

Abstract
Germline mis-sense mutations affecting a single BRCA2 allele predispose humans to cancer. Here, we identify a protein-targeting mechanism disrupted by the cancer-associated mutation, BRCA2D2723H that controls the nuclear localization of BRCA2 and its cargo, the recombination enzyme RAD51. A nuclear export signal (NES) in BRCA2 is masked by its interaction with a partner protein, DSS1, such that point mutations impairing BRCA2-DSS1 binding render BRCA2 cytoplasmic. In turn, cytoplasmic mis-localization of mutant BRCA2 inhibits the nuclear retention of RAD51, by exposing a similar NES in RAD51 usually obscured by the BRCA2-RAD51 interaction. Thus, a series of NES-masking interactions localizes BRCA2 and RAD51 in the nucleus. Interestingly, BRCA2D2723H decreases RAD51 nuclear retention even when wildtype BRCA2 is present. Our findings suggest a mechanism for regulation of the nucleo-cytoplasmic distribution of BRCA2 and RAD51, and for its impairment by a heterozygous disease-associated mutation.
Keywords: BRCA2, RAD51, nuclear localization, protein targeting, cancer predisposition, germline mutation
Introduction
Inherited germline mutations in a single copy of the BRCA2 tumor suppressor predispose to breast, ovarian, pancreatic and other cancers (The Breast Cancer Linkage Consortium)1. Somatic loss of the second allele occurs frequently in the tumors that develop in mutation carriers2,3, as in the case of other tumor suppressors4, but is not always essential for tumorigenesis5. Much evidence suggests that BRCA2 exerts its tumor suppressive function in the cell nucleus through its role in the repair of DNA breaks by homologous recombination6,7. BRCA2 controls the accumulation of the RAD51 recombinase enzyme at sites of DNA breakage in the nucleus8, nucleates RAD51 filament formation at single-strand (ss)-double-strand (ds) DNA junctions9, and promotes RAD51 binding on ssDNA whilst inhibiting dsDNA binding10-15. However, defective homologous recombination occurs typically in cells lacking both BRCA2 alleles7, and so it remains unclear how heterozygous cancer-associated mutations may compromise BRCA2’s function.
To address this issue, we have investigated the cellular effects of a common cancer-associated BRCA2 mis-sense mutation (BRCA2 D2723H) in which Asp2723 is replaced by His16. Unexpectedly, our studies reveal an unreported mechanism wherein the localization of BRCA2 and RAD51 to the cell nucleus is governed by the masking of nuclear exclusion signals (NESs) via protein-protein interactions. The BRCA2 D2723H mutant impairs this mechanism, triggering decreased nuclear accumulation of RAD51, even when wildtype BRCA2 is present, contributing to its deleterious effects in the heterozygous state. Thus, our results link the nucleo-cytoplasmic translocation of the BRCA2 tumor suppressor to the cellular mechanisms of disease following its germline inactivation.
Results
DSS1 binding to BRCA2 is critical for nuclear localization
Cancer-associated mis-sense mutations in human BRCA2 frequently occur in the region spanning residues ~2500 to 285017, which mediates the interaction (Supplementary Fig 1a) of BRCA2 with the small (70 amino acids (aa)), acidic protein DSS118,19. One of these mutations, BRCA2 D2723H, which alters a single Asp residue at position 2723 in the 3418 aa BRCA2 protein to His, has been reported >30 times in the Breast Cancer Information Core database (http://research.nhgri.nih.gov/bic/). Evidence from linkage analysis of human kindreds16 and from functional studies on murine embryonic stem cells suggests that the inheritance of BRCA2 D2723H is functionally deleterious20. Because the BRCA2 D2723H mutation affects a key conserved residue (Supplementary Fig 1a) that directly contacts DSS117, we first determined whether it affects the BRCA2-DSS1 interaction. We generated a fragment of BRCA2 spanning the entire DSS1-binding domain (DBD) across residues 2461-2975, fused to a yellow fluorescent protein (SYFP), plus a nuclear localization signal (NLS), and co-expressed it in human 293T cells with an mCherry-tagged form of full length DSS1. The SYFP-DBD fragment is >70KDa and therefore requires a heterologous NLS to allow nuclear localization. While the wildtype (WT) form of NLS-SYFP-DBD co-immunoprecipitatedwith mCherry-DSS1 (Figure 1a), the D2723H mutant did not, despite higher expression levels. Similarly, a mutant form of the DBD (W2725A), in which an evolutionarily conserved Trp residue at position 2725 implicated in DSS1 binding21 is altered to Ala, immunoprecipitated poorly with mCherry-DSS1.
Figure 1. DSS1 binding correlates with localization of BRCA2.
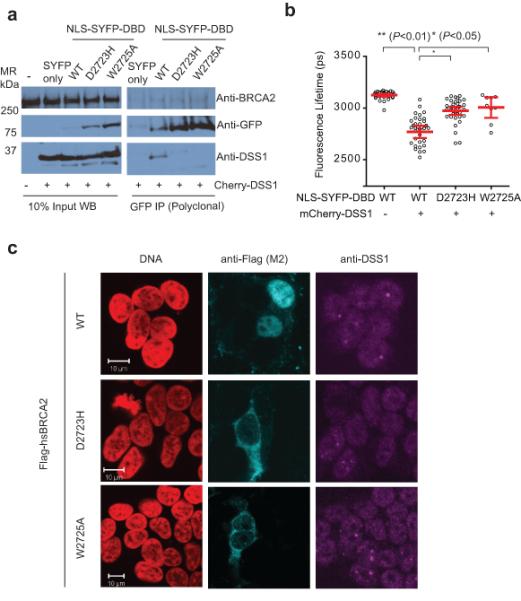
a) is an immunoprecipitation experiment of YFP tagged NLS-DBD fragments with Cherry-DSS1 in 293T cells, using an anti-GFP polyclonal serum. The left panels are of the whole cell lysate (10% of the input), and the panels on the right are of the immunoprecipitate. Transfection of free SYFP with cherry-DSS1 serves as the negative control for the western blot as well as for the immunoprecipitation, and endogenous BRCA2 as the loading control for the experiment. Endogenous DSS1 is difficult to visualize on standard Western blots due to its acidity and small size, requiring unique gel and transfer conditions sub-optimal for the relatively large DBD fragment. Uncropped blots are in Supplementary Fig 8. b) is a dot plot of the mean SYFP lifetime per cell from a FRET-FLIM experiment with NLS-SYFP-DBD and Cherry-DSS1. Each dot represents a single cell; the lines and error bars represent the mean and 95% confidence interval for the population studied in the experiment. U2OS cells co-transfected with the indicated constructs were analyzed by TCSPC. NLS-SYFP DBD forms have a lifetime of about 3200ps, which drops to about 2900ps when mCherry-DSS1 is co-transfected (p<0.01 by two-tailed Student’s t-test, n~30), in contrast to the mutant forms of the DBD. c) shows representative immunofluorescent confocal micrographs of 293T cells nucleofected with Flag-tagged full-length versions of BRCA2, WT or mutant D2723H and W2725A. DNA is colored in red, BRCA2 (anti-Flag) in Cyan, and endogenous DSS1 (anti-DSS1) in Magenta.
Measurement of fluorescence resonance energy transfer (FRET) (Supplementary Fig 1b), a near-field coupling effect, reports on specific intermolecular interactions when the interacting fluorophores are approximately <5nm apart22. SYFP and mCherry are an optimized FRET pair, with a Forster Radius of >5nm, and thus exhibit an enhanced FRET efficiency compared to traditional blue-shifted fluorophore pairs23. We used Fluorescence Lifetime Imaging Microscopy (FLIM) to measure FRET on the donor SYFP molecule, which has a mono-exponential decay of fluorescence lifetime23 (Supplementary Fig 1c). Co-transfection of mCherry-DSS1 with NLS-SYFP-DBD caused a significant decrease in SYFP lifetime from 3200 to 2900 picoseconds on average, evidenced by time-correlated single photon counting, suggestive of FRET through the direct interaction of these molecules (Figure 1b and Supplementary Fig 1d). In contrast, the lifetime of NLS-SYFP-tagged D2723H or W2725A mutants remained close to 3100 picoseconds, confirming that the mutations directly inhibit the BRCA2 DBD-DSS1 interaction (Figure 1b). Human BRCA2 normally localizes to the cell nucleus via tandem nuclear localization signals (NLSs) at its extreme C-terminus24. In contrast, full-length BRCA2 harboring the D2723H mutation mis-localized predominantly to the cytoplasm in transfected cells (Figure 1c) as previously shown25. Interestingly, BRCA2 W2725A was similarly mis-localized (Figure 1c), suggesting a correlation between the loss of DSS1 binding and cytoplasmic mis-localization.
DSS1 masks a Nuclear Exclusion Sequence in BRCA2
Neither the D2723H nor the W2725A mutations is predicted to alter the canonical NLSs positioned in exon 27 of BRCA224. This prompted us to survey BRCA2 for NESs using the NetNES algorithm, which employs neural networks to predict consensus motifs fitting the pattern ϕ-x(1-3)- ϕ-x(1-3)- ϕ -x-ϕ, where ϕ is a hydrophobic amino acid (M,F,I,V or L), and x is any amino acid 26. We detected 4 such potential NES motifs in the DSS1-binding region of BRCA2 (Supplementary Fig 2a). One motif (aa 2682-2698, encoded in human BRCA2 exon 18) shows strong evolutionary conservation, particularly of the critical hydrophobic residues (Figure 2a). This motif, when fused to SYFP was enough to induce nuclear exclusion, in contrast to a control BRCA2-derived peptide of similar size (Figure 2b), substantiating its function.
