

Abstract
The observation of aerobic glycolysis by tumor cells in 1924 by Otto Warburg, and subsequent innovation of imaging glucose uptake by tumors in patients with PET-CT has incited a renewed interest in the altered metabolism of tumors. As tumors grow in situ, a fraction of it is further away from their blood supply, leading to decreased oxygen concentrations (hypoxia), which induces the hypoxia response pathways of HIF1α, mTOR and UPR. In normal tissues, these responses mitigate hypoxic stress and induce neo-angiogenesis. In tumors, these pathways are dysregulated and lead to decreased perfusion and exacerbation of hypoxia as a result of immature and chaotic blood vessels. Hypoxia selects for a glycolytic phenotype and resultant acidification of the tumor microenvironment, facilitated by upregulation of proton transporters. Acidification selects for enhanced metastatic potential and reduced drug efficacy through ion trapping. In this review, we provide a comprehensive summary of pre-clinical and clinical drugs under development for targeting aerobic glycolysis, acidosis, hypoxia and hypoxia-response pathways. Hypoxia and acidosis can be manipulated, providing further therapeutic benefit for cancers that feature these common phenotypes.
Keywords: Warburg Effect, tumor microenvironment, tumor hypoxia, tumor acidosis, tumor metabolism
I. Introduction
Otto Warburg first described an increased rate of aerobic glycolysis followed by lactic acid fermentation in cancer cells in 1924, later termed the Warburg Effect (Warburg, et al., 1927). Almost a century of research has confirmed Warburg’s initial observation, solidifying increased glycolytic flux as a common cancer phenotype (Hanahan & Weinberg, 2011). Increased expression of glycolytic genes are observed in ~70% of human cancers (Altenberg & Greulich, 2004). Warburg had hypothesized the metabolic shift away from oxidative phosphorylation was due to mitochondrial dysfunction, yet this has not been substantiated (Warburg, 1956). While interest in cancer metabolism peaked in the middle part of the 20th century, interest waned with the advent of molecular biological techniques in the 70s. In 1976, Sidney Weinhouse famously declared that “Since our perspectives have broadened over the years, the burning issues of glycolysis and respiration in cancer now flicker only dimly" (Weinhouse, 1976). The development of 18F-fluorodeoxyglucose (18FDG) -PET imaging to visualize increased glucose uptake in tumors and metastasis has rekindled interest in cancer metabolism, and is commonly used clinically for diagnosis and disease monitoring (Kelloff, et al., 2005). An important characteristic of the tumor microenvironment commonly found in cancers and a selection force for the glycolytic phenotype is hypoxia. Tumor hypoxia can be transient or chronic either spatially or temporally, leading to significant heterogeneity and stress. Hypoxia is a challenge clinically due to its correlation with poor prognosis and association with resistance to chemotherapy and radiation therapy (Dewhirst, et al., 2008).
We have previously proposed a series of microenvironment barriers that must be overcome for a tumor to develop during carcinogenesis (Gatenby & Gillies, 2008; Gillies, et al., 2008). As carcinogenesis begins, inadequate growth promotion and loss of contact with the basement membrane are encountered first which are commonly overcome by developing an insensitivity to anti-growth signals and self-sufficiency in growth signals, two Hallmarks of Cancer defined by Hanahan and Weinberg (Hanahan & Weinberg, 2011). As in situ cancers grow further away from the vasculature and beyond the diffusion limit of oxygen, the available concentration of oxygen is reduced, leading to hypoxic conditions. In locally invasive and metastatic lesions, hypoxia is exacerbated when neoangiogenesis creates a chaotic and immature vasculature network resulting in inconsistent oxygen delivery (Gillies, et al., 1999). Cancer cells upregulate glycolysis to maintain energy production in the absence of oxygen (The Pasteur Effect), eventually becoming the preferred energy production pathway even during reoxygenation (The Warburg Effect). Aerobic glycolysis is accompanied by lactic acid fermentation, creating significant amounts of free protons (H+) which are shuttled to the extracellular tumor microenvironment to maintain intracellular pH (pHi) at physiologic levels. Increasing amounts of H+ being pumped into the extracellular space creates an acidic microenvironment, which is known to select for cells with enhanced metastatic potential as well as provide resistance to chemotherapy (Moellering, et al., 2008; Raghunand & Gillies, 2000; Rofstad, et al., 2006; Schlappack, et al., 1991; Wojtkowiak, et al., 2011).
The tumor microenvironmental characteristics described above are heterogeneous within a tumor and are found in virtually all human solid tumors. Furthermore, while there are common metabolic phenotypes, these can arise by a multitude of genetic changes, otherwise known as the “functional equivalence principle” (Gillies, et al., 2008). Hence, targeting the causes and consequences of the tumor microenvironment is an effective way to reach a large population of patients and potentially inhibit overcome tumor growth and metastasis. In this review we describe techniques used clinically for imaging the tumor metabolic microenvironment, as well as developmental drugs to target various aspects of tumor metabolism. Finally, we detail methods that are currently being investigated pre-clinically and clinically to manipulate the tumor microenvironment for therapeutic benefit.
II. IMAGING THE TUMOR MICROENVIRONMENT
Imaging approaches to characterize the metabolic microenvironment of tumors provide useful biomarkers for diagnosis and monitoring therapy response. In the future, it is expected that imaging will be able to be the most beneficial therapy for a particular patient. Below, we will detail some of the most common MRS, MRI, and PET clinical imaging methods of imaging tumor pH and hypoxia (for more detailed review see (Hashim, et al., 2011; Pacheco-Torres, et al., 2011)).
A. MRS and MRI
Magnetic resonance spectroscopy (MRS) imaging techniques depend on differences in chemical shifts of either endogenous or exogenous nuclear MR-active compounds based on pH-dependent or independent resonances (Gillies & Morse, 2005). pH measurements with 31P-MRS can compare the chemical shifts of endogenous inorganic phosphate (Pi) to measure pHi with that of exogenous 3-aminopropyl phosphonate (3-APP) to measure extracellular pH (Shepherd & Kahn). Hyperpolarized 13C bicarbonate enters into a Henderson-Hasselbalch equilibrium which can be used to spatially image a tumor pHe (F. A. Gallagher, et al., 2008). While imaging with hyperpolarized 13C bicarbonate is more sensitive than imaging with 3-APP, the main limitation lies with the rapid (within 1–2 minutes) decrease in hyperpolarization of bicarbonate. An alternative MRI technique is to use pH-dependent relaxation, such as with gadolinium-DOTA-4AmP5− in mixture with dysprosium-DOTP5− (Garcia-Martin, et al., 2006; Raghunand, et al., 2003).
B. PET
Positron emission tomography (PET) imaging of tumors with 18F-2-deoxyglucose (FDG) has had the most impact clinically in diagnosis, analysis of cancer staging and monitoring response to therapy ((Kelloff, et al., 2005). FDG is taken up via glucose transporters (GLUT1 or GLUT3) and is phosphorylated by hexokinase, effectively trapping FDG in the cytoplasm unable to be further metabolized. PET imaging measures the annihilation reaction between a positron released from FDG during decay with a neighboring electron. Computer analysis of the signals received from annihilation reactions can reconstruct the location and quantity of positron-emitting radionucleotides, giving an accurate description of a tumor and metastasis.
A number of PET tracers for hypoxia have been developed. 18F-fluoromisonidazole (FMISO) has been the most widely developed and has been used to image hypoxia in tumors (Valk, et al., 1992). FMISO is a nitroimidazole derivative which enters cells through passive diffusion and undergoes a reduction reaction. Once reduced, FMISO becomes trapped and concentrated in cells in the absence of oxygen, allowing for PET imaging to detect regions of hypoxia within a tumor. FMISO has been studied extensively, and is available through an IND for detection of hypoxia in patients on clinical trials. Clinical studies suggest that uptake of FMISO by a tumor is predictive of its resistance to treatment radiation therapy (Thorwarth, et al., 2006).
Electron paramagnetic resonance imaging (EPRI), an imaging technique similar to nuclear magnetic resonance, measures the interactions between molecular oxygen and a non-toxic stable radical tracer (Matsumoto, et al., 2010). EPRI is able to measure the pO2 of tumors without radioisotopes and is capable of measuring dynamic pO2 changes, allowing for the measurement of intermittent hypoxia in tumors (Bennewith, et al., 2002). Although it has only been applied pre-clinically, EPRI is able to measure tumor hypoxia quickly generating 3-dimensional pO2 maps from data obtained during imaging.
III. TARGETING GLUCOSE METABOLISM
Aerobic glycolysis has long been known to be a common hallmark of solid tumors. This metabolic switch has been proposed to provide an advantage to growing tumors by allowing adaptation to low oxygen environments. This leads to increased acidification of the local tumor microenvironment, allowing for evasion of the immune system and increased metastatic potential (Gillies, et al., 2008; Kroemer & Pouyssegur, 2008). Below we describe drugs that are in pre-clinical or clinical studies that target glucose metabolism of tumors (Figure 1).
Figure 1. Inhibitors of glucose metabolism.

The figure depicts the glycolytic pathway from glucose entry into cells through production of pyruvate, which is converted either to lactate or to acetyl coA for entry into the TCA cycle. Movement of metabolic intermediates through the pathway is designated by arrows. Enzymes in the glycolytic pathway are placed next to the arrow leading from their substrate to their product. Inhibitors of glycolytic enzymes or glucose transporters appear in boxes.
A. Targeting glucose transporters
Glucose, a major carbon source for cells, is a 6-carbon ring structure converted to pyruvate canonically along the Embden-Meyerhoff glycolytic pathway. Entry of glucose into cells occurs by facilitated diffusion through a family of 14 membrane bound proteins called glucose transporters (GLUTs). GLUT1, the founding member of the GLUT family, was isolated from erythrocytes in 1977 (Kasahara & Hinkle, 1977). Upregulation of GLUT1 and GLUT3 expression has been described in many cancers, and may be a key step in tumor progression. Increased expression of GLUTs correlate with poor prognosis and short survival of patients with ovarian, breast and squamous cell carcinomas (Ayala, et al., 2010; Cantuaria, et al., 2001; Pinheiro, et al., 2011). GLUT1 (Km=6.9 mM) and GLUT3 (Km=1.8 mM) each have a high affinity for glucose, and are thought to be the main transport mechanisms for glucose into cells (Burant & Bell, 1992; Gould, et al., 1991; Shepherd & Kahn, 1999). Importantly, Hatanaka showed in 1974 that glucose uptake by cells is a rate-limiting step in glycolysis. Subsequent work by other groups determined that transformed cells with increased expression of glucose transporters at the plasma membrane is a strong independent prognostic indicator for FDG uptake and glucose consumption (Birnbaum, et al., 1987; Bos, et al., 2002; Flier, et al., 1987; Hatanaka, 1974).
Increased expression of GLUT1 and GLUT3 during tumor progression allows for unregulated metabolism of glucose, making it an intriguing therapeutic target. Recent research described the cytotoxic and chemosensitizing properties of anti-GLUT1 antibodies in numerous lung and breast cancer cell lines reconfirming the importance of glucose uptake for survival (Rastogi, et al., 2007). Decades of research have resulted in the discovery of many other GLUT inhibitors, including Cyotochalasin B and select tyrosine kinase inhibitors (Taverna & Langdon, 1973; Vera, et al., 2001).
High throughput screening for drugs capable of sensitizing cells that evade FAS-ligand induced apoptosis have identified fasentin, a small molecule inhibitor that binds to the intracellular channel of GLUT1, reducing glucose transport (Schimmer, et al., 2006). Further studies uncovered altered expression of genes involved in glucose metabolism following treatment of FAS-resistant prostate and leukemia cells with fasentin and FAS-ligand (Wood, et al., 2008). Ultimately, fasentin alone was unable to induce cell death in FAS-ligand resistant cells, despite a rapid, albeit, partial reduction in glucose uptake following fasentin treatment.
Renal Cell Carcinoma (RCC), known for harboring inactivating mutations in the von Hippel-Lindau (VHL) ubiquitin ligase gene, was identified as a candidate for chemical synthetic lethality screening for GLUT inhibitors. (Chan, et al., 2011). VHL mutations often coincide with a reorganized metabolic profile, wherein the tumor becomes highly glycolytic and relies on high levels of GLUT1 expression. One class of compounds, led by STF-31, caused necrotic cell death in RCC cells lacking functional VHL. In silico modeling revealed a potential docking site for STF-31 located in the central channel of GLUT1, and further functional studies confirmed inhibition of GLUT1 by STF-31. FDG-PET scans confirm reduced glucose uptake in RCC tumors treated with STF-31, corresponding with retarded tumor growth. Lack of toxicities resulting from treatment with STF-31 encourages further research into its therapeutic potential and widespread efficacy in other tumors overexpressing GLUT1.
B. Targeting Hexokinase
As glucose enters the cystol, hexokinase phosphorylates the sixth carbon, effectively trapping glucose intracellularly and priming it for catabolism. Hexokinase 2 is frequently overexpressed in cancers, overcoming silencing methylation found on its promoter in normal tissues (A. Goel, et al., 2003). Expression of hexokinase is transcriptionally regulated by both p53 and HIF1α (Mathupala, et al., 1997). Glucose analogs, specifically 2-deoxyglucose, can be radiolabeled to image tumors with increased glucose uptake (18FDG), and have also been studied as inhibitors of glycolysis (Kurtoglu, et al., 2007; Lampidis, et al., 2006). These analogs enter cells normally through GLUT1 or GLUT3 transporters and are phosphorylated by hexokinase. As with glucose, the 6-phospho form of these analogs are unable to exit cells and are feedback inhibitors of hexokinase activity. However, unlike glucose, the phosphorylated glucose analogs are unable to be rapidly catabolized through the remainder of the glycolytic pathway, i.e., phosphofructokinase, and can build up to high levels intracellularly, where they prevent further glucose metabolism. Although there have been some successes using deoxyglucose in vitro and in animal models as a glycolytic inhibitor, clinical successes have not extended past utilization as an imaging contrast agent to visualize tumors or as a radio-sensitizing agent (Ramirez-Peinado, et al., 2011; Song, et al., 1976).
3-bromopyruvate (3-BrPA) has been identified as a potent inhibitor of glycolysis through its promiscuous inhibition of hexokinase 2 as well as glyceraldehyde-3-phosphate dehydrogenase (GAPdH). 3-BrPA has been widely studied as an alkylating agent, but its first anticancer properties were identified in 2001 as an inhibitor of hexokinase 2 (Ko, et al., 2001; Meloche, et al., 1972). Selectivity appears to depend on its uptake by overexpressed monocarboxylate transporter, SLC5A8 (Thangaraju, et al., 2009). In addition to its use as a single agent, recent research has focused on combining 3-BrPA with other chemotherapies to overcome ATP-requiring multi-drug resistance (MDR) mechanisms. Nakano et al used 3-BrPA to sensitize MDR-expressing tumors to daunorubicin or doxorubicin treatment (Nakano, et al., 2011). Similar work by Zhou et al confirms that intracellular ATP is essential for drug resistance, and that disruption of cellular energy levels through inhibition of hexokinase 2 by 3-BrPA resensitized MDR cells to therapy (Y. Zhou, et al., 2012).
Lonidamine was first identified as an inhibitor of aerobic glycolysis through inhibition of hexokinase-2 in tumor cells in 1981 (Floridi & Lehninger, 1983; Floridi, et al., 1981). As with 3-BrPA, inhibition of hexokinase 2 by lonidamine induced apoptosis (Brawer, 2005). Lonidamine acts as a single agent and has been extensively studied as a treatment for multi-drug resistance (MDR) (Y. C. Li, et al., 2002; Ravagnan, et al., 1999). Already approved for use as an anti-cancer chemotherapy in Europe, phase II clinical trials began in the United States in 2005 treating patients with benign prostatic hyperplasia (BPH) (Brawer, 2005; Ditonno, et al., 2005). Despite reports of some cancer patients receiving 40 times the dose than patients in the U.S. trial, and indications that prostate volumes were reduced during treatment, the U.S. phase II trial was terminated due to liver toxicities and no subsequent trials have begun (Ditonno, et al., 2005; Milane, et al., 2011b). In an effort to harness the therapeutic efficacy of lonidamine against MDR and reduce toxicities due to dosage, Milane et al have developed epidermal growth factor receptor (EGFR) targeted nanoparticles encapsulating lonidamine and paclitaxel (Milane, et al., 2011a, 2011b). Orthotopic MDR-positive breast cancer xenografts treated with targeted drug-containing nanoparticles showed reduced tumor growth compared to treatment with blank nanoparticles. Transient weight losses were observed in all groups. Liver toxicities were highest in animals treated with soluble paclitaxel alone or soluble paclitaxel + lonidamine, and were less severe when drugs were bound to nanoparticles. Hematologic analyses also revealed reduced toxicity following treatment with drug combinations encapsulated within nanoparticles. Overall, lonidamine is a promising hexokinase-2 inhibitor that may show clinical benefit either alone or in combination with other chemotherapies.
