

Abstract
Escherichia coli dihydrofolate reductase (DHFR) provides a paradigm for the integrated study of the role of protein dynamics in enzyme function. Previous studies of backbone and side chain dynamics have yielded unprecedented insights into the mechanism by which DHFR progresses through the structural changes that occur during its catalytic cycle. Here we report a comprehensive study of the χ1 rotamer populations of the aromatic and γ-methyl containing residues for complexes of the catalytic cycle, based on NMR measurement of 3JCγCO and 3JCγN coupling constants. We report conformational and dynamic information for eight distinct complexes, where transitions between rotamer wells may occur on a broad picosecond to millisecond timescale. This large volume of 3J data has allowed us to fit new Karplus parameterizations for aromatic side chains, and to select the best available of previously determined parameters for Ile, Thr, and Val. The 3JCγCO and 3JCγN coupling constants are found to be extremely sensitive measures of side chain χ1 rotamers, and to give important insights into the extent of conformational averaging. For a subset of residues in DHFR, the extent of rotamer averaging is invariant to the nature of the bound ligand, while for other residues the rotamer averaging differs in one or more complexes of the enzymatic cycle. These variable-averaging residues are generally located near the active site, but the phenomenon extends into the adenosine binding domain. For several residues, the rotamer populations in different DHFR complexes appear to depend on whether the complex is in the closed or occluded state, and some residues are exquisitely sensitive to small changes in the nature of the bound ligand.
Keywords: NMR, coupling constant, rotamer, Karplus parameters
Dihydrofolate reductase (DHFR) is an essential enzyme found in all organisms and many viruses. It catalyzes the reduction of 7,8-dihydrofolate (DHF) to (6S)-5,6,7,8-tetrahydrofolate (THF) using reduced nicotinamide adenine dinucleotide phosphate (NADPH) as a cofactor. E. coli DHFR is a 159-residue protein composed of an 8-stranded β-sheet (designated βA-βH), four α-helical regions (αB, αC, αE, αF), and connecting loops.1–3 The active site lies in a deep hydrophobic cleft that divides the protein into two subdomains: the adenosine binding subdomain (residues 38–106) and the loop subdomain (Figure 1A). The loop subdomain contains residues from the C- and N-termini and includes three functionally important loop regions: the Met20 loop (residues 9–24), the F-G loop (residues 116–132), and the G-H loop (residues 142–150).4 The Met20 loop has been observed in three distinct configurations: closed, occluded, and open. Figure 1A shows a structural superposition with the position of the Met20 loop in the closed configuration shown in blue (corresponding to the fully-bound NADP+ in light blue), while the occluded configuration is shown in red (corresponding to the red NADP+ with the nicotinamide ring flipped out of the pocket). The catalytic cycle of the enzyme (E), illustrated in Figure 1B, proceeds through five kinetically observable intermediates: the Michaelis complex E:DHF:NADPH (modeled by E:FOL:NADP+, containing the pseudosubstrate folate), the product ternary complex E:THF:NADP+, the product binary complex E:THF, the product release complex E:THF:NADPH, and the holoenzyme E:NADPH. Additionally, the complex with the drug methotrexate (E:MTX:NADPH) has been proposed to represent a model for the transition state between the Michaelis complex and the product ternary complex.3 The Met20 loop is in a closed configuration in the holoenzyme, the Michaelis complex, and the transition state model, and is in an occluded conformation in the product complexes. E:FOL has frequently been used as a model for E:THF, but results described here and elsewhere,5 increasingly demonstrate the exquisite sensitivity of E. coli DHFR to the bound substrate, and we therefore use the product (THF) binary complex itself, despite the experimental difficulties caused by the instability of THF towards air and light.
Figure 1.

A. Structure of E. coli DHFR: adenosine binding domain is shown in pink. Two different structures are superimposed, E:FOL:NADP+ (1RX2),3 with the Met20 loop in the closed configuration (shown in blue) and E:THF:NADP+ (1RX4)3 with the Met20 loop in the occluded configuration (shown in red). The F–G loop is shown in green, and the G–H loop in purple. Folate (from 1RX2) is shown in yellow; NADP+ is in blue for the closed state and red for the occluded state, with the nicotinamide ring in a position out of the active site pocket. B. Enzymatic cycle for DHFR, colored according to the superimposed structures in part A. Top row shows the closed complexes; bottom row, occluded. In addition to the intermediates of the catalytic cycle and analogues, 3J measurements also were obtained for the closed N23PP/S148A E:FOL:NADP+ complex. C. Schematic showing each of the χ1 rotamers (here for isoleucine), with the measured couplings indicated; rotamer populations are calculated based on the measured 3JCγCO and 3JCγN values using Eqs 1–3.
In traversing the catalytic cycle, E. coli DHFR undergoes significant loop and backbone motions.6 Side chain motions also contribute substantially to the configurational entropy of the system.7–9 Primary methods for probing backbone and side chain dynamics include NMR relaxation studies, x-ray crystallography (through temperature factors and room temperature structure determination10), and computer simulations.3,6,11–13 Although side chain rotamer populations do not provide direct information on the timescale at which the rotamer averaging takes place, the observation of differences in rotamer populations between complexes in the DHFR catalytic cycle gives important information on a possible cross section of motions that occur with timescales spanning from picosecond to millisecond.14
For amino acids with side chains that extend beyond Cβ, different orientations of the side chain, associated with variations in the χ1 dihedral angle, can result in substantial changes in local structure. The 3-bond coupling constants between the Cγ of an amino acid side chain and the backbone carbonyl carbon or amide nitrogen of the same residue (3JCγCO and 3JCγN respectively) describe the orientation of the side chain with respect to the backbone, determined by the χ1 dihedral angle (Figure 1C). NMR measurements of the 3JCγCO and 3JCγN coupling constants therefore provide information on the χ1 dihedral angle sampled in solution. Extensive rotamer libraries based on x-ray structures show that side chain χ1 dihedral angles have non-overlapping distributions about the staggered rotamer conformations centered at trans (180°), gauche+ (+60°), and gauche− (−60°).15 The assumption of a 3-site jump model is further supported by x-ray crystallographic studies which show that with increasing resolution, there is a systematic tendency for χ1 angles to approach the three canonical rotamer angles.16,17 Non-canonical rotamers are only rarely observed in high resolution x-ray structures (<1%) and those that are observed are due to a highly restricted conformational state where clashes would occur in each of the trans, gauche+, and gauche− conformations, with such situations most likely to occur for bulky aromatic side chains.17 Comparison of the experimental 3J couplings to expected values described by Karplus curves gives information on the dominant χ1 rotamers.18,19 Deviations from the expected values arise from sampling of multiple rotamers, which can be described using a 3-site jump model20,21 or from restricted conformational sampling within a single rotamer energy well. The propensity of a side chain to undergo either rotamer hopping or motions within a rotamer well is dependent on the packing density around the side chain and the conformational dynamics of neighboring residues or ligands. When rotamer hopping is the dominant contribution to conformational averaging, Karplus parameters allow the 3JCγCO and 3JCγN couplings to be converted into rotamer populations for each of the staggered rotamers. Minor rotamer populations can have functional significance. For proline isomerase, for example, room temperature crystallography has shown that minor rotamers of active site residues are important for promotion of catalysis.22
In the work presented here, the solution state χ1 rotamer populations for 20 aromatic residues (Phe, Tyr, Trp, His) and 29 γ-methyl containing residues (Ile, Thr, Val) were determined for the five intermediates formed in the steady state E. coli DHFR enzymatic cycle, together with a putative transition state model (E:MTX:NADPH) and the pseudo-substrate binary complex E:FOL, which is included for comparison with the true product binary complex E:THF. Finally, to determine the possible effect of μs-ms timescale backbone motions on the measured 3J values, and therefore on the calculated rotamer populations, the coupling constants for the wild type E:FOL:NADP+ complex, which undergoes ms timescale fluctuations in the active site loops, are compared to those for the mutant N23PP/S148A in which these fluctuations are abrogated.23 Additional information is obtained from estimates of Leu χ2 side chain rotamer averaging, predicted from the 13Cmethyl chemical shifts for each complex.24
Although the degree of rotamer averaging for most residues is unchanged throughout the enzymatic cycle, a number of side chains display different rotamer averaging behavior in the various complexes of DHFR. Differences in the extent of rotamer averaging are observed primarily near the active site, but also at sites in the adenosine binding domain. For several residues, variation of the rotamer populations between complexes depends on whether the complex is in the closed or occluded state, whereas for other side chains the variation of rotamer populations between complexes is more dependent on the presence or absence of ligands. Overall, methyl side chains tend to have a basal level of conformational averaging at room temperature, whereas buried aromatic side chains are much more likely to populate a single predominant rotamer conformation. These differences reflect the greater volume of the aromatic side chains compared to the methyl-containing side chains, and reveal important insights into the role of side chain heterogeneity in the mechanism of enzyme action.
