi: 10.1074/jbc.M704601200. [DOI] [PubMed] [Google Scholar]
- 44.Goate A, Chartier-Harlin M-C, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, Mant R, Newton P, Rooke K, Roques P, Talbot C, Pericak-Vance M, Roses A, Williamson R, Rossor M, Owen M, Hardy J. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 45.Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 46.Levy Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K, et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science. 1995;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- 47.Shen J, Kelleher RJ., 3rd The presenilin hypothesis of Alzheimer’s disease: evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci U S A. 2007;104:403–409. doi: 10.1073/pnas.0608332104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tanzi RE. The genetics of Alzheimer disease. Cold Spring Harbor perspectives in medicine. 2012;2 doi: 10.1101/cshperspect.a006296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, Stefansson H, Sulem P, Gudbjartsson D, Maloney J, Hoyte K, Gustafson A, Liu Y, Lu Y, Bhangale T, Graham RR, Huttenlocher J, Bjornsdottir G, Andreassen OA, Jonsson EG, Palotie A, Behrens TW, Magnusson OT, Kong A, Thorsteinsdottir U, Watts RJ, Stefansson K. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature. 2012;488:96–99. doi: 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- 50.Wong PC, Zheng H, Chen H, Becher MW, Sirinathsinghji DJ, Trumbauer ME, Chen HY, Price DL, Van der Ploeg LH, Sisodia SS. Presenilin 1 is required for Notch1 and DII1 expression in the paraxial mesoderm. Nature. 1997;387:288–292. doi: 10.1038/387288a0. [DOI] [PubMed] [Google Scholar]
- 51.Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S. Skeletal and CNS defects in Presenilin-1-deficient mice. Cell. 1997;89:629–639. doi: 10.1016/s0092-8674(00)80244-5. [DOI] [PubMed] [Google Scholar]
- 52.De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, Goate A, Kopan R. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398:518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- 53.Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 54.Louvi A, Artavanis-Tsakonas S. Notch signalling in vertebrate neural development. Nat Rev Neurosci. 2006;7:93–102. doi: 10.1038/nrn1847. [DOI] [PubMed] [Google Scholar]
- 55.Ables JL, Breunig JJ, Eisch AJ, Rakic P. Not(ch) just development: Notch signalling in the adult brain. Nature reviews Neuroscience. 2011;12:269–283. doi: 10.1038/nrn3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lobry C, Oh P, Aifantis I. Oncogenic and tumor suppressor functions of Notch in cancer: it’s NOTCH what you think. J Exp Med. 2011;208:1931–1935. doi: 10.1084/jem.20111855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216–233. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kopan R, Goate A. A common enzyme connects notch signaling and Alzheimer’s disease. Genes Dev. 2000;14:2799–2806. doi: 10.1101/gad.836900. [DOI] [PubMed] [Google Scholar]
- 59.Saxena MT, Schroeter EH, Mumm JS, Kopan R. Murine notch homologs (N1-4) undergo presenilin-dependent proteolysis. J Biol Chem. 2001;276:40268–40273. doi: 10.1074/jbc.M107234200. [DOI] [PubMed] [Google Scholar]
- 60.Haapasalo A, Kovacs DM. The many substrates of presenilin/gamma-secretase. J Alzheimers Dis. 2011;25:3–28. doi: 10.3233/JAD-2011-101065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eli Lilly and Company. Lilly Halts Development of Semagacestat for Alzheimer’s Disease Based on Preliminary Results of Phase III Clinical Trials. 2010 Aug 17; http://newsroom.lilly.com/releasedetail.cfm?releaseid=499794.
- 62.Xia X, Qian S, Soriano S, Wu Y, Fletcher AM, Wang XJ, Koo EH, Wu X, Zheng H. Loss of presenilin 1 is associated with enhanced beta-catenin signaling and skin tumorigenesis. Proc Natl Acad Sci U S A. 2001;98:10863–10868. doi: 10.1073/pnas.191284198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nicolas M, Wolfer A, Raj K, Kummer JA, Mill P, Van Noort M, Hui CC, Clevers H, Dotto GP, Radtke F. Notch1 functions as a tumor suppressor in mouse skin. Nat Genet. 2003;33:416–421. doi: 10.1038/ng1099. [DOI] [PubMed] [Google Scholar]
- 64.Fleisher AS, Raman R, Siemers ER, Becerra L, Clark CM, Dean RA, Farlow MR, Galvin JE, Peskind ER, Quinn JF, Sherzai A, Sowell BB, Aisen PS, Thal LJ. Phase 2 safety trial targeting amyloid beta production with a gamma-secretase inhibitor in Alzheimer disease. Arch Neurol. 2008;65:1031–1038. doi: 10.1001/archneur.65.8.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang R, Tang P, Wang P, Boissy RE, Zheng H. Regulation of tyrosinase trafficking and processing by presenilins: partial loss of function by familial Alzheimer’s disease mutation. Proc Natl Acad Sci U S A. 2006;103:353–358. doi: 10.1073/pnas.0509822102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Takasugi N, Tomita T, Hayashi I, Tsuruoka M, Niimura M, Takahashi Y, Thinakaran G, Iwatsubo T. The role of presenilin cofactors in the gamma-secretase complex. Nature. 2003;422:438–441. doi: 10.1038/nature01506. [DOI] [PubMed] [Google Scholar]