C. Targeting Phosphofructokinases
Phosphofructokinase-1 (PFK-1) catalyzes the phosphorylation of fructose-6-phosphate to fructose-1,6-bisphosphate in a rate-limiting step in the glycolytic pathway. Regulation of PFK-1 activity is reduced as a result of oncogene activation, such as Ras or Src, through elevated levels of fructose-2,6-bisphosphate, a physiologic activator of PFK-1 (Bosca, et al., 1986; Kole, et al., 1991). Phosphofructokinase-2 (PFK-2), as well as the p53 target TIGAR, is as a regulator of the steady state level of intracellular fructose-2,6-bisphospate, and the PFKFB3 isozyme has been identified to be overexpressed in leukemias and solid tumors (Atsumi, et al., 2002; Bensaad, et al., 2006). Small molecule inhibitors targeting the substrate-binding domain of PFKFB3 have been identified as antineoplastic agents (Clem, et al., 2008). In vitro inhibition of recombinant PFKFB3 revealed 3PO (3-(3-Pyrindinyl)-1-(4-Pyridinyl)-2-Propen-1-one) as a lead compound that inhibits PFKFB3 but does not affect activity of PFK-1. 3PO was further shown to inhibit normal cell cycling in several solid tumor and hematologic cell lines further inhibiting tumor growth in xenograft models of lung, breast and leukemia by suppression of glycolytic flux (Clem, et al., 2008).
To improve upon clinical limitations of 3PO, such as solubility and high pre-clinical doses, Akter et al has engineered nanoparticle drug delivery systems for 3PO (Akter, et al., 2011, 2012). Encapsulating 3PO within a hydrophilic shell through conjugation to block copolymers improved 3PO bioavailability. 3PO conjugated block copolymers were also engineered with a hydrazone bond that is cleaved in acidic conditions (pH < 7.0) to preferentially target acidic tumor microenvironments. In vitro experiments with 3PO containing micelles resulted in significant cell death across several cell lines providing encouragement for future work in pre-clinical models.
In a separate study, N4A and YN1 were identified to be a competitive inhibitors of PFKFB3 (Seo, et al., 2011). While treatment of cells with these novel compounds resulted in decreased glycolytic flux followed by cell death, selectivity of the drugs was not ideal, and further optimization of the drug scaffold is currently underway.
D. Targeting Pyruvate Kinase M2
Pyruvate kinase (PK) catalyzes the transfer of a phosphate from phosphoenolpyruvate to ADP in the final step of aerobic glycolysis, resulting in one molecule each of ATP and pyruvate. Of the four pyruvate kinase isoforms, PKM1 is expressed in most tissues. PKM2 is a splice variant of PKM1 that is primarily expressed in embryonic development, but is also reported to be the main isoform expressed in tumors (Christofk, et al., 2008). PKM2 expression has been associated with the Warburg Effect, carcinogenesis and tumor growth. Due to increased expression of PKM2, cancer patients typically have higher levels of PKM2 in plasma and saliva, and this is being investigated in a clinical trial to determine if salivary levels of PKM2 can be used as a biomarker for malignancy (NCT01130584).
TT-232 (TLN-232/CAP-232) is a somatostatin structural analog that has been shown to significantly reduce tumor growth in murine models and has entered clinical trials for refractory metastatic renal cell carcinoma and melanoma (NCT0042278 and NCT00735332). TT-232 has anti-inflammatory effects through its interaction with somatostatin receptor 4 (SSTR4), a G protein-coupled receptor, and anti-tumor effects mediated through its inhibition of PKM2 (Elekes, et al., 2008; Stetak, et al., 2007). Unlike somatostatin, TT-232 is able to exhibit anti-tumor effects without the antisecretory activity that is required for somatostatin’s efficacy in neuroendocrine tumors and pancreatitis (Greenberg, et al., 2000). In addition to inhibition of PKM2, treatment of cells with TT-232 inhibits proliferation, induces cell cycle arrest and initiates apoptosis (Stetak, et al., 2001; Vantus, et al., 2001). Phase I clinical trials of TT-232 were successfully completed without significant adverse events, allowing entry into phase II trials.
E. Targeting Pyruvate dehydrogenase kinase (PDK)-
Following the conversion of phosphoenolpyruvate to pyruvate by PK, further oxidation of pyruvate is enabled by mitochondrial pyruvate dehydrogenase (PdH), which catalyzes the oxidative decarboxylation of pyruvate to acetyl-CoA, which can then enter the TCA (tricarboxylic acid) cycle. PdH is negatively regulated at three serine phosphorylation sites by pyruvate dehydrogenase kinase (PDK), which shifts glucose from oxidative to glycolytic metabolism (Holness & Sugden, 2003).
Dichloroacetate (DCA) has been used clinically over the past several decades for the treatment of lactic acidosis and mitochondrial disorders (Stacpoole, et al., 1988). DCA is an inexpensive, orally available drug that targets PDK (Bowker-Kinley, et al., 1998; Knoechel, et al., 2006; Stacpoole, 1989), and has recently been shown to have anticancer effects both in vitro and in vivo (Bonnet, et al., 2007; Wong, et al., 2008; J. Xie, et al., 2011). The Michelakis group hypothesized that inhibition of PDK with DCA could shift glucose metabolism from glycolytic to oxidative, eliminating excessive lactic acid production observed in cancer cells (Bonnet, et al., 2007). Indeed, treatment of lung, glioblastoma and breast cancer cells reversed cell metabolism from glycolytic to oxidative; and in doing so increased ROS production, decreased mitochondrial membrane potential and sensitized cells to apoptosis. In vivo rodent studies demonstrated the anti-tumor properties of DCA by reducing overall tumor volumes and inducing apoptosis in a lung cancer xenograft model (Bonnet, et al., 2007). Further preclinical studies have shown DCA to have similar pro-apoptotic effects on endometrial cancer cells as well as sensitizing prostate cancer cells to radiation therapy (Cao, et al., 2008; Wong, et al., 2008). Numerous clinical trials are currently recruiting, or underway, to administer DCA as a single agent, or in combination with other chemotherapies or radiation, in a wide range of cancers. The first published data from clinical trials with DCA as an anti-cancer therapy was recently published (Michelakis, et al., 2010). Resected glioblastoma tissue from 49 patients treated with DCA confirmed mitochondrial depolarization in vivo. Five patients with either newly diagnosed or recurrent glioblastoma were placed on a treatment regimen of DCA with standard therapies, temozolomide (TMZ) and radiation therapy, after surgical tumor debulking. During a 15 month follow-up, toxicities were moderate, with peripheral neuropathy being the only toxicity noted with ~80% of patients remaining clinically stable 15 months after the onset of therapy.
F. Targeting Lactate dehydrogenase (LDH5)-
Lactate dehydrogenase (LDH) catalyzes the interconversion of pyruvate and lactate. LDH is a tetrameric protein made from two different (heart and muscle) subunits. LDH5 (a.k.a. LDH-A or M4) is usually expressed in muscle tissue and has a low Km for pyruvate, while LDH1 (a.k.a. H4) is more ubiquitously expressed and has a lower Km for lactate. During the redox reaction of pyruvate to lactate, NADH is oxidized to NAD+, replenishing intracellular levels of NAD+ and allowing glycolysis to become self-sufficient. LDH M subunits are transcriptionally regulated by HIF1α and, hence levels of LDH5 are increased in HIF1α –positive cancers (Firth, et al., 1995; Semenza, et al., 1996). Recently LDH5 has been shown to be important for tumor initiation, although the exact mechanism is currently unclear (Fantin, et al., 2006; Goldman, et al., 1964; H. Xie, et al., 2009).
Gossypol, a cotton seed extract, has been studied as an anti-fertility drug that inhibits sperm LDH, and further experimentation has revealed cross inhibition of gossypol analogs to LDH5 (Kim, et al., 2009). More recent gossypol analog studies focusing on 8-deoxyhemigossylic derivates that target the NADH and pyruvate binding sites of LDH identified 3-dihydroxy-6-methyl-7-(phenylmethyl)-4-propylnaphthalene-1-carboxylic acid, or FX11, as a preferential inhibitor of LDH5 (Yu, et al., 2001). Treatment of human lymphoma cells, P493, with FX11 correlated with knock-down of LDH5 by siRNA by increasing oxygen consumption, ROS production, decreased ATP levels and cell death (A. Le, et al., 2010). Similar results were observed in renal cell carcinoma and breast cell lines, with the sensitivity to FX11 being highest in cells with a more glycolytic phenotype. In vivo studies also indicated that FX11 inhibits both carcinogenesis and tumor progression of lymphoma and pancreatic tumors (A. Le, et al., 2010). It was notable that these treatments were not myelosuppressive or toxic, despite the presence of LDH-A in normal tissues. Although a promising candidate drug to target the glycolytic phenotype of tumors, FX11 is not yet in clinical trials.
The most recent research for novel LDH5 inhibitors began in an attempt to fabricate a drug suitable for entry into the clinic. From this research, a series of N- hydroxyindole based inhibitors were generated to have specificity for LDH5 over LDH1 (Granchi, et al., 2011). In vitro experiments showed promising Ki values in the low micromolar range for some of the compounds synthesized. Additionally, cellular assays resulted in reduced lactate production and retarded cellular proliferation. Virtual screening of the NCI Diversity Set by another group identified galloflavin as a novel LDH inhibitor (Kim, et al., 2009). Galloflavin was further characterized and shown to bind preferentially to free enzyme without blocking either the pyruvate or NADH binding sites. Enzymatic assays using purified LDH1 and LDH5 showed that galloflavin acts as an inhibitor of both isoforms. Cellular assays confirmed in vivo activity of galloflavin with reduced lactate production, a reduction of cellular ATP levels, and decreased cellular proliferation. Preliminary murine experiments suggest that galloflavin could be a well-tolerated drug that should be developed further.
IV. TARGETING HYPOXIA
Hypoxia is another common phenotype of solid tumors. As tumors grow, pro-angiogenic factors stimulate new vessel growth within a tumor. However, these new vessels tend to be immature and chaotic and hence lead to poor perfusion (Gillies, et al., 1999). Tumors found to contain hypoxic regions typically respond poorly to therapy in the clinic (Dewhirst, et al., 2008). Hypoxia can be difficult to target due to its spatial and temporal heterogeneity within tumors and the fact that hypoxic volumes are the most poorly perfused. Nonetheless, successful approaches to target hypoxia have been developed, and some of these are in clinical trials. These approaches can be broadly described as: 1) targeting hypoxia response pathways; 2) drugs that require hypoxia for their activity and thus efficacy; 3) and methods to manipulate hypoxia to our advantage to increase efficacy of hypoxia activated prodrugs (Table 1).
Table 1.
Drugs targeting hypoxia or hypoxia response pathways
| Drug | Target | Stage of Development |
|---|---|---|
| Topotecan | Topo I/HIF1α expression | FDA approved (ovarian, cervical, SCLC) |
| EZN-2968 | HIF1α expression | Phase I/Pilot study |
| PX-478 | HIF1α expression/protein stability | Phase I |
| Rapamycin | mTOR | FDA approved for non-oncogenic indications |
| CCI779(temsirolimus) | mTOR | FDA approved (renal cell carcinoma, mantle cell lymphoma) |
| RAD001(everolimus) | mTOR | FDA approved (renal cell carcinoma, pancreatic neuroendocrine tumors & non-oncogenic indications) |
| Metformin | AMPK/mTOR/cell cycle | FDA approved for non-oncogenic indications |
| Bortezomib(PS-341) | Proteasome/UPR | FDA approved (mantle cell lymphoma, multiple myeloma) |
| STF-083010 | IRE1/UPR | Pre-clinical |
| Salicaldehydes | IRE1/UPR | Pre-clinical |
| Tirapazamine(TPZ) | Hypoxia | Clinical trials completed |
| TH-302 | Hypoxia | Phase I-III |
| Banoxantrone(AQ4N) | Hypoxia | Phase I |
| Apaziquone(E09) | Hypoxia | Phase I-III |
| PR-104 | Hypoxia | Phase I-II |
A. Targeting Hypoxia Response Pathways
Tumors typically have lower oxygen concentrations (pO2) than levels detected in normal tissue (Hockel & Vaupel, 2001). As a tumor grows outward, away from blood vessels, the ability to receive oxygen from diffusion through tissue diminishes quickly leading to diffusion-limited (or chronic) hypoxia. Additionally, perfusion-limited (or acute) hypoxia can result from variable blood flow through chaotic and immature vessels that are characteristic of tumors. Hypoxia can be a significant source of stress for cancer cells and several survival and response pathways have been identified that allows cancer cells to overcome oxygen stress.
1. Targeting the HIF1α pathway-
Modulation of the hypoxia response in cells is orchestrated by transcription factors, Hypoxia inducible transcription factors, HIF1α and/or HIF2α. Under normoxic conditions, HIF1α is inactivated via proteosomal degradation, regulated by the von Hippel Lindau (VHL) ubiquitin ligase (Jaakkola, et al., 2001; Ohh, et al., 2000). In response to hypoxia, HIF1α is not degraded and the resulting stabilized protein will heterodimerize with HIF-1β (a.k.a. the aryl hydrocarbon receptor nuclear translocator, ARNT) and activate promoters containing hypoxia response elements (HREs). Transcriptional targets of HIF1α can be found in glycolytic, angiogenic, survival and migration pathways (Semenza, 2003). Constitutive HIF1α stabilization has been observed in many cancers and is correlated with aggressive disease, poor prognosis and drug resistance, making HIF1α an attractive drug target (Birner, et al., 2000; Bos, et al., 2003; Giatromanolaki, et al., 2001; Osada, et al., 2007). This is an active area of research and there are numerous investigational drugs aimed at inhibiting HIF1α with a number of approaches: e.g. targeting HIF1α mRNA expression, protein translation, protein stability and transcriptional activity. Following, we illustrate some of these approaches. More exhaustive discussion of this subject can be found at (Vaupel, 2004).
Topotecan is an FDA-approved drug that is indicated for ovarian, cervical cancers and small cell lung carcinoma. The primary mechanism of action is through inhibition of topoisomerase I which induces genotoxic stress through DNA double strand breaks (Hsiang, et al., 1985). Screening of the National Cancer Institute “Diversity Set” of chemical compounds for small molecule inhibitors led to the discovery of a second mechanism of topotecan activity through inhibition of HIF1α expression (Rapisarda, et al., 2002). Further topotecan studies confirmed inhibition of HIF1α expression, concluding that translation of HIF1α is inhibited in a topoisomerase 1-dependent mechanism by topotecan (RapisardaUranchimeg, et al., 2004). Tumor xenograft models treated with topotecan have decreased HIF1α levels, diminished angiogenesis and reduced tumor growth (RapisardaZalek, et al., 2004). Furthermore, patients treated with topotecan had low to undetectable levels of HIFα in tumor biopsies, correlating with decreased levels of VEGF and GLUT1 (Kummar, et al., 2011). Seven of ten patients treated with topotecan to receive dynamic contrast enhanced (DCE)-MR imaging exhibited decreased blood flow and permeability through their tumors after one treatment.
Abolishing expression of HIF1α has been shown to be an effective way to inhibit tumor growth, inspiring the development of methods to target mRNA expression of HIF1α as an alternative to targeting HIF1α stability. An antisense oligonucleotide designed to inhibit HIF1α expression has moved into clinical trials (L. Li, et al., 2005). EZN-2968 was developed by Enzon Pharmaceuticals Inc. using locked nucleic acid (LNA) oligonucleotide technology to reduce HIF1α expression (Greenberger, et al., 2008; Vester & Wengel, 2004). EZN-2968 was confirmed to selectively inhibit HIF1α mRNA expression in vitro, resulting in a lasting decrease in HIF1α protein levels, followed by a reduction in expression of HIF1α target genes. EZN-2968 also showed activity in a tumor xenograft model by repressing tumor growth. Phase 1 clinical studies treating hematologic patients with EZN-2968 have recently concluded (NCT00466583) and have been followed by a pilot trial that is currently recruiting patients with liver metastasis (NCT01120288).
PX-478 is an orally available small molecule that has been shown to inhibit HIF1α activity by reducing HIF1α levels (Welsh, et al., 2004). Tumor xenograft experiments using a variety of tumor cell lines showed that treatment with PX-478 reduced tumor growth or tumor regression which correlated with decreased levels of HIF1α and its target genes GLUT1 and VEGF. The half-life of PX-478 in murine plasma is short at 50 minutes, although concentrations capable of inhibiting HIF1α expression can be found for 8 hours. Imaging of tumor xenografts with DCE and diffusion-weighted (DW)-MRI showed that treatment with PX-478 reduced tumor blood vessel permeability within 2 hours of treatment and returned to normal 48 hours after treatment (Jordan, et al., 2005). Mechanistic studies have revealed that PX-478 may have multiple mechanisms of action in the inhibition of HIF1α by hindering both transcription and stability of HIF1α protein (Koh, et al., 2008). PX-478 can also contribute to clinical efficacy by acting as a radiosensitizer in prostate cancer cell lines and in in vivo tumor models (Palayoor, et al., 2008; D. L. Schwartz, et al., 2009). Recently, phase I clinical trials investigating the safety and preliminary efficacy of PX-478 in patients with advanced solid tumor or lymphomas were completed (NCT00522652). Results from the phase I trial, presented at the 2010 ASCO Annual meeting, showed stable disease (SD) in ~40% of participants with mild toxicities (Tibes R., 2010).
2. Targeting mTOR-
The mammalian target of rapamycin (mTOR) is a kinase that is activated during cell stresses, including nutrient and energy depletion, triggering a signaling cascade regulating metabolism and many cell survival mechanisms (Dazert & Hall, 2011; Jung, et al., 2010). mTORC1, a subunit of a complex nucleated by mTOR, has been shown to be important for tumorigenesis following activation of AKT (Skeen, et al., 2006). Exposure to hypoxia in normal cells promotes activation of the tuberous sclerosis protein 1 complex (TSC1/2) which in turn negatively regulates the mTOR complex (Liu, et al., 2006). Additional evidence indicates that inhibition of the mTOR complex due to hypoxia can be accomplished through interaction with promyelocytic leukemia tumor suppressor (PML) or disruption of mTORC1 binding to RHEB (Bernardi, et al., 2006; Y. Li, et al., 2007). It is hypothesized that hypoxia-mediated inhibition of mTOR is a selective mechanism for mutations that are beneficial for cell growth in hostile environments (Graeber, et al., 1996). Alternatively, constitutively active mTOR has been observed in advanced breast cancer. In addition, loss of mTOR repressors, such as PTEN and TSC1/2, can result in unregulated mTOR activity (Connolly, et al., 2006; Kaper, et al., 2006). While the exact role mTOR plays in carcinogenesis is not fully understood, mTOR inhibitors have been successful on the bench, and have moved into the clinic.