Figure 2. DSS1 regulates a functional NES in BRCA2.
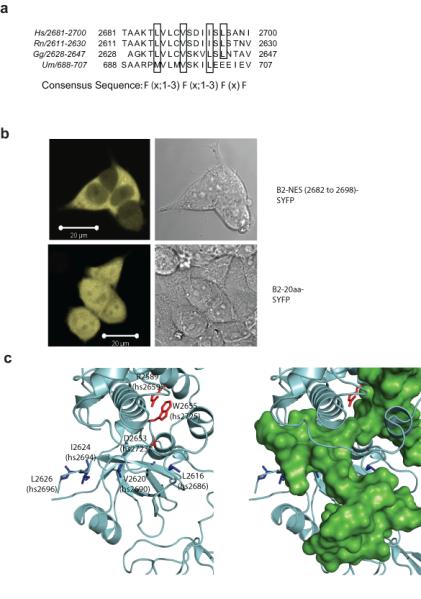
a) is a clustal W alignment of the nuclear exclusion signal within the DBD of BRCA2, with the critical hydrophobic residues that potentially contribute to the NES consensus marked by black boxes. b) is a representative fluorescent micrograph of 293T cells transfected with a BRCA2 NES-SYFP construct (upper panel) and SYFP tagged to a similarly sized sequence within BRCA2 (lower panel), which serves as a negative control. c) is a rendering of the 1YIJ structure of rnBRCA2 DBD. The DBD is shown in grey, and the critical hydrophobic NES residues highlighted in blue. Mutations that affect DSS1 binding are depicted in red. The right panel shows a superimposition of DSS1 (green).
The cytoplasmic mis-localization of BRCA2 mutants carrying alterations that affect DSS1 binding, but not intrinsic NLS or NES motifs, raises the possibility that the BRCA2-DSS1 interaction ‘masks’ an NES of BRCA2. Consistent with this idea, the structure of a BRCA2-DSS1 complex19 suggested that the residues critical for binding to exportins in the potential NES spanning human BRCA2 aa 2682-2698, are obscured by DSS1 (Figure 2c). Moreover, over-expression of DSS1 in cells expressing wildtype forms of either NLS-SYFP-DBD, or full-length BRCA2, enhanced nuclear localization, whereas the D2723H or W2725A mutants were not similarly affected (Supplementary Figs 2b-d). Together, these findings speak to a model wherein the binding of DSS1 to BRCA2 is essential for its nuclear retention, via the masking of an NES within the DBD.
BRCA2 masks a Nuclear Exclusion Sequence in RAD51
The RAD51 recombinase, a 37 kDa protein central to DNA repair by homologous recombination27, binds directly to the BRC repeats of BRCA215,28, an interaction that is essential for the efficient completion of recombination reactions10-14. Eight BRC repeats are present in human BRCA2, and the structural basis of their interaction with RAD51 has been characterized by crystallography as well as by structure-function analyses in mammalian cells28,29. Interestingly, a NetNES analysis of human RAD51 identified a putative NES spanning aa 245-260 (Figure 3a), with strong conservation across species (Figure 3b). This NES lies within a recently identified binding site for the BRC repeats of BRCA229, and an examination of the crystal structure reveals that key exportin-binding residues within this RAD51 NES are likely to be masked when bound to BRCA2 (Figure 3c). The RAD51 NES was sufficient to exclude YFP from the nucleus when fused to the fluorophore (Figure 3d). Mutation of the S208-A209 residues in RAD51 abolished its binding to the BRC repeats in BRCA2, while retaining its self-interaction30. This mutant localized to the cytoplasm when expressed in DT40 cells lacking endogenous Rad51 (Figure 3e). These observations not only confirm that the nuclear retention of RAD51 requires binding to BRCA2, but also corroborate that a RAD51 NES is masked by interaction with the BRC repeats of BRCA2.
Figure 3. BRCA2 regulates a functional NES in RAD51.
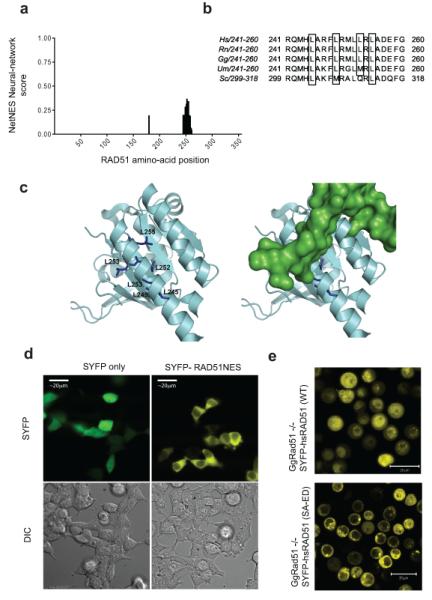
a) is a histogram of the NES scores for amino acids in the HsRAD51 sequence, as determined by the NetNES algorithm, showing the potential NES sequence in the C-terminus of the protein. b) is a sequence alignment of the residues comprising the RAD51 NES from yeast to human. The hydrophobic residues are marked by black boxes. c) is a rendering of the crystal structure 1N0W, in the same color scheme as Figure 2c. NES residues in RAD51 are shown in blue. The right panel shows a superimposition of the BRC repeat (green). d) shows representative micrographs of 293T cells transfected with a RAD51-NES tagged version of SYFP (right), in comparison to free SYFP alone. e) is a fluorescence micrograph of SYFP-tagged wildtype RAD51, compared with a mutant form lacking BRCA2 binding (SYFP-RAD51 (SA-ED)). SYFP-tagged proteins were expressed in Rad51−/−DT40 cells that conditionally express Tet-regulated untagged RAD51. Micrographs were taken 12h after the depletion of untagged RAD51 using doxycycline 30.
In vitro interaction of CRM1 with NESs in BRCA2 and RAD51
We have demonstrated here that putative NESs identified in the DSS1-binding domain of BRCA2and in the BRCA2-binding region of RAD51share the expected consensus characteristics, and can direct the nuclear exclusion of SYFP when fused to this heterologous protein. However, Leu residues in NES-like motifs frequently contribute to the buried hydrophobic cores of protein folds, and may therefore be inaccessible for interaction with the nuclear exportin CRM1 in their native context31,32.To address this issue, we tested the ability of GST-tagged forms of the NES-containing domains of BRCA2 or RAD51 to bind in vitro to recombinant CRM1 in the presence of an active GTP-bound Q69L form of the essential co-factor Ran, under the experimental conditions described previously33. We found (Figure 4a) that a GST-tagged recombinant protein encoding the DBD region of BRCA2 bound to recombinant CRM1 in the presence of Ran-GTP (lane 3), whereas GST alone did not (lane 1). Moreover, the addition of DSS1 inhibited this interaction (Figure 4a, lane 4) in a dose-dependent manner (Figure 4b). These results prompted us to test the effect of the D2723H mutation, which is expected to prevent DSS1 binding. Indeed, the D2723H mutant form of the BRCA2 DBD interacted constitutively with CRM1, whether or not DSS1 was present (Figure 4a, lanes 5 and 6). Thus, our findings provide biochemical evidence that the DBD region of BRCA2 contains authentic NES motifs capable of binding CRM1, that this interaction is lost when the NESs are ‘masked’ by the binding of DSS1, and that loss of this interaction in the D2723H mutant BRCA2 protein promotes constitutive CRM1 binding, consistent with the nuclear export and cytoplasmic mis-localization of the mutant protein.
Figure 4. CRM1 binding to NESs in BRCA2 or RAD51 is masked by DSS1 or BRC4 respectively.
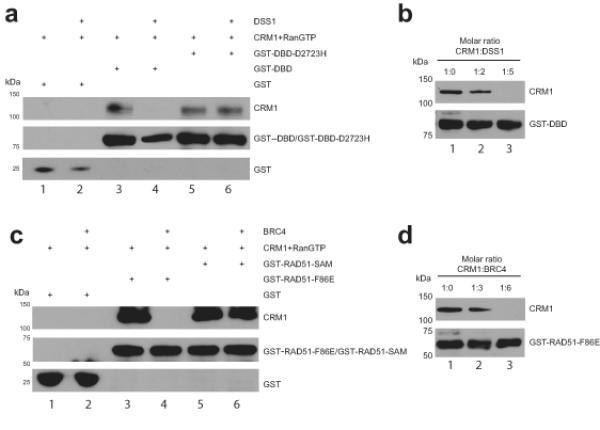
a) Immunoblots of GST pull-down assessing binding of CRM1 to GST-DBD and GST-DBD-D2723H immobilized on a glutathione sepharose matrix in the absence (lanes 3 and 5) or presence (lanes 4 and 6) of DSS1 at the 5-fold molar excess optimized from the representative titration shown in panel b). GST was used as a specificity control. DSS1 inhibits the binding of CRM1 to DBD (lane 4) but not to DBD-D2723H (lane 6). c) Immunoblots of GST pull-down assessing binding of CRM1 to GST-RAD51-F86E and GST-RAD51-SAM immobilized on a glutathione sepharose matrix in the absence (lanes 3 and 5) and presence of (lanes 4 and 6) of BRC4 peptide at the 6-fold molar excess optimized from the representative titration shown in panel d). GST was used as a specificity control. BRC4 peptide disrupts the binding of CRM1 to RAD51-F86E (lane 4) but not to RAD51-SAM (lane 6). Proteins were visualized with antibodies against His or GST. CRM1 protein was His-tagged. Uncropped blots are in Supplementary Fig 8.
