

Abstract
Although acquired bone marrow failure (BMF) is considered a T cell-mediated autoimmune disease, possible innate immune defects as a cause for systemic immune deviations in response to otherwise innocuous infections, have not been extensively explored. In this regard we recently demonstrated an important role of type-I-IFNs in protecting hematopoiesis during systemic stress responses to the opportunistic fungal pathogen Pneumocystis in lymphocyte-deficient mice. Mice deficient in both lymphocytes and type-I-IFN-receptor (IFrag−/− mice) develop rapidly progressing BMF due to accelerated bone marrow cell apoptosis associated with innate immune deviations in the bone marrow in response to Pneumocystis lung infection. However, the communication pathway between lung and bone marrow eliciting the induction of BMF in response to this strictly pulmonary infection has been unclear.
Here we report that absence of an intact type-I-IFN-system during Pneumocystis lung infection not only causes BMF in lymphocyte-deficient mice but also transient bone marrow stress in lymphocyte-competent mice. This is associated with an exuberant systemic IFN-γ response. IFNγ neutralization prevented Pneumocystis lung infection-induced bone marrow depression in type-I-IFN-receptor-deficient (IFNAR−/−) mice, and prolonged neutrophil survival time in bone marrow from IFrag−/− mice. IL-1β and upstream regulators of IFNγ, IL-12 and IL-18, were also upregulated in lung and serum of IFrag−/− mice. In conjunction there was exuberant inflammasome-mediated caspase-1-activation in pulmonary innate immune cells required for processing of IL-18 and IL-1β. Thus, absence of type-I-IFN-signaling during Pneumocystis lung infection may result in deregulation of inflammasome-mediated pulmonary immune activation causing systemic immune deviations triggering BMF in this model.
Introduction
Bone marrow failure can occur in the context of inherited and acquired conditions and manifests in its extreme form as aplastic anemia with severe peripheral cytopenias and acellular bone marrow spaces (1). While most acquired aplastic anemias are thought to be the result of an T cell mediated autoimmune response to an unknown, likely infectious trigger, inherited forms are defined by gene defects often affecting the viability of hematopoietic stem cells in response to inflammatory stimuli (2-5). Furthermore, peripheral cytopenias due to bone marrow suppression can also occur as a complication of severe inflammatory syndromes such as rheumatoid diseases, severe sepsis and AIDS (6-8). Thus, while pathomechanistically complex and multifactorial, a common theme appears to be the presence of inflammatory stimuli accompanied by immune deviations.
Pneumocystis is an extracellular, opportunistic fungal pathogen of the lung that causes a life threatening pneumonia in severely immune compromised individuals with e.g HIV infection or immunosuppressive therapy (9). Although the infection often resolves unnoticed in otherwise healthy individuals, there is increasing evidence that low grade pulmonary Pneumocystis colonization/infection can exacerbate the symptoms of chronic pulmonary diseases such as COPD (10, 11) and thus may also exacerbate systemic complications associated with it (12).
As with many other fungal pathogens, immune protection from Pneumocystis infection critically depends on CD4-T cell mediated immune responses (13, 14). However, while immunity to many other pulmonary fungal pathogens appears to involve inflammasome-mediated immune-activation following innate pattern recognition and activation of a Th-1/TH-17-driven adaptive immune response (15, 16), there is increasing evidence that successful immunity to Pneumocystis lung infection involves TH-2-mediated immune responses including alternative macrophage activation and B cell-mediated clearance (17-22).
Type-I-IFNs have long been known as antiviral (reviewed in (23)), and their role as mediators of immunity to bacterial and some fungal infections has just been recognized (reviewed in (24, 25). Type-I-IFNs activate macrophages, promote DC maturation, enhance TH-1-and NK-cell-mediated immunity (26-28) but also support B cell-differentiation to antibody-secreting plasma cells (29). While type-I-IFN-mediated responses have been implicated in immune-mediated damage to specific pathogens (30) and autoimmune diseases (31, 32), they are also immune modulators. In this regard, type-I-IFNs induce IL-10 production in LPS stimulated macrophages (33) and in antigen-specific T cells leading to the suppression of a Th17-associated autoimmune inflammation in a mouse model of multiple sclerosis (MS) (34, 35). In addition, type-I-IFNs induce transcriptional repression of TNF-α (36), inhibition of inflammasome activation and subsequent IL-1β processing (37), and are thus therapeutically utilized in patients with MS (38-40) and evaluated for patients with chronic inflammatory bowel diseases (41-43).
Therefore, type-I-IFNs are pleiotropic and their activity is likely dose-dependent and determined by the immunological microenvironment. Indeed, low amounts of IFN-β accumulate in tissue in the absence of infection maintaining a wide variety of signaling molecules important for immunity and tissue homeostasis (44). While in high dosages myelosuppressive (45, 46), type-I-IFNs act as neutrophil survival factors similar to G-CSF (47). Furthermore, type-I-IFNs are critical regulators of bone homeostasis (reviewed in (48, 49)) and thus may also protect the bony hematopoietic stem cell niche and hematopoiesis (50, 51). However, excessive IFNα production during inflammatory responses can also induce and exhaust the proliferative capacity of the hematopoietic stem cell (HSC) and impact self renewal (52).
The role of type-I-IFN-responses during HIV infection has also been controversially discussed (53). While early excessive type-I-IFN-production may contribute to immune activation and accelerated loss of CD4 T cells (54), over the course of HIV infection CD4 T cells in conjunction with dendritic cells (DCs), including type-I-IFN-producing plasmacytoid dendritic cells (pDCs), are sequentially deleted and/or their functions are impaired (55-57). The loss of these cell types contributes together and independently to the progression of immune deregulation and immunodeficiency in HIV+ patients (58, 59).
We recently evaluated the role of type-I-IFNs during Pneumocystis lung infection in lymphocyte-competent and lymphocyte-deficient mice and also observed type-I-IFN-mediated pleiotropic effects on local and systemic immune responses to the infection. In a CD4 T cell-depleted mouse model of Pneumocystis pneumonia (PCP), type-IFNs promote CD8-T-cell-induced lung damage. However, absence of type-I-IFN-signaling in lymphocyte-competent mice (IFNAR−/−), induces prolonged pulmonary inflammation despite pathogen clearance eliciting fibrosis (18). In addition, we found that type-I-IFN-signaling plays an essential role in maintaining hematopoiesis during the systemic acute phase response to this fungal infection (60). Combined lymphocyte- and IFNAR-deficient mice (IFrag−/−), but not lymphocyte-deficient but IFNAR-competent mice (RAG−/−), develop rapidly progressing bone marrow failure in response to Pneumocystis lung infection. This is due to accelerated apoptosis of maturing granulocytes combined with lack of replenishment from early progenitors (61). Induction of bone marrow failure in IFrag−/− mice is associated with the deregulated expression of pro-inflammatory and pro-apoptotic cytokines (e.g TNF-a, IL-1, TRAIL) and exuberant ROS-induction in the bone marrow (61, 62). However, the communication pathway initiated between lung and bone marrow resulting in the induction of bone marrow failure in response to this strictly pulmonary infection has been unclear.
Here we report that absence of an intact type-I-IFN-system elicits an innate and exuberant systemic IFN-γ (type II IFN) response to Pneumocystis lung infection that contributes to induction of bone marrow stress in both IFNAR−/− and IFrag−/− mice, respectively. IFNγ–inducing cytokines such as interleukin -12 (IL-12) and interleukin-18 (IL-18), in concert with IL-1β, were concomitantly upregulated in lung and serum of IFrag−/− mice and was associated with exuberant inflammasome-mediated caspase-1 activation in pulmonary innate immune cells in response to Pneumocystis lung infection when compared to RAG−/− mice. Our data point to an important role of type-I-IFNs in regulating of inflammasome-mediated immune activation in the lung in preventing systemic immune deviations and complications in this model of Pneumocystis lung infection-induced bone marrow failure.
Material and Methods
Mice
All mice listed below were bred and maintained at Montana State University Animal Resource Center. Mice were housed in ventilator cages and received sterile food and water. C.B17 SCID mice, as a source for Pneumocystis murina (PC) organism, and RAG1−/− mice (C57/BL6 background) were initially purchased from Jackson Laboratories (Stock no. 001803 and 002096, respectively). IFrag−/− mice were generated by crossing IFNAR KO mice (129SvEv/C57/Bl6 background) with RAG1−/− mice (C57/BL6 background) as previously described (60) and have since been backcrossed 2 more times on a C57/BL6 background. Lymphocyte competent IFNAR−/− mice initially used in experiments were on a 129SvEv/C57BL6 mixed background. However, subsequently all experiments were repeated in IFNARnull/null mice, which are on a C57/BL6 background, and were kindly gifted to us by Dr. Ed Schmidt, MSU and compared to C57/BL6 wildtype mice purchased at Charles Rivers Laboratory (Stock no. 027).
Pneumocystis lung infections and treatments
Most experimental animals were intratracheally infected with 107 Pneumocystis nuclei in 100 μl of lung homogenate diluted in PBS-buffer from infected source mice. Pneumocystis lung-burden was assessed microscopically by enumeration of trophozoid and cyst nuclei count in lung homogenates in 10-50 oil immersion fields as previously described (63). The limit of detection for this technique is log10 4.43. For some experiments mice received limiting dilutions of 105 and 103 nuclei. Some control groups mice received an inoculum of 107 heat inactivated nuclei or clean lung homogenate from uninfected CB17 source mice. Anti-interferon gamma (anti-IFNγ) treatment was performed by intraperitoneal injection of purified 250μg of anti-IFNγ antibody (clone R4-6A2, ATCC, 3× per week). Some mice were placed on antibiotic medicated food (Sulfatrim rodent pellets, LabDiet) Mice were euthanized by exsanguination following induction of deep pentobarbital narcosis. All mouse studies conformed to NIH guidelines and were approved by the IACUC at Montana State University.
Cryptococcus lung infection and evaluation of lung burden
In some experiments mice were intratracheally infected with 5 ×103 Cryptococcus neoformans yeast (strain H99, kindly provided to us by Dr Robert Cramer, MSU) in 100μl PBS. For this, yeast was streaked from frozen stocks onto YPD agar plates and incubated at 37°C for 48 hours. To generate a liquid culture a single colony was picked and inoculated into 10 ml of YPD media and further incubated at 37°C. For infection yeast were enumerated, washed and adjusted to the appropriate concentration in sterile PBS. Infected mice were euthanized at 7 and 10 days post infection, the right lung removed, homogenized in 5 ml of PBS and 100μl were plated onto YPD agar plates at 1:10, 1:100 and 1:1000 dilutions in PBS, incubated for three days and colonies counted.