MATERIALS AND METHODS
Protein Purification and Sample Preparation
Wild type and mutant N23PP/S148A E. coli DHFR were overexpressed in BL21 (DE3) (DNAY) cells in M9 minimal medium. Samples for assignments and 3J coupling experiments were uniformly 13C, 15N-labeled and were purified as described previously.25 NADPH and THF are light and air sensitive, and all samples containing these ligands were prepared in an argon-equilibrated glove box. The NMR buffer was thoroughly degassed under vacuum through a freeze-pump-thaw cycle and the protein was exchanged into NMR buffer [pH 7.6, 70 mM KPi, 25 mM KCl, 1 mM EDTA, 1 mM DTT, 10% D2O]. Final sample concentrations ranged from 1–3 mM. Complexes were formed by addition of 6-fold excess of substrate [FOL, THF, or MTX] and 10-fold excess of cofactor [NADP+ or NADPH]. The sample was further degassed on a vacuum line, overlaid with argon, and flame sealed in an amberized tube. E:THF:NADP+, E:THF:NADPH, and E:NADPH samples are only stable for 1–2 weeks under experimental conditions; several fresh samples were required to complete the three or more trials for measuring 3J coupling constants. Since DHFR is extremely sensitive to the bound ligands, it is essential to use highly pure ligand of the appropriate stereochemistry. (6S)-THF was purchased from Schircks Laboratories, and high purity NADPH and NADP+ were purchased from Sigma.
Side Chain and Backbone Resonance Assignments
Methyl resonance assignments for wild type E:FOL:NADP+ and E:FOL were reported previously.9 Assignments for other complexes were made using 13C-HSQC26 and (H)C(CO)NH/H(CCO)NH27 experiments. Backbone amide assignments were completed using standard HNCA and HN(CO)CA experiments.28 All spectra for assignments and coupling constant measurements were recorded at 301 K at a 1H spectrometer frequency of 600 MHz.
Measurement of Methyl 3JCγCO and 3JCγN
Experiments to measure 3JCγCO and 3JCγN coupling constants for Ile, Thr, and Val γ-methyl groups were adapted from previously developed spin-echo difference 13C-HSQC experiments29,30 by addition of a shaped 180° 13CO pulse so that the same reference experiment could be used for both 3JCγCO and 3JCγN measurements. The modified pulse sequence is shown in Supplementary Figure S1. Data were acquired in an interleaved manner, in triplicate or greater for each complex. The values for the 3JCγCO or 3JCγN couplings were calculated for each residue by comparing the intensity of cross peaks in the spin-echo difference spectrum with the intensity of the corresponding cross peak in a reference spectrum. Coupling constants for methyl-containing residues were calculated using the relationship (Ia−Ib)/Ia = 2 sin2(πJCγXT), where Ia is the reference spectrum intensity, Ib is the intensity in the spin-echo difference spectrum and the constant time evolution T was 28.6 ms. In a previous application of these coupling constant measurements to the E:FOL:NADP+ and E:FOL complexes,9 there was a systematic reduction in the magnitude of 3JCγCO, apparently due to an improperly calibrated 13C carbonyl inversion pulse, leading to errors in rotamer populations for some residues. In the current experiments, this pulse has been replaced by a Q3 shaped pulse, which is far less sensitive to small errors in calibration. The accuracy of the modified pulse scheme has been verified using human ubiquitin, where it faithfully reproduced published coupling constants.31
Measurement of Aromatic 3JCγCO and 3JCγN
Experiments to measure 3JCγCO and 3JCγN coupling constants for His, Phe, Trp, and Tyr residues were adapted from previously developed spin-echo difference 15N-HSQC experiments,32 with coupling and reference experiments collected in an interleaved manner. All 3J measurements were made in triplicate or more. The coupling constants were calculated from the relationship Ib/Ia = cos(2πJCγXT), where Ia is the reference spectrum intensity and Ib is the intensity for the amide of the i+1 residue in the 3JCγCO experiment or for the i residue in the 3JCγN experiment. The constant time evolution T was 25 ms for 3JCγCO experiments and 50 ms for 3JCγN experiments. Since the 3JCγCO measurement for aromatic side chains depends on the amide resonance of the next residue, 3JCγCO values are not available when the i+1 residue is a proline.
Derivation of Karplus Parameters from DHFR Aromatic Couplings
The only available published Karplus parameters for aromatic residues19,33 gave poor fits to the 3JCγCO and 3JCγN measured for the complexes of DHFR. Specifically, the maximum and minimum values, which in many cases should represent the coupling constant expected for a rotamer populated at ~100%, were poorly estimated. The availability of a series of excellent x-ray crystal structures, representing all of the complexes studied in this report, together with nearly complete coupling constant data for the aromatic residues in each complex, suggested the calculation of a new set of Karplus parameters more suited to the tightly-packed aromatic side chains encountered in the DHFR complexes (see Results).
The aromatic 3JCγCO and 3JCγN coupling constants used for the calculation were selected on the basis of an inspection of the structures. Only side chains tightly packed in the interior of the complexes were used, and these were further selected to contain only those residues where the two coupling constants were both consistent with the presence of a single rotamer. The Karplus parameters A, B and C and the phase offset δ calculated by least-squares fit to the Karplus equation (see Eq. 5) are shown in Table 1A.
Table 1.
| Table 1A. Karplus parameters used for rotamer averaging analysisa
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Residue Type | 3JCγN | 3JCγCO | ||||||
| A | B | C | δ | A | B | C | δ | |
| His | 1.33 | −0.84 | 0.57 | 0 | 3.70 | −0.74 | 0.65 | 0 |
| Phe, Trp, Tyr | 1.33 | −0.84 | 0.47 | 0 | 3.70 | −0.74 | −0.01 | 0 |
| Ile | 2.24 | 0.15 | −0.03 | −9 | 3.30 | −0.51 | 0.04 | 4 |
| Thr | 2.01 | 0.21 | −0.12 | 7 | 2.76 | −0.67 | 0.19 | 17 |
| Val Cγ1 | 2.22 | 0.15 | −0.06 | 3 | 3.30 | −0.51 | 0.04 | 4 |
| Val Cγ2 | 2.24 | 0.15 | −0.03 | −9 | 3.30 | −0.51 | 0.04 | 4 |
| Table 1B. Predicted values for 3J couplings at 180°, +60°, and −60° χ1 rotamer anglesb
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Residue Type | 3JCγN (Hz) | 3JCγCO (Hz) | ||||||
| Jt | Jg | Jh | Jt | Jg | Jh | |||
| His | 2.73 | 0.50 | 0.50 | 1.09 | 1.09 | 4.97 | ||
| Phe, Trp, Tyr | 2.62 | 0.38 | 0.38 | 0.55 | 0.55 | 4.43 | ||
| Ile | 0.95 | 0.31 | 2.01 | 0.79 | 3.83 | 0.45 | ||
| Thr | 0.27 | 0.73 | 1.65 | 1.18 | 3.35 | 0.18 | ||
| Val Cγ1 | 2.00 | 0.47 | 0.68 | 0.45 | 0.79 | 3.83 | ||
| Val Cγ2 | 0.31 | 2.01 | 0.95 | 3.83 | 0.45 | 0.79 | ||
Karplus parameters for the relationship: J = A cos2 (θ + δ) + B cos (θ + δ) + C.
Expected 3J values for fully populated staggered rotamers. All values are given with respect to the χ1 angle. Values for the aromatic residues are derived from fits of Karplus curves to the DHFR 3J data versus the χ1 angle in the relevant x-ray structure. Values for the γ-methyl residues are from the parameterizations of Chou et al.14.
Calculation of χ1 Rotamer Populations from Coupling Constants
For each residue, the χ1 rotamer populations were calculated from the values of the 3JCγCO and 3JCγN coupling constants, assuming a 3-site jump model, using the following equations:34
| (1) |
| (2) |
| (3) |
where 3Jexp,CγN and 3Jexp,CγCO are the experimentally measured coupling constants; p−60, p60, and p180 are the populations of the respective χ1 rotamer states; and Jt, Jg, and Jh for CγCO and CγN are the expected coupling values for the fully populated 180°, +60°, and −60° χ1 rotamers, respectively. Choice of Karplus parameters has a large impact on the calculated rotamer populations, and the new Karplus parameters derived from the aromatic couplings measured in this study were used throughout the calculation. The residue dependent Jt, Jg, and Jh values chosen are given in Table 1B, with the appropriate phase shifts made so that all values are with respect to the χ1 angle (e.g., 3JCγN for Ile has a maximum when θ(N-CA-CB-Cγ2) is 180°; this corresponds to the −60° χ1 angle).