The results in Figure 4c similarly provided biochemical evidence that RAD51 contains CRM1-binding NESs that are ‘masked’ by interaction with the BRC4 region of BRCA2. The F86E form of RAD51 which we have previously characterized28,30 was used in these experiments to enable its purification without the spontaneous in vitro aggregation that is typical of wildtype recombinant RAD5128,34. A GST-tagged version of this protein bound to CRM1 in the presence of Ran-GTP (Figure 4c, compare lanes 3 and 4). The addition of BRCA2 BRC4 prevented CRM1 binding (Figure 4c, lane 4) in a dose-dependent manner (Figure 4d). Notably, a mutant form of RAD51 (SAM208-210LEA, denoted RAD51-SAM), which we have previously shown28,30 to be incapable of BRC4 binding, interacted constitutively with CRM1 whether or not BRC4 was present (Figure 4d, lanes 5 and 6). Thus collectively, these in vitro results recapitulating exportin-cargo interactions using recombinant proteins provided strong biochemical evidence for a model wherein CRM1-dependent nuclear export – and its ‘masking’ by protein-protein interactions - is a critical determinant of BRCA2 and RAD51 localization.
Our attempt at en-bloc replacement of consensus hydrophobic residues within the proposed NESs (by Ala substitution of Leu2686, 2688, 2696/Ile 2694 in the BRCA2 stretch from aa 2682-2698, and Leu245, 249, 253, 255 in the RAD51 stretch from aa 245-260) proved beyond the scope of this work, because the BRCA2 and RAD51 domains encoding the putative NESs exhibited poor yield and solubility in bacterial expression. Moreover, further mutational analysis of contributions made by residues located outside these motifs is difficult because the exact length and nature of the NES sequences required for CRM1 binding are reported to be variable33,35 as are the potential binding modes between CRM1 and its various substrates36. Future structural work will be required to precisely define the residues contributing to NES activity.
A heterozygous D2723H mutation in BRCA2 mislocalizes RAD51
Our findings suggest that the nuclear retention of BRCA2 and RAD51 depends on NES-masking interactions between BRCA2 and DSS1, and BRCA2 and RAD51. To investigate whether these interactions are inter-dependent, we first tested the effect on RAD51 localization of BRCA2 depletion using RNAi (Figure 5a). We used an automated microscopic assay, wherein the nucleo-cytoplasmic difference is calculated by subtracting the amount of RAD51 in a cytoplasmic ring surrounding the nucleus from the amount of RAD51 in a circular area within the nuclear boundary (as defined by DNA staining) (Figure 5a, inset). We found that depletion of BRCA2 by RNA interference decreased the nuclear localization of RAD51 (Figure 5a and Supplementary Figs 3a-c), consistent with a role for BRCA2 in the nuclear retention of RAD51.
Figure 5. RAD51 mislocalization by BRCA2 depletion or mutation.
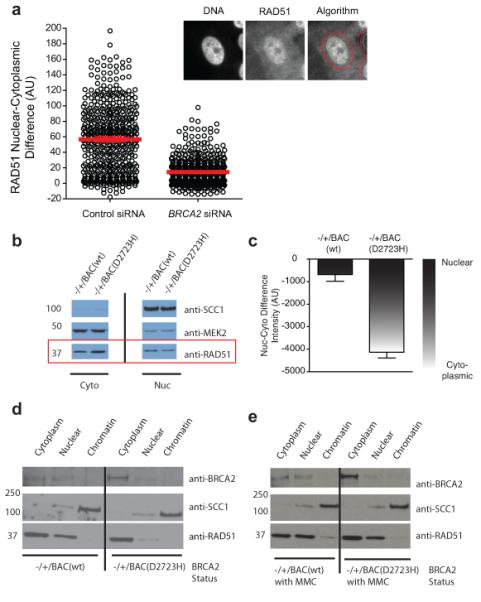
a) is a dot plot of the mean nucleo-cytoplasmic difference of RAD51 per cell, determined for a population of cells with and without exposure to BRCA2 siRNA. The figures in the inset represent the algorithm used for determining the nucleo-cytoplasmic difference by automated microscopy. The left image is of the nucleus as defined by DAPI, the middle image is that of RAD51 staining. The algorithm for nucleo-cytoplasmic intensity difference calculation (see Methods) is overlayed on RAD51 in the right panel. b) is a cell fractionation experiment from heterozygous mouse ES cells, with Western blotting for RAD51 to assess localization. MEK2 (predominantly cytoplasmic) and SCC1 (predominantly nuclear) serve as loading controls and controls for the efficiency of fractionation. c) is a bar graph quantitating the results from three independent fractionation experiments. The nucleo-cytoplasmic intensity difference for RAD51 in each experiment was obtained by generating densitometric profiles for each band (identical exposure) using ImageJ, and then subtracting the cytoplasmic value from the nuclear value. Lower (more negative) values indicate more cytoplasmic protein (n=3, error bars represent SEM). d) shows a cell fractionation experiment of ES cells carrying BACs expressing either the WT or D2723H mutant of BRCA2, in the absence or presence (E) of DNA damage induced by exposure to 100 ng/ml of MMC for 20 hrs. SCC1 is used as a control for fractionation. One experiment representative of 3 independent repeats is shown. Uncropped blots are in Supplementary Fig 8.
Since humans who inherit the D2723H mutation on a single allele of BRCA2 are predisposed to cancer, this raises the possibility that the mutant protein exerts a trans-dominant effect on RAD51 localization when in the heterozygous state. Accordingly, we examined RAD51 localization in murine embryonic stem (ES) cells engineered to be heterozygous for the human BRCA2 D2723H mutant, in comparison to an appropriate WT control in an identical genetic background20. In this system, one allele of murine (mm) Brca2 was disrupted by gene targeting, and the cells complemented with a bacterial artificial chromosome (BAC) containing a segment of human chromosome 13, encoding human (hs)BRCA2. The BAC is reported to drive near-endogenous expression of BRCA2, and suffices to replace mmBrca2 function in these cells, evidenced by rescue of lethality when the second endogenous mmBrca2 allele is also disrupted20. The total levels of BRCA2 and RAD51 expression are comparable in ES cells expressing either wildtype or D2723H forms of BRCA2 (Supplementary Figs 4a-b). We examined the distribution of RAD51 in these cells by biochemical fractionation and quantitative western blotting (Figure 5b), due to technical difficulties in performing accurate microscopic analyses of protein localization in the ES cells.. We observed an increase in cytoplasmic RAD51 in cells carrying a single allele of D2723H BRCA2, with a corresponding decrease in nuclear levels of the protein (Figure 5b-c), whether in the absence (Figure 5d) or presence (Figure 5e) of DNA damage induced by mitomycin C (MMC). Mutant D2723H BRCA2 expressed from the BAC in these cells was also predominantly cytoplasmic, unlike WT BRCA220 (Figure 5d-e). Together, these findings suggest a mechanism underlying the deleterious effects of the BRCA2 D2723H mutant when present in the heterozygous state co-expressed with wildtype BRCA2.
BRCA2 and DSS1 promote RAD51 nuclear enrichment after DNA damage
There is evidence that RAD51 is enriched in the nucleus during the response to genotoxic lesions repaired by homologous recombination37,38. We characterized this further by analyzing the nucleo-cytoplasmic distribution of RAD51 following DNA damage in human cells using automated microscopy. Exposure to the DNA damaging agents etoposide and MMC caused a significant increase in the relative amount of nuclear RAD51, as evidenced by changes in the nuclear-cytoplasmic difference of average pixel intensities within the respective regions (Figure 6a, and Supplementary Fig S5). Moreover, genotoxins (eg., MMC, carboplatin, the PARP inhibitor Ku59848, hydroxyurea or aphidicolin) generating DNA lesions known to engage pathways for repair by HR enhanced the nuclear enrichment of RAD51 to a greater degree than other genotoxins (Figure 6b). Depletion of the damage response kinases39 ataxia-telangiectasia mutated (ATM), ATM-and-Rad53-like (ATR), or CHK1 diminished the nuclear enrichment of RAD51 following MMC exposure, confirming that this response depends on the cellular pathways that sense and signal DNA damage (Figure 6c, Supplementary Fig S6). Notably, depletion of BRCA2 or DSS1 also affected the nuclear enrichment of RAD51 after genotoxic stress (Figure 6d, Supplementary Fig S6). Finally, using Flag-BRCA2 complemented EUFA-423 cells (Supplementary Fig S7), we showed that DNA damage also increases the levels of nuclear BRCA2, as measured with the Cellomics ArrayScan (Supplementary Fig S7c), and that the depletion of DSS1 causes a decrease in nuclear Flag-BRCA2 (Supplementary Fig S7d), Collectively, these results suggest that the enhanced nuclear distribution of RAD51 is a DNA damage response dependent on BRCA2 and DSS1, which may be compromised by cancer-associated mutations that disrupt the DSS1-BRCA2 interaction.