Collection and differentiation of BM cells
BM cells from femur and tibia was collected as previously described by flushing 2 mls of PBS through the BM canal using a 26 ½ g needle and brought into a single cell suspension (64) . BM cells were diluted 1:10 in PBS. Cell numbers were enumerated, spun onto glass slides, and stained with Diff-Quick solution (Dade/Behring). Cell-differentiation was performed based on morphology and staining pattern to distinguish myeloid (including myeloblast-myelocyte and metamyelocyte stage), banded versus segmented neutrophils, eosinophils and others (including erythroid, megakaryocytic and lymphoid cells) (64).
FACS analysis of cellular subsets in bone marrow and lung
FACS analysis was also applied to lung and bone marrow cells, combining cell surface staining in combination with assessment for Caspase 1 activities at the single cell level or expression of intracellular IFNγ. Prior to FACS staining, red blood cell lysis of BM and lung cell samples was performed using ACK lysis buffer. Cells were then suspended at 1×107/ml in FACS buffer (PBS/10% calf serum) containing Fc-block (mouse clone 24G2, Pharmingen) at a 1:800 dilution. Sets of 5×105 cells were stained with specific antibodies: MHCII (PerCP-Cy5-5, clone:M5/114.15.2, eBioscience), anti-CD11b (AlexaFluor700, clone:M1/70, BioLegend), anti-Ly-6G/6C (APC-Cy7, clone:RB6-8C5, Pharmingen), anti-NK1.1. (Pe-Cy7, clone PK136, Pharmingen), anti-CD11c (APC, clone N418, eBioscience), anti-IFNγ (V450, clone XMG1.2, BD) or FAM Flica Caspase-1 (Immunochemistry Technologies) according to the manufactuer’s instructions. To obtain single cell suspensions of lung cells for FACS analysis of lung cells, right lung lobes were removed, diced with scissors and placed into 10 mls of PBS containing 1 mg/ml Collagenase D (Roche) and incubated for 30 minutes at 37°C. Lungs were then placed into a 100um mesh filter screen and carefully pushed through into a clean tube. For the evaluation of IFNγ production of cellular subsets, cells were incubated for 4 hours in the presence of 40ng/ml PMA and 1μg/ml ionomycin in MEM in the presence of 1mg/ml brefeldin A. following cell surface staining with the above mentioned surface markers, cells were fixed and permeabilized using Cytofix/Cytoperm reagents from BD following the manufacturers protocol and either stained with 0.5μg e450-labeled isotype-control antibody or anti- IFNγ. Following repeated wash steps, samples were acquired using a LSR BD FACS machine and analyzed using FlowJo Software.
Lung cell analysis
Broncho-alveolar lavages (BAL) were performed as previously described (65). Briefly, mice were intraperitoneally (i.p.) injected with 90mg/kg sodium pentobarbital solution and exsanguinated. BALs were performed by intratracheal cannulation, and instillation and extraction of 5 × 1 ml HBSS containing 3mM EDTA. Lung lavage cells were enumerated using a hemocytometer, spun onto glass slides and stained with Diff-Quick (Siemens) for differential counting. FACS analysis was performed as described above to evaluate Caspase-1 activity as indicator of inflammasome activation in cellular subsets.
Serum samples
Serum samples were collected throughout the course of infection by harvesting full blood samples at each time point, which were spun and the buffy coat separated from the serum. Samples were s frozen until further analysis.
Cytokine analysis
Cytokine analysis from BAL, serum and bone marrow cell lysate were performed using multiplex assay plates from either Multiplex or Bio-Rad Life Sciences using a Bioplex (Bio-Rad) analysis system. Bone marrow cell lysates were generate from a cell concentrations of 2.5×107/ml using cell lysis buffer from BioVison The following cytokines were evaluated: IFNγ, IL-1β, IL-18, IL-12p70, IL-5, IL-10, IL-15, and TNF-α using this methodology. In some experiments IFNγ protein concentrations were separately measured using ELISA reagents from R&D Systems (Mouse IFNγ Duoset). Furthermore, in some experiments TRAIL concentrations were also evaluated in bone marrow cell lysates using ELISA reagents from R&D Systems (Mouse TRAIL Duoset).
Colony-Forming Cell Assay for mouse bone marrow cells (CFC)
Hematopoietic precursor cell activity in BM from IFrag−/− and RAG−/− mice was assessed by hematopoietic colony forming counts (CFC-U) in Methylcellulose media as previously described (61). For this 105 BM cells per animal and group at each time point were plated in MethCultR GF M3534 media (StemCell Technologies) which has been formulated to support the optimal growth of granulocyte and macrophage precursor cells. Cells from each sample were plated in duplicate according to the manufacturer’s protocol in 35mm sterile culture dishes (StemCell Technologies), placed in 100mm petri dishes in the presence of one 35 mm dish containing sterile water. Cultures were incubated for 7 days in a water jacketed incubator maintained with 5% CO2. Colony recognition (GM-, G-, M- forming colonies) and enumeration was performed according to StemCell Technologies-guidelines.
Microscopy
Microscopic evaluations of cytospin slides were performed using a Nikon Eclipse E200 microscope at 1000× magnification under oil immersion. Bone marrow colony count analysis (CFC-U assays) was performed using a Zeiss Axiovert microscope under 25 and 50× magnification.
Statistical analysis
Data were plotted using Prism Graphpad Software, Inc. and statistical analysis was performed using either a one-way or two-way ANOVA analysis of variance, followed by a Tukey or Bonferroni post hoc test, respectively
Results
Lymphocyte-competent IFNAR−/− mice develop a transient bone marrow crisis in response to Pneumocystis lung infection
Complete bone marrow failure develops in IFrag−/− but not RAG−/− mice in response to Pneumocystis lung infection within 16-21 days post infection. It is reflected by loss of total bone marrow numbers and hematopoietic progenitor/stem cell activity (CFU count) (Figure 1, Panel A) and subsequently leads to premature death of the animal. In contrast, lymphocyte-competent IFNAR−/− mice appear clinically unimpaired, although previously observed significant splenomegaly suggested extramedullary hematopoiesis (60).
Figure 1. Type-I-IFN-signaling is critical in regulating hematopoiesis during systemic responses to Pneumocystis lung infection in both lymphocyte-deficient and lymphocyte competent mice.

Panel A. Shown are total bone marrow cell numbers, hematopoietic colony forming activity (CFU-counts) of bone marrow cells and PC lung burden from IFrag−/− (IFNAR−/−/RAG−/−) and RAG−/− mice (IFNAR+/+/RAG−/−) over the course of Pneumocystis lung infection at day 0, 7, 10 and 16 post infection. Panel B. Comparative analysis of total bone marrow cell numbers, hematopoietic colony forming activity (CFU-counts) of bone marrow cells and PC lung burden from lymphocyte-competent IFNAR−/− (IFNAR−/−/RAG+/+) mice and wildtype mice (IFNAR+/+/RAG+/+) over the course of Pneumocystis lung infection at day 0, 7, 10 and 16 post infection. Panel C. Demonstrated are total spleen counts and hematopoietic colony forming activity (CFU-counts) of spleen cells from IFNAR−/− and wildtype mice as a measure for extramedullary hematopoiesis at day 0, 7 10 and 16 post infection. Statistical analysis was performed using a 2-way ANOVA. Comparisons were made between groups at the same time point. P values are marked as * p< 0.05, ** p<0.01, *** p<0.001.
We now have found that IFNAR−/− mice (not wildtype mice) also develop significant but transient bone marrow depression in response to Pneumocystis lung infection which is maximal at day 10 post infection (Figure 1 Panel B). This was largely due to loss of neutrophils and thus mirrored the early phenotype previously observed in IFrag−/− mice (61). In contrast to IFrag−/− mice, IFNAR−/− mice retained significant hematopoietic progenitor cell activity within the bone marrow which was reflected by high CFU counts of plated bone marrow cells in vitro. In fact, bone marrow CFU counts in IFNAR−/− mice increased when bone marrow depression was maximal. However, consistent with evidence of bone marrow stress, and different from IFrag−/− mice, extramedullary hematopoiesis in the spleen was suggested by increased spleen size and could be confirmed by increased hematopoietic progenitor activity when splenocytes were placed in CFU assays (Figure 1 Panel C). Furthermore, and consistent with bone marrow depression, IFNAR−/− mice demonstrated a deviated, infection-induced bone marrow cytokine profile previously shown to be associated with bone marrow failure in IFrag−/− mice (62). This included a transient up-regulation of pro-inflammatory and pro-apoptotic cytokines such as TNF-α, IL-1β and TRAIL (Figure 2 A-C).
Figure 2. Transient bone marrow depression during Pneumocystis lung infection in IFNAR−/− mice is associated with deviated cytokine responses as previously seen in IFrag−/− mice during progression of bone marrow failure.
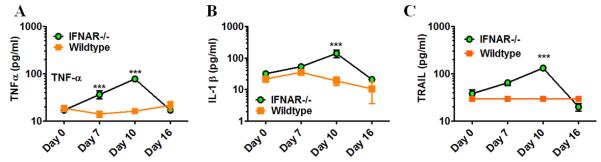
Comparative cytokine analysis was performed in bone marrow lysates isolated from IFNAR−/− and wildtype mice at day 0, 7, 10 and 16 post Pneumocystis lung infection using an ELISA-based multiplex assay system. Shown are data for TNF-α (A), IL-1β (B) and TRAIL (C). Statistical analysis was performed using a 2-way ANOVA. Comparisons were made between groups at the same time point. P values are marked as * p< 0.05, ** p<0.01, *** p<0.001.
Induction of bone marrow failure by Pneumocystis lung infection is dependent on the infectious dose and requires live pathogen
Pneumocystis does not appear to disseminate from the lung to the bone marrow in our model (60). Thus we hypothesized that deviated cytokine responses observed in the bone marrow of IFrag−/− and IFNAR−/− mice in response to Pneumocystis lung infection and associated with bone marrow pathology were caused by actions of immune mediators released from the lung into the system.
Therefore, we first evaluated whether Pneumocystis-induced systemic immune deviations are dependent on the infectious dose and whether it required live pathogen. For this we evaluated total bone marrow cell numbers and composition of six groups of IFrag−/− mice at day 16 post infection that had received a variety of PC dosages: 1) standard dosage of 107 live Pneumocystis nuclei, 2) 105 live Pneumocystis nuclei, 3) 103 live Pneumocystis nuclei, 4) 107 nuclei of heat-inactivated Pneumocystis, 5) clean lung homogenate from an uninfected SCID source mice, and 6) uninfected and unmanipulated.