Rotamer populations were calculated using Eqs. 1–3 and the Jt, Jg, and Jh values of Table 1B. The final rotamer populations were normalized to fall between 0 and 1. For valine, when coupling measurements for both Cγ1 and Cγ2 methyl groups are available, the rotamer populations represent an average of the populations obtained. Population ranges were determined using 3J ± σ, where σ is the standard deviation based on the three or more measurements of 3J; this range is, in general, not symmetric about the average population. Note that small values of 3J are prone to larger percent errors when the primary contribution to error is from noise in the NMR spectra. For some residues, particularly the aromatic side chains, the 3J values clearly do not report on rotamer jumps, but instead reflect either a non-classical rotamer, or more likely are indicative of conformational sampling within a single rotamer well. Since the coupling constants alone do not allow us to distinguish local fluctuations within a single rotamer well from a process that involves jumps between two or more rotamer wells, it is necessary to interpret the 3J coupling values within the context of the three-dimensional structure of each complex [E:NADPH (1RX1), E:FOL:NADP+ (3QL3 and 1RX2), E:MTX:NADPH (1RX3), E:THF:NADP/H (1RX4), E:THF (1RX5), E:FOL (1RX7), and N23PP/S148A E:FOL:NADP+ (3QL0).3,23] The propensity of a side chain to sample a certain χ1 rotamer is dependent on the residue type and the backbone conformation. For reference, Figure S2 shows plots of the DHFR ϕ, ψ values for the E:FOL:NADP+ complex (3QL3) overlaid on plots of the backbone dependent rotamer populations according to Shapovalov and Dunbrack15 for the aromatic, Val, Thr, and Ile residues.
Calculation of Leucine χ2 Rotamer Populations from Chemical Shifts
For leucines, a simple relationship between the chemical shifts of the methyl carbons has been reported as
| (4) |
where p180 is the population of the χ2 = 180° rotamer.24 Despite the nine possible χ1, χ2 pairs for leucine, only two of these are significantly sampled in the PDB: (180, +60) and (−60, 180).15 Therefore, knowledge of the χ2 rotamer is largely predictive of the χ1 rotamer conformation. To minimize the influence of neighboring aromatic side chains or ligand, we subtract the aromatic contribution to each methyl shift as determined from Shifts-4.335 using each of the applicable x-ray structures.
RESULTS
Measurement of 3JCγCO and 3JCγN Coupling Constants
The coupling constants used to estimate χ1 rotamer populations are derived from the intensities of cross peaks in spin-echo difference NMR spectra. A representative data set is shown in Figure 2. Comparison of the intensities of cross peaks in the CγCO or the CγN spectra with the corresponding cross peaks in a reference HSQC spectrum yields values of the 3JCγCO or 3JCγN coupling constants that are used to calculate populations of the three canonical χ1 rotamers according to Equations 1–3, or in some cases, are correlated to side chain motions within a single rotamer well. A total of 8 different complexes of E. coli DHFR were studied, each yielding data for 11 valines, 12 isoleucines, 6 threonines, 6 phenylalanines, 4 tyrosines, 5 tryptophans, and 5 histidines. The 3JCγCO and 3JCγN coupling constants measured for each complex are given in Supplementary Tables S2–S9 for Val, Ile, and Thr, and Supplementary Tables S11–S18 for Phe, Tyr, Trp, and His. Average values over all complexes, which are useful in gauging the extent of rotamer differences between complexes for each residue, are given in Supplementary Tables S1 and S10.
Figure 2.
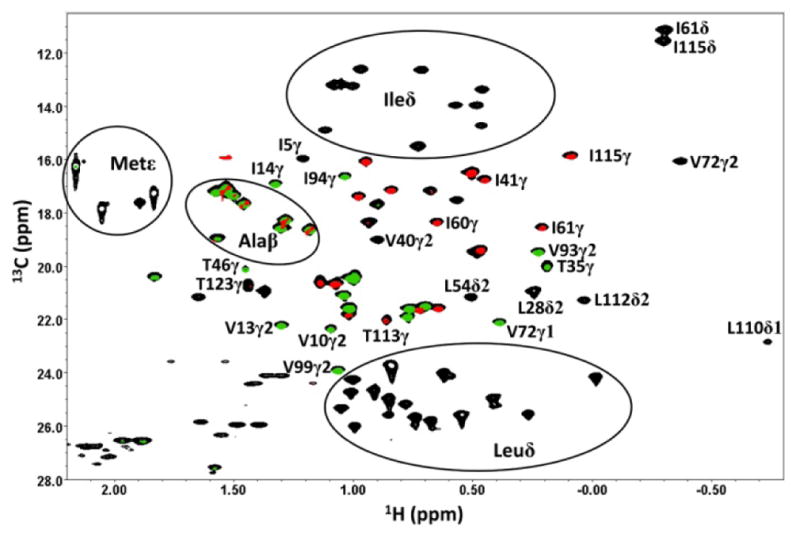
Portion of the 13C HSQC spectrum of N23PP/S148A E:FOL:NADP+ (black), overlaid with the corresponding portions of the CγCO (green) and CγN (red) difference spectra. Only the CγH3 cross peaks for Ile, Thr, and Val give information in these spectra, although the CβH3 cross peaks of Ala have detectable intensity in the difference spectra. The relative contour levels are 1:0.5:0.25 for the reference, 3JCγCO difference, and 3JCγN difference spectra, respectively.
Re-Parameterization of Karplus Curves Using Experimental Data
The general form of the Karplus relationship is
| (5) |
where A, B, and C are the Karplus parameters; θ is the dihedral angle; and δ is a phase offset. When δ is zero, the curves are symmetric about the maximum value. However, since many amino acid side chains are chiral (or in the case of residues such as valine, prochiral, with two methyl groups that are chemically equivalent but not sterically equivalent), the configuration of atoms for the dihedral angles giving minima in the Karplus relationship are not mirror images of each other, and so different minimum coupling values are to be expected, resulting in asymmetric Karplus curves with a non-zero phase shift δ.19 In the absence of motional averaging, the Karplus curve would describe the 3J coupling value for any given dihedral angle. However, Karplus relationships are empirically derived from experimental 3J values that necessarily reflect dynamic effects arising from the thermal energy in a system, as demonstrated by Case and coworkers.36
Two sets of Karplus parameters for χ1 are available in the literature. One set uses 3J measurements of D. vulgaris flavodoxin to determine Karplus parameters for each of the 3J couplings defining the χ1 angle for all residue types.19,33 However, based on our numerous 3J measurements for E. coli DHFR complexes, we find that the flavodoxin-based parameters significantly underestimate the maxima in the Karplus curves, particularly for aromatic residues. This is likely due to the location of many of the flavodoxin aromatic residues in loosely packed regions of the protein, where more motional averaging can be expected.
Since the agreement between our data and the published Karplus parameters for aromatic side chains is so poor, we decided to utilize a limited set of our own coupling constant data, excluding residues that could potentially be subject to rotamer averaging, to determine new Karplus parameters for which the maximum 3J values better estimate those observed in our experiments. Many of the aromatic side chains in DHFR are in tightly packed locations where sampling of multiple rotamers is not expected to occur. Based on the 3J couplings of these motionally restricted aromatic side chains, we have determined new Karplus parameters according to Eq. 5, by performing a least squares fit using the measured couplings and the corresponding x-ray χ1 angle (Figure 3). Residues were excluded from the fit if the error in either coupling constant measurement was > 1.0 Hz or if both 3JCγCO and 3JCγN couplings constants were less than 0.9*max[J(180), J(−60)]. Data for residues with +60° rotamers were included except for Trp22, which appears to undergo fluctuations within its major χ1 rotamer well (see Supplemental Information). Additionally, His45, which has exceptionally large 3JCγCO values, was excluded from the fits for histidine Karplus parameters. Excluded pairs of couplings are shown as open circles in Figure 3; filled circles indicate data used in the re-parameterization. The large number of data points at each of the staggered rotamers for Phe, Trp, and Tyr residues (FWY) in DHFR were fit together. While the Karplus curves for aromatic residues are expected to be at least somewhat asymmetric, we could not justify fitting a non-zero δ in the absence of additional data. Fewer reliable 3J couplings were available for histidine residues in E. coli DHFR complexes, and since these all have an expected −60° χ1 rotamer, we used additional His 3J data from human DHFR, which included 180° χ1 rotamers. Due to the limited His data, we restrained the A and B Karplus parameters to those determined from the FWY fit, varying only C, as was done by Schmidt and coworkers.19,33 The resultant Karplus parameters are given in Table 1A. An illustration of the agreement of our newly derived parameters with available published χ1 data is shown in Figure S3.