Figure 6. RAD51 nuclear enrichment is a BRCA2-dependent DNA damage response.
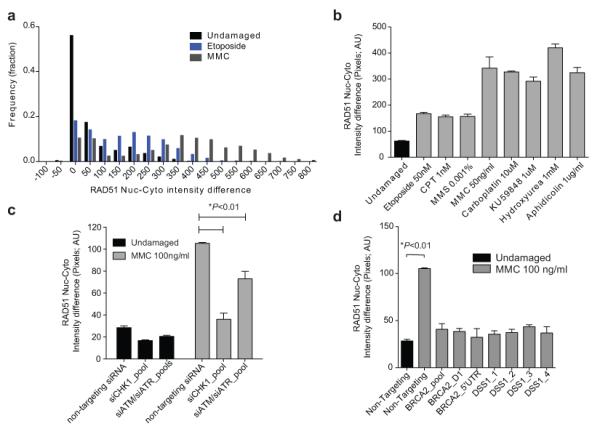
a) represents the frequency distribution of the RAD51 nuclear-cytoplasmic pixel intensity difference (NC-difference) from different populations of MCF10A cells analyzed by automated immunofluorescence microscopy. Results for untreated cells are in black, and for cells treated with etoposide or MMC in blue or grey respectively (n=2000 each). b) shows the mean NC-difference for RAD51 in MCF10A cells treated with the indicated forms of DNA damage. Each bar represents the mean ± standard error of 4 wells in a 96-well plate, with 500 cells counted per well. c) shows the mean NC-difference for RAD51 in MCF10A cells pre-treated with the indicated siRNAs, with (grey) or without (black) exposure to MMC. The increase in the NC-difference after DNA damage is significantly affected (Student’s two tailed t-test, p<0.05, n=500 cells, 3 wells) by depletion of ATM, ATR or CHK1. d) shows the RAD51 NC-difference in MCF10A cells following knockdown of BRCA2 and DSS1 using multiple independent siRNA’s. The black bar on the left represents undamaged cells. The nuclear enrichment seen after DNA damage is significantly affected (p<0.01, Student’s two tailed t-test, n=500 cells, 3 wells) by depletion of both BRCA2 and DSS1. Supplementary Fig 6a-d shows the depletion efficiency with the siRNAs used.
Discussion
The findings we report here suggest a new mechanism controlling the intracellular distribution of the RAD51 recombinase (Figure 7), wherein NESs present in BRCA2 and its cargo RAD51 must be masked by complex formation between BRCA2, DSS1 and RAD51 in order to permit nuclear localization. The NES-masking mechanism suggested by our findings reveals an additional layer of regulation not recognized in earlier work reporting that RAD51 enters the nucleus only when bound to a partner, RAD51C38, which contains an NLS.
Figure 7. A hypothetical model for BRCA2 and RAD51 nuclear localization through masking of nuclear export sequences.
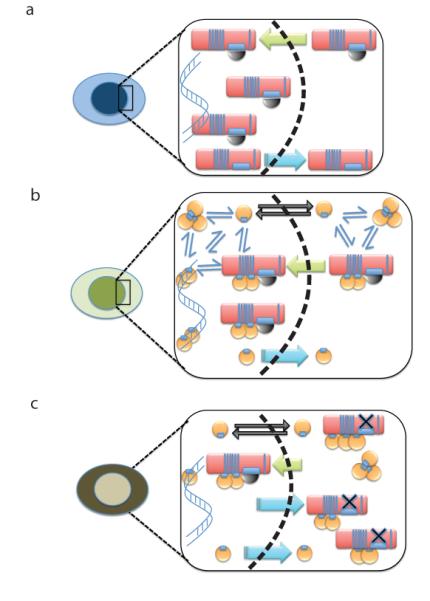
a) depicts the proposed mechanism wherein the nuclear retention of BRCA2 (red oblong) is allowed when its binding to DSS1 (black semi-circle) obscures an NES motif. Nuclear transport (green arrow) is presumably directed by NLSs previously identified in the C-terminal region of BRCA2, which are not shown. Dissociation of BRCA2 from DSS1 may permit nuclear export (blue arrow). b) depicts the proposed mechanism wherein the nuclear retention of RAD51 (yellow circle) is allowed when its binding to the BRC repeats of BRCA2 obscures an NES motif. Nuclear transport (green arrow) of the BRCA2-RAD51 complex is presumably directed by the NLSs in BRCA2. In addition, cytosolic RAD51 may exist in an equilibrium (thin blue arrows) between free monomers and oligomers. Momomeric RAD51 is small enough to diffuse freely across the nuclear membrane (black arrows), whereas oligomers are not. Dissociation of RAD51 from BRCA2 may permit nuclear export (blue arrow). The net result of these different processes is to localize RAD51 predominantly in the nucleus. c) depicts how the processes depicted in the preceding panels may be affected by the BRCA2 D2723H mutation, which prevents DSS1 binding (black X). Mutant BRCA2 is exported from the nucleus (blue arrows) resulting in predominant cytoplasmic localization, and this is proposed to shift the balance towards cytoplasmic localization of RAD51, despite the presence of wildtype BRCA2.
The location of the proposed NESs near the hydrophobic protein cores (Figures 2c and 3c) raises the possibility that these motifs are exposed for CRM1 binding by unfolding in the absence of the masking partner protein, suggesting a mechanism to ensure that only functional BRCA2-DSS1-RAD51 complexes are retained in the nucleus. Moreover, RAD51 oligomerization is also likely to regulate its nuclear localization. Both in vitro and in vivo30,34, an equilibrium is believed to exist between RAD51 monomers and oligomers of varying stoichiometry, whose abundance is closely regulated by interaction with factors like RAD52 or the BRC repeats of BRCA240. Although RAD51 monomers (37kDa) may pass freely into the nucleus, RAD51 oligomers (≥74kDa) are unlikely to equilibrate passively across nuclear pores, which generally accommodate substrates ≤60kDa (reviewed in41). There is structural28, biochemical34 and cell biological28,30 evidence that RAD51 oligomerization is suppressed by its interaction with the BRC repeats of BRCA2. This may help create a pool of monomeric RAD51 in the cytoplasm for transport into the nucleus through free nuclear pore entry, or for binding to partner proteins (including RAD51C) that contain NLSs. The RAD51 monomer-oligomer equilibrium could also exist within the nucleus, but is likely to be biased towards DNA-bound oligomers, or monomers in which BRC binding obscures the NES, thus promoting nuclear retention.
Depletion of DSS1, which not only binds to BRCA2 but is also a component of the 19S proteasome42 and RNA processing complexes43, impairs the assembly of RAD51 into foci at sites of DNA damage44 to mediate homologous DNA recombination (HR). This has previously been ascribed to a direct function of the BRCA2-DSS1 complex in binding to ssDNA via the formation of OB folds19, which may not only localize RAD51 to these substrates but also displace the ssDNA-binding protein RPA11. Our findings suggest an additional function for DSS1 in HR, through regulation of the nuclear localization of BRCA2 and indirectly, RAD51. In this light, it is notable that mutant forms of BRCA2 in which the DSS1 binding domain has been entirely deleted, are still capable of supporting HR45-47. Moreover, DSS1 is apparently required for HR only when the DBD is present48. These observations are consistent with our findings: DBD deletion presumably removes the strong NESs identified in this work, obviating the necessity for their ‘masking’ by DSS1, and allowing mutant forms of BRCA2 lacking the DBD to at least partly function in the nucleus even without DSS1.
We surmise that DBD deletion and loss of DSS1 binding in human BRCA2 may lead to subtle defects in HR, reminiscent of the phenotypes observed in mutants of the BRCA2 homologue Brh2 in the fungus U. maydis that lack DSS1 binding49. On the other hand, the NESs we have identified in human BRCA2 are not well conserved in Brh2 (Figure 2a), raising the possibility that the mechanisms which regulate BRCA2 nuclear localization are not directly comparable between these species. However, U. maydis RAD51 contains a conserved NES (Figure 3a), suggesting its localization may depend on the BRC repeat found in Brh2, whereas S. cerevesiae RAD51 does not (Figure 3a), consistent with the absence of a BRCA2 homologue in yeast.
Several lines of evidence suggest that the DSS1-masked NES we have identified in BRCA2 is dominant in its function over other motifs within the protein that may control nuclear localization24,50. For instance, both the D2723H and the W2725A point mutants of BRCA2, which lack DSS1 binding to mask the NES, mis-localize to the cytosol, and the D2723H variant is embryonic lethal in mice when homozygous20, suggesting it is non-functional. Moreover, our findings suggest that these point mutations are unlikely to exert their effects simply through BRCA2 destabilization, because protein expression is not markedly decreased. Instead, they speak to a critical role for the DSS1-BRCA2 interaction in the control of BRCA2-RAD51 localization. These findings raise questions regarding the expected clinical effects of the cancer-associated BRCA2D2723H allele when expressed in the heterozygous state in germline mutation carriers. Although our findings suggest that BRCA2D2723H heterozygosity may suffice to mislocalize RAD51, we are unaware that carriers of this mutation exhibit radiosensitivity or other clinical features associated with profound defects in DNA repair, consistent with the lack of radiosensitivity of D2723H cell lines in vitro (Supplementary Fig 4c). However, patients heterozygous for BRCA2D2723H do appear to be at risk of developing early-onset cancers, suggesting that this alteration causes a cumulative rather than acute effect on genome stability, which acts over years to promote carcinogenesis. Indeed, BRCA2 is often cytoplasmic in cancer cells24, and mislocalization of RAD51 has been reported in both familial breast cancer51 and sporadic cases of prostate cancer52.