Figure 3A shows Pneumocystis lung burden at day 16 for the respective experimental group, demonstrating lower pathogen load in those mice receiving lower infectious dosages and no Pneumocystis growth in those receiving heat-inactivated pathogen or clean lung homogenate. Figure 3B shows respective bone marrow cell numbers. Bone marrow failure could still be induced at the same rate of 16 days with a 100 fold reduced Pneumocystis dosage of 105 nuclei. In contrast, inoculation with 103 live nuclei, as well as inoculation with heat inactivated pathogen (107 dead nuclei) and clean lung homogenate left the bone marrow unaffected within the observed time frame. Interestingly, initiation of antibiotic treatment of IFrag−/− mice with SMX/TMP chow either beginning prior to (day -3) or at day 0, 3, or 7 post inoculation with 107 (or 105) PC nuclei, could not prevent progression of bone marrow failure, despite subsequent suppression of PC burden near detection limit at day 16 post infection (Figure 3 C and D).
Figure 3. Induction of bone marrow failure in lymphocyte-deficient IFrag−/− mice depends on the exposure to a critical dose of 105 live pathogen nuclei and cannot be prevented by antibiotic treatment.
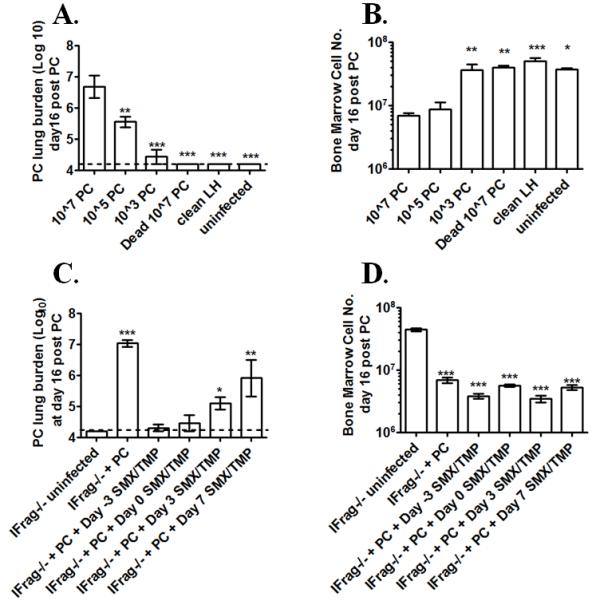
Groups of IFrag−/− mice were intratracheally inoculated with either 107, 105, 103 live Pneumocystis (PC) nuclei, or 107 dead PC nuclei and bone marrow responses compared to animals either inoculated with clean lung homogenate at day 16 post infection and uninfected animals. Figure 3A shows Pneumocystis (PC) lung burden at day 16 post infection following microscopic enumeration. Figure 3 B shows total bone marrow counts of all IFrag−/− comparison groups at day 16 post infection. Antibiotic treatment was initiated in groups of IFrag−/− mice three days prior to (day -3) and at day 0, 3 and 7 post inoculations with 107 PC nuclei. Bone marrow cell numbers and PC lung burden were evaluated and compared to uninfected mice at day 16 post infection. Figure 3C shows PC lung burden and Figure 3D shows total bone marrow counts at day 16 post infection. Statistical analysis was performed using a 1-way ANOVA. All groups were compared to those mice receiving 107 PC nuclei. P values are marked as * p< 0.05, ** p<0.01, *** p<0.001.
IFNAR−/− and IFrag−/−, but not wildtype and RAG−/− mice, demonstrate an exuberant serum IFNγ response to Pneumocystis lung infection that is associated with induction of bone marrow pathology
As previously published, Pneumocystis lung infection elicits a strong but transient systemic type-I-IFN-response with elevated serum IFNα levels in wildtype and RAG−/− mice (62). Here we further evaluated whether specific immune mediators were elicited systemically and uniquely in IFrag−/− and IFNAR−/− mice in response to Pneumocystis lung infection that could be linked to the induction of bone marrow stress.
Interferon gamma (IFNγ), a type-II-IFN cytokine, is produced by an array of innate and adaptive immune cells (66, 67) and has been implicated in the pathogenesis of bone marrow failure in other models of the disease(68, 69). Thus, we compared IFNγ serum levels during early responses to Pneumocystis lung infection in IFrag−/− and RAG−/− mice as well as in IFNAR−/− and wildtype mice over the course of infection and found significantly higher IFNγ serum levels in both IFNAR−/− and in IFrag−/− mice, when compared to wildtype and RAG−/− mice (Figure 4A and B). While in some experiments IFNγ serum responses appeared transient, peaking at day 7, in other experiments IFNγ levels remained high following induction throughout day 16 post infections (see also Figure 8). Regardless, induction of high serum IFNγ levels coincided with or preceded the induction of bone marrow depression /failure, respectively.
Figure 4. IFNAR−/− and IFrag−/− show elevated serum IFNγ levels during early responses to Pneumocystis lung infection which correlate with induction of bone marrow stress.
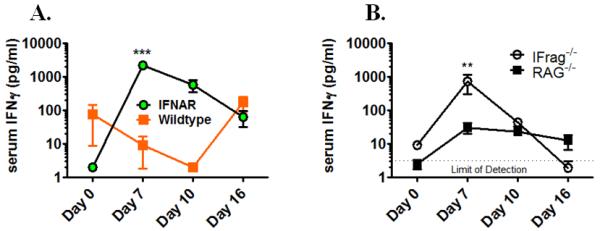
IFNγ serum evaluated over the course of Pneumocystis lung infection in IFNAR−/−, IFrag−/−, wildtype and RAG−/− mice at day 0, 7 10 and 16 post infection using a plate based ELISA assay. Figure 4A compares serum IFNγ responses between lymphocyte-competent IFNAR−/− and wildtype mice, Figure 4B compares serum IFNγ responses between lymphocyte-competent IFrag−/− and RAG−/− mice. Statistical analysis was performed using a 2-way ANOVA. Comparisons were made between groups at the same time point. P values are marked as * p< 0.05, ** p<0.01, *** p<0.001.
Figure 8. Cytokines considered inducers and regulators of IFNγ are concomitantly upregulated in the serum of IFrag−/− but not RAG−/− mice in response to Pneumocystis lung infection.
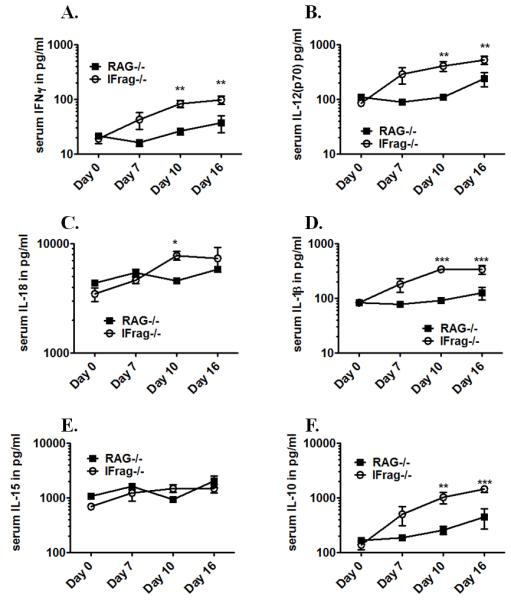
Comparative serum cytokine analysis was performed on IFrag−/− and RAG−/− mice at day 0, 7 10 and 16 post Pneumocystis lung infection using a multiplex ELISA-based assay to evaluate if other pro-inflammatory and IFNγ-regulatory cytokines are uniquely induced in IFrag−/− mice. Shown are comparative data for IFNγ (A), IL-12p70 (B), IL-18 (C), IL-1β (D), IL-15 (E) and IL-10 (F). Statistical analysis was performed using a 2-way ANOVA. Comparisons reflect differences between the groups at each individual time point. P values are marked as * p< 0.05, ** p<0.01, *** p<0.001.
The induction of bone marrow failure in IFrag−/− mice appears specific to responses to Pneumocystis lung infection (60). Cryptococcus neoformans (strain H99) does not induce bone marrow failure in IFrag−/− mice (Figure 5A), although mice succumb to the infection within 21 days if intratracheally infected with a low infectious dose of 5×103 yeast cells. Thus, we also asked if high serum IFNγ levels were also unique to responses Pneumocystis lung infection in our model, and compared serum IFNγ levels of IFrag−/− mice infected with Pneumocystis or Cryptococcus over a course of 10 days. Indeed, we found that IFNγ serum levels were uniquely elevated in response to Pneumocystis, but not to Cryptococcus lung infection (Figure 5B), although the pathogen lung burden for both Pneumocystis and Cryptococcus (Figure 5C) continuously increased in both animal groups over the tested time period.
Figure 5.
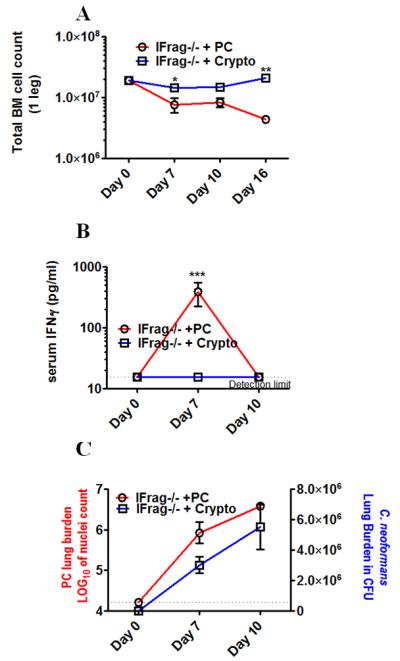
Crypotococcus neoformans lung infection does not induce bone marrow failure in IFrag−/− mice and also does not induce a systemic IFNγ serum response.
Figure 5A compares total bone marrow cell numbers of IFrag−/− mice either infected with Cryptococcus neoformans (strain H99) or Pneumocystis at day 0, 7, 10 and 16 post infection. Figure 5B compares serum IFNγ levels between Cryptococcus and Pneumocystis-infected IFrag−/− mice at day 0, 7 and 10 post infection. Statistical analysis was performed using a 2-way ANOVA. Comparisons were made between groups at the same time point. P values are marked as * p< 0.05, ** p<0.01, *** p<0.001.
Figure 5C shows the respective pulmonary fungal burden for Pneumocystis (left axis) and Cryptococcus (right axis) over the respective time course. Statistical analysis was performed using a 1-way ANOVA comparing fungal burdens within the respective groups. P values are marked as * p< 0.05, ** p<0.01, *** p<0.001.