- 67.Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C. Reconstitution of gamma-secretase activity. Nat Cell Biol. 2003;5:486–488. doi: 10.1038/ncb960. [DOI] [PubMed] [Google Scholar]
- 68.Li YM, Xu M, Lai MT, Huang Q, Castro JL, DiMuzio-Mower J, Harrison T, Lellis C, Nadin A, Neduvelil JG, Register RB, Sardana MK, Shearman MS, Smith AL, Shi XP, Yin KC, Shafer JA, Gardell SJ. Photoactivated gamma-secretase inhibitors directed to the active site covalently label presenilin 1. Nature. 2000;405:689–694. doi: 10.1038/35015085. [DOI] [PubMed] [Google Scholar]
- 69.Esler WP, Kimberly WT, Ostaszewski BL, Diehl TS, Moore CL, Tsai JY, Rahmati T, Xia W, Selkoe DJ, Wolfe MS. Transition-state analogue inhibitors of gamma-secretase bind directly to presenilin-1. Nat Cell Biol. 2000;2:428–434. doi: 10.1038/35017062. [DOI] [PubMed] [Google Scholar]
- 70.Ahn K, Shelton CC, Tian Y, Zhang X, Gilchrist ML, Sisodia SS, Li Y-M. Activation and intrinsic γ-secretase activity of presenilin 1. Proc Natl Acad Sci U S A. 2010;107:21435–21440. doi: 10.1073/pnas.1013246107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature. 1999;398:513–517. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- 72.Steiner H, Kostka M, Romig H, Basset G, Pesold B, Hardy J, Capell A, Meyn L, Grim ML, Baumeister R, Fechteler K, Haass C. Glycine 384 is required for presenilin-1 function and is conserved in bacterial polytopic aspartyl proteases. Nat Cell Biol. 2000;2:848–851. doi: 10.1038/35041097. [DOI] [PubMed] [Google Scholar]
- 73.Li X, Dang S, Yan C, Gong X, Wang J, Shi Y. Structure of a presenilin family intramembrane aspartate protease. Nature. 2013;493:56–61. doi: 10.1038/nature11801. [DOI] [PubMed] [Google Scholar]
- 74.Thinakaran G, Borchelt DR, Lee MK, Slunt HH, Spitzer L, Kim G, Ratovitsky T, Davenport F, Nordstedt C, Seeger M, Hardy J, Levey AI, Gandy SE, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron. 1996;17:181–190. doi: 10.1016/s0896-6273(00)80291-3. [DOI] [PubMed] [Google Scholar]
- 75.Lai MT, Chen E, Crouthamel MC, DiMuzio-Mower J, Xu M, Huang Q, Price E, Register RB, Shi XP, Donoviel DB, Bernstein A, Hazuda D, Gardell SJ, Li YM. Presenilin-1 and Presenilin-2 Exhibit Distinct yet Overlapping {gamma}-Secretase Activities. J Biol Chem. 2003;278:22475–22481. doi: 10.1074/jbc.M300974200. [DOI] [PubMed] [Google Scholar]
- 76.Shirotani K, Edbauer D, Prokop S, Haass C, Steiner H. Identification of Distinct {gamma}-Secretase Complexes with Different APH-1 Variants. J Biol Chem. 2004;279:41340–41345. doi: 10.1074/jbc.M405768200. [DOI] [PubMed] [Google Scholar]
- 77.Shirotani K, Tomioka M, Kremmer E, Haass C, Steiner H. Pathological activity of familial Alzheimer’s disease-associated mutant presenilin can be executed by six different gamma-secretase complexes. Neurobiol Dis. 2007;27:102–107. doi: 10.1016/j.nbd.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 78.Niimura M, Isoo N, Takasugi N, Tsuruoka M, Ui-Tei K, Saigo K, Morohashi Y, Tomita T, Iwatsubo T. Aph-1 contributes to the stabilization and trafficking of the gamma-secretase complex through mechanisms involving intermolecular and intramolecular interactions. J Biol Chem. 2005;280:12967–12975. doi: 10.1074/jbc.M409829200. [DOI] [PubMed] [Google Scholar]
- 79.Shah S, Lee SF, Tabuchi K, Hao YH, Yu C, LaPlant Q, Ball H, Dann CE, 3rd, Sudhof T, Yu G. Nicastrin functions as a gamma-secretase-substrate receptor. Cell. 2005;122:435–447. doi: 10.1016/j.cell.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 80.Capell A, Beher D, Prokop S, Steiner H, Kaether C, Shearman MS, Haass C. Gamma-secretase complex assembly within the early secretory pathway. J Biol Chem. 2005;280:6471–6478. doi: 10.1074/jbc.M409106200. [DOI] [PubMed] [Google Scholar]
- 81.Prokop S, Shirotani K, Edbauer D, Haass C, Steiner H. Requirement of PEN-2 for stabilization of the presenilin N-/C-terminal fragment heterodimer within the gamma-secretase complex. J Biol Chem. 2004;279:23255–23261. doi: 10.1074/jbc.M401789200. [DOI] [PubMed] [Google Scholar]
- 82.Luo WJ, Wang H, Li H, Kim BS, Shah S, Lee HJ, Thinakaran G, Kim TW, Yu G, Xu H. PEN-2 and APH-1 coordinately regulate proteolytic processing of presenilin 1. J Biol Chem. 2003;278:7850–7854. doi: 10.1074/jbc.C200648200. [DOI] [PubMed] [Google Scholar]
- 83.Sato T, Diehl TS, Narayanan S, Funamoto S, Ihara Y, De Strooper B, Steiner H, Haass C, Wolfe MS. Active gamma-secretase complexes contain only one of each component. J Biol Chem. 2007;282:33985–33993. doi: 10.1074/jbc.M705248200. [DOI] [PubMed] [Google Scholar]
- 84.Li YM, Lai MT, Xu M, Huang Q, DiMuzio-Mower J, Sardana MK, Shi XP, Yin KC, Shafer JA, Gardell SJ. Presenilin 1 is linked with gamma-secretase activity in the detergent solubilized state. Proc Natl Acad Sci U S A. 2000;97:6138–6143. doi: 10.1073/pnas.110126897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gu Y, Sanjo N, Chen F, Hasegawa H, Petit A, Ruan X, Li W, Shier C, Kawarai T, Schmitt-Ulms G, Westaway D, St George-Hyslop P, Fraser PE. The presenilin proteins are components of multiple membrane-bound complexes that have different biological activities. J Biol Chem. 2004;279:31329–31336. doi: 10.1074/jbc.M401548200. [DOI] [PubMed] [Google Scholar]