Rapamycin, a metabolite isolated from bacteria, was first identified in the 1970’s to be a powerful anti-fungal drug (Vezina, et al., 1975). Rapamycin was quickly determined to have anti-tumor activity, and was discovered to selectively target mTOR allosterically in the early 1990’s (Heitman, et al., 1991; Houchens, et al., 1983). Rapamycin also has potent immunosuppressive activity and is approved for transplant patients to prevent organ rejection as well as anti-restenosis after heart surgery due to its anti-angiogenic properties, but is not an approved medication for the treatment of cancer. Analogues of rapamycin, or “rapalogues”, are constantly being designed to be more specific to mTOR and have better pharmacologic properties and have been successful in the clinic. Currently, CCI779, or temsirolimus, is approved for treatment of renal cell carcinoma and mantle cell lymphoma, and is being investigated clinically for the treatment of other cancers such as leukemia, non-small cell lung cancer and breast cancer (Hess, et al., 2009; Hudes, et al., 2007; Rini, 2008). RAD001, or everolimus, has been approved for renal cell carcinoma and pancreatic neuroendocrine tumors, as well as an anti-rejection medication following organ transplant (Gabardi & Baroletti, 2010; Motzer, et al., 2008). In addition to single agent drugs, rapalogues are being investigated in coordination with drugs that target other signaling pathways to improve efficacy, such as PI3K or AKT (Ayral-Kaloustian, et al., 2010; Cirstea, et al., 2010; Ikezoe, et al., 2007).
The antidiabetic drug metformin and its analogs buformin and phenformin have recently been identified as having potential anti-cancer activity. Metformin reduces blood glucose levels through decreasing hepatic gluconeogenesis and activation of AMPK (AMP-activated protein kinase) and is commonly used clinically for the treatment of type 2 diabetes (Hundal, et al., 2000; Stumvoll, et al., 1995; G. Zhou, et al., 2001). AMPK can regulate activity of mTOR through activation of TSC1/2 (Inoki, et al., 2003). Studies of diabetic patients receiving metformin revealed significantly reduced cancer risk compared to cohorts receiving other diabetic medications (Bowker, et al., 2006; J. M. Evans, et al., 2005). In vitro studies later confirmed that metformin represses growth of breast cancer cells through an AMPK-dependent signaling and inhibition of mTOR mechanism (Dowling, et al., 2007; Zakikhani, et al., 2006). Metformin treatment seems to inhibit other cellular processes such as the cell cycle through reduction of cyclin D1 and diminishing the transcription of GRP78, an estrogen receptor chaperone protein that is elevated in cancers and involved in UPR signaling (Ben Sahra, et al., 2008; S. Saito, et al., 2009). Metformin is currently being investigated clinically to determine if it is best used as a treatment or a preventative medication.
3. Targeting UPR-
Hypoxia inhibits the ability of the endoplasmic reticulum (ER) to properly fold and organize proteins. The Unfolded Protein Response (UPR) is activated in the ER under hypoxia stress, which functions to maintain ER homeostasis or initiate apoptosis. Three proteins found at the ER membrane, PERK (PKR like ER kinase), IRE-1 (inositol-requiring 1) and ATF6 (activating transcription factor 6), act independently to signal stresses leading to UPR activation (Koumenis, et al., 2002). Response by the UPR to hypoxia is important for tumor growth, and aberrant UPR signaling due to the absence of PERK or IRE-1results in increased regions of hypoxia and reduced growth rates (Bi, et al., 2005; Romero-Ramirez, et al., 2004). Activation of the UPR response results in both reduction of translation and inhibition of protein maturation pathways as well as a detoxification process known as ER-associated degradation (ERAD) and induction of autophagy (Rouschop, et al., 2010). In addition to activation of UPR in response to hypoxia, other cellular stresses often found in solid tumors can lead to UPR activation. Such stresses include calcium homeostasis, redox status and glucose depravation, making UPR an important cellular response mechanism in cancer, and also an attractive pathway to target clinically.
The ERAD response to cellular stresses is activated by the UPR and results in priming misfolded proteins to be shuttled out to the cytoplasm for proteosomal degradation (Travers, et al., 2000). Blocking the ERAD response through proteasome inhibitors like bortezomib (PS-341) has been a successful strategy for tumors with high ER stress such as multiple myeloma (Lee, et al., 2003; Nawrocki, et al., 2005). Recent research suggests that hypoxia sensitizes cells to ER stress resulting from bortezomib treatment, leading authors to suggest pairing bortezomib with normoxia targeting drugs to improve therapeutic response (Fels, et al., 2008). Such combinations have been investigated in murine models, and have shown to repress tumor growth when bortezomib was used in coordination with a HDAC6 specific inhibitor, ACY-1215, in a multiple myeloma model (Santo, et al., 2012). Clinical trials are also ongoing investigating the efficacy of combining bortezomib treatment with other chemotherapies such as mitoxantrone (topoisomerase II inhibitor), mapatumumab (antibody specific for TRAIL death receptor) and vorinostat (HDAC inhibitor).
IRE1 has two enzymatic domains, a kinase domain and an endonuclease domain (Dong, et al., 2001; Nock, et al., 2001). Crystal structures have shown that IRE1 dimerizes in a juxtaposed configuration that allows for autophosphorylation resulting in increased endonuclease activity (Han, et al., 2009; Korennykh, et al., 2009). Screening for potential inhibitors of IRE1 using a cell based reporter system identified STF-083010 (Papandreou, et al., 2011). Treatment of multiple myeloma cells with ER stresses resulted in mRNA cleavage of XBP1 by IRE1, which was abrogated with treatment of STF-083010 (Back, et al., 2006). STF-083010 was shown to selectively inhibit the endonuclease activity of IRE1 without affecting kinase activity. Although in vivo anti-tumorigenic responses were observed, more research will need to be performed to optimize an IRE1 inhibitor using STF-083010 as a scaffold. Another high-throughput screening search found salicylaldimine analogs to be inhibitors of IRE1 (Volkmann, et al., 2011). Similar to STF-083010, salicaldehydes inhibit IRE1 endonuclease activity in vitro and in vivo, increasing the interest to develop more potent and selective inhibitors targeting IRE1.
B. Using hypoxia to our advantage
1. Use of Bioreductive drugs
Bioreductive prodrugs are a class of drugs that are inert in tissues with normal pO2 but are able to undergo chemical reduction in tissues with severe hypoxia to release cytotoxic warheads, selectively targeting cancer cells within hypoxic regions. In general there are 5 different chemical scaffolds that have been used to generate bioreductive prodrugs (nitro groups, quinones, aromatic N-oxides, aliphatic N-oxides and transition metals), all of which are able to be reduced in the absence of oxygen. One of the earliest reports of the use of bioreductive quinones to selectively target hypoxia is the use of mitomycin C in the 1960’s (Iyer & Szybalski, 1964; H. S. Schwartz, et al., 1963). During the last half-century, bioreductive drugs scaffolds have been improved upon making them more selective and potent in hypoxic tumors.
Tirapazamine, or TPZ, is one of the most advanced bioreductive drugs through the clinical trials process. TPZ is built off of an aromatic N-oxide bioreductive scaffold (Zeman, et al., 1986). During hypoxia, TPZ undergoes an intracellular one-electron reduction to a radical anion then further converted to either a hydroxyl radical or an oxidizing radical, ultimately resulting in DNA damage (Anderson, et al., 2003; Baker, et al., 1988; Zagorevskii, et al., 2003). TPZ creates DNA interstrand cross links which stall replication forks and induce DNA breaks that require homologous recombination repair (J. W. Evans, et al., 2008). TPZ has been extensively studied clinically in combination with cisplatin and radiation in patients with squamous cell carcinoma, head and neck cancer, and lung cancer with moderate to inconclusive results (Q. T. Le, et al., 2004; Rischin, et al., 2005; Rischin, et al., 2010; von Pawel, et al., 2000). Further analysis showed that TPZ was being metabolized too quickly, and was not effectively penetrating tumor tissues (Hicks, et al., 1998; Kyle & Minchinton, 1999). Consequently, TPZ analogs are currently being developed with the goal of improving drug solubility, cytotoxicity, selectivity and tissue penetration characteristics (Hicks, et al., 2010).
TH-302 is built upon a scaffold of a 2-nitroimidazole and is a nitrogen mustard prodrug that is selectively reduced under hypoxia (<0.5% O2) (Duan, et al., 2008). As TH-302 is reduced, the prodrug splits and releases its cytotoxic warhead, Bromo-isophosphoramide mustard (Br-IPM). As Br-IPM is released into hypoxic tissue, it cross-links with DNA, killing cells in the hypoxia compartment as well as neighboring cells with its bystander effect (Sun, et al., 2012; J. Zhang, et al., 2005). TH-302 was shown to have efficacy in vitro and in vivo in a wide subset of cancer cell lines and xenografts and was further found to have favorable drug-like properties and pharmacokinetic profiles (Duan, et al., 2008; Hu, et al., 2010; Meng, et al., 2012). TH-302 entered phase I clinical trials as a single agent drug in patients with advanced solid tumors and has also been tested in combination with doxorubicin in patients with advanced soft-tissue sarcoma, gemcitabine in patents with pancreatic cancer; docetaxel for patients with prostate or lung cancers (Ganjoo, et al., 2011; Weiss, et al., 2011). TH-302 was generally well tolerated, but some patients experienced skin and mucosal dose limiting toxicities. Recently, phase I/II clinical trials of TH-302 as a single agent concluded with stable disease or better detected across a number of cancer types. Current clinical trials are investigating the efficacy of TH-302 as a single-agent or in combination therapy for cancers including melanoma, multiple myeloma, renal cell carcinoma, pancreatic carcinoma and phase III trials have begun in patients with sarcoma.
Banoxantrone, or AQ4N, is a N-oxide bioreductive prodrug that was developed to selectively target hypoxic regions of tumors (Smith, et al., 1997). The reduction under hypoxia releases a cytotoxic alkylaminoanthraquinone metabolite (AQ4) which induces DNA damage through inhibition of topoisomerase II. AQ4N has been shown to be efficacious in murine models of breast cancer when combined with chemotherapy or radiation therapy (R. Gallagher, et al., 2001; Patterson, et al., 2000; Williams, et al., 2009). Phase I clinical trials have investigated the activity of AQ4N either as a single-agent or in combination with radiation therapy (Papadopoulos, et al., 2008; Steward, et al., 2007). AQ4N was well tolerated by patients and is now being tested in clinical trials to evaluate the efficacy of AQ4N (NCT00394628, NCT00109356 and NCT00090727).
Two other bioreductive drugs, apaziquone (E09) and PR-104, have been successful on the bench top and have moved into clinical studies (Hendricksen, et al., 2009; McKeage, et al., 2011). While bioreductive drugs have been especially successful in pre-clinical studies, and have shown some success in the clinic, no bioreductive prodrug has been approved by the FDA to date. Current research is aimed at improving bioreductive prodrug selectivity, stability and cytotoxicity. Additionally, research is ongoing to develop bioreductive prodrugs that are non-genotoxic and instead target other cellular processes. For example, 2-nitroimidazol-5-ylmethyl is a 2-nitroimidazole that releases 5-bromoisoquinolone after reduction, targeting poly(ADP-ribose) polymerase 1 (PARP1) (Parveen, et al., 1999).
2. Manipulating hypoxia
While the data from hypoxia activated prodrugs (HAPs) in the clinic are promising, it can be reasoned that they may be more efficacious if tumor hypoxia can be selectively and transiently increased at the time of treatment. Thus, inducing hypoxia in tumors can be an efficient way of increasing the efficacy of drugs that target hypoxia. There are a number of mechanisms available with which to exacerbate tumor hypoxia selectively, including metabolically (e.g. pyruvate or DCA), or by reducing oxygen delivery (e.g. anti-angiogenic agents or vasodilators).
It has recently been shown that tumor hypoxia can be increased following intravenous injection of pyruvate (K. Saito, et al., 2011), whose mechanism of action may involve inducing cells to increase respiration (Kauppinen & Nicholls, 1986). Electron paramagnetic resonance imaging (EPRI), a spectroscopic imaging technique that measures in-vivo oxygen concentrations, of tumors in mice following an intravenous injection of hyperpolarized 13C pyruvate revealed a significant decrease in tumor oxygenation that reached a maximum at one hour, and returned to normal within five hours (K. Saito, et al., 2011). Knowledge of a tumors oxygenation status is important for treatment plans, as pyruvate induced hypoxia created reduced the ability of radiotherapy to kill cancer cells even after tumor oxygenation had returned to normal levels. DCA, an inhibitor of PDK, has also been reported to initiate a metabolic switch in cancer cells from glycolysis to oxidative phosphorylation (J. Xie, et al., 2011). Induction of oxidative phosphorylation by DCA increased reactive oxygen species, pH and apoptotic proteins in HeLa cells. Additionally, the metabolic switch observed after DCA treatment correlated with an increased sensitivity of HeLa cells to cisplatin, suggesting that manipulation of a tumors metabolism may be therapeutically successful.
Tumor oxygenation can also be manipulated by controlling oxygen delivery with anti-angiogenic or anti-vascular agents. Angiogenesis is a common phenotype (“Hallmark”) of cancer that is regulated by HIF1α signaling (Hanahan & Weinberg, 2011). Tumors support an induction of angiogenesis by producing angiogenic growth factors such as vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF). Several anti-angiogenic inhibitors that target the immature angiogenic vasculature have been approved, including sorafenib, a VEGFR and PDGFR inhibitor, avastin (bevacizumab), an antibody targeting VEGF, and sunitinib, a VEGFR and PDGFR inhibitor (Chung, et al., 2010). Although resistance to anti-angiogenic drugs has become a major obstacle in clinical cancer treatment (Mitchell & Bryan, 2010), their use to acutely increase hypoxia in combination with HAPs has not yet been published. Alternatively, there are agents, such as combetestatin, that will target mature vessels, and these are also known to increase tumor hypoxia (Dachs et al, BMC Cancer 6, 280, 2006). Another characteristic of the immature tumor vasculature is a lack of tone. Thus, vasodilators, such as hydralazine, induce a systemic drop in blood pressure, which is not matched by the tumor vasculature, causing a transient decrease in perfusion within the tumor (Sonveaux, 2008). This “steal” phenomenon has been demonstrated using Doppler Ultrasound to measure decreased tumor blood flow (Horsman, et al., 1992). The decrease in perfusion leads to increases in acidosis and hypoxia; both have been shown using pH electrodes or magnetic resonance spectroscopy, MRS, for acidosis and pO2 electrodes for hypoxia (Adachi & Tannock, 1999; Belfi, et al., 1994; Nordsmark, et al., 1996; Okunieff, et al., 1988).
V. Targeting Acidosis
The microenvironment of solid tumors is known to be more acidic (pH 6.5–6.9) than the physiological pH of normal tissue (pH 7.2–7.5), which can be attributed to a tumor’s increased glycolytic flux and poor vasculature perfusion (Griffiths, 1991; Wike-Hooley, et al., 1984). Acidic microenvironments have been shown to increase the invasiveness of a tumor, leading to increased metastasis (Moellering, et al., 2008; Rofstad, 2000; Rofstad, et al., 2006). In this section we will describe drugs that target acidosis in tumors and systematic approaches to reduce acidosis in the tumor microenvironment (Figure 2).
Figure 2. Proteins that contribute to tumor acidosis and their inhibitors.
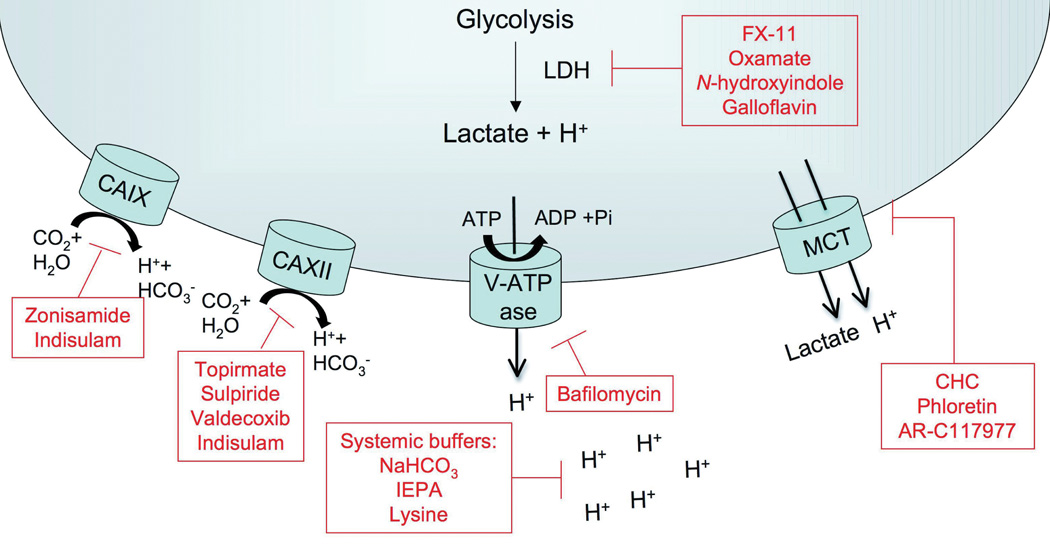
The figure depicts proteins and transporters that contribute to extracellular acidosis in a tumor due to increased lactate production from increased glycolytic flux. Included are CAIX and CAXII, carbonic anhydrases that catalyze the interconversion between carbon dioxide and water to bicarbonate and protons; and V-ATPases and MCTs, which allow transport of H+ into the extracellular environment. Inhibitors of the proteins that contribute to tumor acidosis appear in boxes.