Figure 3.

Karplus correlations (plots of χ1 angle against 3J) for aromatic residues. The χ1 angle values are taken from the x-ray crystal structures of E:NADPH (1RX1), E:FOL:NADP+ (3QL3), E:MTX:NADPH (1RX3), E:THF:NADP/H (1RX4), E:THF (1RX5), E:FOL (1RX7), and N23PP/S148A E:FOL:NADP+ (3QL0).3,23 The 3J values were selected from the data measured in this work for the same complexes in solution. All measured data are plotted. For the least-squares fit to the Karplus equation (J = A cos2 (θ + δ) + B cos (θ + δ) + C), only 3J values for residues without significant rotamer averaging were used. These are indicated by filled circles. Empty circles indicate data with exceptionally large errors or with 3J couplings that are suggestive of rotamer averaging; these data were excluded from the re-parameterization fits. The black curves in each panel are derived from published Karplus parameters.19,33 The red curves are derived from the Karplus parameters calculated for the non-averaging aromatic residues in DHFR. Vertical red lines represent the error bars (standard deviations between multiple measurements) for the experimental 3J values.
A second set of published Karplus parameters for χ1 of the γ-methyl containing residues Ile, Thr, and Val used DFT simulations and comparison to experimental dipolar coupling data on human ubiquitin and protein GB3 to determine the 3JCγCO and 3JCγN Karplus parameters.14 Reasonable agreement is seen between these Karplus curves and the DHFR coupling data for 3JCγCO and 3JCγN of Thr, 3JCγN of Val, and 3JCγCO of Val-Cγ2 (Figures S3 and S4). However, our 3JCγCO data for Val-Cγ1 (Figure S4) do not reflect as large a phase shift as determined by the DFT parameters14 but fit better to the 3JCγCO Val-Cγ2 DFT curve. Given the slight, but systematic differences in the χ1 rotamer populations determined for Val-Cγ1 and Val-Cγ2, it is probable that these probes should have unique 3JCγCO Karplus parameters. The same parameters as were used for Val-Cγ2 also provide a better fit for the Ile 3J data than did the ‘Ile/Val Best Fit’ curves.14 However, our DHFR data suggest that the expectation value for Ile 3JCγN at χ1 = +60° should be somewhat larger than the 0.31 Hz associated with the Val-Cγ2 Karplus parameters. The resulting expected 3J values for the fully populated staggered rotamers (Jt, Jg, and Jh of Eqs. 1–3) for each residue type are given in Table 1B.
The values of the 3JCγCO and 3JCγN coupling constants obtained for γ-methyl and aromatic residues for each of the complexes are plotted in Figures 4 and 5, together with a comparison of the expected values of each of the fully-populated rotamers, calculated according to the Karplus parameters of Table 1.
Figure 4.

Measured Ile, Thr, and Val methyl 3JCγCO and 3JCγN values for the 8 complexes of DHFR, represented by the colored symbols shown. When the values for all complexes have overlapping error ranges, the value is represented by a square halved red and blue, with error bars representing the standard deviation of the values if appropriate. If all occluded or closed complexes have overlapping error ranges, that value is represented by a red or blue triangle respectively. Error bars for individual complexes are left off for clarity (see SI for complete tables with error). NPPSA refers to the N23PP/S148A E:FOL:NADP+ complex. The expectation values Jt, Jg, and Jh (J180, J+60, J−60) for each residue, derived using the new Karplus parameters, are shown as blue, green, and red bars, respectively, with shaded areas showing a ±5° variation about these values. The following residues are excluded from the ‘All’ complex 3J averages due to resonance broadening and/or overlap: Ile5, E:THF:NADP/H; Val13 Cγ1, closed complexes; Trp47, E:FOL:NADP+; Val88 Cγ1, E:NADPH and E:THF:NADPH; Val99 Cγ1, all complexes; Thr113, THF containing complexes. Thr46 couplings that are aberrantly large in the presence of NADP/H are not interpreted in terms of Eqs 1–3.
Figure 5.

Measured Phe, Trp, Tyr, and His 3JCγCO and 3JCγN values for the 8 complexes of DHFR, represented by the colored symbols, groups and expectation values shown at the top of Figure 4. The NPPSA 3JCγN value for His141 is a lower bound, due to resonance overlap.
Determination of Preferred Rotamers and Rotamer Populations
When the 3-site rotamer jumping assumption holds, the 3JCγCO and 3JCγN couplings can be converted into rotamer populations according to Eqs. 1–3. To avoid unduly propagating the inherently larger errors of smaller 3J coupling values, the couplings for each residue were first analyzed to determine if observed differences in coupling constants between complexes are statistically significant and therefore reflect different rotamer averaging. In addition to having larger experimental errors, the smaller 3J couplings are also more sensitive to small changes in dihedral angle, since the smallest of the staggered rotamer 3J values tend to occur where the slope of the Karplus curves are the steepest. This is illustrated in Figure 6A, which shows the FWY Karplus curves with shaded areas indicating the 3J values spanned for regions within ± 5° of the staggered χ1 angles. This 10° range about the expected 3J values is shown on Figures 4 and 5 as blue, green, or red shaded areas for Jt, Jg, and Jh, respectively. Residues that exhibit variation amongst the complexes in the smaller coupling, but that are tightly clustered for the largest coupling are considered to have equivalent couplings (e.g. Val93). The average couplings across all equivalent complexes for each residue are used to determine the rotamer populations, while populations for residues that differ significantly between complexes are considered based on the experimental couplings for that complex alone. The major rotamer population, pmajor = max[p−60, p+60, p180], is a proxy for the extent of rotamer averaging a residue undergoes, whether due to rotamer hopping or due to restricted motions within a rotamer well. In cases where multiple staggered rotamer conformations are unlikely, a reduced value of pmajor indicates increased local rotamer sampling.
Figure 6.

A. 3JCγCO (red) and 3JCγN (blue) Karplus curves for Phe, Trp, and Tyr residues (FWY), determined from fits of DHFR 3J data. Shaded regions span ±5° about the staggered rotamer angles; this variation in 3J is plotted as shaded bars around the Jt, Jg, and Jh values in Figures 4 and 5. B. Effect of local dynamics on Karplus correlations, according to Eq. 6. Dotted curves illustrate a hypothetical Karplus correlation for fixed side chains. Solid and dashed curves show the effect of Gaussian rotamer sampling with (respectively) 15° and 30° standard deviation relative to the fixed Karplus curve.
Local motions can effectively decrease the expected maximum coupling and broaden the wells in the Karplus correlations. If the local motions can be represented by a Gaussian distribution about a single dihedral angle with variance σ2 (in radians), the averaged 3J coupling can be described by an averaged Karplus curve with the following parameters:36
| (6) |
An example of the effect of a σ of 15° and 30° on the 3JCγCO and 3JCγN Karplus curves is shown in Figure 6B, where the Karplus curves in the absence of motion are shown as dotted lines, and the effect of increasing local motions is shown by solid and dashed lines.
A comparison of the major rotamer determined from 3J couplings and the x-ray structure χ1 angles for each complex is shown in Figure 7. The γ-methyl rotamer populations for each complex and the average values over all complexes are given in Supplementary Tables S1–S9 and the aromatic rotamer populations and their average values are given in Supplementary Tables S10–S18.
Figure 7.

Primary χ1 rotamer angle for the aromatic, Ile, Thr, and Val residues from A. the x-ray structures (PDB codes shown) and B. the major rotamer based on 3J couplings.
Estimation of Leucine χ2 Rotamers from Methyl Carbon Chemical Shifts
For Leu side chains, a simple correlation between the 13Cmethyl chemical shifts has been proposed to predict χ2 rotamer averaging.24 The stereochemically assigned Leu methyl chemical shifts for the DHFR complexes have been used to give an estimate of the χ2 values for the leucine residues in the various complexes of DHFR, and the values are compared with those obtained from the x-ray structures in Figure 8. Full data for all complexes are shown in Table S19. These values and their variation from those expected for fully-populated staggered rotamers essentially report on the conformational space available to the side chain, and hence provide insights into the environment surrounding each residue.