- 86.Edbauer D, Winkler E, Haass C, Steiner H. Presenilin and nicastrin regulate each other and determine amyloid beta-peptide production via complex formation. Proc Natl Acad Sci U S A. 2002;99:8666–8671. doi: 10.1073/pnas.132277899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Evin G, Canterford LD, Hoke DE, Sharples RA, Culvenor JG, Masters CL. Transition-state analogue gamma-secretase inhibitors stabilize a 900 kDa presenilin/nicastrin complex. Biochemistry. 2005;44:4332–4341. doi: 10.1021/bi0481702. [DOI] [PubMed] [Google Scholar]
- 88.Zhou S, Zhou H, Walian PJ, Jap BK. CD147 is a regulatory subunit of the gamma-secretase complex in Alzheimer’s disease amyloid beta-peptide production. Proc Natl Acad Sci U S A. 2005;102:7499–7504. doi: 10.1073/pnas.0502768102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chen F, Hasegawa H, Schmitt-Ulms G, Kawarai T, Bohm C, Katayama T, Gu Y, Sanjo N, Glista M, Rogaeva E, Wakutani Y, Pardossi-Piquard R, Ruan X, Tandon A, Checler F, Marambaud P, Hansen K, Westaway D, St George-Hyslop P, Fraser P. TMP21 is a presenilin complex component that modulates gamma-secretase but not epsilon-secretase activity. Nature. 2006;440:1208–1212. doi: 10.1038/nature04667. [DOI] [PubMed] [Google Scholar]
- 90.Teranishi Y, Hur JY, Gu GJ, Kihara T, Ishikawa T, Nishimura T, Winblad B, Behbahani H, Kamali-Moghaddam M, Frykman S, Tjernberg LO. Erlin-2 is associated with active gamma-secretase in brain and affects amyloid beta-peptide production. Biochemical and biophysical research communications. 2012;424:476–481. doi: 10.1016/j.bbrc.2012.06.137. [DOI] [PubMed] [Google Scholar]
- 91.Hur JY, Teranishi Y, Kihara T, Yamamoto NG, Inoue M, Hosia W, Hashimoto M, Winblad B, Frykman S, Tjernberg LO. Identification of novel gamma-secretase-associated proteins in detergent-resistant membranes from brain. J Biol Chem. 2012;287:11991–12005. doi: 10.1074/jbc.M111.246074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Frykman S, Teranishi Y, Hur JY, Sandebring A, Yamamoto NG, Ancarcrona M, Nishimura T, Winblad B, Bogdanovic N, Schedin-Weiss S, Kihara T, Tjernberg LO. Identification of two novel synaptic gamma-secretase associated proteins that affect amyloid beta-peptide levels without altering Notch processing. Neurochem Int. 2012;61:108–118. doi: 10.1016/j.neuint.2012.03.016. [DOI] [PubMed] [Google Scholar]
- 93.Thathiah A, Horre K, Snellinx A, Vandewyer E, Huang Y, Ciesielska M, De Kloe G, Munck S, De Strooper B. beta-arrestin 2 regulates Abeta generation and gamma-secretase activity in Alzheimer’s disease. Nat Med. 2013;19:43–49. doi: 10.1038/nm.3023. [DOI] [PubMed] [Google Scholar]
- 94.Liu X, Zhao X, Zeng X, Bossers K, Swaab DF, Zhao J, Pei G. beta-Arrestin1 regulates gamma-secretase complex assembly and modulates amyloid-beta pathology. Cell Res. 2013;23:351–365. doi: 10.1038/cr.2012.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Vetrivel KS, Zhang X, Meckler X, Cheng H, Lee S, Gong P, Lopes KO, Chen Y, Iwata N, Yin KJ, Lee JM, Parent AT, Saido TC, Li YM, Sisodia SS, Thinakaran G. Evidence that CD147 modulation of beta-amyloid (Abeta) levels is mediated by extracellular degradation of secreted Abeta. J Biol Chem. 2008;283:19489–19498. doi: 10.1074/jbc.M801037200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hussain I, Fabregue J, Anderes L, Ousson S, Borlat F, Eligert V, Berger S, Dimitrov M, Alattia JR, Fraering PC, Beher D. The role of gamma-secretase activating protein (GSAP) and imatinib in the regulation of gamma-secretase activity and amyloid-beta generation. J Biol Chem. 2013;288:2521–2531. doi: 10.1074/jbc.M112.370924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vetrivel KS, Gong P, Bowen JW, Cheng H, Chen Y, Carter M, Nguyen PD, Placanica L, Wieland FT, Li YM, Kounnas MZ, Thinakaran G. Dual roles of the transmembrane protein p23/TMP21 in the modulation of amyloid precursor protein metabolism. Mol Neurodegener. 2007;2:4. doi: 10.1186/1750-1326-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wakabayashi T, Craessaerts K, Bammens L, Bentahir M, Borgions F, Herdewijn P, Staes A, Timmerman E, Vandekerckhove J, Rubinstein E, Boucheix C, Gevaert K, De Strooper B. Analysis of the gamma-secretase interactome and validation of its association with tetraspanin-enriched microdomains. Nat Cell Biol. 2009;11:1340–1346. doi: 10.1038/ncb1978. [DOI] [PubMed] [Google Scholar]
- 99.Placanica L, Chien JW, Li YM. Characterization of an atypical gamma-secretase complex from hematopoietic origin. Biochemistry. 2010;49:2796–2804. doi: 10.1021/bi901388t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Serneels L, Van Biervliet J, Craessaerts K, Dejaegere T, Horre K, Van Houtvin T, Esselmann H, Paul S, Schafer MK, Berezovska O, Hyman BT, Sprangers B, Sciot R, Moons L, Jucker M, Yang Z, May PC, Karran E, Wiltfang J, D’Hooge R, De Strooper B. gamma-Secretase heterogeneity in the Aph1 subunit: relevance for Alzheimer’s disease. Science. 2009;324:639–642. doi: 10.1126/science.1171176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Beher D, Fricker M, Nadin A, Clarke EE, Wrigley JD, Li YM, Culvenor JG, Masters CL, Harrison T, Shearman MS. In vitro characterization of the presenilin-dependent gamma-secretase complex using a novel affinity ligand. Biochemistry. 2003;42:8133–8142. doi: 10.1021/bi034045z. [DOI] [PubMed] [Google Scholar]