A. Targeting Proton transport-
Metabolically-produced hydrogen ions (acid) can be exported from cells by a variety of mechanisms including, inter alia, sodium-hydrogen exchange (NHE), anion exchangers (AE), vacuolar ATPases and membrane-bound carbonic anhydrases (CA) (Neri & Supuran, 2011). NHE and AE are ubiquitously expressed and have proven to be poor anti-cancer drug targets, either through inefficacy or through toxicity, and these have been reviewed (Grinstein, et al., 1989). Following, we will discuss some of the newer, less well-explored members of this class of transporters.
Carbonic anhydrases are metalloenzymes that catalyze the interconversion of carbon dioxide and water to bicarbonate and protons. Mammalian carbonic anhydrases (α-CAs) can be cystolic, mitochondrial, secreted or membrane bound. The primary function of mammalian carbonic anhydrases is to maintain the acid-base balance of cells, tissue, and blood. As aerobic glycolysis becomes the primary means of energy production for a tumor cell, the ability to regulate physiological pHi becomes paramount to maintain cellular processes such as proliferation as well as inhibition of apoptosis (Shen, et al., 2006; Tiseo, et al., 2009). CAIX and CAXII are two transmembrane carbonic anhydrases that have been identified to be associated with tumor progression and metastasis (Aulitzky, et al., 1989; Fantin, et al., 2006). As a transcriptional target of HIF1α, CAIX expression is up-regulated in hypoxic tissue and has been shown to be a poor prognostic marker in several cancer types, including breast cancer (Lou, et al., 2011). CAXII is also overexpressed in tumors and is associated with disease progression and response to therapy (Supuran, 2008; Tureci, et al., 1998). As carbon dioxide is hydrated, HCO3− is moved intracellularly to maintain intracellular pH (pHi) while protons are pumped into the extracellular environment of a tumor, decreasing the extracellular pH (Shepherd & Kahn) promoting an aggressive metastatic environment (Jaakkola, et al., 2001; H. Xie, et al., 2009). Members of α-CA require zinc for activity, making them susceptible to inhibition by sulfonamides, which coordinates with the zinc ion found in the active sites of carbonic anhydrases. Sulfonamide analogs such as topirmate, sulpiride and valdecoxib have been shown to potently inhibit CAXII, while zonisamide has been identified to be an effective inhibitor of CAIX (Greenberger, et al., 2008; L. Li, et al., 2005). Perhaps the most studied sulfonamide analog, indisulam, has high affinity for CAIX and CAXII, in addition to seven other carbonic anhydrases (Greenberger, et al., 2008; L. Li, et al., 2005). Indisulam inhibits CAIX in nanomolar quantities and shows efficacy against tumor xenografts in vivo. In addition to CAIX inhibition, indisulam induced sequelae, such as disruption of the G1/G2 phases of the cell cycle and expression changes of genes related to cell adhesion, cell signaling, and altered glucose metabolism (Owa, et al., 1999; Rapisarda, et al., 2002; RapisardaZalek, et al., 2004; Vester & Wengel, 2004).
Clinical trials for the treatment of solid tumors with indisulam have been ongoing for the past decade. Five phase I clinical trials have been conducted focusing on optimizing the dosing regimen of indisulam to patients with solid tumors (Birner, et al., 2000; Bos, et al., 2003; Giatromanolaki, et al., 2001; Kummar, et al., 2011; Welsh, et al., 2004). Fatigue and mucositis were noted as adverse events during the trial, and reversible neutropenia and thrombocytopenia were dose-limiting toxicities. Phase II trials have been completed on patients with platinum-pretreated NSCLC in a multi-center study (Jordan, et al., 2005). While some patients experienced a positive response to indisulam, the effect was not long-term. Objective responses to indisulam therapy were not achieved during this trial, which may be attributed to inherent difficulties of being a second-line therapy to platinum-pretreated NSCLC (Koh, et al., 2008). Further trials are being conducted using indisulam as both a single agent or as combination therapy for different tumor types.
Another membrane bound transporter involved with acidification of the tumor microenvironment is V-ATPase (Palayoor, et al., 2008; D. L. Schwartz, et al., 2009). In tumor cells, V-ATPases can prevent intracellular acidification by transporting protons into lysosomal compartments that are released into extracellular space, or by directly pumping protons into the tumor microenvironment (Skeen, et al., 2006). In addition to promoting tumor metastasis by acidifying the tumor microenvironment, overexpression of V-ATPases following chemotherapy treatment appears to be a drug resistance mechanism (Y. Li, et al., 2007; Liu, et al., 2006). In 1988, bafilomycins were identified to be potent inhibitors of V-ATPases (Bernardi, et al., 2006). Since this discovery, several generations of V-ATPase inhibitors have been developed and investigated and can be classified into 5 families of V-ATPase inhibitors (Perez-Sayans, et al., 2009). While targeting V-ATPases is desirable as an anti-cancer target to reduce metastatic potential and drug resistance, clinical relevance is unknown due to likely toxicities (Bi, et al., 2005; Connolly, et al., 2006; Kaper, et al., 2006; Koumenis, et al., 2002).
The monocarboxylate transporter 1, MCT1, a membrane bound transporter is required for lactate (coupled with a proton) to move across the plasma membrane. MCT1 has been documented to have dysregulated expression in colorectal, breast, and cervical carcinomas (Asada, et al., 2003; PinheiroLongatto-FilhoFerreira, et al., 2008; PinheiroLongatto-FilhoScapulatempo, et al., 2008). Inhibition of MCT1 reduces intracellular pH and induces apoptosis, making it an attractive target for anti-tumorigenic therapy (Sonveaux, et al., 2008). Several small molecule inhibitors of MCT1 have been identified including α-cyano-4-hydroxycinnamate (CHC), phloretin and AR-C117977 (Bueno, et al., 2007; Sonveaux, et al., 2008). Currently, no MCT1 inhibitors are being investigated clinically.
B. Manipulating tumor microenvironment pH
Orally distributed systemic buffers have been shown to be an effective way to increase extracellular pH of a tumor (Silva, et al., 2009). Continuous oral delivery of sodium bicarbonate to tumor bearing mice have been shown to increase selectively the pHe of a tumor and are effective at reducing the rate and size of metastasis, without changing the volume of the primary tumor (Jahde, et al., 1990; Robey, et al., 2009). In addition to reducing metastasis, buffering with sodium bicarbonate increased breast tumors sensitivity to doxorubicin and mitoxantrone, chemotherapies known to be ineffective in acidic tumor environments (Jahde, et al., 1990; Raghunand, et al., 2001; Wojtkowiak, et al., 2011). A similar reduction in metastasis was achieved using orally available imidazole (IEPA) or lysine buffers in murine experimental metastasis models (Ibrahim Hashim, et al., 2011; J., 2011).
VI. Manipulating the microenvironment for therapeutic benefit
Combination therapy has been a long standing strategy for the treatment of cancer patients. Drug resistance to single agent regimens is a major obstacle in the clinic and combination therapy aims to target more of a heterogeneous tumor, reducing the ability of a tumor to develop resistance. The commonality of phenotypic characteristics of the tumor microenvironment between patients encourages the targeting of the microenvironment in combination with other cytotoxic chemotherapies. In the above sections, we detailed a number of approaches to target the tumor metabolic phenotype as well as describing strategies to manipulate hypoxia (exacerbation of hypoxia metabolically or by reducing oxygen delivery) and acidosis (buffer therapy) for therapeutic benefit. In this section, we will describe additional combination therapies that manipulate the metabolic or physiologic phenotype of cancers.
2DG, the glucose analog hexokinase inhibitor, has been unsuccessful as a single agent chemotherapy in the clinic, but has recently been of interest as a sensitizer of cancer cells to other chemotherapies or radiation therapy (Coleman, et al., 2008; Lin, et al., 2003; Simons, et al., 2007; F. Zhang & Aft, 2009). Targeting metabolic pathways or DNA integrity through ionizing radiation (IR) or treatment with drugs like metformin in coordination with 2DG treatment can lead to significant antitumor effects (Ben Sahra, et al., 2010; Cheong, et al., 2011). Clinical studies have verified that co-treatment of 2DG with IR is safe for patients, and reduced toxicity associated with IR in some patients (Mohanti, et al., 1996; Singh, et al., 2005). Preclinical studies using 2DG as a sensitizer are promising; however clinical studies investigating the efficacy need to be completed before 2DG sensitizing treatment becomes routine.
VEGF inhibitors, and antiangiogenic inhibitors in general, have similarly unintended effects on the tumor microenvironment, resulting in normalization of the tumor vasculature. Vascular normalization, first described by Rakesh K. Jain, is a maturation of existing immature vessels within a tumor when neoangiogenesis is inhibited (S. Goel, et al., 2011; Jain, 2001, 2005). Vascular maturation results in better oxygen delivery and tumor perfusion, relieving interstitial tumor pressure which is hypothesized to provide better drug delivery to patients and reduce resistance to chemotherapy (Jain, 2005). Treatment of tumor bearing mice with VEGF inhibitor DC101 resulted in tumor vascular remodeling, where vasculature became non-leaky and more organized (Tong, et al., 2004). Further studies have been conducted to study the timing of vascular normalization with optimal sensitivity to radiation treatment (Matsumoto, et al., 2011; Winkler, et al., 2004). Vascular normalization has been observed in patients with non-metastatic rectal adenocarcinoma receiving bevacizumab (Willett, et al., 2004; Willett, et al., 2010; Willett, et al., 2009). Although tumor reduction was not observed, microvessel density and vascular permeability decreased and histological analysis confirmed the presence of mature vasculature within tumors. Pre-clinical and clinical studies have provided support for the vascular normalization hypothesis, however more studies need to be completed to fully optimize the normalization window to improve efficacy of this treatment.
VII. Conclusion
Initially a barrier during carcinogenesis, the tumor microenvironment during the later stages of carcinogenesis provides an advantage for a tumor to outcompete normal tissue, becoming more aggressive and metastatic. Additionally, common characteristics of a tumor microenvironment provide a haven of protection for a tumor against chemotherapies. The immature and chaotic vasculature that exacerbates hypoxia within a tumor also provides minimal perfusion through a tumor for effective drug therapy, and extracellular acidosis due to preferential metabolism through aerobic glycolysis creates an environment that effectively traps weakly basic drugs from moving intracellularly. Extensive research has been focused on targeting the tumor microenvironment, providing clinicians with chemotherapies that target the glycolytic pathway, acidosis, hypoxia and hypoxia response pathways (Table 2). Manipulation of the tumor microenvironment has been an effective strategy for the treatment of a wide range of patients and will continue to be an important area of drug discovery in the future.
Table 2.
Clinical Trials*
| Drug | Clinicaltrials.gov Identifier | Site | Phase | Sponsor |
|---|---|---|---|---|
| Biomarker Study | NCT01130584 | Salivary levels of PKM2 | Observational | National University Hospital, Singapore |
| TT-232 | NCT00422786 | Renal Cell Carcinoma | II | Thallion Pharmaceuticals |
| TT-232 | NCT00735332 | Melanoma | II | Thallion Pharmaceuticals |
| EZN-2968 | NCT00466583 | Carcinoma/Lymphoma | I | Enzon Pharmaceuticals, Inc. |
| EZN-2968 | NCT01120288 | Neoplasms/Liver Metastases | I | National Cancer Institute (Luciani, et al.) |
| PX-478 | NCT00522652 | Advanced solid tumors/Lymphoma | I | Oncothyreon Inc. |
| AQ4N | NCT00394628 | Glioblastoma multiforme | Ib/IIa | Novacea |
| AQ4N | NCT00109356 | Lymphoma/Leukemia | I/II | Novacea |
| AQ4N | NCT00090727 | Solid tumors/Non-Hodgkin’s Lymphoma | I | Novacea |
Table describes clinical trials mentioned in review. Additional trials can be found at www.clinicaltrials.gov.
Acknowledgements
Work conducted in the authors’ laboratory was supported by NIH grants R01 CA077575 (“Causes and Consequences of Acid pH in Tumors”) and R01 CA125627 (“Imaging biomarkers for response to anti-cancer therapies”).
Non-standard abbreviations
- pHi
intracellular pH
- MRS
magnetic resonance spectroscopy
- 3-APP
3-aminopropyl phosphate
- pHe
extracellular pH
- PET
positron emission tomography
- FDG
18F-2-deoxyglucose
- FMISO
18F-fluoromisonidazole
- EPRI
Electron paramagnetic resonance imaging
- GLUT
glucose transporters
- RCC
renal cell carcinoma
- VHL
von Hippel-Lindau tumor suppressor
- 2DG
2-deoxyglucose
- 3-BrPA
3-bromopyruvate
- GAPdH
glyceraldehyde-3-phosphate dehydrogenase
- MDR
multi-drug resistance
- BPH
benign prostate hyperplasia
- EGFR
epidermal growth factor receptor
- ALT
alanine aminotransferase
- LDH
lactate dehydrogenase
- PFK
1 phosphofructokinase-1
- 3PO
3-(3-Pyrindinyl)-1-(4-Pyridinyl)-2-Propen-1-one
- PK
pyruvate kinase
- SSTR4
somatostatin receptor 4
- PdH
pyruvate dehydrogenase
- TCA
tricarboxylic acid cycle
- PDK
pyruvate dehydrogenase kinase
- DCA
dichloroacetate
- TMZ
temozolomide
- pO2
partial oxygen pressure
- HIF1α
hypoxia inducible factor 1α
- HRE
hypoxia response element
- DCE-MRI
dynamic contrast enhanced MRI
- LNA
locked nucleic acid
- DW-MRI
diffusion-weighted MRI
- SD
stable disease
- mTOR
mammalian target of rapamycin
- TSC1/2
tuberous sclerosis protein 1 complex
- PML
promyelocytic leukemia tumor suppressor
- AMPK
AMP-activated protein kinase
- ER
endoplasmic reticulum
- UPR
unfolded protein response
- PERK
PKR like ER kinase
- IRE-1
inositol requiring 1
- AFT6
activating transcription factor 6
- ERAD
ER-associated degradation
- TPZ
tirapazamine
- Br-IPM
bromo-isophosphoramide mustard
- AQ4
alkylaminoanthraquinone
- PARP1
poly(ADP-ribose) polymerase 1
- EPRI
electron paramagnetic resonance imaging
- VEGF
vascular endothelial growth factor
- PDGF
platelet-derived growth factor
- FX11
3-dihydroxy-6-methyl-7-(phenylmethyl)-4-propylnaphthalene-1-carboxylic acid
- CA
carbonic anhydrase
- MCT1
monocarboxylate transporter 1
- CHC
α-cyano-4-hydroxycinnamate
- IEPA
imidazole
- IR
ionizing radiation
- Pi
inorganic phosphate
Footnotes
Conflict of Interest Statement: The authors have no conflicts of interest to declare.