Anti-IFNγ–treatment prevented bone marrow depression in IFNAR−/− mice and ameliorated progression of bone marrow failure in IFrag−/− mice
To determine if a cause and effect relationship existed between elevated systemic IFNγ levels and bone marrow pathology in our systems, we first treated lymphocyte-competent IFNAR−/− mice with neutralizing anti-IFNγ antibody (clone R4-6A2) and compared their bone marrow response to those that were left untreated over a time course of 16 days post Pneumocystis lung infection. As demonstrated in Figure 6, treatment of Pneumocystis-infected IFNAR−/− mice averted the transient bone marrow crisis normally present at day 10 post infection (Figure 6A) by preventing accelerated loss of band and segmented neutrophils (Figure 6B and C). Amelioration of bone marrow stress was associated with reduced extramedullary hematopoiesis (Figure 6D) and specific reduction in CFU-G formation as a measure of extramedullary granulopoiesis (Figure 6E). Furthermore, anti-IFNγ treatment reduced up-regulation of TRAIL-receptor DR-5 (Figure 6 F) and TRAIL expression in the bone marrow (Figure 6G) normally associated with bone marrow depression in Pneumocystis-infected IFNAR−/− mice (see Figure 2C).
Figure 6. Anti-IFNγ treatment prevents the transient bone marrow crisis in lymphocyte-competent IFNAR−/− mice.

IFNAR−/− mice were infected with Pneumocystis and received either neutralizing anti-IFNγ (3× weekly 250μg, clone R4-6A2) or remained untreated. Bone marrow and spleen responses were analyzed at day 0, 10 and 16 post infections. Shown in Figure 6A-C are comparative analyses of total bone marrow cell numbers (A), and bone marrow cell differentiation with percentage of band neutrophils (B) and segmented neutrophils (C) between treated and untreated IFNAR−/− mice. Figure 6D shows comparative total CFU-counts of spleen cells as a measure of extramedullary hematopoiesis. Figure 6E shows the percentage of G-CFU within total CFUs counted as a measure for extramedullary granulopoiesis. Figure 6 F shows the percentage of TRAIL-receptor (DR5+) bone marrow cells plotted as % positive cells over the course of infection. Figure 6G shows TRAIL-protein levels in bone marrow cell lysates of comparison groups over the course of infection (Figure 6G). Statistical analysis was performed using a 2-way ANOVA. Comparisons were made between the groups at the same time point. P values are marked as * p< 0.05, ** p<0.01, *** p<0.001.
Despite the profound effects of anti-IFNγ treatment on hematopoiesis in lymphocyte-competent IFNAR−/− mice, the same treatment had only a partial effect when performed in Pneumocystis-infected, lymphocyte-deficient IFrag−/− mice. In IFrag−/− mice, anti-IFNγ treatment ameliorated the progression of bone marrow failure (Figure 7A), seemingly by prolonging the survival time of segmented and band neutrophils in the bone marrow (Figure 7B and C). However, neutrophils subsequently disappeared and remained unreplenished, similar to what was seen in untreated IFrag−/− mice. This suggested an additional mechanism responsible for the full progression of the disease in lymphocyte-deficient mice.
Figure 7. Anti-IFNγ treatment ameliorates loss of mature neutrophils but cannot prevent the progression of bone marrow failure in lymphocyte-deficient IFrag−/− mice.
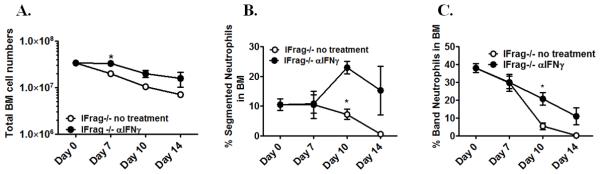
IFrag−/− mice were infected with Pneumocystis and received either neutralizing anti-IFNγ (3× weekly 250μg, clone R4-6A2) or no treatment. Total bone marrow cell numbers and cell differentiation was performed and compared between the treated (open circle) and untreated group (closed circle) at day 0, 7, 10 and 16 post infection. Figure 7A shows total bone marrow cell numbers, Figure 7B and C show the percentage of band and segmented neutrophils in the bone marrow of the comparison groups. Statistical analysis was performed using a 2-way ANOVA. Comparisons between the groups were made at each time point. P values are marked as * p< 0.05, ** p<0.01, *** p<0.001.
Induction of a systemic IFNγ response to Pneumocystis lung infection in IFrag−/− mice is associated with increased serum and BAL levels of IL-18 and IL-12
Neutralization experiments suggested a partial role of systemically upregulated IFNγ in the pathogenesis of bone marrow failure in IFrag−/− mice. however, understanding mechanisms causing its deregulation could lend insight into the global mechanisms by which type-I-IFNs act to prevent systemic immune deviation associated with bone marrow failure in response to Pneumocystis lung infection. Therefore, we assessed serum IFNγ responses of IFrag−/− compared to RAG−/− mice over the course of Pneumocystis lung infection (day 0, 7, 10 and 16 post infection) in conjunction with an array of cytokines that are known to induce or regulate IFNγ production (IL-12p70, IL-15, IL-18, IL-1β and IL-10) (67). As shown in Figure 8, serum analysis demonstrated increased levels of IFNγ in conjunction with IL-12, IL-18 and also IL-1β, uniquely in samples from IFrag−/− mice starting at day 7 with maximum concentrations at day 10 post infection (Figure 8A-D), while IL-15 was not differentially regulated. Surprisingly, the immunosuppressive cytokine IL-10 was also uniquely increased in serum IFrag−/− mice despite high levels of IFNγ and other pro-inflammatory cytokines.
IL-12 and IL-18, as well as IL-1β and IFNγ can be produced by a variety of innate immune cells, including macrophages and dendritic cells following TLR-mediated activation (70, 71). To assess whether the source of elevated serum cytokines was primarily located in the lung, we measured BAL concentrations for IL-12, IL-18, IL-1b and IFNγ in BAL fluid from IFrag−/− and RAG−/− mice at day 6 and 10 post infection. While we found uniquely elevated levels for IL-12p70, IL18 and IL-1β in BAL fluid of IFrag−/− mice in response to Pneumocystis lung infection at day 6 and 10 post infection (Figure 9A-C), we were repeatedly unable to detect elevated IFNγ in the BAL of IFrag−/− mice (data not shown), suggesting either an interstitially located cellular source or a source located outside of the lung.
Figure 9. Cytokine profile of broncho alveolar lavage (BAL) fluid in part mirrors serum cytokine profile.
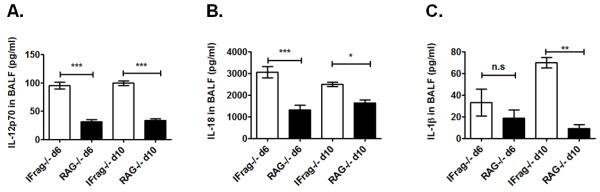
Comparative cytokine analysis of BAL fluid from IFrag−/− and RAG−/− mice with focus on pro-inflammatory cytokines and upstream regulators of IFNγ using a multiplex assay system. Demonstrated are comparative results for IL12p70 (A), IL-18, (B) and IL-1β (C). Statistical analysis was performed using a 1-way ANOVA. Comparisons were made between the groups at each time point. P values are marked as * p< 0.05, ** p<0.01, *** p<0.001.
Deviated local and systemic immune activation to Pneumocystis lung infection in IFrag−/− mice is associated with increased inflammasome-mediated Caspase-1 activity in the lung
While IL-12p70 is directly secreted as a heterodimer from its source (72), IL-18 and IL-1β require inflammasome-activated caspase-1 processing in order to be released in its bioactive form (73-75). To evaluate if elevated local and systemic cytokine levels in IFrag−/− mice were associated with increased inflammasome-mediated immune activation in the lung, we assessed caspase-1 activity in cells from BAL fluid and lung digests of IFrag−/− compared to RAG−/− mice at day 0, 7, and 10 post Pneumocystis lung infection using a FACS-based fluorescent assay (FAM-FLICA) in conjunction with cell surface marker staining. As demonstrated in Figure 10, we found a significantly higher percentage of active Caspase-1+ cells in BAL fluid (Figure 10A) and lung digest (Figure 10B) of IFrag−/− mice when compared to RAG−/− mice in a kinetic consistent with high BAL and serum IL-18 and IL-1β concentrations (Figure 9B-C and 8D-F) and induction of bone marrow failure. The majority of caspase-1+ cells in the BAL were CD11c+ MHCII+ cells macrophage-type cells, and CD11b+Gr-1+ neutrophils in lung digest (data no shown). These data implicate exuberant inflammasome-mediated immune activation in pulmonary innate immune cells as a contributor to the systemic immune deregulation in IFrag−/− mice.
Figure 10. Increased BAL and serum IL-18 and IL-1β levels in IFrag−/− mice are associated with increased induction of inflammasome-mediated Caspase-1 activity in cellular sources of BAL and lung digest.
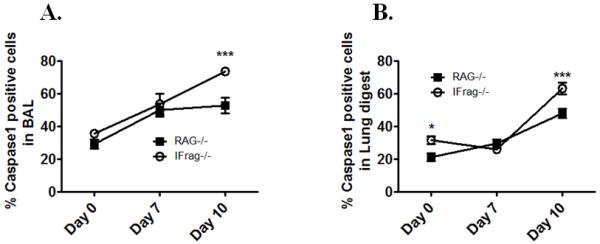
Caspase-1 activity was evaluated on a single cell level in cellular sources of BAL fluid and lung digest of IFrag−/− and RAG−/− mice at day 0, 7, 10 and 16 of Pneumocystis lung infection using a live cell assay in which fluorescently-labeled Caspase-1 inhibitor (FAM-Flicka) enters the cell and binds irreversibly to activated caspase-1, allowing subsequent FACS analysis in combination with cell surface marker staining. Figure 10 A shows the percentage of active Caspase-1+ cells in BAL fluid and Figure 10B shows the percentage of caspase-1+ cells in lung digest single cell suspensions. Statistical analysis was performed using a 2-way ANOVA. Comparisons were made between groups at each time point. P values are marked as * p< 0.05, ** p<0.01, *** p<0.001.
Discussion
We recently found type-I-IFNs to be critical in supporting on-demand hematopoiesis during systemic acute phase responses to Pneumocystis lung infection, especially in lymphocyte-deficient mice (60, 61). In the present study we further evaluated the systemic communication pathway between lung and bone marrow and how type-I-IFNs modulate early innate pulmonary responses to prevent the induction of bone marrow failure in our model.