- 102.Placanica L, Tarassishin L, Yang G, Peethumnongsin E, Kim SH, Zheng H, Sisodia SS, Li YM. Pen2 and Presenilin-1 Modulate the Dynamic Equilibrium of Presenilin-1 and Presenilin-2 {gamma}-Secretase Complexes. J Biol Chem. 2009;284:2967–2977. doi: 10.1074/jbc.M807269200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Placanica L, Zhu L, Li YM. Gender- and age-dependent gamma-secretase activity in mouse brain and its implication in sporadic Alzheimer disease. PLoS ONE. 2009;4:e5088. doi: 10.1371/journal.pone.0005088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Teranishi Y, Hur JY, Welander H, Franberg J, Aoki M, Winblad B, Frykman S, Tjernberg LO. Affinity pulldown of gamma-secretase and associated proteins from human and rat brain. J Cell Mol Med. 2010;14:2675–2686. doi: 10.1111/j.1582-4934.2009.00907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Eriksen JL, Sagi SA, Smith TE, Weggen S, Das P, McLendon DC, Ozols VV, Jessing KW, Zavitz KH, Koo EH, Golde TE. NSAIDs and enantiomers of flurbiprofen target gamma-secretase and lower Abeta 42 in vivo. J Clin Invest. 2003;112:440–449. doi: 10.1172/JCI18162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wilcock GK, Black SE, Hendrix SB, Zavitz KH, Swabb EA, Laughlin MA Tarenflurbil Phase IISi. Efficacy and safety of tarenflurbil in mild to moderate Alzheimer’s disease: a randomised phase II trial. Lancet neurology. 2008;7:483–493. doi: 10.1016/S1474-4422(08)70090-5. [DOI] [PubMed] [Google Scholar]
- 107.Green RC, Schneider LS, Amato DA, Beelen AP, Wilcock G, Swabb EA, Zavitz KH Tarenflurbil Phase 3 Study G. Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. JAMA : the journal of the American Medical Association. 2009;302:2557–2564. doi: 10.1001/jama.2009.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Peretto I, Radaelli S, Parini C, Zandi M, Raveglia LF, Dondio G, Fontanella L, Misiano P, Bigogno C, Rizzi A, Riccardi B, Biscaioli M, Marchetti S, Puccini P, Catinella S, Rondelli I, Cenacchi V, Bolzoni PT, Caruso P, Villetti G, Facchinetti F, Del Giudice E, Moretto N, Imbimbo BP. Synthesis and biological activity of flurbiprofen analogues as selective inhibitors of beta-amyloid(1)(−)(42) secretion. J Med Chem. 2005;48:5705–5720. doi: 10.1021/jm0502541. [DOI] [PubMed] [Google Scholar]
- 109.Imbimbo BP, Frigerio E, Breda M, Fiorentini F, Fernandez M, Sivilia S, Giardino L, Calza L, Norris D, Casula D, Shenouda M. Pharmacokinetics and Pharmacodynamics of CHF5074 After Short-term Administration in Healthy Subjects. Alzheimer disease and associated disorders. 2012 doi: 10.1097/WAD.0b013e3182622ace. [DOI] [PubMed] [Google Scholar]
- 110.Kretner B, Fukumori A, Gutsmiedl A, Page RM, Luebbers T, Galley G, Baumann K, Haass C, Steiner H. Attenuated Abeta42 responses to low potency gamma-secretase modulators can be overcome for many pathogenic presenilin mutants by second-generation compounds. J Biol Chem. 2011;286:15240–15251. doi: 10.1074/jbc.M110.213587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mullard A. Sting of Alzheimer’s failures offset by upcoming prevention trials. Nat Rev Drug Discov. 2012;11:657–660. doi: 10.1038/nrd3842. [DOI] [PubMed] [Google Scholar]
- 112.Miller G. Stopping Alzheimer’s Before It Starts. Science. 2012;337:790–792. doi: 10.1126/science.337.6096.790. [DOI] [PubMed] [Google Scholar]
- 113.Page RM, Gutsmiedl A, Fukumori A, Winkler E, Haass C, Steiner H. Beta-amyloid precursor protein mutants respond to gamma-secretase modulators. J Biol Chem. 2010;285:17798–17810. doi: 10.1074/jbc.M110.103283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hall A, Elliott RL, Giblin GM, Hussain I, Musgrave J, Naylor A, Sasse R, Smith B. Piperidine-derived gamma-secretase modulators. Bioorg Med Chem Lett. 2010;20:1306–1311. doi: 10.1016/j.bmcl.2009.08.072. [DOI] [PubMed] [Google Scholar]
- 115.Hawkins J, Harrison DC, Ahmed S, Davis RP, Chapman T, Marshall I, Smith B, Mead TL, Medhurst A, Giblin GM, Hall A, Gonzalez MI, Richardson J, Hussain I. Dynamics of Abeta42 reduction in plasma, CSF and brain of rats treated with the gamma-secretase modulator, GSM-10h. Neurodegener Dis. 2011;8:455–464. doi: 10.1159/000324511. [DOI] [PubMed] [Google Scholar]
- 116.Li T, Huang Y, Jin S, Ye L, Rong N, Yang X, Ding Y, Cheng Z, Zhang J, Wan Z, Harrison DC, Hussain I, Hall A, Lee DH, Lau LF, Matsuoka Y. Gamma-secretase modulators do not induce Abeta-rebound and accumulation of beta-C-terminal fragment. J Neurochem. 2012;121:277–286. doi: 10.1111/j.1471-4159.2011.07560.x. [DOI] [PubMed] [Google Scholar]