References
- Adachi E, Tannock IF. The effects of vasodilating drugs on pH in tumors. [Research Support, Non-U.S. Gov't] Oncol Res. 1999;11(4):179–185. [PubMed] [Google Scholar]
- Akter S, Clem BF, Lee HJ, Chesney J, Bae Y. Block Copolymer Micelles for Controlled Delivery of Glycolytic Enzyme Inhibitors. Pharm Res. 2011 doi: 10.1007/s11095-011-0613-4. [DOI] [PubMed] [Google Scholar]
- Akter S, Clem BF, Lee HJ, Chesney J, Bae Y. Block copolymer micelles for controlled delivery of glycolytic enzyme inhibitors. Pharm Res. 2012;29(3):847–855. doi: 10.1007/s11095-011-0613-4. [DOI] [PubMed] [Google Scholar]
- Altenberg B, Greulich KO. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. [Research Support, Non-U.S. Gov't] Genomics. 2004;84(6):1014–1020. doi: 10.1016/j.ygeno.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Anderson RF, Shinde SS, Hay MP, Gamage SA, Denny WA. Activation of 3-amino-1,2,4-benzotriazine 1,4-dioxide antitumor agents to oxidizing species following their one-electron reduction. J Am Chem Soc. 2003;125(3):748–756. doi: 10.1021/ja0209363. [DOI] [PubMed] [Google Scholar]
- Asada K, Miyamoto K, Fukutomi T, Tsuda H, Yagi Y, Wakazono K, et al. Reduced expression of GNA11 and silencing of MCT1 in human breast cancers. [Research Support, Non-U.S. Gov't] Oncology. 2003;64(4):380–388. doi: 10.1159/000070297. [DOI] [PubMed] [Google Scholar]
- Atsumi T, Chesney J, Metz C, Leng L, Donnelly S, Makita Z, et al. High expression of inducible 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (iPFK-2; PFKFB3) in human cancers. Cancer Res. 2002;62(20):5881–5887. [PubMed] [Google Scholar]
- Aulitzky WE, Aulitzky W, Gastl G, Lanske B, Reitter J, Frick J, et al. Acute effects of single doses of recombinant interferon-gamma on blood cell counts and lymphocyte subsets in patients with advanced renal cell cancer. J Interferon Res. 1989;9(4):425–433. doi: 10.1089/jir.1989.9.425. [DOI] [PubMed] [Google Scholar]
- Ayala FR, Rocha RM, Carvalho KC, Carvalho AL, da Cunha IW, Lourenco SV, et al. GLUT1 and GLUT3 as potential prognostic markers for Oral Squamous Cell Carcinoma. [Research Support, Non-U.S. Gov't] Molecules. 2010;15(4):2374–2387. doi: 10.3390/molecules15042374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayral-Kaloustian S, Gu J, Lucas J, Cinque M, Gaydos C, Zask A, et al. Hybrid inhibitors of phosphatidylinositol 3-kinase (PI3K) and the mammalian target of rapamycin (mTOR): design, synthesis, and superior antitumor activity of novel wortmannin-rapamycin conjugates. J Med Chem. 2010;53(1):452–459. doi: 10.1021/jm901427g. [DOI] [PubMed] [Google Scholar]
- Back SH, Lee K, Vink E, Kaufman RJ. Cytoplasmic IRE1alpha-mediated XBP1 mRNA splicing in the absence of nuclear processing and endoplasmic reticulum stress. [Research Support, N.I.H., Extramural] J Biol Chem. 2006;281(27):18691–18706. doi: 10.1074/jbc.M602030200. [DOI] [PubMed] [Google Scholar]
- Baker MA, Zeman EM, Hirst VK, Brown JM. Metabolism of SR 4233 by Chinese hamster ovary cells: basis of selective hypoxic cytotoxicity. Cancer Res. 1988;48(21):5947–5952. [PubMed] [Google Scholar]
- Belfi CA, Paul CR, Shan S, Ngo FQ. Comparison of the effects of hydralazine on tumor and normal tissue blood perfusion by MRI. [Comparative Study Research Support, U.S. Gov't, P.H.S.] Int J Radiat Oncol Biol Phys. 1994;29(3):473–479. doi: 10.1016/0360-3016(94)90441-3. [DOI] [PubMed] [Google Scholar]
- Ben Sahra I, Laurent K, Giuliano S, Larbret F, Ponzio G, Gounon P, et al. Targeting cancer cell metabolism: the combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. 2010;70(6):2465–2475. doi: 10.1158/0008-5472.CAN-09-2782. [DOI] [PubMed] [Google Scholar]
- Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, et al. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008;27(25):3576–3586. doi: 10.1038/sj.onc.1211024. [DOI] [PubMed] [Google Scholar]
- Bennewith KL, Raleigh JA, Durand RE. Orally administered pimonidazole to label hypoxic tumor cells. Cancer Res. 2002;62(23):6827–6830. [PubMed] [Google Scholar]
- Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. [Research Support, Non-U.S. Gov't] Cell. 2006;126(1):107–120. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- Bernardi R, Guernah I, Jin D, Grisendi S, Alimonti A, Teruya-Feldstein J, et al. PML inhibits HIF-1alpha translation and neoangiogenesis through repression of mTOR. [Research Support, N.I.H., Extramural] Nature. 2006;442(7104):779–785. doi: 10.1038/nature05029. [DOI] [PubMed] [Google Scholar]
- Bi M, Naczki C, Koritzinsky M, Fels D, Blais J, Hu N, et al. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. [Comparative Study Research Support, N.I.H., Extramural Research Support, U.S. Gov't, P.H.S.] EMBO J. 2005;24(19):3470–3481. doi: 10.1038/sj.emboj.7600777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum MJ, Haspel HC, Rosen OM. Transformation of rat fibroblasts by FSV rapidly increases glucose transporter gene transcription. Science. 1987;235(4795):1495–1498. doi: 10.1126/science.3029870. [DOI] [PubMed] [Google Scholar]
- Birner P, Schindl M, Obermair A, Plank C, Breitenecker G, Oberhuber G. Overexpression of hypoxia-inducible factor 1alpha is a marker for an unfavorable prognosis in early-stage invasive cervical cancer. Cancer Res. 2000;60(17):4693–4696. [PubMed] [Google Scholar]
- Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11(1):37–51. doi: 10.1016/j.ccr.2006.10.020. [DOI] [PubMed] [Google Scholar]
- Bos R, van der Groep P, Greijer AE, Shvarts A, Meijer S, Pinedo HM, et al. Levels of hypoxia-inducible factor-1alpha independently predict prognosis in patients with lymph node negative breast carcinoma. Cancer. 2003;97(6):1573–1581. doi: 10.1002/cncr.11246. [DOI] [PubMed] [Google Scholar]
- Bos R, van Der Hoeven JJ, van Der Wall E, van Der Groep P, van Diest PJ, Comans EF, et al. Biologic correlates of (18)fluorodeoxyglucose uptake in human breast cancer measured by positron emission tomography. [Research Support, Non-U.S. Gov't] J Clin Oncol. 2002;20(2):379–387. doi: 10.1200/JCO.2002.20.2.379. [DOI] [PubMed] [Google Scholar]
- Bosca L, Mojena M, Ghysdael J, Rousseau GG, Hue L. Expression of the v-src or v-fps oncogene increases fructose 2,6-bisphosphate in chick-embryo fibroblasts. Novel mechanism for the stimulation of glycolysis by retroviruses. Biochem J. 1986;236(2):595–599. doi: 10.1042/bj2360595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J. 1998;329(Pt 1):191–196. doi: 10.1042/bj3290191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care. 2006;29(2):254–258. doi: 10.2337/diacare.29.02.06.dc05-1558. [DOI] [PubMed] [Google Scholar]
- Brawer MK. Lonidamine: basic science and rationale for treatment of prostatic proliferative disorders. Rev Urol. 2005;7(Suppl 7):S21–S26. [PMC free article] [PubMed] [Google Scholar]
- Bueno V, Binet I, Steger U, Bundick R, Ferguson D, Murray C, et al. The specific monocarboxylate transporter (MCT1) inhibitor, AR-C117977, a novel immunosuppressant, prolongs allograft survival in the mouse. [Research Support, Non-U.S. Gov't] Transplantation. 2007;84(9):1204–1207. doi: 10.1097/01.tp.0000287543.91765.41. [DOI] [PubMed] [Google Scholar]
- Burant CF, Bell GI. Mammalian facilitative glucose transporters: evidence for similar substrate recognition sites in functionally monomeric proteins. [Comparative Study Research Support, Non-U.S. Gov't Research Support, U.S. Gov't, P.H.S.] Biochemistry. 1992;31(42):10414–10420. doi: 10.1021/bi00157a032. [DOI] [PubMed] [Google Scholar]
- Cantuaria G, Fagotti A, Ferrandina G, Magalhaes A, Nadji M, Angioli R, et al. GLUT-1 expression in ovarian carcinoma: association with survival and response to chemotherapy. Cancer. 2001;92(5):1144–1150. doi: 10.1002/1097-0142(20010901)92:5<1144::aid-cncr1432>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, et al. Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate. 2008;68(11):1223–1231. doi: 10.1002/pros.20788. [DOI] [PubMed] [Google Scholar]
- Chan DA, Sutphin PD, Nguyen P, Turcotte S, Lai EW, Banh A, et al. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci Transl Med. 2011;3(94) doi: 10.1126/scitranslmed.3002394. 94ra70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong JH, Park ES, Liang J, Dennison JB, Tsavachidou D, Nguyen-Charles C, et al. Dual inhibition of tumor energy pathway by 2-deoxyglucose and metformin is effective against a broad spectrum of preclinical cancer models. Mol Cancer Ther. 2011;10(12):2350–2362. doi: 10.1158/1535-7163.MCT-11-0497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452(7184):230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- Chung AS, Lee J, Ferrara N. Targeting the tumour vasculature: insights from physiological angiogenesis. Nat Rev Cancer. 2010;10(7):505–514. doi: 10.1038/nrc2868. [DOI] [PubMed] [Google Scholar]
- Cirstea D, Hideshima T, Rodig S, Santo L, Pozzi S, Vallet S, et al. Dual inhibition of akt/mammalian target of rapamycin pathway by nanoparticle albumin-bound-rapamycin and perifosine induces antitumor activity in multiple myeloma. Mol Cancer Ther. 2010;9(4):963–975. doi: 10.1158/1535-7163.MCT-09-0763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clem B, Telang S, Clem A, Yalcin A, Meier J, Simmons A, et al. Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol Cancer Ther. 2008;7(1):110–120. doi: 10.1158/1535-7163.MCT-07-0482. [DOI] [PubMed] [Google Scholar]
- Coleman MC, Asbury CR, Daniels D, Du J, Aykin-Burns N, Smith BJ, et al. 2-deoxy-D-glucose causes cytotoxicity, oxidative stress, and radiosensitization in pancreatic cancer. Free Radic Biol Med. 2008;44(3):322–331. doi: 10.1016/j.freeradbiomed.2007.08.032. [DOI] [PubMed] [Google Scholar]
- Connolly E, Braunstein S, Formenti S, Schneider RJ. Hypoxia inhibits protein synthesis through a 4E–BP1 and elongation factor 2 kinase pathway controlled by mTOR and uncoupled in breast cancer cells. [Comparative Study Research Support, Non-U.S. Gov't Research Support, U.S. Gov't, Non-P.H.S.] Mol Cell Biol. 2006;26(10):3955–3965. doi: 10.1128/MCB.26.10.3955-3965.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dazert E, Hall MN. mTOR signaling in disease. Curr Opin Cell Biol. 2011;23(6):744–755. doi: 10.1016/j.ceb.2011.09.003. [Research Support, Non-U.S. Gov't Review] [DOI] [PubMed] [Google Scholar]
- Dewhirst MW, Cao Y, Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer. 2008;8(6):425–437. doi: 10.1038/nrc2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditonno P, Battaglia M, Selvaggio O, Garofalo L, Lorusso V, Selvaggi FP. Clinical Evidence Supporting the Role of Lonidamine for the Treatment of BPH. Rev Urol. 2005;7(Suppl 7):S27–S33. [PMC free article] [PubMed] [Google Scholar]
- Dong B, Niwa M, Walter P, Silverman RH. Basis for regulated RNA cleavage by functional analysis of RNase L and Ire1p. [Comparative Study Research Support, U.S. Gov't, P.H.S.] RNA. 2001;7(3):361–373. doi: 10.1017/s1355838201002230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007;67(22):10804–10812. doi: 10.1158/0008-5472.CAN-07-2310. [DOI] [PubMed] [Google Scholar]
- Duan JX, Jiao H, Kaizerman J, Stanton T, Evans JW, Lan L, et al. Potent and highly selective hypoxia-activated achiral phosphoramidate mustards as anticancer drugs. J Med Chem. 2008;51(8):2412–2420. doi: 10.1021/jm701028q. [DOI] [PubMed] [Google Scholar]
- Elekes K, Helyes Z, Kereskai L, Sandor K, Pinter E, Pozsgai G, et al. Inhibitory effects of synthetic somatostatin receptor subtype 4 agonists on acute and chronic airway inflammation and hyperreactivity in the mouse. Eur J Pharmacol. 2008;578(2–3):313–322. doi: 10.1016/j.ejphar.2007.09.033. [DOI] [PubMed] [Google Scholar]
- Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330(7503):1304–1305. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans JW, Chernikova SB, Kachnic LA, Banath JP, Sordet O, Delahoussaye YM, et al. Homologous recombination is the principal pathway for the repair of DNA damage induced by tirapazamine in mammalian cells. Cancer Res. 2008;68(1):257–265. doi: 10.1158/0008-5472.CAN-06-4497. [DOI] [PubMed] [Google Scholar]
- Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9(6):425–434. doi: 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]
- Fels DR, Ye J, Segan AT, Kridel SJ, Spiotto M, Olson M, et al. Preferential cytotoxicity of bortezomib toward hypoxic tumor cells via overactivation of endoplasmic reticulum stress pathways. Cancer Res. 2008;68(22):9323–9330. doi: 10.1158/0008-5472.CAN-08-2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth JD, Ebert BL, Ratcliffe PJ. Hypoxic regulation of lactate dehydrogenase A. Interaction between hypoxia-inducible factor 1 and cAMP response elements. [Research Support, Non-U.S. Gov't] J Biol Chem. 1995;270(36):21021–21027. doi: 10.1074/jbc.270.36.21021. [DOI] [PubMed] [Google Scholar]
- Flier JS, Mueckler MM, Usher P, Lodish HF. Elevated levels of glucose transport and transporter messenger RNA are induced by ras or src oncogenes. Science. 1987;235(4795):1492–1495. doi: 10.1126/science.3103217. [DOI] [PubMed] [Google Scholar]
- Floridi A, Lehninger AL. Action of the antitumor and antispermatogenic agent lonidamine on electron transport in Ehrlich ascites tumor mitochondria. Arch Biochem Biophys. 1983;226(1):73–83. doi: 10.1016/0003-9861(83)90272-2. [DOI] [PubMed] [Google Scholar]
- Floridi A, Paggi MG, Marcante ML, Silvestrini B, Caputo A, De Martino C. Lonidamine, a selective inhibitor of aerobic glycolysis of murine tumor cells. J Natl Cancer Inst. 1981;66(3):497–499. [PubMed] [Google Scholar]
- Gabardi S, Baroletti SA. Everolimus: a proliferation signal inhibitor with clinical applications in organ transplantation, oncology, and cardiology. Pharmacotherapy. 2010;30(10):1044–1056. doi: 10.1592/phco.30.10.1044. [DOI] [PubMed] [Google Scholar]
- Gallagher FA, Kettunen MI, Day SE, Hu DE, Ardenkjaer-Larsen JH, Zandt R, et al. Magnetic resonance imaging of pH in vivo using hyperpolarized 13C–labelled bicarbonate. Nature. 2008;453(7197):940–943. doi: 10.1038/nature07017. [DOI] [PubMed] [Google Scholar]
- Gallagher R, Hughes CM, Murray MM, Friery OP, Patterson LH, Hirst DG, et al. The chemopotentiation of cisplatin by the novel bioreductive drug AQ4N. [Comparative Study Research Support, Non-U.S. Gov't] Br J Cancer. 2001;85(4):625–629. doi: 10.1054/bjoc.2001.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganjoo KN, Cranmer LD, Butrynski JE, Rushing D, Adkins D, Okuno SH, et al. A phase I study of the safety and pharmacokinetics of the hypoxia-activated prodrug TH-302 in combination with doxorubicin in patients with advanced soft tissue sarcoma. Oncology. 2011;80(1–2):50–56. doi: 10.1159/000327739. [DOI] [PubMed] [Google Scholar]
- Garcia-Martin ML, Martinez GV, Raghunand N, Sherry AD, Zhang S, Gillies RJ. High resolution pH(e) imaging of rat glioma using pH-dependent relaxivity. Magn Reson Med. 2006;55(2):309–315. doi: 10.1002/mrm.20773. [DOI] [PubMed] [Google Scholar]
- Gatenby RA, Gillies RJ. A microenvironmental model of carcinogenesis. Nat Rev Cancer. 2008;8(1):56–61. doi: 10.1038/nrc2255. [Review] [DOI] [PubMed] [Google Scholar]
- Giatromanolaki A, Koukourakis MI, Sivridis E, Turley H, Talks K, Pezzella F, et al. Relation of hypoxia inducible factor 1 alpha and 2 alpha in operable non-small cell lung cancer to angiogenic/molecular profile of tumours and survival. Br J Cancer. 2001;85(6):881–890. doi: 10.1054/bjoc.2001.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillies RJ, Morse DL. In vivo magnetic resonance spectroscopy in cancer. Annu Rev Biomed Eng. 2005;7:287–326. doi: 10.1146/annurev.bioeng.7.060804.100411. [DOI] [PubMed] [Google Scholar]
- Gillies RJ, Robey I, Gatenby RA. Causes and consequences of increased glucose metabolism of cancers. J Nucl Med. 2008;49(Suppl 2):24S–42S. doi: 10.2967/jnumed.107.047258. [DOI] [PubMed] [Google Scholar]