Notably, our findings also indicate that alterations in the normal intracellular distribution of RAD51 may mark defects in the cellular response to DNA damage arising at several distinct steps (Figure 6c-d). We therefore suggest that quantitative measurement of the nucleo-cytoplasmic levels of RAD51 may serve as a surrogate marker for these defects, which may be of value in assessments of cancer diagnosis or therapeutic responsiveness.
OnlineMethods
Antibodies and siRNA
Antibodies used in this work were:
| Antibody | Application and dilution | Comments | |
|---|---|---|---|
| 1 | anti-RAD51 [14B4] (Genetex) |
WB (1:2000) | |
| 2 | Anti-RAD51 B01P (Abnova) |
IF (1:2000) | |
| 3 | anti-RAD51 (Ab1; Calbiochem) |
IF (1:1000) | Polyclonal serum (batches prior to 2010) |
| 4 | anti-Flag (M2;Sigma F1804) |
WB (1:3000), IF (1:2000) | |
| 5 | anti-DSS1 (goat polyclonal; Abcam, Ab5649) |
WB (1:1000), IF (1:500) | |
| 6 | anti-GFP Clontech 632460 |
WB 1: 2000 | |
| 7 | anti-GFP Clontech 632381 |
IP (1ul per mg of lysate) | Polyclonal serum, suitable only for IP |
| 8 | anti-GFP (MBL-598) |
IF (1:1000) | |
| 9 | anti-SCC1 (rabbit polyclonal –abcam Ab992 |
WB (1:2000) | |
| 10 | anti-MEK2(BD Transduction LaboratoriesCat.No. 610236) |
WB 1:3000 | |
| 11 | Anti BRCA2 (Ab1/OP95, Merck) |
WB 1:500 | Human BRCA2 |
| 12 | Anti BRCA2 H-300 (Santa Cruz), |
WB 1:500 | Pan-species |
siRNA sequences targeting the following mRNAs were:
| BRCA2 (Dharmacon siGenome) | GAAACGGACUUGCUAUUUA GUAAAGAAAUGCAGAAUUC GGUAUCAGAUGCUUCAUUA GAAGAAUGCAGGUUUAAUA* (* also used as single siRNA) |
| BRCA2 (Qiagen, 5′UTR) | TTGGAGGAATATCGTAGGTAA |
| Chk1 (Dharmacon siGenome) | GCAACAGUAUUUCGGUAUA GGACUUCUCUCCAGUAAAC AAAGAUAGAUGGUACAACA CCACAUGUCCUGAUCAUAU |
| ATM (Dharmacon siGenome) |
GCAAAGCCCUAGUAACAUA GGGCAUUACGGGUGUUGAA UCGCUUAGCAGGAGGUGUA UGAUGAAGAGAGACGGAAU |
| ATR (Dharmacon siGenome) | GAACAACACUGCUGGUUUG GCAACUCGCCUAACAGAUA UCUCAGAAGUCAACCGAUU GAAUUGUGUUGCAGAGCUU |
| DSS1 (Eurofins MWG Operon) | GCAGCCGGUAGACUUAGGU GAGUUCCCUGCCGAAGACU UGUAGAGGAUGACUUCUCU GAUGAAGAUGCACAUGUCU |
Plasmids and Cloning
NLS-SYFP-DBD
The BRCA2 DBD was cloned between nt7382 and 8926, to generate fragments spanning the residues from 2461 to 2975 in HsBRCA2, using the primers:
FOR 5′- AAGCCCAGAAAGGGTGCTTCTTCAAC
REV 5′- GCACCCTTTCTGGGCTTAGGCATC
These fragments were cloned into the vectors pEF-Myc-Nuc, and pEGFP-C1-NotI (generated previously in our lab to contain an extra Not1 site in the MCS). The EGFP-DBD plasmid was then digested to release the EGFP tag, which was replaced with an NLS-containing version of SYFP for nuclear localization, using the primers
FOR (including NLS tag)
5′-GGCCGCTAGCATGGATCCAAAAAAGAAGAGAAAGGTAGATCCAAAAA AGAAGAGAAAGGTAGATCCAAAAAAGAAGAGAAAGGTAGTGAGCAAGG GCGAGGAGCTGTTCA-3′
REV 5′- GAATCTCGAGTCTTGTACAGCTCGTCCATGCCGAGAGT- 3′
from the plasmid pSYFP2-C1 (kind gift of Dr. Theodorus Gadella, Amsterdam, Netherlands)
The W2725A and D2723H mutation for disruption of DSS1 binding was introduced into these plasmids using the QuickChange mutagenesis kit (Stratagene) and the primer NdeI-W2725A, and NcoI-D2723H.
5′- TTGAACTTACAGATGGGGCATATGCTGTTAAGGCCCAGTTAG- 3′
5′- GCCATTATTGAACTTACCCATGGGTGGTATGCTGTTAAGG- 3′
Mutations were generated with the Quikchange XL site-directed mutagenesis kit (Stratagene) according to the manufacturer’s instructions, with the exception that the digestion of the parental DNA template with DpnI was for 4 h.
Cloning of the RAD 51 Nuclear Exclusion signal
Rad 51 F
GATCCAGGCAGATGCACTTGGCCAGGTTTCTGCGGATGCTTCTGCGACTCGCTGATGAGTTTGGTTGAG
RAD51 R
AATTCTCAACCAAACTCATCAGCGAGTCGCAGAAGCATCCGCAGAAACCTGGCCAAGTGCATCTGCCTG
pSYFP-C1 was digested with BglI and EcoRI, before ligation with the annealed product of RAD51 F & R primers. The annealed primers spanned aa 241-260 of human RAD51 and resulted in in-frame fusion with SYFP.
Cloning of the BRCA2 NES
BRCA2 NES-F
GATCCGCAAAAACACTTGTTCTCTGTGTTTCTGACATAATTTCATTGAGCGCAAATATATGAG
BRCA2 NES-R
AATTCTCATATATTTGCGCTCAATGAAATTATGTCAGAAACACAGAGAACAAGTGTTTTTGCG
BRCA2 Control-F
GATCCGTTAAGGCCCAGTTAGATCCTCCCCTCTTAGCTGTCTTAAAGAATGGCAGACTGACAGTTGGTCAGTGAG
BRCA2 Control-R
AATTCTCACTGACCAACTGTCAGTCTGCCATTCTTTAAGACAGCTAAGAGGGGAGGATCTAACTGGGCCTTAACG
pSYFP-C1 was digested with BglI and EcoRI and ligated with the annealed product of the primers described above to yield the SYFP-NES and SYFP-control sequences.
Flag-BRCA2 and Flag-BRCA2 D2723H
The pCAG-FLAG-BRCA2-IRES-Neo plasmid was generated in a 2-step procedure. p3xFLAG-BRCA21 was modified by introducing IRES fragment from pIRES-Neo vector (Clontech) to generate a FLAG-tagged BRCA2 and Neomycin resistant gene in a single transcript. Then, the 3xFLAG -BRCA2 - IRES-Neo fragment was inserted to the pCAG-GFP vector (Addgene) and the GFP and SV40 early promoter regions were removed to obtain the stable expression of the Flag-BRCA2 protein in SV40 transformed cells. The D2723H mutation was obtained using the quickchange mutagenesis kit and the primer (NcoI-D2723H):
5′- GCCATTATTGAACTTACCCATGGGTGGTATGCTGTTAAGG- 3′
Flag-DSS1 and Cherry-DSS1
To make the Flag-DSS1 construct, full-length (70 amino acids) hDSS1 sequences were cloned into HindIII and BamHI sites of 3xFlag CMV10 vector (Sigma). The primers used are:
For 5′-GTCAAGCTTTCAGAGAAAAAGCAGCCGGTAGAC-3′
Rev 5′-GGCGGATCCCTATGAAGTCTCCATCTTATAAC-3′
To make the mCherry-DSS1 construct, full-length hDSS1 sequences were cloned between BstB1 and BamHI sites, excising RAD51 from a previously described2 mCherry-RAD51 vector. The primers used were:
For 5′-GCGTTCGAATATGTCAGAGAAAAAGCAGCCGGTAG-3′
Rev 5′-CGCGGATCCCTATGAAGTCTCCATCTTATAAC-3′
SYFP-RAD51, SAED
To make the SYFP-RAD51 and SYFP-RAD51-SAED constructs, full-length SYFP sequences (from pSYFP-C1) were cloned into AgeI and SacI sites, excising EGFP from EGFP-RAD51 and EGFP-RAD51 SAED (previously described). The primers used are:
For 5′ ATACCGGTATGGTGAGCAAGGGCGAGGAGCTGTT 3′
Rev 5′ ATGAGCTCGCTTGTACAGCTCGTCCATGCCGAGAGT 3′
GST-DBD/RAD51
To make GST-DBD construct for in vitro binding assay, BRCA2 DBD (from amino acid residue 2461 to 2975 in hsBRCA2) was PCR-amplified from the NLS-SYFP-DBD construct and cloned into SalI and NotI sites of the vector pGEX-4T3 (GE Healthcare). For GST-DBD D2723H, the same region was PCR-amplified from the NLS-SYFP-DBD D2723H construct.