- 117.Rogers K, Felsenstein KM, Hrdlicka L, Tu Z, Albayya F, Lee W, Hopp S, Miller MJ, Spaulding D, Yang Z, Hodgdon H, Nolan S, Wen M, Costa D, Blain JF, Freeman E, De Strooper B, Vulsteke V, Scrocchi L, Zetterberg H, Portelius E, Hutter-Paier B, Havas D, Ahlijanian M, Flood D, Leventhal L, Shapiro G, Patzke H, Chesworth R, Koenig G. Modulation of gamma-secretase by EVP-0015962 reduces amyloid deposition and behavioral deficits in Tg2576 mice. Mol Neurodegener. 2012;7:61. doi: 10.1186/1750-1326-7-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Van Broeck B, Chen JM, Treton G, Desmidt M, Hopf C, Ramsden N, Karran E, Mercken M, Rowley A. Chronic treatment with a novel gamma-secretase modulator, JNJ-40418677, inhibits amyloid plaque formation in a mouse model of Alzheimer’s disease. Br J Pharmacol. 2011;163:375–389. doi: 10.1111/j.1476-5381.2011.01207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Peng H, Talreja T, Xin Z, Cuervo JH, Kumaravel G, Humora MJ, Xu L, Rohde E, Gan L, Jung M-y, Shackett MN, Chollate S, Dunah AW, Snodgrass-Belt PA, Arnold HM, Taveras AG, Rhodes KJ, Scannevin RH. Discovery of BIIB042, a Potent, Selective, and Orally Bioavailable γ-Secretase Modulator. ACS Med Chem Lett. 2011;2:786–791. doi: 10.1021/ml200175q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kukar TL, Ladd TB, Bann MA, Fraering PC, Narlawar R, Maharvi GM, Healy B, Chapman R, Welzel AT, Price RW, Moore B, Rangachari V, Cusack B, Eriksen J, Jansen-West K, Verbeeck C, Yager D, Eckman C, Ye W, Sagi S, Cottrell BA, Torpey J, Rosenberry TL, Fauq A, Wolfe MS, Schmidt B, Walsh DM, Koo EH, Golde TE. Substrate-targeting gamma-secretase modulators. Nature. 2008;453:925–929. doi: 10.1038/nature07055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Narlawar R, Baumann K, Czech C, Schmidt B. Conversion of the LXR-agonist TO-901317--from inverse to normal modulation of gamma-secretase by addition of a carboxylic acid and a lipophilic anchor. Bioorg Med Chem Lett. 2007;17:5428–5431. doi: 10.1016/j.bmcl.2007.07.044. [DOI] [PubMed] [Google Scholar]
- 122.Zall A, Kieser D, Hottecke N, Naumann EC, Thomaszewski B, Schneider K, Steinbacher DT, Schubenel R, Masur S, Baumann K, Schmidt B. NSAID-derived gamma-secretase modulation requires an acidic moiety on the carbazole scaffold. Bioorg Med Chem. 2011;19:4903–4909. doi: 10.1016/j.bmc.2011.06.062. [DOI] [PubMed] [Google Scholar]
- 123.Ohki Y, Higo T, Uemura K, Shimada N, Osawa S, Berezovska O, Yokoshima S, Fukuyama T, Tomita T, Iwatsubo T. Phenylpiperidine-type gamma-secretase modulators target the transmembrane domain 1 of presenilin 1. EMBO J. 2011;30:4815–4824. doi: 10.1038/emboj.2011.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Oborski CE, Iyer R, Maguire BA, Bora G, Atchison K, Pozdnyakov N, Wood K, Parker C, Subashi TA, Pettersson M, Johnson DS, Bales KR. Pharmacological assessment of gamma-secretase activity from rodent and human brain. Neuroscience & Medicine. 2012;3:149–161. [Google Scholar]
- 125.Hussain I, Harrison DC, Hawkins J, Chapman T, Marshall I, Facci L, Ahmed S, Brackenborough K, Skaper SD, Mead TL, Smith BB, Giblin GM, Hall A, Gonzalez MI, Richardson JC. TASTPM mice expressing amyloid precursor protein and presenilin-1 mutant transgenes are sensitive to gamma-secretase modulation and amyloid-beta(4)(2) lowering by GSM-10h. Neurodegener Dis. 2011;8:15–24. doi: 10.1159/000313903. [DOI] [PubMed] [Google Scholar]
- 126.Mitani Y, Yarimizu J, Akashiba H, Shitaka Y, Ni K, Matsuoka N. Amelioration of cognitive deficits in plaque-bearing Alzheimer’s disease model mice through selective reduction of nascent soluble Abeta42 without affecting other Abeta pools. J Neurochem. 2012 doi: 10.1111/jnc.12125. [DOI] [PubMed] [Google Scholar]
- 127.Kounnas MZ, Danks AM, Cheng S, Tyree C, Ackerman E, Zhang X, Ahn K, Nguyen P, Comer D, Mao L, Yu C, Pleynet D, Digregorio PJ, Velicelebi G, Stauderman KA, Comer WT, Mobley WC, Li YM, Sisodia SS, Tanzi RE, Wagner SL. Modulation of gamma-secretase reduces beta-amyloid deposition in a transgenic mouse model of Alzheimer’s disease. Neuron. 2010;67:769–780. doi: 10.1016/j.neuron.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Portelius E, Van Broeck B, Andreasson U, Gustavsson MK, Mercken M, Zetterberg H, Borghys H, Blennow K. Acute effect on the Abeta isoform pattern in CSF in response to gamma-secretase modulator and inhibitor treatment in dogs. J Alzheimers Dis. 2010;21:1005–1012. doi: 10.3233/JAD-2010-100573. [DOI] [PubMed] [Google Scholar]
- 129.Borgegard T, Jureus A, Olsson F, Rosqvist S, Sabirsh A, Rotticci D, Paulsen K, Klintenberg R, Yan H, Waldman M, Stromberg K, Nord J, Johansson J, Regner A, Parpal S, Malinowsky D, Radesater AC, Li T, Singh R, Eriksson H, Lundkvist J. First and second generation gamma-secretase modulators (GSMs) modulate amyloid-beta (Abeta) peptide production through different mechanisms. J Biol Chem. 2012;287:11810–11819. doi: 10.1074/jbc.M111.305227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hashimoto T, Ishibashi A, Hagiwara H, Murata Y, Takenaka O, Miyagawa T. E2012: A novel gamma-secretase modulator - pharmacology. Alzheimers Dement. 2010;6(Suppl):S242. [Google Scholar]
- 131.Nagy C, Schuck E, Ishibashi A, Nakatani Y, Rege B, Logovinsky V. E2012, a novel gamma-secretase modulator, decreases plasma amyloid-beta (Aβ) levels in humans. Alzheimers Dement. 2010;6(Suppl):S574. [Google Scholar]
- 132.(http://www.neurogeneticpharmaceuticals.com/about-ngp.html).