Figure 8.

Primary χ2 rotamer angle for Leu residues. A. from the x-ray structures (PDB codes shown in Figure 7). B. the chemical shift based population of the 180° rotamer.
DISCUSSION
In unfolded proteins, all side chains undergo χ1 rotamer averaging, with populations of trans, gauche+, and gauche− rotamers that are consistent with the distribution in backbone-dependent rotamer libraries.37,38 The populations vary between different side chains, reflecting differences in bulk and potential for steric clash with backbone heavy atoms, principally the carbonyl oxygen. In the core of well-structured proteins, the extent of rotamer averaging diminishes in a manner proportional to the size of the side chain due to a reduction in the available conformational space. Rotamer populations of folded proteins report on the extent to which a particular rotamer is stabilized and/or that other rotamers are destabilized due to steric clash with nearby groups or hydrogen bonding interactions. Due to the large size of aromatic side chains, this usually translates to the presence of one predominant rotamer. The smaller methyl-containing side chains frequently maintain some rotamer averaging in the core of a folded protein, though generally to a lower extent than in the random coil state. Information on the rotameric states of side chains can be obtained by measuring three-bond couplings, 3JCγCO and 3JCγN (Figure 1C). By combining this experimental information with empirical analysis of rotamer states obtained from Karplus curves, an estimate of the populations of χ1 rotamers can be obtained.
The extent of rotamer averaging is plotted on the structure of E:FOL:NADP+ (PDB: 3QL3) in Figure 9. Aromatic and methyl side chains with the same rotamer averaging across all DHFR complexes, are shown by spheres colored with a white to red gradient, where more red indicates a greater extent of averaging (i.e., a smaller pmajor). Residues that populate different rotamers or display different rotamer averaging in one or more of the DHFR complexes are colored in green; these will be discussed in greater detail below. This approach to the analysis is especially suited to comparison of extent of averaging for different residues within each complex, or between the same residue in different complexes.
Figure 9.

Extent of rotamer averaging plotted on the E:FOL:NADP+ structure (3QL3). Going clockwise, each quadrant shows a 90° rotation about the vertical axis in the plane of the page. FOL is shown as yellow sticks; NADP+ is orange sticks. Spheres representing side chain heavy atoms are shown for all aromatic, Ile, Thr, and Val residues. Residues with statistically significant variations in rotamer averaging, with uncertainties in 3J values taken into account, in one or more DHFR complexes are colored green (with green residue labels). Residues that show similar rotamer averaging for each of the DHFR complexes studied are labeled in red and colored white to red to indicate the extent of averaging, with red coloring denoting the most rotamer averaging. The gradient goes from white (pmajor = 1.0) to red (pmajor = 0.6), where pmajor = max[p180, p−60, p+60] as determined according to Eqs 1–3. In cases where rotamer hopping is not expected (e.g. Trp22 and other aromatics), this pmajor is considered a measure of the extent of local motions within a single rotamer well (see Figure 6B for an illustration of the effect this has on 3J values).
Side Chains with the Same Rotamer Averaging across all Complexes
A comparison of the extent of side chain rotamer averaging for each of the E. coli DHFR complexes shows that the majority of residues for which data are available are invariant to the nature of the bound ligands. These include numerous methyl residues in the adenosine binding domain that show basal levels of averaging between different rotamers. When bulky methyl-containing amino acids such as Ile and Val are located in close proximity in the hydrophobic core, they show similar levels of rotamer averaging. The aromatic residues within this invariant subset are almost all in one predominant rotamer; however many of the 3J measurements for the aromatics indicate some level of in-well averaging.
Rotamer Averaging of Ile, Val, and Thr Side Chains
Ile and Val residues that are in close proximity to each other – and are not in flexible loop regions – have similar levels of rotamer averaging. The average pmajor of all Ile, Thr, and Val residues in the adenosine domain is 88 ± 6%. Notably, the two residues in the adenosine domain with the least rotamer averaging (Val88 and Val99), are somewhat isolated from these other methyl residues. In particular, Val88 shows very little rotamer averaging, with average pmajor across all complexes of 97% (range: 94–98%). The Val99 side chain lines part of the binding pocket for the adenosine moiety of NADP/H and so may be expected to display different rotamer averaging in the presence or absence of cofactor; however, very little rotamer averaging is observed in any of the complexes (pmajor 95%, range 79–98%). Amongst the other Leu and Val residues, Ile60 and Ile41, located in the core of the adenosine domain, have the least rotamer averaging with pmajor of 90% (82–90%) and 89% (86–90%), respectively. Ile2, Ile91, Ile82, Val78, and Val93 are located around these core residues and have pmajor values ranging from 81 to 85%. Similar levels of rotamer averaging are also seen for proximal residues in the loop subdomain. In particular, Val136 and Ile155, which are in adjacent beta strands, have major populations of 84% (81–88%) and 88% (82–90%), respectively.
Rotamer Averaging of Aromatic Side Chains
Of the residues with invariant rotamer averaging across the series of complexes, Phe140 is the only aromatic side chain that clearly averages over multiple staggered rotamer conformations, as indicated by the intermediate values of both 3JCγCO and 3JCγN couplings. It is solvent exposed, and the local structure is such that any of the staggered conformations could be accommodated, as is demonstrated by the different χ1 conformations built in the x-ray structures of the DHFR complexes (Figure 7). The Phe 140 coupling constants vary little between complexes and their average indicates a major rotamer population of 51% χ1=180° with the remainder split between the ±60° rotamers. For Phe137, the intermediate and tightly clustered 3JCγN values for the various complexes indicates significant rotamer averaging, with the χ1 = 180° rotamer dominant. Inspection of the structure indicates that there would be significant steric clashes in the +60° rotamer. Additionally, the 3JCγCO coupling for Phe137 does not increase in a way to suggest significant populations of −60° rotamer. The region between 180° and −60° rotamers is solvent exposed, and therefore conformationally accessible, but as the side chain approaches the classical −60° rotamer, steric clashing would occur with the neighboring Val136 carbonyl oxygen. The decreased 3JCγN (relative to Jt), combined with the lack of an increase in 3JCγCO (relative to Jh), suggests a dominant 180° rotamer with asymmetric conformational sampling in the region between a χ1 of 180° and −60°; this is consistent with the shape of the Karplus curves at 180° (see Figure 6).
The other aromatic side chains show much less rotamer averaging than Phe137 or Phe140. Trp74, Tyr111, and Tyr128 appear to be the most conformationally restricted of the aromatic side chains. The remaining aromatic side chains with similar rotamer behavior in all complexes show some small amount of rotamer averaging, most likely due to conformational sampling within a single rotamer well. For Trp22, 3JCγCO and 3JCγN are both very small (0.71 ± 0.19 and 0.68 ± 0.21 Hz, respectively), indicating a predominantly +60° conformation, but these couplings are somewhat larger than expected for a fully +60° rotamer (Jg of 0.55 and 0.38 Hz, respectively). Given that Trp22 is deeply buried in the active site and is involved in a conserved, water mediated hydrogen bond network, averaging with alternative χ1 rotamers is highly improbable, since both +60° and 180° conformations would result in extreme steric clashes and would expose the Trp side chain to solvent. A more likely explanation for the increased couplings is conformational sampling by fluctuations within the χ1 = +60° rotamer well; examination of x-ray structures for the wild type DHFR complexes studied suggests that the Trp22 side chain can move substantially and without steric clash by averaging about χ1. The major rotamer population determined from Eqs. 1–3 is still a reasonable measure of this extent of this restricted conformational sampling, since it is a measure of the deviation of the couplings from the expected maximum and minimum values. Therefore, the increased red coloring in Figure 9 of, for example, Trp22 and Phe137, is indicative of more extensive rotamer averaging within a single well than for the lighter colored aromatic side chains.
Side Chains with Complex-dependent Rotamer Averaging
The residues colored in green in Figure 9 have 3JCγCO and 3JCγN values that are unique in one or more of the E. coli DHFR complexes studied or have Leu 13Cmethyl chemical shifts that indicate differences in rotamer averaging between complexes. These residues are primarily near the ligand binding sites, with most of the residues with complex-dependent averaging clustering around the active site. Variable methyl rotamer averaging is seen for Ile14, Leu28, Val40, Thr46, Ile50, Leu54, Leu62, Val75, Ile94, Val119, and Thr123, and for the aromatic residues His45, Tyr100, His124, and His149. The following paragraphs provide a brief summary of the behavior of some of these side chains. A more comprehensive discussion of the results for individual side chains is given in the Supplementary Material.