Although we previously showed that lymphocyte-competent IFNAR−/− mice are protected from the progression to complete bone marrow failure by yet unclear mechanisms provided by B cells (60), we now know that lymphocyte-competent IFNAR−/− mice nevertheless develop a transient bone marrow crisis with significant loss of bone marrow neutrophils. This crisis is associated with similar bone marrow cytokine-deviations previously noted in IFrag−/− mice (62) that are consistent with neutrophil apoptosis (76, 77) and include up-regulation of TNF-α and TRAIL. However, in contrast to IFrag−/− mice, lymphocyte-competent IFNAR−/− mice retain hematopoietic progenitor activity with the ability to replenish transiently lost cells, although significant extramedullary hematopoiesis serves as additional evidence of bone marrow stress. These distinct findings between lymophocyte-competent IFNAR−/− and lymphocyte-deficient IFrag−/− mice suggest a “two-hit” process in the pathogenesis of bone marrow failure in our model. A first “hit” that affects survival time of mature and maturing neutrophils, while a second “hit” affects the health of early progenitors/HSC resulting in lack of replenishment.
While possible, Pneumocystis has rarely been shown to disseminate to other organ sites, especially to the bone marrow (78, 79). We show here that in IFrag−/− mice, bone marrow failure still progresses unchanged when 100-fold less pathogen (105 nuclei) was administered compared to the initial infection strategy, but did not develop within the time frame of 16 days when a 10,000 fold reduced inoculum was used. Surprisingly, antibiotic treatment with SMX/TMP prior to or at an early time point following infection with 105 or 107 PC nuclei, could not prevent the progression of bone marrow failure in IFrag−/− mice. This experiment suggested that a specific level of pathogen inoculum is required to reach a threshold of immunological detection that when surpassed, in the absence of type-I-IFN-signaling, sets off a series of responses that initiate bone marrow failure in our model.
Pneumocystis, like a variety of other fungal pathogens, is recognized by innate immune cells via engagement of Dectin-1 receptor with fungal cell wall β-glucans (80-82) which sets off a strong innate inflammatory program. While we never had evidence that Pneumocystis disseminates from the lung to the bone marrow in our system, in some experiments we were able to detect soluble fungal β-glucans in the serum of IFrag−/− mice around day 5-6 post infection (data not shown). This was of interest as Cryptococcus neoformans, due to its capsule, does not shed β-glucans systemically (83) or induces bone marrow failure in our system. While this could be a mechanism by which this strictly pulmonary infection elicits significant systemic effects, recent work by Goodrich et al demonstrated that innate immune cells distinguish soluble β-glucans from microbe-bound β-glucans and dectin-1-signaling is only activated by microbe-bound particulate and not by soluble β-glucans (84). Therefore, we concluded that abnormal immune responses in the lung, induced in the absence of type-I-IFN-signaling specifically in response to Pneumocystis infection, must cause systemic immune deviations that initiate aberrant bone marrow responses in IFrag−/− and IFNAR−/− mice, leading to bone marrow failure or stress, respectively.
Responses to IFNγ (and TNF-α) have been implicated in the pathogenesis of acquired and some inherited bone marrow failure syndromes (2, 68, 69, 85-88) and cross-regulation between these cytokines and type-I-IFNs exists (26, 89, 90). We previously demonstrated that absence of type-I-IFN-signaling results in an early exuberant, pulmonary TNF-α and IFNγ response in IFNAR−/− mice (91). Indeed, here we also found high IFNγ serum concentrations uniquely in IFNAR−/− and IFrag−/− mice, but not in wildtype and RAG−/− mice specifically induced in response to Pneumocystis lung infection. Importantly, the induction of high serum IFNγ levels between day 7 and 10 coincided with the induction of bone marrow stress in these mice and was not induced when mice were infected with Cryptococcus neoformans.
Interferon gamma (IFNγ) is a powerful pro-inflammatory mediator produced by an array of innate and adaptive immune cells that coordinates a diverse array of cellular programs via transcriptional regulation, including pro- and anti-apoptotic pathways (66). While both, type-I-IFNs and IFNγ prolong neutrophil survival time at inflammatory sites via the induction of anti-apoptotic regulators (47), IFNγ also promotes apoptosis e.g via induction of TRAIL-expression in a variety of tissue types (92-95). The regulation of TRAIL by IFNγ is of interest as TRAIL may provide the first “hit” in the progression of bone marrow failure in our model. Indeed TRAIL has been shown to accelerate neutrophil apoptosis and aid in eliminating senescent neutrophils form the bone marrow (76). Furthermore, we previously demonstrated that partial neutralization of TRAIL activity in IFrag−/− mice ameliorates the progression of bone marrow failure by seemingly prolonging neutrophil survival time with no effects on the progressive loss of progenitor cell activity (62). Consistent with this observation, we show here that anti-IFNγ treatment of lymphocyte-competent IFNAR−/− mice protected from transient bone marrow depression by preventing neutrophil loss and concomitant up-regulation of TRAIL and TRAIL receptor (DR5). This was also associated with reduced extramedullary hematopoiesis. In lymphocyte-deficient IFrag−/− mice however, the same treatment only ameliorated the progression of complete bone marrow failure by seemingly prolonging neutrophils survival times, but with no effects on the concomitant loss of progenitor activity. These data support our “two-hit” hypothesis in the pathogenesis of bone marrow failure in our model: The first hit includes a deregulated IFNγ systemic response, possibly communicating the upregulation of TRAIL in the bone marrow. The second hit however, appears to affect early hematopoietic progenitor cell activity and their ability to replenish lost cells. This seems to be specifically protected by B cell-mediated mechanisms in lymphocyte-competent IFNAR−/− mice and may be facilitated via protection of the hematopoietic stem cell niche (61, 62).
A deregulated systemic IFNγ response appears to be only one contributing aspect mediating the progression of bone marrow failure in IFrag−/− mice (“first-hit”). However, by understanding upstream events of its deregulation we thought to gain insights into early innate pulmonary deregulation in IFNAR-deficient mice ultimately responsible for initiation of all systemic immune-deviations contributing to the induction (“first hit”) and progression (“second hit”) of bone marrow failure in our model.
IL-18 in combination with IL-12 are potent inducers of IFNγ (96-99) while IL-10 is considered an important negative-regulator of IFNγ-transcription (100). In a recent study we found high IFN-γ protein levels in BAL-fluid, in conjunction with a significant up-regulation of IL-18 and IL-12 mRNA in pulmonary CD11c+ cells uniquely from lymphocyte-competent IFNAR−/− mice, but not wildtype mice, during early time points of Pneumocystis lung infection (day 7 and 14, respectively) (91). This suggested a distinctive pulmonary immune activation pathway in response to Pneumocystis lung infection in the absence of type-I-IFN-signaling. When these cytokines were evaluated in lymphocyte-deficient mice, we also found elevated IL-18 and IL-12 in conjunction with IL1β protein levels uniquely present in the serum of IFrag−/− mice, and not in RAG−/− mice, which also coincided with elevated serum IFNγ during early responses (starting at day 7) to Pneumocystis lung infection. Furthermore, we found IL-12, IL-18 and IL1β cytokine levels uniquely elevated in BAL fluid from IFrag−/− mice. However, we were repeatedly unable to detect IFNγ in BAL samples of these mice. This was also the case when time points were chosen as early as day 3 and 5 post infection (data not shown). This suggested that the source of early serum IFNγ was either not the lung itself or was systemically released from the lung by polarized secretion from an interstitially located cellular source (101). In this regard, intracellular cytokine staining of phorbol-ester stimulated lung cells isolated from both IFrag−/− and RAG−/− mice over the course of infection demonstrated presence of an IFNγ-producing NK-like innate cell subset co-expressing CD49b/CD11c/CD11b and MHCII in both animal groups (data not shown). Interestingly, NK-cells have recently been shown to provide immunity to Pneumocystis lung infection (102) and type-I-IFNs contribute to NK cell activation and their homeostatic control (103). Thus, given a cellular composition with similar potential for IFNγ-secretion within the lung of IFrag−/− and RAG−/− mice, it is possible that these cells may be differentially regulated depending on whether innate immune responses to Pneumocystis are elicited in the absence or presence of type-I-IFN-signaling. This is currently under further investigation.
IL-12, IL-18 and IL-1β are produced by innate immune cells such as macrophages in response to activation by pattern recognition receptors such as TLRs (72, 75, 104). However, while IL-12 can be immediately secreted in its active form, IL-18 and IL-1β require the cleavage of their precursor to their active form via inflammasome-induced caspase-1 activation (75). Interestingly, type-I-IFNs have been shown to suppress the processing of IL-1β (and IL-18) by repressing NLRP1 and NLRP3 inflammasome activity via STAT-1-mediated mechanisms (37). To evaluate a potential for differential inflammasome activity in IFrag−/− versus RAG−/− mice during immune responses to Pneumocystis lung infection, we screened the pulmonary compartment of IFrag−/− and RAG−/− mice for caspase-1 activity on single cell level. Indeed, we found a higher percentage of cells positive for caspase-1 activity in cells from BAL and lung digest of IFrag−/− mice compared to RAG−/− mice in response to the infection. This coincided with the highest serum IL-18, IL-1β and IFNγ levels. In addition, we also found induction of caspase-1-activity in CD11b+Gr-1low bone marrow cells of IFrag−/− mice (data not shown), which may potentially be a secondary response to cell stress and induction of apoptosis during progression of bone marrow failure as previously described (37, 105). These data also suggested an important role of type-I-IFNs in modulating innate immune pathways leading to inflammasome-mediated immune activation during responses to Pneumocystis lung infection.
HIV infection causes a complex immunodeficiency that can impact innate immune functions such as type-I-IFN-responses (106-108) and NK-cell-mediated immune deviations (), in concert with the loss of CD4+ T cells causing high susceptibility to Pneumocystis lung infection (109). In addition, often unexplained complications such global pancytopenia due to regenerative bone marrow failure occurs (7, 8, 110). Furthermore, while acquired aplastic anemias of previously healthy individuals are thought to be T cell-mediated (2), the possibility of an underlying defect in innate immune mechanisms as a cause for systemic immune deviations in response to otherwise innocuous infections, such as Pneumocystis, has not been extensively explored. Therefore, our findings may have implications in understanding the mechanisms underlying regenerative bone marrow failure as it may occur associated with autoimmune-mediated diseases or AIDS.
While many questions still remain our mouse model highlights the important role of type-I-IFNs in the regulation of communications between organ systems, such as lung and bone marrow, during systemic responses to Pneumocystis lung infection. Importantly, our data point to a role of type-I-IFN-signaling in modulating Pneumocystis lung-infection-induced inflammasome-activation in innate immune cells to prevent systemic immune deviations and innate immunity-mediated pathologies at distant organ sites, such as the bone marrow.