- 133.Huang X, Zhou W, Liu X, Li H, Sun G, Mandal M, Vicarel M, Zhu X, Bennett C, McCraken T, Pissarnitski D, Zhao Z, Cole D, Gallo G, Zhu Z, Palani A, Aslanian R, Clader J, Czarniecki M, Greenlee W, Burnett D, Cohen-Williams M, Hyde L, Song L, Zhang L, Chu I, Buevich A. Synthesis and SAR Studies of Fused Oxadiazines as γ-Secretase Modulators for Treatment of Alzheimer’s Disease. ACS Med Chem Lett. 2012;3:931–935. doi: 10.1021/ml300209g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sun ZY, Asberom T, Bara T, Bennett C, Burnett D, Chu I, Clader J, Cohen-Williams M, Cole D, Czarniecki M, Durkin J, Gallo G, Greenlee W, Josien H, Huang X, Hyde L, Jones N, Kazakevich I, Li H, Liu X, Lee J, Maccoss M, Mandal MB, McCracken T, Nomeir A, Mazzola R, Palani A, Parker EM, Pissarnitski DA, Qin J, Song L, Terracina G, Vicarel M, Voigt J, Xu R, Zhang L, Zhang Q, Zhao Z, Zhu X, Zhu Z. Cyclic hydroxyamidines as amide isosteres: discovery of oxadiazolines and oxadiazines as potent and highly efficacious gamma-secretase modulators in vivo. J Med Chem. 2012;55:489–502. doi: 10.1021/jm201407j. [DOI] [PubMed] [Google Scholar]
- 135.Huang X, Aslanian R, Zhou W, Zhu X, Qin J, Greenlee W, Zhu Z, Zhang L, Hyde L, Chu I, Cohen-Williams M, Palani A. The Discovery of Pyridone and Pyridazone Heterocycles as γ-Secretase Modulators. ACS Med Chem Lett. 2010;1:184–187. doi: 10.1021/ml1000799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ebke A, Luebbers T, Fukumori A, Shirotani K, Haass C, Baumann K, Steiner H. Novel gamma-secretase enzyme modulators directly target presenilin protein. J Biol Chem. 2011;286:37181–37186. doi: 10.1074/jbc.C111.276972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lu Y, Riddell D, Hajos-Korcsok E, Bales K, Wood KM, Nolan CE, Robshaw AE, Zhang L, Leung L, Becker SL, Tseng E, Barricklow J, Miller EH, Osgood S, O’Neill BT, Brodney MA, Johnson DS, Pettersson M. Cerebrospinal fluid amyloid-beta (Abeta) as an effect biomarker for brain Abeta lowering verified by quantitative preclinical analyses. J Pharmacol Exp Ther. 2012;342:366–375. doi: 10.1124/jpet.112.192625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bischoff F, Berthelot D, De Cleyn M, Macdonald G, Minne G, Oehlrich D, Pieters S, Surkyn M, Trabanco AA, Tresadern G, Van Brandt S, Velter I, Zaja M, Borghys H, Masungi C, Mercken M, Gijsen HJ. Design and synthesis of a novel series of bicyclic heterocycles as potent gamma-secretase modulators. J Med Chem. 2012;55:9089–9106. doi: 10.1021/jm201710f. [DOI] [PubMed] [Google Scholar]
- 139.Borghys H, Tuefferd M, Van Broeck B, Clessens E, Dillen L, Cools W, Vinken P, Straetemans R, De Ridder F, Gijsen H, Mercken M. A canine model to evaluate efficacy and safety of gamma-secretase inhibitors and modulators. J Alzheimers Dis. 2012;28:809–822. doi: 10.3233/JAD-2011-111475. [DOI] [PubMed] [Google Scholar]
- 140.Gijsen HJ, Mercken M. gamma-Secretase Modulators: Can We Combine Potency with Safety? International journal of Alzheimer’s disease. 2012;2012:295207. doi: 10.1155/2012/295207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hahn S, Bruning T, Ness J, Czirr E, Baches S, Gijsen H, Korth C, Pietrzik CU, Bulic B, Weggen S. Presenilin-1 but not amyloid precursor protein mutations present in mouse models of Alzheimer’s disease attenuate the response of cultured cells to gamma-secretase modulators regardless of their potency and structure. J Neurochem. 2011;116:385–395. doi: 10.1111/j.1471-4159.2010.07118.x. [DOI] [PubMed] [Google Scholar]
- 142.Rivkin A, Ahearn SP, Chichetti SM, Kim YR, Li C, Rosenau A, Kattar SD, Jung J, Shah S, Hughes BL, Crispino JL, Middleton RE, Szewczak AA, Munoz B, Shearman MS. Piperazinyl pyrimidine derivatives as potent gamma-secretase modulators. Bioorg Med Chem Lett. 2010;20:1269–1271. doi: 10.1016/j.bmcl.2009.11.101. [DOI] [PubMed] [Google Scholar]
- 143.Findeis MA, Schroeder F, McKee TD, Yager D, Fraering PC, Creaser SP, Austin WF, Clardy J, Wang R, Selkoe D, Eckman CB. Discovery of a novel pharmacological and structural class of gamma secretase modulators derived from the extract of Actaea racemosa. ACS Chem Neurosci. 2012;3:941–951. doi: 10.1021/cn3000857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hubbs JL, Fuller NO, Austin WF, Shen R, Creaser SP, McKee TD, Loureiro RM, Tate B, Xia W, Ives J, Bronk BS. Optimization of a natural product-based class of gamma-secretase modulators. J Med Chem. 2012;55:9270–9282. doi: 10.1021/jm300976b. [DOI] [PubMed] [Google Scholar]
- 145.Pfizer. WO 2012/131539
- 146.Hoffmann-La Roche AG. WO 2012/116965
- 147.Bristol-Myers Squibb Company. WO 2012/103297
- 148.Geoghegan KF, Johnson DS. Chemical proteomic technologies for drug target identification. Annu Rep Med Chem. 2010;45:345–360. [Google Scholar]
- 149.Seiffert D, Bradley JD, Rominger CM, Rominger DH, Yang F, Meredith JE, Jr, Wang Q, Roach AH, Thompson LA, Spitz SM, Higaki JN, Prakash SR, Combs AP, Copeland RA, Arneric SP, Hartig PR, Robertson DW, Cordell B, Stern AM, Olson RE, Zaczek R. Presenilin-1 and -2 are molecular targets for gamma-secretase inhibitors. J Biol Chem. 2000;275:34086–34091. doi: 10.1074/jbc.M005430200. [DOI] [PubMed] [Google Scholar]