One of the most variable of the side-chains is Ile14, which shows near-100% population of the +60° χ1 rotamer in all complexes except E:NADPH and E:FOL and near-50:50 mixture of the +60° and −60° rotamers in the E:NADPH and E:FOL complexes. The backbone structure of Ile14 in the occluded DHFR x-ray structures favors the +60° χ1 rotamer15 (blue point in Figure S2). The 3J couplings of E:NADPH and E:FOL are consistent with either averaging with the −60° rotamer or local averaging within the +60° rotamer well. Based on the x-ray structure of E:NADPH (1RX1), the −60° rotamer would result in steric clash between Cγ2 of Ile14 and the nicotinamide ring of NADPH and some motions must occur to accommodate this minor rotamer. The greater extent of averaging for Ile14 in E:NADPH likely arises from decreased crowding in the active site in the absence of substrate, and from motions of the active site loops. While for most residues rotamer averaging is similar for E:THF and the analogue E:FOL, there is significant difference for Ile14. Although the x-ray structures of E:FOL and E:THF are essentially identical in the active site loops, there are significant differences in the backbone dynamics: numerous residues in the E:FOL Met20 loop show relaxation dispersion over a broad range of temperatures, while E:THF shows dispersion for only Ala19 at 303 K and higher.5 These differences in loop motions are expected to contribute to the rotamer averaging observed in E:FOL for Ile14. Contrary to our previous report,9 neither Ile94 nor Ile14 are found to have any significant χ1 = 180° rotamer subpopulation in the E:FOL:NADP+ complex. Errors in the previous measurements appear to have arisen from a miscalibrated 13C carbonyl inversion shaped pulse, which led to the systematic underestimation of the 3JCγCO values and the subsequent impact on calculated rotamer populations.
For Val119, the differences in the extent of rotamer averaging can be directly ascribed to the presence of a closed or occluded complex. Little rotamer averaging occurs in closed complexes, with average pmajor = 92% in the χ1 = −60° rotamer. The occluded complexes exhibit extensive rotamer averaging, with an average population of 41% in χ1 = 180° rotamer and with the remaining population split between the ±60° rotamers. Large amplitude motions on a ps/ns timescale were observed for residues within the Met20 and F-G loops in occluded complexes, motions that are attenuated for closed complexes.11 The greater degree of available space, together with loss if crucial hydrogen bonds in the occluded complexes, likely contribute to the enhanced backbone dynamics in the active site loops, leading to the extensive rotamer averaging seen in occluded complexes.
The side chain of Tyr100 is tightly packed in the active site, forming hydrogen bonds through its hydroxyl group to Ile5-CO, Ile94-CO, the C4 atom of the nicotinamide ring for closed complexes (this is the proton donor for the reduction of DHF to THF), and the N8 atom of the pterin ring of THF when THF is bound. The large 3JCγCO and small 3JCγN show that χ1 lies within the gauche− (−60°) rotamer well. Variations in coupling constants amongst complexes likely reflect differences in χ1 fluctuations within the gauche− well, adoption of non-canonical χ1 angles, or some combination of both. Rotamer averaging with the gauche+ or trans wells is extremely unlikely; the x-ray structures indicate that these alternate rotamers would lead to severe steric clashes of the Tyr100 side chain. We note that Tyr100 has skewed χ1 angles (between −79° and −90°) in the x-ray structures of all of the wild type E. coli DHFR complexes (Tables S11–S18).
Changes in Rotamer Averaging through the DHFR Catalytic cycle
The changes in rotamer states and the extent of rotamer averaging between the various DHFR complexes can be rationalized with reference to the changes that occur between adjacent complexes in the catalytic cycle, as illustrated in Figure 10.
Figure 10.

Summary of differences in rotamer averaging between complexes of E. coli DHFR. Differences can arise from changes in the extent of rotamer hopping or from changes in local in-well rotamer motions. The corresponding x-ray structure is shown for each complex [E:NADPH (1RX1), E:FOL:NADP+ (3QL3), E:MTX:NADPH (1RX3), E:THF:NADP/H (1RX4), E:THF (1RX5), E:FOL (1RX7), and N23PP/S148A E:FOL:NADP+ (3QL0)3,23]. Spheres are shown for Ile, Thr, Val, Leu, and aromatic residues. Ligands are shown as sticks with FOL/THF in yellow and NADP/H in orange. A. The difference in pmajor between the given complex and the next complex in the enzymatic cycle is shown for the residues that show variable averaging between complexes (green residues in Figure 9). Side chains with the same rotamer averaging are shown in white, residues with more (less) rotamer averaging than the next complex in the cycle are shown as red (blue), with more red or blue coloring indicating a larger population difference. B. Difference in rotamer averaging between E:MTX:NADPH and E:FOL:NADP+ complexes. C. Difference in rotamer averaging between N23PP/S148A and wild type E:FOL:NADP+ complexes. D. Difference in rotamer averaging between E:FOL and E:THF. A–D Leu62, Ile94, and Val119 have a different major rotamer depending on the complex, so differences in rotamer averaging are given with respect to the 180° χ2, +60o χ1 and −60° χ1 rotamers, respectively. The color gradient in panels A–D runs from Δpmajor = −0.5 (blue) < 0 (white) < 0.5 (red), where Δpmajor is the difference in major rotamer population between the displayed complex and the next complex in the catalytic cycle (A) or the complex indicated in parentheses (B–D).
E:NADPH to E:FOL:NADP+
Relative to the E:FOL:NADP+ complex, E:NADPH shows strikingly greater motional averaging in residues located along the plane dividing the adenosine binding domain and active site loop regions. Increased averaging about χ1 is observed for Ile14, Ile50, Tyr100, and His124, and about χ2 for Leu54 (red residues in Figure 10). The coupling constants suggest that Ile94 exhibits a comparable extent of χ1 averaging in both complexes but the major rotamer differs (χ1 = −60° for E:NADPH in solution, χ1 = +60° for E:FOL:NADP+ and also in the x-ray structure of the E:NADPH complex). We note that the χ1 = −60° rotamer would cause the Ile94 γ2 methyl to clash with the pterin ring in the E:FOL:NADP+ complex so this state is only favorable when the substrate/product binding site is empty; it is likely that Ile94 undergoes averaging within the +60° rotamer well in complexes with bound folate or THF, rather than averaging with the χ1 = −60° rotameric state. The nicotinamide ring of NADPH, which bridges the two domains of DHFR, and the backbone of the Met20 loop also show an increased level of disorder in E:NADPH relative to the E:FOL:NADP+, as evidenced by x-ray B-factors.3 For the residues in direct contact with the cofactor (Ile14 and Tyr100), the apparent increase in rotamer averaging (decreased 3JCγCO) in the E:NADPH complex most likely reflects increased fluctuations within the dominant χ1 rotamer well rather than jumps to one of the alternative rotameric states, which the x-ray structures suggest would be sterically unfavorable. However, Ile50 (χ1) and Leu54 (χ2), which line one face of the substrate/product binding site, exhibit increased rotamer averaging in the E:NADPH complex where this binding site is vacant. Increased χ2 rotamer averaging is also observed for Leu28 on the opposite side of the empty substrate binding pocket. The flexibility of certain side chains that line the walls of the empty binding pocket in the E:NADPH holoenzyme may play a functional role in facilitating capture and binding of incoming substrate.
E:FOL:NADP+ to E:THF:NADP+
The most notable difference in rotamer averaging between the Michaelis model complex and the product ternary complex is for Val119, which reflects the closed to occluded transition as discussed above. Most residues display a very similar level of rotamer averaging in these two complexes. However, His45 and Ile50 exhibit slightly higher flexibility in the E:THF:NADP+ product complex, while Leu28 and Leu62 show a slight decrease in χ2 rotamer averaging. The rotamer averaging for these residues appears to be sensitive to the nature of the bound ligand; the Leu62 χ1 angle in the x-ray structures of E:FOL:NADP+ and E:MTX:NADPH (Table S19) differs from that in the other complexes and allows averaging about χ2, which is disfavored due to steric clash with the adenosine ring in the E:NADPH and E:THF:NADP/H complexes.
E:THF:NADP+ to E:THF and E:THF to E:THF:NADPH
The ternary product complexes are essentially indistinguishable in their NMR spectra and measured 3J couplings. Comparison of these complexes to the product binary complex E:THF primarily reveals an increase in flexibility in the E:THF complex in the absence of cofactor. This is most significant for His45 which can sample multiple rotamers when the cofactor binding site is vacant. His124 is expected to show in-well rotamer averaging as for E:NADPH, and Leu62 changes major χ2 rotamer from 180° for the ternary complexes to +60° in E:THF.