The primers used are:
For 5′-GCGTCGACAACTCCAATCAAGCAGCAGCT-3′
Rev 5′-GCGCGGCCGCTCATACAATACGCAACTTCCACAC-3′
To make GST-RAD51 F86E and GST-RAD51 SAM constructs, the respective RAD51 mutant sequences were PCR-amplified from GFP-RAD51 F86E and GFP-RAD51 SAM constructs3 and cloned into SalI and NotI sites of the vector pGEX-4T3 using the following primers.
For 5′-GCGTCGACATGGCAATGCAGATGCAGCTT-3′
Rev 5′-GCGCGGCCGCTCAGTCTTTGGCATCTCCCACT-3′
His-DSS1
To make His-DSS1 construct, full-length hDSS1 sequences were cloned into NdeI and BamHI sites of modified pET28a Vector (Novagene). The primers used were:
For 5′-GCGCCATATGTCAGAGAAAAAGCAGCCG-3′
Rev 5′-GCGCGGATCCCTATGAAGTCTCCATCTTATAACC-3′
hCRM1 and hRanQ69L constructs for bacterial overexpression are gifts from Dr Dirk Gorlich4,5 (MPI, Gottingen)
BRC4 peptides were synthesized by Cambridge Research Biochemicals Ltd with an N-terminal biotin moiety attached through a 6-aminohexanoic acid spacer and a C-terminal amide. The sequence of the peptide is:
KEPTLLGFHT ASGKKVKIAK ESLDKVKNLF DEKEQ
Cell culture and transfection
293T and U2OS were cultured in sterile-filtered growth medium (Dulbecco’s Modified Eagle Medium (D-MEM) with GlutaMAX-1, 4500 mg/L D-glucose, sodium pyruvate and pyridoxine (Gibco), supplemented with 10% fetal calf serum (FCS) (Gibco) and 2% penicillin streptomycin (Gibco)) in a 37°C humidified incubator in the presence of 5% CO2. Cells were split every 2-3 days and maintained at 50% confluency. MCF10A cells were grown in D-MEM/F12 (1:1) with 15mM HEPES buffer (Gibco) supplemented with 5% horse serum (Invitrogen), 10 μg/ml insulin (Sigma), 20 ng/ml EGF (Sigma), 100 ng/ml cholera toxin (Sigma), 500 ng/ml hydrocortisone (Sigma) and 1% penicillin streptomycin. ES cells were grown in gelatinized plates with GMEM medium (Sigma) supplemented with 15% FBS (Hyclone), 1mM sodium pyruvate, 1x non-essential amino acids (Invitrogen) 1xglutamate-penicillin-Streptomycin, (Invitrogen), 100uM β-mercaptoethanol and 1000 units per ml of Leukemia inhibitory factor (ESGRO Chemicon).
Transient transfection of DBD plasmids was performed by lipofection using lipfectamine 2000 (Invitrogen) using 1μg DNA and 3μl Lipofectamine in OptiMEM (Invitrogen) for 106 cells. For high content microscopy, U2OS cells were plated in a 24-well plate. One day post plating cells were transfected with NLS-SYFP-DBD construct, WT or D2723H mutant (0.25μg), together with an empty mCherry plasmid or mCherry-DSS1 plasmid (0.125μg), using lipofectamine. Full length BRCA2 was introduced into U2OS cells using Amaxa Cell Line Nucleofector Kit V and Nucleofector I Device with the Program X-01 (Lonza). For high-content microscopy experiments, MCF10A cells were reverse transfected with indicated siRNAs with final concentration of 25nM using Dharmafect I transfection reagent (Fermentas GmbH, Thermo Fisher Scientific), in a 96-well plate. EUFA423BRCA2 (EUFA-BRCA2#34) cell lines were established by stably transfecting EUFA423 cells (SV40 immortalized BRCA2 deficient human fibroblasts) with the pCAG-FLAG-BRCA2-IRES-Neo construct in a standard DMEM 10% FBS with P/S supplemented with 750 μg/ml G418.
Cell viability assay
Cells were plated into 96-well plates at a density of 8000 cells per well. Different doses of mitomycin C (Sigma) were added (4 wells per dose/cell line), and the plates were incubated at 37°C for five doubling times. CellTiter-Blue reagents (Promega) were added to each well of the 96-well plate according to the manufacturer’s guidelines. The plates were incubated at 37°C for approximately 1-2 hour in a humidified 5% CO2 atmosphere. Number of viable cells was determined using the Fusion plate readerat A590 nm. A similar procedure was followed for the cell viability assay on ES cells exposed to different doses of ionizing radiation using a Faxitron X-ray unit.
Lifetime measurement
DBD and DSS1 transfected cells were plated on MatTek poly-d-lysine coated, No.1 glass bottomed 35mm dishes and media replaced with phenol red free L15. Experiments were performed on a Time-Correlated Single Photon Counting (TCSPC) system (Becker and Hickl Inc., SPC 830) connected to the de-scanned and fibre-coupled output of an Olympus FV300 confocal scanner (Laser Analytics Group, Department of Chemical Engineering). A supercontinuum laser (Fianium Inc, SC400) was used as the illumination source. An excitation wavelength of 436nm (+/− 1nm) was selected by an Acousto Optic Tunable Filter (AOTF) and coupled into the confocal scanner. The fluorescence output was filtered between 480nm and 550nm before being detected by the TCSPC photomultiplier tube. TCSPC data were analyzed to generate lifetime information on dedicated Matlab (Mathworks, inc) and Becker and Hickl software by Simon Schlachter (Cambridge, UK).
Immunofluorescence analysis
Cell staining and immunofluorescence analyses were carried out as described previously6. Briefly, transfected and untransfected cells were grown on coverslips and subjected to different treatments as indicated in the text prior to fixation. Cells for IF experiments were grown on coverslips to 70–80% confluence, washed in PBS and fixed in 4% paraformaldehyde or 95% Ethanol+5% Acetic acid. The cells were permeablized by adding TBS with 0.1% Triton and 0.2%Tween for 5 min, blocked in TBS-Triton-Tween + 2% BSA) and incubated with the primary antibody and secondary antibody in a humidified chamber with TBS-Tween-Triton washes in between. The coverslip is then mounted onto a slide for viewing using a mounting medium with DAPI. Immunofluorescence images were captured using a Zeiss Axiovert200 LSM510meta confocal microscope using a 40x objective with fixed optical slice, laser power and detector/amplifier settings for all samples across each individual experiment to allow comparison.
High content microscopy for adherent cells
High content microscopy experiments were performed on the Cellomics VTI-Arrayscan Instrument (Thermofisher) as described previously6; 96 well plates (Nunc) with cells seeded at 3000/well, or in 24 well plates. Transfections were performed using 0.25μg or 0.125μg of DNA and lipofectamine. The cells were treated with specified DNA damaging agents, fixed with 4% formaldehyde for 10 min, immunostained as per conventional protocols, and incubated with 0.1% Triton X-100 with Hoechst 33342 DNA dye for 10 min, followed by PBS washes. Cellomics VTI ArrayScan was used for image acquisition using the Compartmental Analysis BioApplication, using a 40x non-immersion objective. Hoechst staining was used for object identification and the average intensity of fluorescent signal per nucleus was estimated using the target activation Cellomics bio-application. 300-500 cells were analyzed per well, and standard errors calculated from an average of the means of multiple wells.
Immunoprecipitation
Whole cell extracts were prepared from 15×106 293T cells 24 hours after transfection. The trypsinized or scraped cells were spun down in a Beckman Centrifuge at 1000 rpm for 3 minutes, washed once with PBS, and resuspended in 1ml of ice-cold immunoprecipitation lysis buffer (50mM HEPES, pH7.4, 100mM NaCl, 0.5% Nonidet P-40 (NP-40), 10mM EDTA, 2mM β-glycerophosphate, 1mM DTT, 1mM PMSF supplemented with protease inhibitors (Roche)) for 15 minutes on ice. Extracts were then spun down in an Eppendorf centrifuge for 15 minutes at 12,000 rpm and the supernatant was collected and quantified for protein concentration by the biocinchoninic acid (BCA) assay (Sigma). 1mg of whole cell extract was precleared with 30μl of a 50% slurry of protein A-Sepharose (Sigma) for 20 minutes at 4°C with gentle shaking to reduce non-specific protein-bead interactions, followed by immunoprecipitation using 1-2μg of antibody and 30μl of a 50% slurry of protein A-Sepharose overnight in the cold room with gentle shaking. The immune complexes were then washed 4 times in ice-cold immunoprecipitation lysis buffer and 1 time in PBS before resuspension in 4xloading buffer (Invitrogen) and 50mM DTT.