- 150.Kornilova AY, Bihel F, Das C, Wolfe MS. The initial substrate-binding site of gamma-secretase is located on presenilin near the active site. Proc Natl Acad Sci U S A. 2005;102:3230–3235. doi: 10.1073/pnas.0407640102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Fuwa H, Takahashi Y, Konno Y, Watanabe N, Miyashita H, Sasaki M, Natsugari H, Kan T, Fukuyama T, Tomita T, Iwatsubo T. Divergent synthesis of multifunctional molecular probes to elucidate the enzyme specificity of dipeptidic gamma-secretase inhibitors. ACS Chem Biol. 2007;2:408–418. doi: 10.1021/cb700073y. [DOI] [PubMed] [Google Scholar]
- 152.Brunner J. New photolabeling and crosslinking methods. Annu Rev Biochem. 1993;62:483–514. doi: 10.1146/annurev.bi.62.070193.002411. [DOI] [PubMed] [Google Scholar]
- 153.Crump CJ, am Ende CW, Ballard TE, Pozdnyakov N, Pettersson M, Chau DM, Bales KR, Li YM, Johnson DS. Development of clickable active site-directed photoaffinity probes for gamma-secretase. Bioorg Med Chem Lett. 2012;22:2997–3000. doi: 10.1016/j.bmcl.2012.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Crump CJ, Fish BA, Castro SV, Chau DM, Gertsik N, Ahn K, Stiff C, Pozdnyakov N, Bales KR, Johnson DS, Li YM. Piperidine acetic acid based gamma-secretase modulators directly bind to Presenilin-1. ACS chemical neuroscience. 2011;2:705–710. doi: 10.1021/cn200098p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lleo A, Berezovska O, Herl L, Raju S, Deng A, Bacskai BJ, Frosch MP, Irizarry M, Hyman BT. Nonsteroidal anti-inflammatory drugs lower Abeta42 and change presenilin 1 conformation. Nat Med. 2004;10:1065–1066. doi: 10.1038/nm1112. [DOI] [PubMed] [Google Scholar]
- 156.Uemura K, Farner KC, Hashimoto T, Nasser-Ghodsi N, Wolfe MS, Koo EH, Hyman BT, Berezovska O. Substrate docking to gamma-secretase allows access of gamma-secretase modulators to an allosteric site. Nat Commun. 2010;1:130. doi: 10.1038/ncomms1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Chau DM, Crump CJ, Villa JC, Scheinberg DA, Li YM. Familial Alzheimer Disease Presenilin-1 Mutations Alter the Active Site Conformation of gamma-secretase. J Biol Chem. 2012;287:17288–17296. doi: 10.1074/jbc.M111.300483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Shelton CC, Zhu L, Chau D, Yang L, Wang R, Djaballah H, Zheng H, Li YM. Modulation of gamma-secretase specificity using small molecule allosteric inhibitors. Proc Natl Acad Sci U S A. 2009;106:20228–20233. doi: 10.1073/pnas.0910757106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Crump CJ, Castro SV, Wang F, Pozdnyakov N, Ballard TE, Sisodia SS, Bales KR, Johnson DS, Li YM. BMS-708,163 Targets Presenilin and Lacks Notch-Sparing Activity. Biochemistry. 2012;51:7209–7211. doi: 10.1021/bi301137h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Xu M, Lai MT, Huang Q, DiMuzio-Mower J, Castro JL, Harrison T, Nadin A, Neduvelil JG, Shearman MS, Shafer JA, Gardell SJ, Li YM. gamma-Secretase: characterization and implication for Alzheimer disease therapy. Neurobiol Aging. 2002;23:1023–1030. doi: 10.1016/s0197-4580(02)00126-4. [DOI] [PubMed] [Google Scholar]
- 161.Chun J, Yin YI, Yang G, Tarassishin L, Li YM. Stereoselective Synthesis of Photoreactive Peptidomimetic gamma-Secretase Inhibitors. J Org Chem. 2004;69:7344–7347. doi: 10.1021/jo0486948. [DOI] [PubMed] [Google Scholar]
- 162.Yang G, Yin YI, Chun J, Shelton CC, Ouerfelli O, Li YM. Stereo-controlled synthesis of novel photoreactive gamma-secretase inhibitors. Bioorg Med Chem Lett. 2009;19:922–925. doi: 10.1016/j.bmcl.2008.11.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Takahashi Y, Hayashi I, Tominari Y, Rikimaru K, Morohashi Y, Kan T, Natsugari H, Fukuyama T, Tomita T, Iwatsubo T. Sulindac sulfide is a noncompetitive gamma-secretase inhibitor that preferentially reduces Abeta 42 generation. J Biol Chem. 2003;278:18664–18670. doi: 10.1074/jbc.M301619200. [DOI] [PubMed] [Google Scholar]
- 164.Beher D, Clarke EE, Wrigley JD, Martin AC, Nadin A, Churcher I, Shearman MS. Selected non-steroidal anti-inflammatory drugs and their derivatives target gamma-secretase at a novel site. Evidence for an allosteric mechanism. J Biol Chem. 2004;279:43419–43426. doi: 10.1074/jbc.M404937200. [DOI] [PubMed] [Google Scholar]
- 165.Clarke EE, Churcher I, Ellis S, Wrigley JD, Lewis HD, Harrison T, Shearman MS, Beher D. Intra- or intercomplex binding to the gamma-secretase enzyme. A model to differentiate inhibitor classes. J Biol Chem. 2006;281:31279–31289. doi: 10.1074/jbc.M605051200. [DOI] [PubMed] [Google Scholar]
- 166.Iben LG, Olson RE, Balanda LA, Jayachandra S, Robertson BJ, Hay V, Corradi J, Prasad CV, Zaczek R, Albright CF, Toyn JH. Signal peptide peptidase and gamma-secretase share equivalent inhibitor binding pharmacology. J Biol Chem. 2007;282:36829–36836. doi: 10.1074/jbc.M707002200. [DOI] [PubMed] [Google Scholar]
- 167.Uemura K, Farner KC, Hashimoto T, Nasser-Ghodsi N, Wolfe MS, Koo EH, Hyman BT, Berezovska O. Substrate docking to gamma-secretase allows access of gamma-secretase modulators to an allosteric site. Nat Commun. 2010;1:130. doi: 10.1038/ncomms1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Botev A, Munter LM, Wenzel R, Richter L, Althoff V, Ismer J, Gerling U, Weise C, Koksch B, Hildebrand PW, Bittl R, Multhaup G. The amyloid precursor protein C-terminal fragment C100 occurs in monomeric and dimeric stable conformations and binds gamma-secretase modulators. Biochemistry. 2011;50:828–835. doi: 10.1021/bi1014002. [DOI] [PubMed] [Google Scholar]