E:THF:NADPH to E:NADPH
The product release complex E:THF:NADPH shows significantly less rotamer averaging than the holoenzyme E:NADPH. This is particularly true for residues in the plane dividing the adenosine and active site loop domains, which with the exception of Ile50 also exhibited altered rotamer averaging in E:NADPH relative to E:FOL:NADP+.
E:MTX:NADPH to E:FOL:NADP+
The transition state model E:MTX:NADPH has very similar levels of rotamer averaging as the Michaelis model complex E:FOL:NADP+ (Figure 10). The most significant difference is for Tyr100, which is highly sensitive to the nature of the bound ligands and shows more in-well rotamer averaging or a differently skewed rotamer for the E:MTX:NADPH complex.
N23PP/S148A to WT E:FOL:NADP+
Mutant and wildtype E:FOL:NADP+ exhibit highly similar levels of rotamer averaging throughout the protein (Figure 10). Slight differences for Tyr100, Leu28, and Trp30 may be indicative of minor changes in the active site packing for the mutant relative to the wild-type E:FOL:NADP+ complex.
E:FOL to E:THF
The extent of rotamer averaging between the substrate analogue and product binary complexes is most different for residues near the cofactor binding site (Figure 10). E:FOL shows an increase in rotamer averaging for His124, Ile14, Tyr100, and His45, which are all proximal to the cofactor binding site, and for Ile61 in the adenosine binding domain. There are additional subtle differences around FOL/THF, which further demonstrates the sensitivity of E. coli DHFR to the nature of the bound ligand.
Comparison with S2axis
Correlations between pmajor and the side chain order parameter S2axis have been proposed previously.39,40 However, we find no significant correlation between the pmajor values and published methyl order parameters for E:FOL and E:FOL:NADP+.9 Most of the S2axis values reported are larger than 0.7 and are not predictive of the degree of rotamer averaging found. In the case of Val119 in the E:FOL complex, S2axis is small (0.27 for Cγ1 and 0.25 for Cγ2) which correlates well with the high degree of rotamer averaging observed from the coupling constants, with each staggered rotamer significantly populated. In contrast, however, Val119 also has a low S2axis value (0.45 for Cγ2) in E:FOL:NADP+, yet the side chain exhibits a very low level of χ1 rotamer averaging (pmajor = 0.88). The lack of a correlation between S2axis and pmajor likely reflects the mismatch of timescales on which the methods report (picosecond timescale motions for S2axis versus a broad ps-ms timescale for 3J couplings) and also the fact that S2axis is sensitive to fast timescale motions that may not involve change in the χ1 dihedral angle, which is the primary contribution to the 3J value.
CONCLUSIONS
To our knowledge, the current set of 3JCγCO and 3JCγN coupling constants for 8 E.coli DHFR complexes is the most complete set of data available for any protein. These data provide valuable insights into determinants of side chain rotamer averaging and into the uncertainties inherent in interpretation of coupling constant data using rotamer jump models. From the point of view of protein structure, it is striking that the rotameric states of the vast majority of side chains probed (12 Ile, 11 Val, 6Thr, 11 Leu (at χ2), 5 Trp, 6 Phe, 4 Tyr, and 5 His) populate a dominant rotameric state that is determined primarily by the protein fold and packing of the hydrophobic core (Figure 5). Nevertheless, a subset of side chains do undergo changes in rotamer or exhibit rotamer averaging in response to binding of different ligands or backbone conformational change.
The current data indicate the need for considerable caution in the interpretation of 3JCγCO and 3JCγN coupling constants and demonstrate emphatically that the assumption of a simple 3-site rotamer jump model is not always valid. Firstly, the availability of such a large number of independently measured coupling constants for a series of related complexes of known 3D structure allowed us to reevaluate existing Karplus parameterizations for both aliphatic and aromatic side chains. Secondly, the uncertainties inherent in measurement of 3JCγCO and 3JCγN coupling constants, at least for a protein as large as DHFR (18 kDa), suggest a need for caution in interpreting small differences in terms of changes in rotamer averaging, especially in the case where both couplings are small and the errors are correspondingly large. Thirdly, we frequently observed substantial variation in the magnitude of the coupling constants for a given residue in different complexes, even when it was clear that the side chain was predominantly in the same rotamer well in each complex and when steric considerations made averaging over other rotameric states improbable, e.g. for Trp22 or Tyr100. Indeed, we found that the 3J couplings can be extremely sensitive to small variations in the structure of both the protein and the nature of the bound ligands. It is important to interpret the experimental coupling constants for each residue within the context of its environment within the protein: does a rotamer averaging model make sense, with only minimal steric clash in one or both of the alternative rotameric states, or are the data better interpreted in terms of averaging within a single rotamer well? In this regard we note that certain side chains may be constrained to non-classical rotamer angles (e.g. Tyr100 is tightly packed in the active site, with χ1 between −79 and −90° in the x-ray structures of the wild type E. coli complexes) and may undergo asymmetric conformational averaging within their energy wells. All of these factors need to be taken into consideration for meaningful interpretation of NMR coupling constants.
Supplementary Material
Acknowledgments
Funding
This work was supported by grant GM75995 from the National Institutes of Health.
We thank Gerard Kroon for his assistance in setting up the required NMR coupling experiments and Maria Yamout-Martinez and Gira Bhabha for their assistance in sample preparation.
ABBREVIATIONS
- DHFR
dihydrofolate reductase
- NADP+
nicotinamide adenine dinucleotide phosphate
- NADPH
reduced nicotinamide adenine dinucleotide phosphate
- DHF
7,8-dihydrofolate
- THF
(6S)-5,6,7,8-tetrahydrofolate
- FOL
folate
- MTX
methotrexate
- NMR
nuclear magnetic resonance
- HSQC
heteronuclear single quantum correlation
Footnotes
The authors declare no competing financial interest.
SUPPORTING INFORMATION AVAILABLE
Tables showing complete rotamer populations and 3JCγCO and 3JCγN coupling constants for each of the complexes, supplementary figures and detailed descriptions of the behavior of a number of residues. Supporting materials may be accessed free of charge online at http://pubs.acs.org.