In-vitro pull-down assays
The in-vitro CRM1 binding assays were performed using an assay described previously33. For each assay condition, 1 μg of GST tagged protein (DBD or DBD-D2723H or RAD51-F86E or RAD51-SAM) bound to glutathione-separose 4B beads was incubated with 1 μM CRM1/RanGTP for 1 h at 4°C in 500 μL of binding buffer (50 mM Hepes (pH7.5), 150 mM NaCl, 25 mM KCl, 50 mM LiCl, 2 mM Mg(OAc)2, 5 mM DTT and 20 μM GTPγS (a non-hydrolysable GTP analogue). DSS1 proteins or BRC4 peptides (or binding buffer alone for GST control) were then added to individual tubes as indicated, followed by incubation at 4°C for 2 h. Beads were washed three times with 500 μL of binding buffer, followed by elution of proteins and SDS-PAGE electrophoresis. DBD, DBD-D2723H, RAD51-F86E and RAD51-SAM were detected by immunoblotting with anti-GST antibody and CRM1 was detected by immunoblotting with anti-His antibody.
Cell Fractionation
Cells were washed in PBS and resuspended in solution A (10 mM Hepes [pH 7.9], 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM DTT, protease and phosphatase inhibitors). Cells were incubated on ice for 5 min, and the cytoplasmic (S1) and nuclear fractions (P1) were harvested by centrifugation at 1,300 × g for 4 min. Isolated nuclei were then washed in solution A, lysed in solution B (0.1% Triton X-100, 3 mM EDTA, 0.2 mM EGTA, 1 mM DTT, protease and phosphatase inhibitors), and incubated on ice for 10 min. The soluble nuclear (S3) and chromatin fractions were harvested by centrifugation at 1,700 × g for 4 min. To release chromatin-bound proteins by nuclease treatment, the P2 fraction was incubated in buffer A plus 1 mM CaCl2 and 5 U micrococcal nuclease for 15 min at 37°C, after which the reaction was stopped by the addition of 1 mM EGTA and spun down at 10,000 × g to collect the supernatant enriched for chromatin proteins (P3). The S3 and P3 fractions were pooled to obtain a nuclear extract.
Proteins were resolved on 3-8% Tris-Acetate SDS-PAGE (Invitrogen) for BRCA2 experiments, 4-12% MES SDS-PAGE (Invitrogen) for most other experiments, and 4-20% Tris Glycine gels for DSS1 experiments. Transfer was performed to PVDF membranes (Immobilon-P, Millipore) at 30V for 2hours. Western blots were blocked in 5% milk (Marvel) and 0.5% Tween-20 in TBS for 30min at room temperature before probing with the appropriate primary antibodies. Western blots were developed with HRP-conjugated secondary antibodies against rabbit and mouse at 1/15000 and 1/10000 respectively, and detected with either ECL or ECL Plus detection reagents (Amersham) and exposed to CL-Xposure™ Film (Pierce).
Quantitative RT-PCR
| Gene | Sequence |
|---|---|
| GAPDH | FW: -TGAGCTTGACAAAGTGGTCG; REV: –GTCAGTGGTGGACCTGACCT; |
| CHK1 | FW: -CCAGATGCTCAGAGATTCTTCCA; REV: - TGTTCAACAAACGCTCACGATTA; |
| ATM | FW:–GGCTATTCAGTGTGCGAGACA; REV: –TGGCTCCTTTCGGATGATGGA; |
| ATR | FW:-TCCCTTGAATACAGTGGCCTA; REV: -TCCTTGAAAGTACGGCAGTTC. |
| DSS1 | FW:-GAAAAAGCAGCCGGTAGACTT, REV: ATCCCAATTATCCTCCCAGACA |
| BRCA2 | FW:-TTGGCTGATGGTGGATGGCTCATA, REV: TTTGGATCCACACCTGGAGTGTCA |
To analyse the effects of siRNA knockdown, 0.5 μg of total RNA, extracted with Rneasy Plus Mini Kit (Qiagen), was used for cDNA synthesis using Cloned AMV First-Strand Syntehesis kit (Invitrogen). Q-PCR was performed using the LightCycler 480 SYBR Green I Master mix (Roche) with the primers listed above. The Ct values were obtained from the LightCycler 480 and referenced to the expression of the GAPDH housekeeping gene. All samples were then normalized to Non-Targeting siRNA pool (Fermentas GmbH).
For the RT-PCR comparison between MmBrca2 and HsBRCA2, the following primers were used:
| GAPDH Primers | |
| F | GAAGGTCGGTGTGAACGGATTT |
| R | CATTTGATGTTAGTGGGGTCTC |
| Mouse Brca2 primers | |
| 5022F | TTATGCAGGAATCTTTGGACA |
| 5173R | AGTTCTGCATTTCTTCACATTTT |
| Human BRCA2 primers | |
| 731F | AAAGTTTGTGAAGGGTCGTC |
| 840R | TAAGGGTGGGTGGTGTAGC |
RNA made from exponentially growing ES cells using Qiagen total RNA kit. 2ug RNA was converted into cDNA using Roche Transcriptor High Fidelity cDNA synthesis Kit, and Real time PCR performed as described above. The Cp values were directly represented in the figure to provide an unbiased assessment, using GAPDH as a control.
Original images of blots used in this study can be found in Supplementary Figure 8.
Supplementary Material
Acknowledgements
We acknowledge the generous gifts of BRCA2 WT and D2723H ES cells from S Sharan (NIH, Bethesda, USA), and SYFP cDNA from T Gadella (University of Amsterdam, Netherlands). We thank D Gorlich (MPI Gottingen, Germany), not only for generously providing constructs encoding RanQ69L and CRM1, but also for information concerning assay conditions. We thank members of the Venkitaraman laboratory for technical assistance and constructive discussions, and M. Goode for help with feeder free culture of ES cell cultures. ADJ was supported by a career development fellowship from the UK Medical Research Council Cancer Cell Unit. Work in CK’s laboratory was funded by the UK Biosciences & Biotechnology Research Council, and in ARV’s laboratory, by the UK Medical Research Council.
Footnotes
The authors declare no competing financial interests.
References
- 1.The Breast Cancer Linkage Consortium Cancer risks in BRCA2 mutation carriers. The Breast Cancer Linkage Consortium. J Natl Cancer Inst. 1999;91:1310–6. doi: 10.1093/jnci/91.15.1310. [DOI] [PubMed] [Google Scholar]
- 2.Collins N, et al. Consistent loss of the wild type allele in breast cancers from a family linked to the BRCA2 gene on chromosome 13q12-13. Oncogene. 1995;10:1673–5. [PubMed] [Google Scholar]
- 3.King TA, et al. Heterogenic loss of the wild-type BRCA allele in human breast tumorigenesis. Ann Surg Oncol. 2007;14:2510–8. doi: 10.1245/s10434-007-9372-1. [DOI] [PubMed] [Google Scholar]
- 4.Knudson AG., Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68:820–3. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Skoulidis F, et al. Germline Brca2 heterozygosity promotes Kras(G12D) -driven carcinogenesis in a murine model of familial pancreatic cancer. Cancer Cell. 2010;18:499–509. doi: 10.1016/j.ccr.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 6.Xia F, et al. Deficiency of human BRCA2 leads to impaired homologous recombination but maintains normal nonhomologous end joining. Proceedings of the National Academy of Sciences. 2001;98:8644–8649. doi: 10.1073/pnas.151253498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell. 2001;7:263–72. doi: 10.1016/s1097-2765(01)00174-5. [DOI] [PubMed] [Google Scholar]
- 8.Chen J, et al. Stable interaction between the products of the BRCA1 and BRCA2 tumor suppressor genes in mitotic and meiotic cells. Mol Cell. 1998;2:317–28. doi: 10.1016/s1097-2765(00)80276-2. [DOI] [PubMed] [Google Scholar]
- 9.Yang H, Li Q, Fan J, Holloman WK, Pavletich NP. The BRCA2 homologue Brh2 nucleates RAD51 filament formation at a dsDNA-ssDNA junction. Nature. 2005;433:653–7. doi: 10.1038/nature03234. [DOI] [PubMed] [Google Scholar]
- 10.Thorslund T, et al. The breast cancer tumor suppressor BRCA2 promotes the specific targeting of RAD51 to single-stranded DNA. Nat Struct Mol Biol. 2010;17:1263–5. doi: 10.1038/nsmb.1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu J, Doty T, Gibson B, Heyer WD. Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat Struct Mol Biol. 2010;17:1260–2. doi: 10.1038/nsmb.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jensen RB, Carreira A, Kowalczykowski SC. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature. 2010;467:678–83. doi: 10.1038/nature09399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carreira A, et al. The BRC repeats of BRCA2 modulate the DNA-binding selectivity of RAD51. Cell. 2009;136:1032–43. doi: 10.1016/j.cell.2009.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shivji MK, et al. The BRC repeats of human BRCA2 differentially regulate RAD51 binding on single- versus double-stranded DNA to stimulate strand exchange. Proc Natl Acad Sci U S A. 2009;106:13254–9. doi: 10.1073/pnas.0906208106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen P-L, et al. The BRC repeats in BRCA2 are critical for RAD51 binding and resistance to methyl methanesulfonate treatment. Proceedings of the National Academy of Sciences. 1998;95:5287–5292. doi: 10.1073/pnas.95.9.5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldgar DE, et al. Integrated evaluation of DNA sequence variants of unknown clinical significance: application to BRCA1 and BRCA2. Am J Hum Genet. 2004;75:535–44. doi: 10.1086/424388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karchin R, Agarwal M, Sali A, Couch F, Beattie MS. Classifying Variants of Undetermined Significance in BRCA2 with protein likelihood ratios. Cancer Inform. 2008;6:203–16. doi: 10.4137/cin.s618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marston NJ, et al. Interaction between the product of the breast cancer susceptibility gene BRCA2 and DSS1, a protein functionally conserved from yeast to mammals. Mol Cell Biol. 1999;19:4633–42. doi: 10.1128/mcb.19.7.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang H, et al. BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science. 2002;297:1837–48. doi: 10.1126/science.297.5588.1837. [DOI] [PubMed] [Google Scholar]