- 169.Richter L, Munter LM, Ness J, Hildebrand PW, Dasari M, Unterreitmeier S, Bulic B, Beyermann M, Gust R, Reif B, Weggen S, Langosch D, Multhaup G. Amyloid beta 42 peptide (Abeta42)-lowering compounds directly bind to Abeta and interfere with amyloid precursor protein (APP) transmembrane dimerization. Proc Natl Acad Sci U S A. 2010;107:14597–14602. doi: 10.1073/pnas.1003026107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Beel AJ, Barrett P, Schnier PD, Hitchcock SA, Bagal D, Sanders CR, Jordan JB. Nonspecificity of binding of gamma-secretase modulators to the amyloid precursor protein. Biochemistry. 2009;48:11837–11839. doi: 10.1021/bi901839d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Barrett PJ, Sanders CR, Kaufman SA, Michelsen K, Jordan JB. NSAID-Based γ-Secretase Modulators Do Not Bind to the Amyloid-β Polypeptide. Biochemistry. 2011;50:10328–10342. doi: 10.1021/bi201371j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Espeseth AS, Xu M, Huang Q, Coburn CA, Jones KL, Ferrer M, Zuck PD, Strulovici B, Price EA, Wu G, Wolfe AL, Lineberger JE, Sardana M, Tugusheva K, Pietrak BL, Crouthamel MC, Lai MT, Dodson EC, Bazzo R, Shi XP, Simon AJ, Li Y, Hazuda DJ. Compounds that bind APP and inhibit Abeta processing in vitro suggest a novel approach to Alzheimer disease therapeutics. J Biol Chem. 2005;280:17792–17797. doi: 10.1074/jbc.M414331200. [DOI] [PubMed] [Google Scholar]
- 173.Munter LM, Voigt P, Harmeier A, Kaden D, Gottschalk KE, Weise C, Pipkorn R, Schaefer M, Langosch D, Multhaup G. GxxxG motifs within the amyloid precursor protein transmembrane sequence are critical for the etiology of Abeta42. EMBO J. 2007;26:1702–1712. doi: 10.1038/sj.emboj.7601616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Barrett PJ, Song Y, Van Horn WD, Hustedt EJ, Schafer JM, Hadziselimovic A, Beel AJ, Sanders CR. The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol. Science. 2012;336:1168–1171. doi: 10.1126/science.1219988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Okochi M, Fukumori A, Jiang J, Itoh N, Kimura R, Steiner H, Haass C, Tagami S, Takeda M. Secretion of the Notch-1 Abeta-like peptide during Notch signaling. J Biol Chem. 2006;281:7890–7898. doi: 10.1074/jbc.M513250200. [DOI] [PubMed] [Google Scholar]
- 176.Wanngren J, Ottervald J, Parpal S, Portelius E, Stromberg K, Borgegard T, Klintenberg R, Jureus A, Blomqvist J, Blennow K, Zetterberg H, Lundkvist J, Rosqvist S, Karlstrom H. Second generation gamma-secretase modulators exhibit different modulation of Notch beta and Abeta production. J Biol Chem. 2012;287:32640–32650. doi: 10.1074/jbc.M112.376541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Sagi SA, Lessard CB, Winden KD, Maruyama H, Koo JC, Weggen S, Kukar TL, Golde TE, Koo EH. Substrate sequence influences gamma-secretase modulator activity, role of the transmembrane domain of the amyloid precursor protein. J Biol Chem. 2011;286:39794–39803. doi: 10.1074/jbc.M111.277228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Sato T, Nyborg AC, Iwata N, Diehl TS, Saido TC, Golde TE, Wolfe MS. Signal peptide peptidase: biochemical properties and modulation by nonsteroidal antiinflammatory drugs. Biochemistry. 2006;45:8649–8656. doi: 10.1021/bi060597g. [DOI] [PubMed] [Google Scholar]
- 179.Jumpertz T, Rennhack A, Ness J, Baches S, Pietrzik CU, Bulic B, Weggen S. Presenilin Is the Molecular Target of Acidic gamma-Secretase Modulators in Living Cells. PLoS ONE. 2012;7:e30484. doi: 10.1371/journal.pone.0030484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Pozdnyakov N, Murrey HE, Crump CJ, Pettersson M, Ballard TE, Am Ende CW, Ahn K, Li YM, Bales KR, Johnson DS. gamma-Secretase modulator (GSM) photoaffinity probes reveal distinct allosteric binding sites on presenilin. J Biol Chem DOI. 2013 doi: 10.1074/jbc.M112.398602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Chavez-Gutierrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, Lismont S, Zhou L, Van Cleynenbreugel S, Esselmann H, Wiltfang J, Serneels L, Karran E, Gijsen H, Schymkowitz J, Rousseau F, Broersen K, De Strooper B. The mechanism of gamma-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012;31:2261–2274. doi: 10.1038/emboj.2012.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kukar TL, Ladd TB, Robertson P, Pintchovski SA, Moore B, Bann MA, Ren Z, Jansen-West K, Malphrus K, Eggert S, Maruyama H, Cottrell BA, Das P, Basi GS, Koo EH, Golde TE. Lysine 624 of the amyloid precursor protein (APP) is a critical determinant of amyloid beta peptide length: support for a sequential model of gamma-secretase intramembrane proteolysis and regulation by the amyloid beta precursor protein (APP) juxtamembrane region. J Biol Chem. 2011;286:39804–39812. doi: 10.1074/jbc.M111.274696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Schofield PR, Sperling RA, Salloway S, Morris JC Dominantly Inherited Alzheimer N. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]