Acknowledgements
We would like to thank Dr. Allen Harmsen and Dr. Steve Swain for insight and critical reading of the manuscript
Funding sources: NIH RO1 HL90488 and COBRE 2P20RR020185
References
- 1.Leguit RJ, van den Tweel JG. The pathology of bone marrow failure. Histopathology. 2010;57:655–670. doi: 10.1111/j.1365-2559.2010.03612.x. [DOI] [PubMed] [Google Scholar]
- 2.Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006;108:2509–2519. doi: 10.1182/blood-2006-03-010777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bagby GC, Lipton JM, Sloand EM, Schiffer CA. Marrow failure. Hematology Am Soc Hematol Educ Program. 2004:318–336. doi: 10.1182/asheducation-2004.1.318. [DOI] [PubMed] [Google Scholar]
- 4.Bijangi-Vishehsaraei K, Saadatzadeh MR, Werne A, McKenzie KA, Kapur R, Ichijo H, Haneline LS. Enhanced TNF-alpha-induced apoptosis in Fanconi anemia type C-deficient cells is dependent on apoptosis signal-regulating kinase 1. Blood. 2005;106:4124–4130. doi: 10.1182/blood-2005-05-2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Du W, Adam Z, Rani R, Zhang X, Pang Q. Oxidative stress in Fanconi anemia hematopoiesis and disease progression. Antioxid Redox Signal. 2008;10:1909–1921. doi: 10.1089/ars.2008.2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zheng P, Chang X, Lu Q, Liu Y. Cytopenia and autoimmune diseases: A vicious cycle fueled by mTOR dysregulation in hematopoietic stem cells. J Autoimmun. 2013;41:182–187. doi: 10.1016/j.jaut.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Isgro A, Aiuti A, Leti W, Gramiccioni C, Esposito A, Mezzaroma I, Aiuti F. Immunodysregulation of HIV disease at bone marrow level. Autoimmun Rev. 2005;4:486–490. doi: 10.1016/j.autrev.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 8.Isgro A, Aiuti A, Mezzaroma I, Ruco L, Pinti M, Cossarizza A, Aiuti F. HIV type 1 protease inhibitors enhance bone marrow progenitor cell activity in normal subjects and in HIV type 1-infected patients. AIDS Res Hum Retroviruses. 2005;21:51–57. doi: 10.1089/aid.2005.21.51. [DOI] [PubMed] [Google Scholar]
- 9.Thomas CF, Jr., Limper AH. Pneumocystis pneumonia. N Engl J Med. 2004;350:2487–2498. doi: 10.1056/NEJMra032588. [DOI] [PubMed] [Google Scholar]
- 10.Morris A, Wei K, Afshar K, Huang L. Epidemiology and clinical significance of pneumocystis colonization. J Infect Dis. 2008;197:10–17. doi: 10.1086/523814. [DOI] [PubMed] [Google Scholar]
- 11.Morris A, Netravali M, Kling HM, Shipley T, Ross T, Sciurba FC, Norris KA. Relationship of pneumocystis antibody response to severity of chronic obstructive pulmonary disease. Clin Infect Dis. 2008;47:e64–68. doi: 10.1086/591701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biskobing DM. COPD and osteoporosis. Chest. 2002;121:609–620. doi: 10.1378/chest.121.2.609. [DOI] [PubMed] [Google Scholar]
- 13.Harmsen AG, Stankiewicz M. Requirement for CD4+ cells in resistance to Pneumocystis carinii pneumonia in mice. J Exp Med. 1990;172:937–945. doi: 10.1084/jem.172.3.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shellito J, Suzara VV, Blumenfeld W, Beck JM, Steger HJ, Ermak TH. A new model of Pneumocystis carinii infection in mice selectively depleted of helper T lymphocytes. J Clin Invest. 1990;85:1686–1693. doi: 10.1172/JCI114621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Romani L. Immunity to fungal infections. Nat Rev Immunol. 11:275–288. doi: 10.1038/nri2939. [DOI] [PubMed] [Google Scholar]
- 16.Joly S, Sutterwala FS. Fungal pathogen recognition by the NLRP3 inflammasome. Virulence. 1:276–280. doi: 10.4161/viru.1.4.11482. [DOI] [PubMed] [Google Scholar]
- 17.Lund FE, Schuer K, Hollifield M, Randall TD, Garvy BA. Clearance of Pneumocystis carinii in mice is dependent on B cells but not on P carinii-specific antibody. J Immunol. 2003;171:1423–1430. doi: 10.4049/jimmunol.171.3.1423. [DOI] [PubMed] [Google Scholar]
- 18.Meissner NN, Swain S, Tighe M, Harmsen A. Role of type I IFNs in pulmonary complications of Pneumocystis murina infection. J Immunol. 2005;174:5462–5471. doi: 10.4049/jimmunol.174.9.5462. [DOI] [PubMed] [Google Scholar]
- 19.Swain SD, Meissner N, Han S, Harmsen A. Pneumocystis infection in an immunocompetent host can promote collateral sensitization to respiratory antigens. Infect Immun. 2012;79:1905–1914. doi: 10.1128/IAI.01273-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nelson MP, Christmann BS, Werner JL, Metz AE, Trevor JL, Lowell CA, Steele C. IL-33 and M2a alveolar macrophages promote lung defense against the atypical fungal pathogen Pneumocystis murina. J Immunol. 2011;186:2372–2381. doi: 10.4049/jimmunol.1002558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J, Gigliotti F, Bhagwat SP, George TC, Wright TW. Immune modulation with sulfasalazine attenuates immunopathogenesis but enhances macrophage-mediated fungal clearance during Pneumocystis pneumonia. PLoS Pathog. 2010;6:e1001058. doi: 10.1371/journal.ppat.1001058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gigliotti F, Haidaris CG, Wright TW, Harmsen AG. Passive intranasal monoclonal antibody prophylaxis against murine Pneumocystis carinii pneumonia. Infect Immun. 2002;70:1069–1074. doi: 10.1128/IAI.70.3.1069-1074.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levy DE, Marie IJ, Durbin JE. Induction and function of type I and III interferon in response to viral infection. Curr Opin Virol. 2011;1:476–486. doi: 10.1016/j.coviro.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bogdan C, Mattner J, Schleicher U. The role of type I interferons in non-viral infections. Immunol Rev. 2004;202:33–48. doi: 10.1111/j.0105-2896.2004.00207.x. [DOI] [PubMed] [Google Scholar]
- 25.Ramirez-Ortiz ZG, Lee CK, Wang JP, Boon L, Specht CA, Levitz SM. A nonredundant role for plasmacytoid dendritic cells in host defense against the human fungal pathogen Aspergillus fumigatus. Cell Host Microbe. 2011;9:415–424. doi: 10.1016/j.chom.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 27.Stetson DB, Medzhitov R. Type I interferons in host defense. Immunity. 2006;25:373–381. doi: 10.1016/j.immuni.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 28.de Waal Malefyt R. The role of type I interferons in the differentiation and function of Th1 and Th2 cells. Semin Oncol. 1997;24:S9-94–S99-98. [PubMed] [Google Scholar]
- 29.Jego G, Palucka AK, Blanck JP, Chalouni C, Pascual V, Banchereau J. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity. 2003;19:225–234. doi: 10.1016/s1074-7613(03)00208-5. [DOI] [PubMed] [Google Scholar]
- 30.Xin L, Vargas-Inchaustegui DA, Raimer SS, Kelly BC, Hu J, Zhu L, Sun J, Soong L. Type I IFN receptor regulates neutrophil functions and innate immunity to Leishmania parasites. J Immunol. 2011;184:7047–7056. doi: 10.4049/jimmunol.0903273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Banchereau J, Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity. 2006;25:383–392. doi: 10.1016/j.immuni.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 32.Pascual V, Farkas L, Banchereau J. Systemic lupus erythematosus: all roads lead to type I interferons. Curr Opin Immunol. 2006;18:676–682. doi: 10.1016/j.coi.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 33.Chang EY, Guo B, Doyle SE, Cheng G. Cutting edge: involvement of the type I IFN production and signaling pathway in lipopolysaccharide-induced IL-10 production. J Immunol. 2007;178:6705–6709. doi: 10.4049/jimmunol.178.11.6705. [DOI] [PubMed] [Google Scholar]
- 34.Guo B, Chang EY, Cheng G. The type I IFN induction pathway constrains Th17-mediated autoimmune inflammation in mice. J Clin Invest. 2008;118:1680–1690. doi: 10.1172/JCI33342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Levings MK, Sangregorio R, Galbiati F, Squadrone S, de Waal Malefyt R, Roncarolo MG. IFN-alpha and IL-10 induce the differentiation of human type 1 T regulatory cells. J Immunol. 2001;166:5530–5539. doi: 10.4049/jimmunol.166.9.5530. [DOI] [PubMed] [Google Scholar]
- 36.Sharif MN, Sosic D, Rothlin CV, Kelly E, Lemke G, Olson EN, Ivashkiv LB. Twist mediates suppression of inflammation by type I IFNs and Axl. J Exp Med. 2006;203:1891–1901. doi: 10.1084/jem.20051725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Forster I, Farlik M, Decker T, Du Pasquier RA, Romero P, Tschopp J. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity. 34:213–223. doi: 10.1016/j.immuni.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 38.Barbero P, Bergui M, Versino E, Ricci A, Zhong JJ, Ferrero B, Clerico M, Pipieri A, Verdun E, Giordano L, Durelli L. Every-other-day interferon beta-1b versus once-weekly interferon beta-1a for multiple sclerosis (INCOMIN Trial) II: analysis of MRI responses to treatment and correlation with Nab. Mult Scler. 2006;12:72–76. doi: 10.1191/135248506ms1247oa. [DOI] [PubMed] [Google Scholar]
- 39.Clerico M, Faggiano F, Palace J, Rice G, Tintore M, Durelli L. Recombinant interferon beta or glatiramer acetate for delaying conversion of the first demyelinating event to multiple sclerosis. Cochrane Database Syst Rev. 2008 doi: 10.1002/14651858.CD005278.pub3. CD005278. [DOI] [PubMed] [Google Scholar]