- Gillies RJ, Schornack PA, Secomb TW, Raghunand N. Causes and effects of heterogeneous perfusion in tumors. Neoplasia. 1999;1(3):197–207. doi: 10.1038/sj.neo.7900037. [Research Support, U.S. Gov't, P.H.S. Review] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel A, Mathupala SP, Pedersen PL. Glucose metabolism in cancer. Evidence that demethylation events play a role in activating type II hexokinase gene expression. J Biol Chem. 2003;278(17):15333–15340. doi: 10.1074/jbc.M300608200. [DOI] [PubMed] [Google Scholar]
- Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. 2011;91(3):1071–1121. doi: 10.1152/physrev.00038.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman RD, Kaplan NO, Hall TC. Lactic Dehydrogenase in Human Neoplastic Tissues. Cancer Res. 1964;24:389–399. [PubMed] [Google Scholar]
- Gould GW, Thomas HM, Jess TJ, Bell GI. Expression of human glucose transporters in Xenopus oocytes: kinetic characterization and substrate specificities of the erythrocyte, liver, and brain isoforms. Biochemistry. 1991;30(21):5139–5145. doi: 10.1021/bi00235a004. [DOI] [PubMed] [Google Scholar]
- Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW, et al. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. [Research Support, Non-U.S. Gov't Research Support, U.S. Gov't, P.H.S.] Nature. 1996;379(6560):88–91. doi: 10.1038/379088a0. [DOI] [PubMed] [Google Scholar]
- Granchi C, Roy S, Giacomelli C, Macchia M, Tuccinardi T, Martinelli A, et al. Discovery of N-hydroxyindole-based inhibitors of human lactate dehydrogenase isoform A (LDH-A) as starvation agents against cancer cells. J Med Chem. 2011;54(6):1599–1612. doi: 10.1021/jm101007q. [DOI] [PubMed] [Google Scholar]
- Greenberg R, Haddad R, Kashtan H, Kaplan O. The effects of somatostatin and octreotide on experimental and human acute pancreatitis. J Lab Clin Med. 2000;135(2):112–121. doi: 10.1067/mlc.2000.104457. [DOI] [PubMed] [Google Scholar]
- Greenberger LM, Horak ID, Filpula D, Sapra P, Westergaard M, Frydenlund HF, et al. A RNA antagonist of hypoxia-inducible factor-1alpha, EZN-2968, inhibits tumor cell growth. Mol Cancer Ther. 2008;7(11):3598–3608. doi: 10.1158/1535-7163.MCT-08-0510. [DOI] [PubMed] [Google Scholar]
- Griffiths JR. Are cancer cells acidic? Br J Cancer. 1991;64(3):425–427. doi: 10.1038/bjc.1991.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinstein S, Rotin D, Mason MJ. Na+/H+ exchange and growth factor-induced cytosolic pH changes. Role in cellular proliferation. Biochim Biophys Acta. 988(1989)(1):73–97. doi: 10.1016/0304-4157(89)90004-x. [Research Support, Non-U.S. Gov't Review] [DOI] [PubMed] [Google Scholar]
- Han D, Lerner AG, Vande Walle L, Upton JP, Xu W, Hagen A, et al. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't] Cell. 2009;138(3):562–575. doi: 10.1016/j.cell.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hashim AI, Zhang X, Wojtkowiak JW, Martinez GV, Gillies RJ. Imaging pH and metastasis. NMR Biomed. 2011;24(6):582–591. doi: 10.1002/nbm.1644. [Review] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatanaka M. Transport of sugars in tumor cell membranes. Biochim Biophys Acta. 1974;355(1):77–104. doi: 10.1016/0304-419x(74)90008-0. [DOI] [PubMed] [Google Scholar]
- Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991;253(5022):905–909. doi: 10.1126/science.1715094. [DOI] [PubMed] [Google Scholar]
- Hendricksen K, van der Heijden AG, Cornel EB, Vergunst H, de Reijke TM, van Boven E, et al. Two-year follow-up of the phase II marker lesion study of intravesical apaziquone for patients with non-muscle invasive bladder cancer. World J Urol. 2009;27(3):337–342. doi: 10.1007/s00345-009-0382-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess G, Herbrecht R, Romaguera J, Verhoef G, Crump M, Gisselbrecht C, et al. Phase III study to evaluate temsirolimus compared with investigator's choice therapy for the treatment of relapsed or refractory mantle cell lymphoma. J Clin Oncol. 2009;27(23):3822–3829. doi: 10.1200/JCO.2008.20.7977. [DOI] [PubMed] [Google Scholar]
- Hicks KO, Fleming Y, Siim BG, Koch CJ, Wilson WR. Extravascular diffusion of tirapazamine: effect of metabolic consumption assessed using the multicellular layer model. Int J Radiat Oncol Biol Phys. 1998;42(3):641–649. doi: 10.1016/s0360-3016(98)00268-5. [DOI] [PubMed] [Google Scholar]
- Hicks KO, Siim BG, Jaiswal JK, Pruijn FB, Fraser AM, Patel R, et al. Pharmacokinetic/pharmacodynamic modeling identifies SN30000 and SN29751 as tirapazamine analogues with improved tissue penetration and hypoxic cell killing in tumors. Clin Cancer Res. 2010;16(20):4946–4957. doi: 10.1158/1078-0432.CCR-10-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockel M, Vaupel P. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst. 2001;93(4):266–276. doi: 10.1093/jnci/93.4.266. [Research Support, Non-U.S. Gov't Review] [DOI] [PubMed] [Google Scholar]
- Holness MJ, Sugden MC. Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochem Soc Trans. 2003;31(Pt 6):1143–1151. doi: 10.1042/bst0311143. [Research Support, Non-U.S. Gov't Review] [DOI] [PubMed] [Google Scholar]
- Horsman MR, Christensen KL, Overgaard J. Relationship between the hydralazine-induced changes in murine tumor blood supply and mouse blood pressure. [Research Support, Non-U.S. Gov't] Int J Radiat Oncol Biol Phys. 1992;22(3):455–458. doi: 10.1016/0360-3016(92)90852-9. [DOI] [PubMed] [Google Scholar]
- Houchens DP, Ovejera AA, Riblet SM, Slagel DE. Human brain tumor xenografts in nude mice as a chemotherapy model. Eur J Cancer Clin Oncol. 1983;19(6):799–805. doi: 10.1016/0277-5379(83)90012-3. [DOI] [PubMed] [Google Scholar]
- Hsiang YH, Hertzberg R, Hecht S, Liu LF. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. [Research Support, U.S. Gov't, P.H.S.] J Biol Chem. 1985;260(27):14873–14878. [PubMed] [Google Scholar]
- Hu J, Handisides DR, Van Valckenborgh E, De Raeve H, Menu E, Vande Broek I, et al. Targeting the multiple myeloma hypoxic niche with TH-302, a hypoxia-activated prodrug. Blood. 2010;116(9):1524–1527. doi: 10.1182/blood-2010-02-269126. [DOI] [PubMed] [Google Scholar]
- Hudes G, Carducci M, Tomczak P, Dutcher J, Figlin R, Kapoor A, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007;356(22):2271–2281. doi: 10.1056/NEJMoa066838. [DOI] [PubMed] [Google Scholar]
- Hundal RS, Krssak M, Dufour S, Laurent D, Lebon V, Chandramouli V, et al. Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes. 2000;49(12):2063–2069. doi: 10.2337/diabetes.49.12.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim Hashim A, Cornnell HH, Coelho Ribeiro Mde L, Abrahams D, Cunningham J, Lloyd M, et al. Reduction of metastasis using a non-volatile buffer. Clin Exp Metastasis. 2011;28(8):841–849. doi: 10.1007/s10585-011-9415-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikezoe T, Nishioka C, Bandobashi K, Yang Y, Kuwayama Y, Adachi Y, et al. Longitudinal inhibition of PI3K/Akt/mTOR signaling by LY294002 and rapamycin induces growth arrest of adult T-cell leukemia cells. Leuk Res. 2007;31(5):673–682. doi: 10.1016/j.leukres.2006.08.001. [DOI] [PubMed] [Google Scholar]
- Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115(5):577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- Iyer VN, Szybalski W. Mitomycins and Porfiromycin: Chemical Mechanism of Activation and Cross-Linking of DNA. Science. 1964;145(3627):55–58. doi: 10.1126/science.145.3627.55. [DOI] [PubMed] [Google Scholar]
- J., I.-H. A. W. J. W. R. M. E. V. B. K. M. C. H. H. G. R. A. G. R. Free Base Lysine Increases Survival and Reduces Metastasis in Prostate Cancer Model. J Cancer Sci Ther. 2011:S1. [PMC free article] [PubMed] [Google Scholar]
- Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. [Research Support, Non-U.S. Gov't] Science. 2001;292(5516):468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- Jahde E, Glusenkamp KH, Rajewsky MF. Protection of cultured malignant cells from mitoxantrone cytotoxicity by low extracellular pH: a possible mechanism for chemoresistance in vivo. Eur J Cancer. 1990;26(2):101–106. doi: 10.1016/0277-5379(90)90290-a. [DOI] [PubMed] [Google Scholar]
- Jain RK. Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med. 2001;7(9):987–989. doi: 10.1038/nm0901-987. [DOI] [PubMed] [Google Scholar]
- Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307(5706):58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- Jordan BF, Runquist M, Raghunand N, Baker A, Williams R, Kirkpatrick L, et al. Dynamic contrast-enhanced and diffusion MRI show rapid and dramatic changes in tumor microenvironment in response to inhibition of HIF-1alpha using PX-478. Neoplasia. 2005;7(5):475–485. doi: 10.1593/neo.04628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010;584(7):1287–1295. doi: 10.1016/j.febslet.2010.01.017. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't Review] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaper F, Dornhoefer N, Giaccia AJ. Mutations in the PI3K/PTEN/TSC2 pathway contribute to mammalian target of rapamycin activity and increased translation under hypoxic conditions. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't] Cancer Res. 2006;66(3):1561–1569. doi: 10.1158/0008-5472.CAN-05-3375. [DOI] [PubMed] [Google Scholar]
- Kasahara M, Hinkle PC. Reconstitution and purification of the D-glucose transporter from human erythrocytes. J Biol Chem. 1977;252(20):7384–7390. [PubMed] [Google Scholar]
- Kauppinen RA, Nicholls DG. Synaptosomal bioenergetics. The role of glycolysis, pyruvate oxidation and responses to hypoglycaemia. [In Vitro Research Support, Non-U.S. Gov't] Eur J Biochem. 1986;158(1):159–165. doi: 10.1111/j.1432-1033.1986.tb09733.x. [DOI] [PubMed] [Google Scholar]
- Kelloff GJ, Hoffman JM, Johnson B, Scher HI, Siegel BA, Cheng EY, et al. Progress and promise of FDG-PET imaging for cancer patient management and oncologic drug development. Clin Cancer Res. 2005;11(8):2785–2808. doi: 10.1158/1078-0432.CCR-04-2626. [DOI] [PubMed] [Google Scholar]
- Kim Y, Lin Q, Glazer PM, Yun Z. Hypoxic tumor microenvironment and cancer cell differentiation. Curr Mol Med. 2009;9(4):425–434. doi: 10.2174/156652409788167113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoechel TR, Tucker AD, Robinson CM, Phillips C, Taylor W, Bungay PJ, et al. Regulatory roles of the N-terminal domain based on crystal structures of human pyruvate dehydrogenase kinase 2 containing physiological and synthetic ligands. Biochemistry. 2006;45(2):402–415. doi: 10.1021/bi051402s. [DOI] [PubMed] [Google Scholar]
- Ko YH, Pedersen PL, Geschwind JF. Glucose catabolism in the rabbit VX2 tumor model for liver cancer: characterization and targeting hexokinase. Cancer Lett. 2001;173(1):83–91. doi: 10.1016/s0304-3835(01)00667-x. [DOI] [PubMed] [Google Scholar]
- Koh MY, Spivak-Kroizman T, Venturini S, Welsh S, Williams RR, Kirkpatrick DL, et al. Molecular mechanisms for the activity of PX-478, an antitumor inhibitor of the hypoxia-inducible factor-1alpha. Mol Cancer Ther. 2008;7(1):90–100. doi: 10.1158/1535-7163.MCT-07-0463. [DOI] [PubMed] [Google Scholar]
- Kole HK, Resnick RJ, Van Doren M, Racker E. Regulation of 6-phosphofructo-1-kinase activity in ras-transformed rat-1 fibroblasts. Arch Biochem Biophys. 1991;286(2):586–590. doi: 10.1016/0003-9861(91)90084-v. [DOI] [PubMed] [Google Scholar]
- Korennykh AV, Egea PF, Korostelev AA, Finer-Moore J, Zhang C, Shokat KM, et al. The unfolded protein response signals through high-order assembly of Ire1. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't] Nature. 2009;457(7230):687–693. doi: 10.1038/nature07661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koumenis C, Naczki C, Koritzinsky M, Rastani S, Diehl A, Sonenberg N, et al. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. [Research Support, Non-U.S. Gov't Research Support, U.S. Gov't, P.H.S.] Mol Cell Biol. 2002;22(21):7405–7416. doi: 10.1128/MCB.22.21.7405-7416.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer's Achilles' heel. Cancer Cell. 2008;13(6):472–482. doi: 10.1016/j.ccr.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Kummar S, Raffeld M, Juwara L, Horneffer Y, Strassberger A, Allen D, et al. Multihistology, target-driven pilot trial of oral topotecan as an inhibitor of hypoxia-inducible factor-1alpha in advanced solid tumors. [Clinical Trial, Phase I Research Support, N.I.H., Extramural] Clin Cancer Res. 2011;17(15):5123–5131. doi: 10.1158/1078-0432.CCR-11-0682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtoglu M, Maher JC, Lampidis TJ. Differential toxic mechanisms of 2-deoxy-D-glucose versus 2-fluorodeoxy-D-glucose in hypoxic and normoxic tumor cells. Antioxid Redox Signal. 2007;9(9):1383–1390. doi: 10.1089/ars.2007.1714. [DOI] [PubMed] [Google Scholar]
- Kyle AH, Minchinton AI. Measurement of delivery and metabolism of tirapazamine to tumour tissue using the multilayered cell culture model. Cancer Chemother Pharmacol. 1999;43(3):213–220. doi: 10.1007/s002800050886. [DOI] [PubMed] [Google Scholar]
- Lampidis TJ, Kurtoglu M, Maher JC, Liu H, Krishan A, Sheft V, et al. Efficacy of 2-halogen substituted D-glucose analogs in blocking glycolysis and killing "hypoxic tumor cells". Cancer Chemother Pharmacol. 2006;58(6):725–734. doi: 10.1007/s00280-006-0207-8. [DOI] [PubMed] [Google Scholar]
- Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, et al. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't] Proc Natl Acad Sci U S A. 2010;107(5):2037–2042. doi: 10.1073/pnas.0914433107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le QT, McCoy J, Williamson S, Ryu J, Gaspar LE, Edelman MJ, et al. Phase I study of tirapazamine plus cisplatin/etoposide and concurrent thoracic radiotherapy in limited-stage small cell lung cancer (S0004): a Southwest Oncology Group study. Clin Cancer Res. 2004;10(16):5418–5424. doi: 10.1158/1078-0432.CCR-04-0436. [DOI] [PubMed] [Google Scholar]
- Lee AH, Iwakoshi NN, Anderson KC, Glimcher LH. Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. [Research Support, Non-U.S. Gov't Research Support, U.S. Gov't, P.H.S.] Proc Natl Acad Sci U S A. 2003;100(17):9946–9951. doi: 10.1073/pnas.1334037100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Lin X, Staver M, Shoemaker A, Semizarov D, Fesik SW, et al. Evaluating hypoxia-inducible factor-1alpha as a cancer therapeutic target via inducible RNA interference in vivo. [Research Support, Non-U.S. Gov't] Cancer Res. 2005;65(16):7249–7258. doi: 10.1158/0008-5472.CAN-04-4426. [DOI] [PubMed] [Google Scholar]
- Li Y, Wang Y, Kim E, Beemiller P, Wang CY, Swanson J, et al. Bnip3 mediates the hypoxia-induced inhibition on mammalian target of rapamycin by interacting with Rheb. [Research Support, N.I.H., Extramural Research Support, U.S. Gov't, Non-P.H.S.] J Biol Chem. 2007;282(49):35803–35813. doi: 10.1074/jbc.M705231200. [DOI] [PubMed] [Google Scholar]
- Li YC, Fung KP, Kwok TT, Lee CY, Suen YK, Kong SK. Mitochondrial targeting drug lonidamine triggered apoptosis in doxorubicin-resistant HepG2 cells. Life Sci. 2002;71(23):2729–2740. doi: 10.1016/s0024-3205(02)02103-3. [DOI] [PubMed] [Google Scholar]
- Lin X, Zhang F, Bradbury CM, Kaushal A, Li L, Spitz DR, et al. 2-Deoxy-D-glucose-induced cytotoxicity and radiosensitization in tumor cells is mediated via disruptions in thiol metabolism. Cancer Res. 2003;63(12):3413–3417. [PubMed] [Google Scholar]
- Liu L, Cash TP, Jones RG, Keith B, Thompson CB, Simon MC. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol Cell. 2006;21(4):521–531. doi: 10.1016/j.molcel.2006.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou Y, McDonald PC, Oloumi A, Chia S, Ostlund C, Ahmadi A, et al. Targeting tumor hypoxia: suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors. Cancer Res. 2011;71(9):3364–3376. doi: 10.1158/0008-5472.CAN-10-4261. [DOI] [PubMed] [Google Scholar]