References
- 1.Bystroff C, Oatley SJ, Kraut J. Crystal structures of Escherichia coli dihydrofolate reductase: the NADP+ holoenzyme and the folate. NADP+ ternary complex Substrate binding and a model for the transition state. Biochemistry. 1990;29:3263–3277. doi: 10.1021/bi00465a018. [DOI] [PubMed] [Google Scholar]
- 2.Bystroff C, Kraut J. Crystal structure of unliganded Escherichia coli dihydrofolate reductase. Ligand-induced conformational changes and cooperativity in binding. Biochemistry. 1991;30:2227–2239. doi: 10.1021/bi00222a028. [DOI] [PubMed] [Google Scholar]
- 3.Sawaya MR, Kraut J. Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: crystallographic evidence. Biochemistry. 1997;36:586–603. doi: 10.1021/bi962337c. [DOI] [PubMed] [Google Scholar]
- 4.Schnell JR, Dyson HJ, Wright PE. Structure, dynamics and catalytic function of dihydrofolate reductase. Ann Rev Biophys Biomol Struct. 2004;33:119–140. doi: 10.1146/annurev.biophys.33.110502.133613. [DOI] [PubMed] [Google Scholar]
- 5.Boehr DD, McElheny D, Dyson HJ, Wright PE. Millisecond timescale fluctuations in dihydrofolate reductase are exquisitely sensitive to the bound ligands. Proceedings of the National Academy of Sciences. 2010;107:1373–1378. doi: 10.1073/pnas.0914163107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boehr DD, McElheny D, Dyson HJ, Wright PE. The dynamic energy landscape of dihydrofolate reductase catalysis. Science. 2006;313:1638–1642. doi: 10.1126/science.1130258. [DOI] [PubMed] [Google Scholar]
- 7.Mittermaier A, Kay LE, Forman-Kay JD. Analysis of deuterium relaxation-derived methyl axis order parameters and correlation with local structure. J Biomol NMR. 1999;13:181–185. doi: 10.1023/A:1008387715167. [DOI] [PubMed] [Google Scholar]
- 8.Wand AJ, Urbauer JL, McEvoy RP, Bieber RJ. Internal dynamics of human ubiquitin revealed by 13C-relaxation studies of randomly fractionally labeled protein. Biochemistry. 1996;35:6116–6125. doi: 10.1021/bi9530144. [DOI] [PubMed] [Google Scholar]
- 9.Schnell JR, Dyson HJ, Wright PE. Effect of cofactor binding and loop conformation on side chain methyl dynamics in dihydrofolate reductase. Biochemistry. 2004;43:374–383. doi: 10.1021/bi035464z. [DOI] [PubMed] [Google Scholar]
- 10.Fraser JS, van den Bedem H, Samelson AJ, Lang PT, Holton JM, Echols N, Alber T. Accessing protein conformational ensembles using room-temperature X-ray crystallography. Proc Natl Acad Sci U S A. 2011;108:16247–16252. doi: 10.1073/pnas.1111325108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Osborne MJ, Schnell J, Benkovic SJ, Dyson HJ, Wright PE. Backbone dynamics in dihydrofolate reductase complexes: Role of loop flexibility in the catalytic mechanism. Biochemistry. 2001;40:9846–9859. doi: 10.1021/bi010621k. [DOI] [PubMed] [Google Scholar]
- 12.Radkiewicz JL, Brooks CL. Protein dynamics in enzymatic catalysis: Exploration of dihydrofolate reductase. J Am Chem Soc. 2000;122:225–231. [Google Scholar]
- 13.Agarwal PK, Billeter SR, Rajagopalan PTR, Benkovic SJ, Hammes-Schiffer S. Network of coupled promoting motions in enzyme catalysis. Proc Natl Acad Sci USA. 2002;99:2794–2799. doi: 10.1073/pnas.052005999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chou JJ, Case DA, Bax A. Insights into the mobility of methyl-bearing side chains in proteins from (3)J(CC) and (3)J(CN) couplings. J Am Chem Soc. 2003;125:8959–8966. doi: 10.1021/ja029972s. [DOI] [PubMed] [Google Scholar]
- 15.Shapovalov MV, Dunbrack RL., Jr A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure. 2011;19:844–858. doi: 10.1016/j.str.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.MacArthur MW, Thornton JM. Protein side-chain conformation: a systematic variation of chi 1 mean values with resolution - a consequence of multiple rotameric states? Acta Crystallogr D Biol Crystallogr. 1999;55:994–1004. doi: 10.1107/s0907444999002231. [DOI] [PubMed] [Google Scholar]
- 17.Shapovalov MV, Dunbrack RL., Jr Statistical and conformational analysis of the electron density of protein side chains. Proteins. 2007;66:279–303. doi: 10.1002/prot.21150. [DOI] [PubMed] [Google Scholar]
- 18.Karplus M. Vicinal proton coupling in nuclear magnetic resonance. J Am Chem Soc. 1963;85:2870–2871. [Google Scholar]
- 19.Schmidt JM. Asymmetric Karplus curves for the protein side-chain 3J couplings. J Biomol NMR. 2007;37:287–301. doi: 10.1007/s10858-006-9140-8. [DOI] [PubMed] [Google Scholar]
- 20.Pachler KGR. Nuclear magnetic resonance study of some [alpha]-amino acids--I: Coupling constants in alkaline and acidic medium. Spectrochimica Acta. 1963;19:2085–2092. [Google Scholar]
- 21.Pachler KGR. Nuclear magnetic resonance study of some [alpha]-amino acids--II: Rotational isomerism. Spectrochimica Acta. 1964;20:581–587. [Google Scholar]
- 22.Fraser JS, Clarkson MW, Degnan SC, Erion R, Kern D, Alber T. Hidden alternative structures of proline isomerase essential for catalysis. Nature. 2009;462:669–673. doi: 10.1038/nature08615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhabha G, Lee J, Ekiert DC, Gam J, Wilson IA, Dyson HJ, Benkovic SJ, Wright PE. A Dynamic Knockout Reveals That Conformational Fluctuations Influence the Chemical Step of Enzyme Catalysis. Science. 2011;332:234–238. doi: 10.1126/science.1198542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mulder FAA. Leucine Side-Chain Conformation and Dynamics in Proteins from 13C NMR Chemical Shifts. Chembiochem. 2009;10:1477–1479. doi: 10.1002/cbic.200900086. [DOI] [PubMed] [Google Scholar]
- 25.Bhabha G, Tuttle L, Martinez-Yamout MA, Wright PE. Identification of endogenous ligands bound to bacterially expressed human and E. coli dihydrofolate reductase by 2D NMR. FEBS Letters. 2011;585:3528–3532. doi: 10.1016/j.febslet.2011.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grzesiek S, Anglister J, Bax A. Correlation of backbone amide and aliphatic side chain resonances in 13C/15N-enriched proteins by isotropic mixing of 13C magnetization. J Magn Reson Series B. 1993;101:114–119. [Google Scholar]
- 27.Montelione GT, Lyons BA, Emerson SD, Tashiro M. An efficient triple resonance experiment using carbon-13 isotropic mixing for determining sequence-specific resonance assignments of isotopically-enriched proteins. J Am Chem Soc. 1992;114:10974–10975. [Google Scholar]
- 28.Grzesiek S, Bax A. Improved 3D triple-resonance NMR techniques applied to a 31 kDa protein. J Magn Reson. 1992;96:432–440. [Google Scholar]
- 29.Grzesiek S, Vuister GW, Bax A. A simple and sensitive experiment for measurement of JCC couplings between backbone carbonyl and methyl carbons in isotopically enriched proteins. J Biomol NMR. 1993;3:487–493. doi: 10.1007/BF00176014. [DOI] [PubMed] [Google Scholar]
- 30.Vuister GW, Wang AC, Bax A. Measurement of three-bond nitrogen-carbon J couplings in proteins uniformly enriched in 15N and 13C. J Am Chem Soc. 1993;115:5334–5335. [Google Scholar]
- 31.Hu JS, Bax A. χ1 angle information from a simple two-dimensional NMR experiment that identifies trans 3JNCγ couplings in isotopically enriched proteins. J Biomol NMR. 1997;9:323–328. doi: 10.1023/a:1018691228238. [DOI] [PubMed] [Google Scholar]
- 32.Hu JS, Grzesiek S, Bax A. Two-dimensional NMR methods for determining χ1 angles of aromatic residues in proteins from three-bond JC′Cγ and JNCγ couplings. J Am Chem Soc. 1997;119:1803–1804. [Google Scholar]
- 33.Perez C, Löhr F, Rüterjans H, Schmidt JM. Self-consistent Karplus parametrization of 3J couplings depending on the polypeptide side-chain torsion chi1. J Am Chem Soc. 2001;123:7081–7093. doi: 10.1021/ja003724j. [DOI] [PubMed] [Google Scholar]
- 34.Hennig M, Bermel W, Spencer A, Dobson CM, Smith LJ, Schwalbe H. Side-chain conformations in an unfolded protein: chi1 distributions in denatured hen lysozyme determined by heteronuclear 13C, 15N NMR spectroscopy. J Mol Biol. 1999;288:705–723. doi: 10.1006/jmbi.1999.2722. [DOI] [PubMed] [Google Scholar]
- 35.Xu XP, Case DA. Automated prediction of 15N, 13Cα, 13Cβ and 13C′ chemical shifts in proteins using a density functional database. J Biomol NMR. 2001;21:321–333. doi: 10.1023/a:1013324104681. [DOI] [PubMed] [Google Scholar]
- 36.Case DA, Scheurer C, Brüschweiler R. Static and dynamic effects on vicinal scalar J couplings in proteins and peptides: A MD/DFT analysis. J Am Chem Soc. 2000;122:10390–10397. [Google Scholar]
- 37.Vajpai N, Gentner M, Huang, Blackledge M, Grzesiek S. Side-Chain χ1 Conformations in Urea-Denatured Ubiquitin and Protein G from 3J Coupling Constants and Residual Dipolar Couplings. J Am Chem Soc. 2010;132:3196–3203. doi: 10.1021/ja910331t. [DOI] [PubMed] [Google Scholar]
- 38.Dunbrack RL., Jr Rotamer libraries in the 21st century. Curr Opin Struct Biol. 2002;12:431–440. doi: 10.1016/s0959-440x(02)00344-5. [DOI] [PubMed] [Google Scholar]
- 39.Hu H, Hermans J, Lee AL. Relating side-chain mobility in proteins to rotameric transitions: Insights from molecular dynamics simulations and NMR. J Biomol NMR. 2005;32:151–162. doi: 10.1007/s10858-005-5366-0. [DOI] [PubMed] [Google Scholar]
- 40.Scouras AD, Daggett V. The Dynameomics rotamer library: amino acid side chain conformations and dynamics from comprehensive molecular dynamics simulations in water. Protein Sci. 2011;20:341–352. doi: 10.1002/pro.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.