- 40.Durelli L, Barbero P, Clerico M. A randomized study of two interferon-beta treatments in relapsing-remitting multiple sclerosis. Neurology. 2006;67:2264. doi: 10.1212/01.wnl.0000252724.67789.1e. author reply 2264-2265. [DOI] [PubMed] [Google Scholar]
- 41.Madsen SM, Schlichting P, Davidsen B, Nielsen OH, Federspiel B, Riis P, Munkholm P. An open-labeled, randomized study comparing systemic interferon-alpha-2A and prednisolone enemas in the treatment of left-sided ulcerative colitis. Am J Gastroenterol. 2001;96:1807–1815. doi: 10.1111/j.1572-0241.2001.03875.x. [DOI] [PubMed] [Google Scholar]
- 42.Wirtz S, Neurath MF. Illuminating the role of type I IFNs in colitis. J Clin Invest. 2005;115:586–588. doi: 10.1172/JCI24518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Atreya R, Mudter J, Finotto S, Mullberg J, Jostock T, Wirtz S, Schutz M, Bartsch B, Holtmann M, Becker C, Strand D, Czaja J, Schlaak JF, Lehr HA, Autschbach F, Schurmann G, Nishimoto N, Yoshizaki K, Ito H, Kishimoto T, Galle PR, Rose-John S, Neurath MF. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in crohn disease and experimental colitis in vivo. Nat Med. 2000;6:583–588. doi: 10.1038/75068. [DOI] [PubMed] [Google Scholar]
- 44.Gough DJ, Messina NL, Clarke CJ, Johnstone RW, Levy DE. Constitutive type I interferon modulates homeostatic balance through tonic signaling. Immunity. 2012;36:166–174. doi: 10.1016/j.immuni.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kirkwood JM, Bender C, Agarwala S, Tarhini A, Shipe-Spotloe J, Smelko B, Donnelly S, Stover L. Mechanisms and management of toxicities associated with high-dose interferon alfa-2b therapy. J Clin Oncol. 2002;20:3703–3718. doi: 10.1200/JCO.2002.03.052. [DOI] [PubMed] [Google Scholar]
- 46.Schmitt K, Hompesch BC, Oeland K, von Staehr WG, Thurmann PA. Autoimmune thyroiditis and myelosuppression following treatment with interferon-alpha for hepatitis C. Int J Clin Pharmacol Ther. 1999;37:165–167. [PubMed] [Google Scholar]
- 47.Sakamoto E, Hato F, Kato T, Sakamoto C, Akahori M, Hino M, Kitagawa S. Type I and type II interferons delay human neutrophil apoptosis via activation of STAT3 and up-regulation of cellular inhibitor of apoptosis 2. J Leukoc Biol. 2005;78:301–309. doi: 10.1189/jlb.1104690. [DOI] [PubMed] [Google Scholar]
- 48.David JP. Osteoimmunology: a view from the bone. Adv Immunol. 2007;95:149–165. doi: 10.1016/S0065-2776(07)95005-1. [DOI] [PubMed] [Google Scholar]
- 49.Takayanagi H. Inflammatory bone destruction and osteoimmunology. J Periodontal Res. 2005;40:287–293. doi: 10.1111/j.1600-0765.2005.00814.x. [DOI] [PubMed] [Google Scholar]
- 50.Aguila HL, Rowe DW. Skeletal development, bone remodeling, and hematopoiesis. Immunol Rev. 2005;208:7–18. doi: 10.1111/j.0105-2896.2005.00333.x. [DOI] [PubMed] [Google Scholar]
- 51.Adams GB, Scadden DT. The hematopoietic stem cell in its place. Nat Immunol. 2006;7:333–337. doi: 10.1038/ni1331. [DOI] [PubMed] [Google Scholar]
- 52.Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, Trumpp A. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. 2009;458:904–908. doi: 10.1038/nature07815. [DOI] [PubMed] [Google Scholar]
- 53.Hosmalin A, Lebon P. Type I interferon production in HIV-infected patients. J Leukoc Biol. 2006;80:984–993. doi: 10.1189/jlb.0306154. [DOI] [PubMed] [Google Scholar]
- 54.Sivaraman V, Zhang L, Su L. Type I interferon contributes to CD4+ T cell depletion induced by infection with HIV-1 in the human thymus. J Virol. 2011;85:9243–9246. doi: 10.1128/JVI.00457-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pacanowski J, Develioglu L, Kamga I, Sinet M, Desvarieux M, Girard PM, Hosmalin A. Early plasmacytoid dendritic cell changes predict plasma HIV load rebound during primary infection. J Infect Dis. 2004;190:1889–1892. doi: 10.1086/425020. [DOI] [PubMed] [Google Scholar]
- 56.Pacanowski J, Kahi S, Baillet M, Lebon P, Deveau C, Goujard C, Meyer L, Oksenhendler E, Sinet M, Hosmalin A. Reduced blood CD123+ (lymphoid) and CD11c+ (myeloid) dendritic cell numbers in primary HIV-1 infection. Blood. 2001;98:3016–3021. doi: 10.1182/blood.v98.10.3016. [DOI] [PubMed] [Google Scholar]
- 57.Chehimi J, Campbell DE, Azzoni L, Bacheller D, Papasavvas E, Jerandi G, Mounzer K, Kostman J, Trinchieri G, Montaner LJ. Persistent decreases in blood plasmacytoid dendritic cell number and function despite effective highly active antiretroviral therapy and increased blood myeloid dendritic cells in HIV-infected individuals. J Immunol. 2002;168:4796–4801. doi: 10.4049/jimmunol.168.9.4796. [DOI] [PubMed] [Google Scholar]
- 58.Siegal FP, Fitzgerald-Bocarsly P, Holland BK, Shodell M. Interferon-alpha generation and immune reconstitution during antiretroviral therapy for human immunodeficiency virus infection. Aids. 2001;15:1603–1612. doi: 10.1097/00002030-200109070-00002. [DOI] [PubMed] [Google Scholar]
- 59.Fitzgerald-Bocarsly P, Jacobs ES. Plasmacytoid dendritic cells in HIV infection: striking a delicate balance. J Leukoc Biol. 2010;87:609–620. doi: 10.1189/jlb.0909635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Meissner N, Rutkowski M, Harmsen AL, Han S, Harmsen AG. Type I interferon signaling and B cells maintain hemopoiesis during Pneumocystis infection of the lung. J Immunol. 2007;178:6604–6615. doi: 10.4049/jimmunol.178.10.6604. [DOI] [PubMed] [Google Scholar]
- 61.Taylor D, Wilkison M, Voyich J, Meissner N. Prevention of bone marrow cell apoptosis and regulation of hematopoiesis by type I IFNs during systemic responses to pneumocystis lung infection. J Immunol. 2011;186:5956–5967. doi: 10.4049/jimmunol.1003558. [DOI] [PubMed] [Google Scholar]
- 62.Wilkison M, Gauss K, Ran Y, Searles S, Taylor D, Meissner N. Type 1 IFNs Suppress Accelerated Osteoclastogenesis and Prevent Loss of Bone Mass During Systemic Inflammatory Responses to Pneumocystis Lung Infection. Am J Pathol. 2012 doi: 10.1016/j.ajpath.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Swain SD, Lee SJ, Nussenzweig MC, Harmsen AG. Absence of the macrophage mannose receptor in mice does not increase susceptibility to Pneumocystis carinii infection in vivo. Infect Immun. 2003;71:6213–6221. doi: 10.1128/IAI.71.11.6213-6221.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bolliger AP. Cytologic evaluation of bone marrow in rats: indications, methods, and normal morphology. Vet Clin Pathol. 2004;33:58–67. doi: 10.1111/j.1939-165x.2004.tb00351.x. [DOI] [PubMed] [Google Scholar]
- 65.Harmsen AG. Role of alveolar macrophages in lipopolysaccharide-induced neutrophil accumulation. Infect Immun. 1988;56:1858–1863. doi: 10.1128/iai.56.8.1858-1863.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 67.Hu X, Ivashkiv LB. Cross-regulation of signaling pathways by interferon-gamma: implications for immune responses and autoimmune diseases. Immunity. 2009;31:539–550. doi: 10.1016/j.immuni.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dufour C, Corcione A, Svahn J, Haupt R, Poggi V, Beka’ssy AN, Scime R, Pistorio A, Pistoia V. TNF-alpha and IFN-gamma are overexpressed in the bone marrow of Fanconi anemia patients and TNF-alpha suppresses erythropoiesis in vitro. Blood. 2003;102:2053–2059. doi: 10.1182/blood-2003-01-0114. [DOI] [PubMed] [Google Scholar]
- 69.Zeng W, Miyazato A, Chen G, Kajigaya S, Young NS, Maciejewski JP. Interferon-gamma-induced gene expression in CD34 cells: identification of pathologic cytokine-specific signature profiles. Blood. 2006;107:167–175. doi: 10.1182/blood-2005-05-1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Case records of the Massachusetts General Hospital Weekly clinicopathological exercises. Case 36-1993. A 28-year-old man with AIDS, persistent pancytopenia, and lymphoma. N Engl J Med. 1993;329:792–799. doi: 10.1056/NEJM199309093291109. [DOI] [PubMed] [Google Scholar]
- 71.Sims JE, Smith DE. The IL-1 family: regulators of immunity. Nat Rev Immunol. 2010;10:89–102. doi: 10.1038/nri2691. [DOI] [PubMed] [Google Scholar]
- 72.Watford WT, Moriguchi M, Morinobu A, O’Shea JJ. The biology of IL-12: coordinating innate and adaptive immune responses. Cytokine Growth Factor Rev. 2003;14:361–368. doi: 10.1016/s1359-6101(03)00043-1. [DOI] [PubMed] [Google Scholar]
- 73.Barker BR, Taxman DJ, Ting JP. Cross-regulation between the IL-1beta/IL-18 processing inflammasome and other inflammatory cytokines. Curr Opin Immunol. 2011;23:591–597. doi: 10.1016/j.coi.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schmidt RL, Lenz LL. Distinct licensing of IL-18 and IL-1beta secretion in response to NLRP3 inflammasome activation. PLoS One. 2012;7:e45186. doi: 10.1371/journal.pone.0045186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.van de Veerdonk FL, Netea MG, Dinarello CA, Joosten LA. Inflammasome activation and IL-1beta and IL-18 processing during infection. Trends Immunol. 32:110–116. doi: 10.1016/j.it.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 76.Lum JJ, Bren G, McClure R, Badley AD. Elimination of senescent neutrophils by TNF-related apoptosis-inducing [corrected] ligand. J Immunol. 2005;175:1232–1238. doi: 10.4049/jimmunol.175.2.1232. [DOI] [PubMed] [Google Scholar]