- Luciani F, Spada M, De Milito A, Molinari A, Rivoltini L, Montinaro A, et al. Effect of proton pump inhibitor pretreatment on resistance of solid tumors to cytotoxic drugs. J Natl Cancer Inst. 2004;96(22):1702–1713. doi: 10.1093/jnci/djh305. [DOI] [PubMed] [Google Scholar]
- Mathupala SP, Heese C, Pedersen PL. Glucose catabolism in cancer cells. The type II hexokinase promoter contains functionally active response elements for the tumor suppressor p53. J Biol Chem. 1997;272(36):22776–22780. doi: 10.1074/jbc.272.36.22776. [DOI] [PubMed] [Google Scholar]
- Matsumoto S, Batra S, Saito K, Yasui H, Choudhuri R, Gadisetti C, et al. Antiangiogenic agent sunitinib transiently increases tumor oxygenation and suppresses cycling hypoxia. Cancer Res. 2011;71(20):6350–6359. doi: 10.1158/0008-5472.CAN-11-2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto S, Yasui H, Mitchell JB, Krishna MC. Imaging cycling tumor hypoxia. Cancer Res. 2010;70(24):10019–10023. doi: 10.1158/0008-5472.CAN-10-2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeage MJ, Gu Y, Wilson WR, Hill A, Amies K, Melink TJ, et al. A phase I trial of PR-104, a pre-prodrug of the bioreductive prodrug PR-104A, given weekly to solid tumour patients. BMC Cancer. 2011;11:432. doi: 10.1186/1471-2407-11-432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meloche HP, Luczak MA, Wurster JM. The substrate analog, bromopyruvate, as both a substrate and alkylating agent for 2-keto-3-deoxy-6-phosphogluconic aldolase. Kinetic and stereochemical studies. J Biol Chem. 1972;247(13):4186–4191. [PubMed] [Google Scholar]
- Meng F, Evans JW, Bhupathi D, Banica M, Lan L, Lorente G, et al. Molecular and Cellular Pharmacology of the Hypoxia-Activated Prodrug TH-302. Mol Cancer Ther. 2012 doi: 10.1158/1535-7163.MCT-11-0634. [DOI] [PubMed] [Google Scholar]
- Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010;2(31) doi: 10.1126/scitranslmed.3000677. 31ra34. [DOI] [PubMed] [Google Scholar]
- Milane L, Duan Z, Amiji M. Development of EGFR-targeted polymer blend nanocarriers for combination paclitaxel/lonidamine delivery to treat multi-drug resistance in human breast and ovarian tumor cells. Mol Pharm. 2011a;8(1):185–203. doi: 10.1021/mp1002653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milane L, Duan Z, Amiji M. Therapeutic efficacy and safety of paclitaxel/lonidamine loaded EGFR-targeted nanoparticles for the treatment of multi-drug resistant cancer. PLoS One. 2011b;6(9):e24075. doi: 10.1371/journal.pone.0024075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell DC, Bryan BA. Anti-angiogenic therapy: adapting strategies to overcome resistant tumors. J Cell Biochem. 2010;111(3):543–553. doi: 10.1002/jcb.22764. [Review] [DOI] [PubMed] [Google Scholar]
- Moellering RE, Black KC, Krishnamurty C, Baggett BK, Stafford P, Rain M, et al. Acid treatment of melanoma cells selects for invasive phenotypes. Clin Exp Metastasis. 2008;25(4):411–425. doi: 10.1007/s10585-008-9145-7. [DOI] [PubMed] [Google Scholar]
- Mohanti BK, Rath GK, Anantha N, Kannan V, Das BS, Chandramouli BA, et al. Improving cancer radiotherapy with 2-deoxy-D-glucose: phase I/II clinical trials on human cerebral gliomas. Int J Radiat Oncol Biol Phys. 1996;35(1):103–111. doi: 10.1016/s0360-3016(96)85017-6. [DOI] [PubMed] [Google Scholar]
- Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372(9637):449–456. doi: 10.1016/S0140-6736(08)61039-9. [DOI] [PubMed] [Google Scholar]
- Nakano A, Tsuji D, Miki H, Cui Q, El Sayed SM, Ikegame A, et al. Glycolysis inhibition inactivates ABC transporters to restore drug sensitivity in malignant cells. PLoS One. 2011;6(11):e27222. doi: 10.1371/journal.pone.0027222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrocki ST, Carew JS, Dunner K, Jr, Boise LH, Chiao PJ, Huang P, et al. Bortezomib inhibits PKR-like endoplasmic reticulum (ER) kinase and induces apoptosis via ER stress in human pancreatic cancer cells. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't] Cancer Res. 2005;65(24):11510–11519. doi: 10.1158/0008-5472.CAN-05-2394. [DOI] [PubMed] [Google Scholar]
- Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov. 2011;10(10):767–777. doi: 10.1038/nrd3554. [Research Support, Non-U.S. Gov't Review] [DOI] [PubMed] [Google Scholar]
- Nock S, Gonzalez TN, Sidrauski C, Niwa M, Walter P. Purification and activity assays of the catalytic domains of the kinase/endoribonuclease Ire1p from Saccharomyces cerevisiae. [Research Support, Non-U.S. Gov't Research Support, U.S. Gov't, P.H.S.] Methods Enzymol. 2001;342:3–10. doi: 10.1016/s0076-6879(01)42530-4. [DOI] [PubMed] [Google Scholar]
- Nordsmark M, Maxwell RJ, Wood PJ, Stratford IJ, Adams GE, Overgaard J, et al. Effect of hydralazine in spontaneous tumours assessed by oxygen electrodes and 31P-magnetic resonance spectroscopy. [Research Support, Non-U.S. Gov't] Br J Cancer Suppl. 1996;27:S232–S235. [PMC free article] [PubMed] [Google Scholar]
- Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, et al. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. [Research Support, Non-U.S. Gov't] Nat Cell Biol. 2000;2(7):423–427. doi: 10.1038/35017054. [DOI] [PubMed] [Google Scholar]
- Okunieff P, Kallinowski F, Vaupel P, Neuringer LJ. Effects of hydralazine-induced vasodilation on the energy metabolism of murine tumors studied by in vivo 31P-nuclear magnetic resonance spectroscopy. [Research Support, Non-U.S. Gov't Research Support, U.S. Gov't, P.H.S.] J Natl Cancer Inst. 1988;80(10):745–750. doi: 10.1093/jnci/80.10.745. [DOI] [PubMed] [Google Scholar]
- Osada R, Horiuchi A, Kikuchi N, Yoshida J, Hayashi A, Ota M, et al. Expression of hypoxia-inducible factor 1alpha, hypoxia-inducible factor 2alpha, and von Hippel-Lindau protein in epithelial ovarian neoplasms and allelic loss of von Hippel-Lindau gene: nuclear expression of hypoxia-inducible factor 1alpha is an independent prognostic factor in ovarian carcinoma. [Research Support, Non-U.S. Gov't] Hum Pathol. 2007;38(9):1310–1320. doi: 10.1016/j.humpath.2007.02.010. [DOI] [PubMed] [Google Scholar]
- Owa T, Yoshino H, Okauchi T, Yoshimatsu K, Ozawa Y, Sugi NH, et al. Discovery of novel antitumor sulfonamides targeting G1 phase of the cell cycle. J Med Chem. 1999;42(19):3789–3799. doi: 10.1021/jm9902638. [DOI] [PubMed] [Google Scholar]
- Pacheco-Torres J, Lopez-Larrubia P, Ballesteros P, Cerdan S. Imaging tumor hypoxia by magnetic resonance methods. NMR Biomed. 2011;24(1):1–16. doi: 10.1002/nbm.1558. [Research Support, Non-U.S. Gov't Review] [DOI] [PubMed] [Google Scholar]
- Palayoor ST, Mitchell JB, Cerna D, Degraff W, John-Aryankalayil M, Coleman CN. PX-478, an inhibitor of hypoxia-inducible factor-1alpha, enhances radiosensitivity of prostate carcinoma cells. Int J Cancer. 2008;123(10):2430–2437. doi: 10.1002/ijc.23807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos KP, Goel S, Beeram M, Wong A, Desai K, Haigentz M, et al. A phase 1 open-label, accelerated dose-escalation study of the hypoxia-activated prodrug AQ4N in patients with advanced malignancies. [Clinical Trial, Phase I Research Support, Non-U.S. Gov't] Clin Cancer Res. 2008;14(21):7110–7115. doi: 10.1158/1078-0432.CCR-08-0483. [DOI] [PubMed] [Google Scholar]
- Papandreou I, Denko NC, Olson M, Van Melckebeke H, Lust S, Tam A, et al. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't] Blood. 2011;117(4):1311–1314. doi: 10.1182/blood-2010-08-303099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parveen I, Naughton DP, Whish WJ, Threadgill MD. 2-nitroimidazol-5-ylmethyl as a potential bioreductively activated prodrug system: reductively triggered release of the PARP inhibitor 5-bromoisoquinolinone. Bioorg Med Chem Lett. 1999;9(14):2031–2036. doi: 10.1016/s0960-894x(99)00306-6. [DOI] [PubMed] [Google Scholar]
- Patterson LH, McKeown SR, Ruparelia K, Double JA, Bibby MC, Cole S, et al. Enhancement of chemotherapy and radiotherapy of murine tumours by AQ4N, a bioreductively activated anti-tumour agent. Br J Cancer. 2000;82(12):1984–1990. doi: 10.1054/bjoc.2000.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Sayans M, Somoza-Martin JM, Barros-Angueira F, Rey JM, Garcia-Garcia A. V-ATPase inhibitors and implication in cancer treatment. Cancer Treat Rev. 2009;35(8):707–713. doi: 10.1016/j.ctrv.2009.08.003. [DOI] [PubMed] [Google Scholar]
- Pinheiro C, Longatto-Filho A, Ferreira L, Pereira SM, Etlinger D, Moreira MA, et al. Increasing expression of monocarboxylate transporters 1 and 4 along progression to invasive cervical carcinoma. [Research Support, Non-U.S. Gov't] Int J Gynecol Pathol. 2008;27(4):568–574. doi: 10.1097/PGP.0b013e31817b5b40. [DOI] [PubMed] [Google Scholar]
- Pinheiro C, Longatto-Filho A, Scapulatempo C, Ferreira L, Martins S, Pellerin L, et al. Increased expression of monocarboxylate transporters 1, 2, and 4 in colorectal carcinomas. Virchows Arch. 2008;452(2):139–146. doi: 10.1007/s00428-007-0558-5. [DOI] [PubMed] [Google Scholar]
- Pinheiro C, Sousa B, Albergaria A, Paredes J, Dufloth R, Vieira D, et al. GLUT1 and CAIX expression profiles in breast cancer correlate with adverse prognostic factors and MCT1 overexpression. [Research Support, Non-U.S. Gov't] Histol Histopathol. 2011;26(10):1279–1286. doi: 10.14670/HH-26.1279. [DOI] [PubMed] [Google Scholar]
- Raghunand N, Gillies RJ. pH and drug resistance in tumors. Drug Resist Updat. 2000;3(1):39–47. doi: 10.1054/drup.2000.0119. [DOI] [PubMed] [Google Scholar]
- Raghunand N, Howison C, Sherry AD, Zhang S, Gillies RJ. Renal and systemic pH imaging by contrast-enhanced MRI. Magn Reson Med. 2003;49(2):249–257. doi: 10.1002/mrm.10347. [DOI] [PubMed] [Google Scholar]
- Raghunand N, Mahoney B, van Sluis R, Baggett B, Gillies RJ. Acute metabolic alkalosis enhances response of C3H mouse mammary tumors to the weak base mitoxantrone. Neoplasia. 2001;3(3):227–235. doi: 10.1038/sj.neo.7900151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Peinado S, Alcazar-Limones F, Lagares-Tena L, El Mjiyad N, Caro-Maldonado A, Tirado OM, et al. 2-deoxyglucose induces Noxa-dependent apoptosis in alveolar rhabdomyosarcoma. Cancer Res. 2011 doi: 10.1158/0008-5472.CAN-11-0759. [DOI] [PubMed] [Google Scholar]
- Rapisarda A, Uranchimeg B, Scudiero DA, Selby M, Sausville EA, Shoemaker RH, et al. Identification of small molecule inhibitors of hypoxia-inducible factor 1 transcriptional activation pathway. [Research Support, U.S. Gov't, P.H.S.] Cancer Res. 2002;62(15):4316–4324. [PubMed] [Google Scholar]
- Rapisarda A, Uranchimeg B, Sordet O, Pommier Y, Shoemaker RH, Melillo G. Topoisomerase I-mediated inhibition of hypoxia-inducible factor 1: mechanism and therapeutic implications. [Research Support, U.S. Gov't, P.H.S.] Cancer Res. 2004;64(4):1475–1482. doi: 10.1158/0008-5472.can-03-3139. [DOI] [PubMed] [Google Scholar]
- Rapisarda A, Zalek J, Hollingshead M, Braunschweig T, Uranchimeg B, Bonomi CA, et al. Schedule-dependent inhibition of hypoxia-inducible factor-1alpha protein accumulation, angiogenesis, and tumor growth by topotecan in U251-HRE glioblastoma xenografts. [Research Support, U.S. Gov't, P.H.S.] Cancer Res. 2004;64(19):6845–6848. doi: 10.1158/0008-5472.CAN-04-2116. [DOI] [PubMed] [Google Scholar]
- Rastogi S, Banerjee S, Chellappan S, Simon GR. Glut-1 antibodies induce growth arrest and apoptosis in human cancer cell lines. Cancer Lett. 2007;257(2):244–251. doi: 10.1016/j.canlet.2007.07.021. [DOI] [PubMed] [Google Scholar]
- Ravagnan L, Marzo I, Costantini P, Susin SA, Zamzami N, Petit PX, et al. Lonidamine triggers apoptosis via a direct, Bcl-2-inhibited effect on the mitochondrial permeability transition pore. Oncogene. 1999;18(16):2537–2546. doi: 10.1038/sj.onc.1202625. [DOI] [PubMed] [Google Scholar]
- Rini BI. Temsirolimus, an inhibitor of mammalian target of rapamycin. Clin Cancer Res. 2008;14(5):1286–1290. doi: 10.1158/1078-0432.CCR-07-4719. [DOI] [PubMed] [Google Scholar]
- Rischin D, Peters L, Fisher R, Macann A, Denham J, Poulsen M, et al. Tirapazamine, Cisplatin, and Radiation versus Fluorouracil, Cisplatin, and Radiation in patients with locally advanced head and neck cancer: a randomized phase II trial of the Trans-Tasman Radiation Oncology Group (TROG 98.02) J Clin Oncol. 2005;23(1):79–87. doi: 10.1200/JCO.2005.01.072. [DOI] [PubMed] [Google Scholar]
- Rischin D, Peters LJ, O’Sullivan B, Giralt J, Fisher R, Yuen K, et al. Tirapazamine, cisplatin, and radiation versus cisplatin and radiation for advanced squamous cell carcinoma of the head and neck (TROG 02.02, HeadSTART): a phase III trial of the Trans-Tasman Radiation Oncology Group. J Clin Oncol. 2010;28(18):2989–2995. doi: 10.1200/JCO.2009.27.4449. [DOI] [PubMed] [Google Scholar]
- Robey IF, Baggett BK, Kirkpatrick ND, Roe DJ, Dosescu J, Sloane BF, et al. Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res. 2009;69(6):2260–2268. doi: 10.1158/0008-5472.CAN-07-5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rofstad EK. Microenvironment-induced cancer metastasis. Int J Radiat Biol. 2000;76(5):589–605. doi: 10.1080/095530000138259. [DOI] [PubMed] [Google Scholar]
- Rofstad EK, Mathiesen B, Kindem K, Galappathi K. Acidic extracellular pH promotes experimental metastasis of human melanoma cells in athymic nude mice. [Research Support, Non-U.S. Gov't] Cancer Res. 2006;66(13):6699–6707. doi: 10.1158/0008-5472.CAN-06-0983. [DOI] [PubMed] [Google Scholar]
- Romero-Ramirez L, Cao H, Nelson D, Hammond E, Lee AH, Yoshida H, et al. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. [Research Support, Non-U.S. Gov't Research Support, U.S. Gov't, P.H.S.] Cancer Res. 2004;64(17):5943–5947. doi: 10.1158/0008-5472.CAN-04-1606. [DOI] [PubMed] [Google Scholar]
- Rouschop KM, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. [Research Support, Non-U.S. Gov't] J Clin Invest. 2010;120(1):127–141. doi: 10.1172/JCI40027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Matsumoto S, Devasahayam N, Subramanian S, Munasinghe JP, Morris HD, et al. Transient decrease in tumor oxygenation after intravenous administration of pyruvate. Magn Reson Med. 2011 doi: 10.1002/mrm.23065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito S, Furuno A, Sakurai J, Sakamoto A, Park HR, Shin-Ya K, et al. Chemical genomics identifies the unfolded protein response as a target for selective cancer cell killing during glucose deprivation. [Research Support, Non-U.S. Gov't] Cancer Res. 2009;69(10):4225–4234. doi: 10.1158/0008-5472.CAN-08-2689. [DOI] [PubMed] [Google Scholar]
- Santo L, Hideshima T, Kung AL, Tseng JC, Tamang D, Yang M, et al. Preclinical activity, pharmacodynamic and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood. 2012 doi: 10.1182/blood-2011-10-387365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimmer AD, Thomas MP, Hurren R, Gronda M, Pellecchia M, Pond GR, et al. Identification of small molecules that sensitize resistant tumor cells to tumor necrosis factor-family death receptors. Cancer Res. 2006;66(4):2367–2375. doi: 10.1158/0008-5472.CAN-05-1061. [DOI] [PubMed] [Google Scholar]
- Schlappack OK, Zimmermann A, Hill RP. Glucose starvation and acidosis: effect on experimental metastatic potential, DNA content and MTX resistance of murine tumour cells. [Research Support, Non-U.S. Gov't] Br J Cancer. 1991;64(4):663–670. doi: 10.1038/bjc.1991.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz DL, Powis G, Thitai-Kumar A, He Y, Bankson J, Williams R, et al. The selective hypoxia inducible factor-1 inhibitor PX-478 provides in vivo radiosensitization through tumor stromal effects. Mol Cancer Ther. 2009;8(4):947–958. doi: 10.1158/1535-7163.MCT-08-0981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz HS, Sodergren JE, Philips FS. Mitomycin C: Chemical and Biological Studies on Alkylation. Science. 1963;142(3596):1181–1183. doi: 10.1126/science.142.3596.1181. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3(10):721–732. doi: 10.1038/nrc1187. [Review] [DOI] [PubMed] [Google Scholar]
- Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, et al. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. [Research Support, Non-U.S. Gov't Research Support, U.S. Gov't, P.H.S.] J Biol Chem. 1996;271(51):32529–32537. doi: 10.1074/jbc.271.51.32529. [DOI] [PubMed] [Google Scholar]
- Seo M, Kim JD, Neau D, Sehgal I, Lee YH. Structure-Based Development of Small Molecule PFKFB3 Inhibitors: A Framework for Potential Cancer Therapeutic Agents Targeting the Warburg Effect. PLoS One. 2011;6(9):e24179. doi: 10.1371/journal.pone.0024179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen LZ, Wang JH, Wang HQ, Hua YB, Wu WX. Zhonghua Wei Chang Wai Ke Za Zhi. 2006;9(5):425–428. [Efficacy of antitumour colon cancer cell vaccine modified by Escherichia coli cytosine deaminase gene] [PubMed] [Google Scholar]
- Shepherd PR, Kahn BB. Glucose transporters and insulin action--implications for insulin resistance and diabetes mellitus. N Engl J Med. 1999;341(4):248–257. doi: 10.1056/NEJM199907223410406. [Comment Research Support, Non-U.S. Gov't Research Support, U.S. Gov't, P.H.S. Review] [DOI] [PubMed] [Google Scholar]
- Silva AS, Yunes JA, Gillies RJ, Gatenby RA. The potential role of systemic buffers in reducing intratumoral extracellular pH and acid-mediated invasion. Cancer Res. 2009;69(6):2677–2684. doi: 10.1158/0008-5472.CAN-08-2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons AL, Ahmad IM, Mattson DM, Dornfeld KJ, Spitz DR. 2-Deoxy-D-glucose combined with cisplatin enhances cytotoxicity via metabolic oxidative stress in human head and neck cancer cells. Cancer Res. 2007;67(7):3364–3370. doi: 10.1158/0008-5472.CAN-06-3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh D, Banerji AK, Dwarakanath BS, Tripathi RP, Gupta JP, Mathew TL, et al. Optimizing cancer radiotherapy with 2-deoxy-d-glucose dose escalation studies in patients with glioblastoma multiforme. Strahlenther Onkol. 2005;181(8):507–514. doi: 10.1007/s00066-005-1320-z. [DOI] [PubMed] [Google Scholar]
- Skeen JE, Bhaskar PT, Chen CC, Chen WS, Peng XD, Nogueira V, et al. Akt deficiency impairs normal cell proliferation and suppresses oncogenesis in a p53-independent and mTORC1-dependent manner. Cancer Cell. 2006;10(4):269–280. doi: 10.1016/j.ccr.2006.08.022. [DOI] [PubMed] [Google Scholar]
- Smith PJ, Blunt NJ, Desnoyers R, Giles Y, Patterson LH. DNA topoisomerase II-dependent cytotoxicity of alkylaminoanthraquinones and their N-oxides. Cancer Chemother Pharmacol. 1997;39(5):455–461. doi: 10.1007/s002800050598. [Comparative Study] [DOI] [PubMed] [Google Scholar]
- Song CW, Clement JJ, Levitt SH. Preferential cytotoxicity of 5-thio-D-glucose against hypoxic tumor cells. J Natl Cancer Inst. 1976;57(3):603–605. doi: 10.1093/jnci/57.3.603. [DOI] [PubMed] [Google Scholar]
- Sonveaux P. Provascular strategy: targeting functional adaptations of mature blood vessels in tumors to selectively influence the tumor vascular reactivity and improve cancer treatment. Radiother Oncol. 2008;86(3):300–313. doi: 10.1016/j.radonc.2008.01.024. [Research Support, Non-U.S. Gov't Review] [DOI] [PubMed] [Google Scholar]
- Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't] J Clin Invest. 2008;118(12):3930–3942. doi: 10.1172/JCI36843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacpoole PW. The pharmacology of dichloroacetate. Metabolism. 1989;38(11):1124–1144. doi: 10.1016/0026-0495(89)90051-6. [DOI] [PubMed] [Google Scholar]
- Stacpoole PW, Lorenz AC, Thomas RG, Harman EM. Dichloroacetate in the treatment of lactic acidosis. Ann Intern Med. 1988;108(1):58–63. doi: 10.7326/0003-4819-108-1-58. [DOI] [PubMed] [Google Scholar]
- Stetak A, Lankenau A, Vantus T, Csermely P, Ullrich A, Keri G. The antitumor somatostatin analogue TT-232 induces cell cycle arrest through PKCdelta and c-Src. Biochem Biophys Res Commun. 2001;285(2):483–488. doi: 10.1006/bbrc.2001.5199. [DOI] [PubMed] [Google Scholar]
- Stetak A, Veress R, Ovadi J, Csermely P, Keri G, Ullrich A. Nuclear translocation of the tumor marker pyruvate kinase M2 induces programmed cell death. Cancer Res. 2007;67(4):1602–1608. doi: 10.1158/0008-5472.CAN-06-2870. [DOI] [PubMed] [Google Scholar]
- Steward WP, Middleton M, Benghiat A, Loadman PM, Hayward C, Waller S, et al. The use of pharmacokinetic and pharmacodynamic end points to determine the dose of AQ4N, a novel hypoxic cell cytotoxin, given with fractionated radiotherapy in a phase I study. [Clinical Trial, Phase I Research Support, Non-U.S. Gov't] Ann Oncol. 2007;18(6):1098–1103. doi: 10.1093/annonc/mdm120. [DOI] [PubMed] [Google Scholar]
- Stumvoll M, Nurjhan N, Perriello G, Dailey G, Gerich JE. Metabolic effects of metformin in non-insulin-dependent diabetes mellitus. N Engl J Med. 1995;333(9):550–554. doi: 10.1056/NEJM199508313330903. [DOI] [PubMed] [Google Scholar]
- Sun JD, Liu Q, Wang J, Ahluwalia D, Ferraro D, Wang Y, et al. Selective Tumor Hypoxia Targeting by Hypoxia-Activated Prodrug TH-302 Inhibits Tumor Growth in Preclinical Models of Cancer. Clin Cancer Res. 2012;18(3):758–770. doi: 10.1158/1078-0432.CCR-11-1980. [DOI] [PubMed] [Google Scholar]
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov. 2008;7(2):168–181. doi: 10.1038/nrd2467. [Research Support, Non-U.S. Gov't Review] [DOI] [PubMed] [Google Scholar]
- Taverna RD, Langdon RG. Reversible association of cytochalasin B with the human erythrocyte membrane. Inhibition of glucose transport and the stoichiometry of cytochalasin binding. Biochim Biophys Acta. 1973;323(2):207–219. doi: 10.1016/0005-2736(73)90145-4. [DOI] [PubMed] [Google Scholar]
- Thangaraju M, Karunakaran SK, Itagaki S, Gopal E, Elangovan S, Prasad PD, et al. Transport by SLC5A8 with subsequent inhibition of histone deacetylase 1 (HDAC1) and HDAC3 underlies the antitumor activity of 3-bromopyruvate. [Research Support, N.I.H., Extramural] Cancer. 2009;115(20):4655–4666. doi: 10.1002/cncr.24532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorwarth D, Eschmann SM, Holzner F, Paulsen F, Alber M. Combined uptake of [18F]FDG and [18F]FMISO correlates with radiation therapy outcome in head-and-neck cancer patients. Radiother Oncol. 2006;80(2):151–156. doi: 10.1016/j.radonc.2006.07.033. [DOI] [PubMed] [Google Scholar]
- Tibes RFGS, Von Hoff DD, Weiss GJ, Iyengar T, Kurzrock R, Pestano L, Lowe AM, Herbst RS, Virginia G. Piper. Results from a phase I, dose-escalation study of PX-478, an orally available inhibitor of HIF-1α. J Clin Oncol. 2010;28 [Abstract] [Google Scholar]
- Tiseo M, Bartolotti M, Gelsomino F, Ardizzoni A. First-line treatment in advanced non-small-cell lung cancer: the emerging role of the histologic subtype. Expert Rev Anticancer Ther. 2009;9(4):425–435. doi: 10.1586/era.09.3. [DOI] [PubMed] [Google Scholar]
- Tong RT, Boucher Y, Kozin SV, Winkler F, Hicklin DJ, Jain RK. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 2004;64(11):3731–3736. doi: 10.1158/0008-5472.CAN-04-0074. [DOI] [PubMed] [Google Scholar]
- Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. [Research Support, Non-U.S. Gov't Research Support, U.S. Gov't, P.H.S.] Cell. 2000;101(3):249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- Tureci O, Sahin U, Vollmar E, Siemer S, Gottert E, Seitz G, et al. Human carbonic anhydrase XII: cDNA cloning, expression, and chromosomal localization of a carbonic anhydrase gene that is overexpressed in some renal cell cancers. [Research Support, Non-U.S. Gov't Research Support, U.S. Gov't, P.H.S.] Proc Natl Acad Sci U S A. 1998;95(13):7608–7613. doi: 10.1073/pnas.95.13.7608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valk PE, Mathis CA, Prados MD, Gilbert JC, Budinger TF. Hypoxia in human gliomas: demonstration by PET with fluorine-18-fluoromisonidazole. J Nucl Med. 1992;33(12):2133–2137. [PubMed] [Google Scholar]
- Vantus T, Keri G, Krivickiene Z, Valius M, Stetak A, Keppens S, et al. The somatostatin analogue TT-232 induces apoptosis in A431 cells: sustained activation of stress-activated kinases and inhibition of signalling to extracellular signal-regulated kinases. Cell Signal. 2001;13(10):717–725. doi: 10.1016/s0898-6568(01)00194-2. [DOI] [PubMed] [Google Scholar]
- Vaupel P. The role of hypoxia-induced factors in tumor progression. Oncologist. 2004;9(Suppl 5):10–17. doi: 10.1634/theoncologist.9-90005-10. [Review] [DOI] [PubMed] [Google Scholar]
- Vera JC, Reyes AM, Velasquez FV, Rivas CI, Zhang RH, Strobel P, et al. Direct inhibition of the hexose transporter GLUT1 by tyrosine kinase inhibitors. Biochemistry. 2001;40(3):777–790. doi: 10.1021/bi001660j. [DOI] [PubMed] [Google Scholar]
- Vester B, Wengel J. LNA (locked nucleic acid): high-affinity targeting of complementary RNA and DNA. Biochemistry. 2004;43(42):13233–13241. doi: 10.1021/bi0485732. [Research Support, Non-U.S. Gov't Review] [DOI] [PubMed] [Google Scholar]
- Vezina C, Kudelski A, Sehgal SN. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J Antibiot (Tokyo) 1975;28(10):721–726. doi: 10.7164/antibiotics.28.721. [DOI] [PubMed] [Google Scholar]
- Volkmann K, Lucas JL, Vuga D, Wang X, Brumm D, Stiles C, et al. Potent and selective inhibitors of the inositol-requiring enzyme 1 endoribonuclease. [Research Support, Non-U.S. Gov't] J Biol Chem. 2011;286(14):12743–12755. doi: 10.1074/jbc.M110.199737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Pawel J, von Roemeling R, Gatzemeier U, Boyer M, Elisson LO, Clark P, et al. Tirapazamine plus cisplatin versus cisplatin in advanced non-small-cell lung cancer: A report of the international CATAPULT I study group. Cisplatin and Tirapazamine in Subjects with Advanced Previously Untreated Non-Small-Cell Lung Tumors. J Clin Oncol. 2000;18(6):1351–1359. doi: 10.1200/JCO.2000.18.6.1351. [DOI] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Warburg O, Wind F, Negelein E. The Metabolism of Tumors in the Body. J Gen Physiol. 1927;8(6):519–530. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinhouse S. The Warburg hypothesis fifty years later. [Research Support, U.S. Gov't, P.H.S.] Z Krebsforsch Klin Onkol Cancer Res Clin Oncol. 1976;87(2):115–126. doi: 10.1007/BF00284370. [DOI] [PubMed] [Google Scholar]
- Weiss GJ, Infante JR, Chiorean EG, Borad MJ, Bendell JC, Molina JR, et al. Phase 1 study of the safety, tolerability, and pharmacokinetics of TH-302, a hypoxia-activated prodrug, in patients with advanced solid malignancies. Clin Cancer Res. 2011;17(9):2997–3004. doi: 10.1158/1078-0432.CCR-10-3425. [DOI] [PubMed] [Google Scholar]
- Welsh S, Williams R, Kirkpatrick L, Paine-Murrieta G, Powis G. Antitumor activity and pharmacodynamic properties of PX-478, an inhibitor of hypoxia-inducible factor-1alpha. Mol Cancer Ther. 2004;3(3):233–244. [PubMed] [Google Scholar]
- Wike-Hooley JL, Haveman J, Reinhold HS. The relevance of tumour pH to the treatment of malignant disease. Radiother Oncol. 1984;2(4):343–366. doi: 10.1016/s0167-8140(84)80077-8. [DOI] [PubMed] [Google Scholar]
- Willett CG, Boucher Y, di Tomaso E, Duda DG, Munn LL, Tong RT, et al. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat Med. 2004;10(2):145–147. doi: 10.1038/nm988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett CG, Duda DG, Ancukiewicz M, Shah M, Czito BG, Bentley R, et al. A safety and survival analysis of neoadjuvant bevacizumab with standard chemoradiation in a phase I/II study compared with standard chemoradiation in locally advanced rectal cancer. Oncologist. 2010;15(8):845–851. doi: 10.1634/theoncologist.2010-0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett CG, Duda DG, di Tomaso E, Boucher Y, Ancukiewicz M, Sahani DV, et al. Efficacy, safety, and biomarkers of neoadjuvant bevacizumab, radiation therapy, and fluorouracil in rectal cancer: a multidisciplinary phase II study. J Clin Oncol. 2009;27(18):3020–3026. doi: 10.1200/JCO.2008.21.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KJ, Albertella MR, Fitzpatrick B, Loadman PM, Shnyder SD, Chinje EC, et al. In vivo activation of the hypoxia-targeted cytotoxin AQ4N in human tumor xenografts. [Research Support, Non-U.S. Gov't] Mol Cancer Ther. 2009;8(12):3266–3275. doi: 10.1158/1535-7163.MCT-09-0396. [DOI] [PubMed] [Google Scholar]
- Winkler F, Kozin SV, Tong RT, Chae SS, Booth MF, Garkavtsev I, et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell. 2004;6(6):553–563. doi: 10.1016/j.ccr.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Wojtkowiak JW, Verduzco D, Schramm KJ, Gillies RJ. Drug resistance and cellular adaptation to tumor acidic pH microenvironment. Mol Pharm. 2011;8(6):2032–2038. doi: 10.1021/mp200292c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I. Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecol Oncol. 2008;109(3):394–402. doi: 10.1016/j.ygyno.2008.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood TE, Dalili S, Simpson CD, Hurren R, Mao X, Saiz FS, et al. A novel inhibitor of glucose uptake sensitizes cells to FAS-induced cell death. Mol Cancer Ther. 2008;7(11):3546–3555. doi: 10.1158/1535-7163.MCT-08-0569. [DOI] [PubMed] [Google Scholar]
- Xie H, Valera VA, Merino MJ, Amato AM, Signoretti S, Linehan WM, et al. LDH-A inhibition, a therapeutic strategy for treatment of hereditary leiomyomatosis and renal cell cancer. Mol Cancer Ther. 2009;8(3):626–635. doi: 10.1158/1535-7163.MCT-08-1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Wang BS, Yu DH, Lu Q, Ma J, Qi H, et al. Dichloroacetate shifts the metabolism from glycolysis to glucose oxidation and exhibits synergistic growth inhibition with cisplatin in HeLa cells. Int J Oncol. 2011;38(2):409–417. doi: 10.3892/ijo.2010.851. [DOI] [PubMed] [Google Scholar]
- Yu Y, Deck JA, Hunsaker LA, Deck LM, Royer RE, Goldberg E, et al. Selective active site inhibitors of human lactate dehydrogenases A4, B4, and C4. Biochem Pharmacol. 2001;62(1):81–89. doi: 10.1016/s0006-2952(01)00636-0. [DOI] [PubMed] [Google Scholar]
- Zagorevskii D, Song M, Breneman C, Yuan Y, Fuchs T, Gates KS, et al. A mass spectrometry study of tirapazamine and its metabolites. insights into the mechanism of metabolic transformations and the characterization of reaction intermediates. J Am Soc Mass Spectrom. 2003;14(8):881–892. doi: 10.1016/S1044-0305(03)00334-9. [DOI] [PubMed] [Google Scholar]
- Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006;66(21):10269–10273. doi: 10.1158/0008-5472.CAN-06-1500. [DOI] [PubMed] [Google Scholar]
- Zeman EM, Brown JM, Lemmon MJ, Hirst VK, Lee WW. SR-4233: a new bioreductive agent with high selective toxicity for hypoxic mammalian cells. Int J Radiat Oncol Biol Phys. 1986;12(7):1239–1242. doi: 10.1016/0360-3016(86)90267-1. [DOI] [PubMed] [Google Scholar]
- Zhang F, Aft RL. Chemosensitizing and cytotoxic effects of 2-deoxy-D-glucose on breast cancer cells. J Cancer Res Ther. 2009;5(Suppl 1):S41–S43. doi: 10.4103/0973-1482.55140. [DOI] [PubMed] [Google Scholar]
- Zhang J, Tian Q, Yung Chan S, Chuen Li S, Zhou S, Duan W, et al. Metabolism and transport of oxazaphosphorines and the clinical implications. Drug Metab Rev. 2005;37(4):611–703. doi: 10.1080/03602530500364023. [DOI] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108(8):1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Tozzi F, Chen J, Fan F, Xia L, Wang J, et al. Intracellular ATP levels are a pivotal determinant of chemoresistance in colon cancer cells. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't Research Support, U.S. Gov't, Non-P.H.S.] Cancer Res. 2012;72(1):304–314. doi: 10.1158/0008-5472.CAN-11-1674. [DOI] [PMC free article] [PubMed] [Google Scholar]