- 20.Kuznetsov SG, Liu P, Sharan SK. Mouse embryonic stem cell-based functional assay to evaluate mutations in BRCA2. Nat Med. 2008;14:875–81. doi: 10.1038/nm.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou Q, et al. Dss1 interaction with Brh2 as a regulatory mechanism for recombinational repair. Mol Cell Biol. 2007;27:2512–26. doi: 10.1128/MCB.01907-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jares-Erijman EA, Jovin TM. FRET imaging. Nat Biotech. 2003;21:1387–1395. doi: 10.1038/nbt896. [DOI] [PubMed] [Google Scholar]
- 23.Goedhart J, Vermeer JEM, Adjobo-Hermans MJW, van Weeren L, Gadella TWJ., Jr. Sensitive Detection of p65 Homodimers Using Red-Shifted and Fluorescent Protein-Based FRET Couples. PLoS ONE. 2007;2:e1011. doi: 10.1371/journal.pone.0001011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spain BH, Larson CJ, Shihabuddin LS, Gage FH, Verma IM. Truncated BRCA2 is cytoplasmic: implications for cancer-linked mutations. Proc Natl Acad Sci U S A. 1999;96:13920–5. doi: 10.1073/pnas.96.24.13920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu K, et al. Functional evaluation and cancer risk assessment of BRCA2 unclassified variants. Cancer Res. 2005;65:417–26. [PubMed] [Google Scholar]
- 26.la Cour T, et al. Analysis and prediction of leucine-rich nuclear export signals. Protein Engineering Design and Selection. 2004;17:527–536. doi: 10.1093/protein/gzh062. [DOI] [PubMed] [Google Scholar]
- 27.Sonoda E, et al. Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. Embo J. 1998;17:598–608. doi: 10.1093/emboj/17.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pellegrini L, et al. Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature. 2002;420:287–93. doi: 10.1038/nature01230. [DOI] [PubMed] [Google Scholar]
- 29.Rajendra E, Venkitaraman AR. Two modules in the BRC repeats of BRCA2 mediate structural and functional interactions with the RAD51 recombinase. Nucleic Acids Res. 2009;38:82–96. doi: 10.1093/nar/gkp873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu DS, et al. Dynamic control of Rad51 recombinase by self-association and interaction with BRCA2. Mol Cell. 2003;12:1029–41. doi: 10.1016/s1097-2765(03)00394-0. [DOI] [PubMed] [Google Scholar]
- 31.Rittinger K, et al. Structural analysis of 14-3-3 phosphopeptide complexes identifies a dual role for the nuclear export signal of 14-3-3 in ligand binding. Mol Cell. 1999;4:153–66. doi: 10.1016/s1097-2765(00)80363-9. [DOI] [PubMed] [Google Scholar]
- 32.Hantschel O, et al. Structural Basis for the Cytoskeletal Association of Bcr-Abl/c-Abl. Molecular cell. 2005;19:461–473. doi: 10.1016/j.molcel.2005.06.030. [DOI] [PubMed] [Google Scholar]
- 33.Guttler T, et al. NES consensus redefined by structures of PKI-type and Rev-type nuclear export signals bound to CRM1. Nat Struct Mol Biol. 2010;17:1367–1376. doi: 10.1038/nsmb.1931. [DOI] [PubMed] [Google Scholar]
- 34.Davies AA, et al. Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Mol Cell. 2001;7:273–82. doi: 10.1016/s1097-2765(01)00175-7. [DOI] [PubMed] [Google Scholar]
- 35.Xu D, Farmer A, Collett G, Grishin NV, Chook YM. Sequence and structural analyses of nuclear export signals in the NESdb database. Mol Biol Cell. 2012;23:3677–93. doi: 10.1091/mbc.E12-01-0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dong X, et al. Structural basis for leucine-rich nuclear export signal recognition by CRM1. Nature. 2009;458:1136–1141. doi: 10.1038/nature07975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mladenov E, Anachkova B, Tsaneva I. Sub-nuclear localization of Rad51 in response to DNA damage. Genes to Cells. 2006;11:513–524. doi: 10.1111/j.1365-2443.2006.00958.x. [DOI] [PubMed] [Google Scholar]
- 38.Gildemeister OS, Sage JM, Knight KL. Cellular redistribution of Rad51 in response to DNA damage: A novel role for Rad51C. Journal of Biological Chemistry. 2009 doi: 10.1074/jbc.M109.024646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ciccia A, Elledge SJ. The DNA Damage Response: Making It Safe to Play with Knives. Molecular cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem. 2008;77:229–57. doi: 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- 41.Dingwall C, Laskey RA. Protein Import into the Cell Nucleus. Annual Review of Cell Biology. 1986;2:367–390. doi: 10.1146/annurev.cb.02.110186.002055. [DOI] [PubMed] [Google Scholar]
- 42.Krogan NJ, et al. Proteasome Involvement in the Repair of DNA Double-Strand Breaks. Molecular cell. 2004;16:1027–1034. doi: 10.1016/j.molcel.2004.11.033. [DOI] [PubMed] [Google Scholar]
- 43.Mannen T, Andoh T, Tani T. Dss1 associating with the proteasome functions in selective nuclear mRNA export in yeast. Biochemical and Biophysical Research Communications. 2008;365:664–671. doi: 10.1016/j.bbrc.2007.11.024. [DOI] [PubMed] [Google Scholar]
- 44.Gudmundsdottir K, Lord CJ, Witt E, Tutt AN, Ashworth A. DSS1 is required for RAD51 focus formation and genomic stability in mammalian cells. EMBO Rep. 2004;5:989–93. doi: 10.1038/sj.embor.7400255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saeki H, et al. Suppression of the DNA repair defects of BRCA2-deficient cells with heterologous protein fusions. Proc Natl Acad Sci U S A. 2006;103:8768–73. doi: 10.1073/pnas.0600298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Edwards SL, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111–1115. doi: 10.1038/nature06548. [DOI] [PubMed] [Google Scholar]
- 47.Sakai W, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451:1116–1120. doi: 10.1038/nature06633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Siaud N, et al. Plasticity of BRCA2 function in homologous recombination: genetic interactions of the PALB2 and DNA binding domains. PLoS Genet. 2011;7:e1002409. doi: 10.1371/journal.pgen.1002409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kojic M, Zhou Q, Lisby M, Holloman WK. Brh2-Dss1 interplay enables properly controlled recombination in Ustilago maydis. Mol Cell Biol. 2005;25:2547–57. doi: 10.1128/MCB.25.7.2547-2557.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Han X, Saito H, Miki Y, Nakanishi A. A CRM1-mediated nuclear export signal governs cytoplasmic localization of BRCA2 and is essential for centrosomal localization of BRCA2. Oncogene. 2007;27:2969–2977. doi: 10.1038/sj.onc.1210968. [DOI] [PubMed] [Google Scholar]
- 51.Honrado E, et al. Immunohistochemical Expression of DNA Repair Proteins in Familial Breast Cancer Differentiate BRCA2-Associated Tumors. Journal of Clinical Oncology. 2005;23:7503–7511. doi: 10.1200/JCO.2005.01.3698. [DOI] [PubMed] [Google Scholar]
- 52.Mitra A, et al. Overexpression of RAD51 occurs in aggressive prostatic cancer. Histopathology. 2009;55:696–704. doi: 10.1111/j.1365-2559.2009.03448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
References for Methods
- 1.Hattori H, Skoulidis F, Russell P, Venkitaraman AR. Context dependence of checkpoint kinase 1 as a therapeutic target for pancreatic cancers deficient in the BRCA2 tumor suppressor. Mol Cancer Ther. 2011;10:670–8. doi: 10.1158/1535-7163.MCT-10-0781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jeyasekharan AD, et al. DNA damage regulates the mobility of Brca2 within the nucleoplasm of living cells. Proc Natl Acad Sci USA. 2010;107:21937–42. doi: 10.1073/pnas.1009577107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yu DS, et al. Dynamic control of Rad51 recombinase by self-association and interaction with BRCA2. Mol Cell. 2003;12:1029–41. doi: 10.1016/s1097-2765(03)00394-0. [DOI] [PubMed] [Google Scholar]
- 4.Guttler T, et al. NES consensus redefined by structures of PKI-type and Rev-type nuclear export signals bound to CRM1. Nat Struct Mol Biol. 2010;17:1367–1376. doi: 10.1038/nsmb.1931. [DOI] [PubMed] [Google Scholar]
- 5.Frey S, Richter RP, Gorlich D. FG-rich repeats of nuclear pore proteins form a three-dimensional meshwork with hydrogel-like properties. Science. 2006;314:815–7. doi: 10.1126/science.1132516. [DOI] [PubMed] [Google Scholar]
- 6.Ayoub N, Jeyasekharan AD, Bernal JA, Venkitaraman AR. HP1-beta mobilization promotes chromatin changes that initiate the DNA damage response. Nature. 2008;453:682–6. doi: 10.1038/nature06875. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.