- 77.Matsuyama W, Yamamoto M, Higashimoto I, Oonakahara K, Watanabe M, Machida K, Yoshimura T, Eiraku N, Kawabata M, Osame M, Arimura K. TNF-related apoptosis-inducing ligand is involved in neutropenia of systemic lupus erythematosus. Blood. 2004;104:184–191. doi: 10.1182/blood-2003-12-4274. [DOI] [PubMed] [Google Scholar]
- 78.Amin MB, Abrash MP, Mezger E, Sekerak GF. Systemic dissemination of Pneumocystis carinii in a patient with acquired immunodeficiency syndrome. Henry Ford Hosp Med J. 1990;38:68–71. [PubMed] [Google Scholar]
- 79.Corti M, Perez R, de Tezanos M, Cermelj M, Candela M, Schtirbu R. Bone marrow involvement by Pneumocystis carinii in an AIDS patient. Enferm Infecc Microbiol Clin. 1999;17:420–422. [PubMed] [Google Scholar]
- 80.Dennehy KM, Brown GD. The role of the beta-glucan receptor Dectin-1 in control of fungal infection. J Leukoc Biol. 2007;82:253–258. doi: 10.1189/jlb.1206753. [DOI] [PubMed] [Google Scholar]
- 81.Evans SE, Hahn PY, McCann F, Kottom TJ, Pavlovic ZV, Limper AH. Pneumocystis cell wall beta-glucans stimulate alveolar epithelial cell chemokine generation through nuclear factor-kappaB-dependent mechanisms. Am J Respir Cell Mol Biol. 2005;32:490–497. doi: 10.1165/rcmb.2004-0300OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Steele C, Marrero L, Swain S, Harmsen AG, Zheng M, Brown GD, Gordon S, Shellito JE, Kolls JK. Alveolar macrophage-mediated killing of Pneumocystis carinii f. sp. muris involves molecular recognition by the Dectin-1 beta-glucan receptor. J Exp Med. 2003;198:1677–1688. doi: 10.1084/jem.20030932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.O’Meara TR, Alspaugh JA. The Cryptococcus neoformans capsule: a sword and a shield. Clin Microbiol Rev. 2012;25:387–408. doi: 10.1128/CMR.00001-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Goodridge HS, Reyes CN, Becker CA, Katsumoto TR, Ma J, Wolf AJ, Bose N, Chan AS, Magee AS, Danielson ME, Weiss A, Vasilakos JP, Underhill DM. Activation of the innate immune receptor Dectin-1 upon formation of a ‘phagocytic synapse’. Nature. 2011;472:471–475. doi: 10.1038/nature10071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bloom ML, Wolk AG, Simon-Stoos KL, Bard JS, Chen J, Young NS. A mouse model of lymphocyte infusion-induced bone marrow failure. Exp Hematol. 2004;32:1163–1172. doi: 10.1016/j.exphem.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 86.Yu JM, Emmons RV, Hanazono Y, Sellers S, Young NS, Dunbar CE. Expression of interferon-gamma by stromal cells inhibits murine long-term repopulating hematopoietic stem cell activity. Exp Hematol. 1999;27:895–903. doi: 10.1016/s0301-472x(99)00009-0. [DOI] [PubMed] [Google Scholar]
- 87.Zoumbos N, Gascon P, Young N. The function of lymphocytes in normal and suppressed hematopoiesis. Blut. 1984;48:1–9. doi: 10.1007/BF00320711. [DOI] [PubMed] [Google Scholar]
- 88.Pang Q, Fagerlie S, Christianson TA, Keeble W, Faulkner G, Diaz J, Rathbun RK, Bagby GC. The Fanconi anemia protein FANCC binds to and facilitates the activation of STAT1 by gamma interferon and hematopoietic growth factors. Mol Cell Biol. 2000;20:4724–4735. doi: 10.1128/mcb.20.13.4724-4735.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Alexander WS, Starr R, Fenner JE, Scott CL, Handman E, Sprigg NS, Corbin JE, Cornish AL, Darwiche R, Owczarek CM, Kay TW, Nicola NA, Hertzog PJ, Metcalf D, Hilton DJ. SOCS1 is a critical inhibitor of interferon gamma signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell. 1999;98:597–608. doi: 10.1016/s0092-8674(00)80047-1. [DOI] [PubMed] [Google Scholar]
- 90.Fenner JE, Starr R, Cornish AL, Zhang JG, Metcalf D, Schreiber RD, Sheehan K, Hilton DJ, Alexander WS, Hertzog PJ. Suppressor of cytokine signaling 1 regulates the immune response to infection by a unique inhibition of type I interferon activity. Nat Immunol. 2006;7:33–39. doi: 10.1038/ni1287. [DOI] [PubMed] [Google Scholar]
- 91.Meissner N, Swain S, McInnerney K, Han S, Harmsen AG. Type-I IFN signaling suppresses an excessive IFN-gamma response and thus prevents lung damage and chronic inflammation during Pneumocystis (PC) clearance in CD4 T cell-competent mice. Am J Pathol. 2010;176:2806–2818. doi: 10.2353/ajpath.2010.091158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Smyth MJ, Cretney E, Takeda K, Wiltrout RH, Sedger LM, Kayagaki N, Yagita H, Okumura K. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) contributes to interferon gamma-dependent natural killer cell protection from tumor metastasis. J Exp Med. 2001;193:661–670. doi: 10.1084/jem.193.6.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kakagianni T, Giannakoulas NC, Thanopoulou E, Galani A, Michalopoulou S, Kouraklis-Symeonidis A, Zoumbos NC. A probable role for trail-induced apoptosis in the pathogenesis of marrow failure. Implications from an in vitro model and from marrow of aplastic anemia patients. Leuk Res. 2006;30:713–721. doi: 10.1016/j.leukres.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 94.Langaas V, Shahzidi S, Johnsen JI, Smedsrod B, Sveinbjornsson B. Interferon-gamma modulates TRAIL-mediated apoptosis in human colon carcinoma cells. Anticancer Res. 2001;21:3733–3738. [PubMed] [Google Scholar]
- 95.Meng RD, El-Deiry WS. p53-independent upregulation of KILLER/DR5 TRAIL receptor expression by glucocorticoids and interferon-gamma. Exp Cell Res. 2001;262:154–169. doi: 10.1006/excr.2000.5073. [DOI] [PubMed] [Google Scholar]
- 96.Carson WE, Dierksheide JE, Jabbour S, Anghelina M, Bouchard P, Ku G, Yu H, Baumann H, Shah MH, Cooper MA, Durbin J, Caligiuri MA. Coadministration of interleukin-18 and interleukin-12 induces a fatal inflammatory response in mice: critical role of natural killer cell interferon-gamma production and STAT-mediated signal transduction. Blood. 2000;96:1465–1473. [PubMed] [Google Scholar]
- 97.Micallef MJ, Tanimoto T, Kohno K, Ikegami H, Kurimoto M. Interleukin 18 induces a synergistic enhancement of interferon gamma production in mixed murine spleen cell-tumor cell cultures: role of endogenous interleukin 12. Cancer Detect Prev. 2000;24:234–243. [PubMed] [Google Scholar]
- 98.Robak E, Robak T, Wozniacka A, Zak-Prelich M, Sysa-Jedrzejowska A, Stepien H. Proinflammatory interferon-gamma--inducing monokines (interleukin-12, interleukin-18, interleukin-15)--serum profile in patients with systemic lupus erythematosus. Eur Cytokine Netw. 2002;13:364–368. [PubMed] [Google Scholar]
- 99.Torre D. Early production of gamma-interferon in clinical malaria: role of interleukin-18 and interleukin-12. Clin Infect Dis. 2009;48:1481–1482. doi: 10.1086/598508. [DOI] [PubMed] [Google Scholar]
- 100.Mosser DM, Zhang X. Interleukin-10: new perspectives on an old cytokine. Immunol Rev. 2008;226:205–218. doi: 10.1111/j.1600-065X.2008.00706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Stanley AC, Lacy P. Pathways for cytokine secretion. Physiology (Bethesda) 2010;25:218–229. doi: 10.1152/physiol.00017.2010. [DOI] [PubMed] [Google Scholar]
- 102.Kelly MN, Zheng M, Ruan S, Kolls J, D’Souza A, Shellito JE. Memory CD4+ T cells are required for optimal NK cell effector functions against the opportunistic fungal pathogen Pneumocystis murina. J Immunol. 2013;190:285–295. doi: 10.4049/jimmunol.1200861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Swann JB, Hayakawa Y, Zerafa N, Sheehan KC, Scott B, Schreiber RD, Hertzog P, Smyth MJ. Type I IFN contributes to NK cell homeostasis, activation, and antitumor function. J Immunol. 2007;178:7540–7549. doi: 10.4049/jimmunol.178.12.7540. [DOI] [PubMed] [Google Scholar]
- 104.Del Vecchio M, Bajetta E, Canova S, Lotze MT, Wesa A, Parmiani G, Anichini A. Interleukin-12: biological properties and clinical application. Clin Cancer Res. 2007;13:4677–4685. doi: 10.1158/1078-0432.CCR-07-0776. [DOI] [PubMed] [Google Scholar]
- 105.Masters SL, Gerlic M, Metcalf D, Preston S, Pellegrini M, O’Donnell JA, McArthur K, Baldwin TM, Chevrier S, Nowell CJ, Cengia LH, Henley KJ, Collinge JE, Kastner DL, Feigenbaum L, Hilton DJ, Alexander WS, Kile BT, Croker BA. NLRP1 inflammasome activation induces pyroptosis of hematopoietic progenitor cells. Immunity. 37:1009–1023. doi: 10.1016/j.immuni.2012.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kamga I, Kahi S, Develioglu L, Lichtner M, Maranon C, Deveau C, Meyer L, Goujard C, Lebon P, Sinet M, Hosmalin A. Type I interferon production is profoundly and transiently impaired in primary HIV-1 infection. J Infect Dis. 2005;192:303–310. doi: 10.1086/430931. [DOI] [PubMed] [Google Scholar]
- 107.Soumelis V, Scott I, Gheyas F, Bouhour D, Cozon G, Cotte L, Huang L, Levy JA, Liu YJ. Depletion of circulating natural type 1 interferon-producing cells in HIV-infected AIDS patients. Blood. 2001;98:906–912. doi: 10.1182/blood.v98.4.906. [DOI] [PubMed] [Google Scholar]
- 108.Soumelis V, Scott I, Liu YJ, Levy J. Natural type 1 interferon producing cells in HIV infection. Hum Immunol. 2002;63:1206–1212. doi: 10.1016/s0198-8859(02)00760-7. [DOI] [PubMed] [Google Scholar]
- 109.Sepkowitz KA. Opportunistic infections in patients with and patients without Acquired Immunodeficiency Syndrome. Clin Infect Dis. 2002;34:1098–1107. doi: 10.1086/339548. [DOI] [PubMed] [Google Scholar]
- 110.Moses A, Nelson J, Bagby GC., Jr. The influence of human immunodeficiency virus-1 on hematopoiesis. Blood. 1998;91:1479–1495. [PubMed] [Google Scholar]