

Abstract
Objective
Dravet syndrome is a severe form of intractable pediatric epilepsy with a high incidence of SUDEP: Sudden Unexpected Death in epilepsy. Cardiac arrhythmias are a proposed cause for some cases of SUDEP, yet the susceptibility and potential mechanism of arrhythmogenesis in Dravet syndrome remain unknown. The majority of Dravet syndrome patients have de novo mutations in SCN1A, resulting in haploinsufficiency. We propose that, in addition to neuronal hyperexcitability, SCN1A haploinsufficiency alters cardiac electrical function and produces arrhythmias, providing a potential mechanism for SUDEP.
Methods
Postnatal day 15-21 heterozygous SCN1A-R1407X knock-in mice, expressing a human Dravet syndrome mutation, were used to investigate a possible cardiac phenotype. A combination of single cell electrophysiology and in vivo electrocardiogram (ECG) recordings were performed.
Results
We observed a 2-fold increase in both transient and persistent Na+ current density in isolated Dravet syndrome ventricular myocytes that resulted from increased activity of a tetrodotoxin-resistant Na+ current, likely Nav1.5. Dravet syndrome myocytes exhibited increased excitability, action potential duration prolongation, and triggered activity. Continuous radiotelemetric ECG recordings showed QT prolongation, ventricular ectopic foci, idioventricular rhythms, beat-to-beat variability, ventricular fibrillation, and focal bradycardia. Spontaneous deaths were recorded in 2 DS mice, and a third became moribund and required euthanasia.
Interpretation
These data from single cell and whole animal experiments suggest that altered cardiac electrical function in Dravet syndrome may contribute to the susceptibility for arrhythmogenesis and SUDEP. These mechanistic insights may lead to critical risk assessment and intervention in human patients.
Introduction
Dravet Syndrome (DS, previously known as Severe Myoclonic Epilepsy of Infancy) is a devastating, intractable pediatric epileptic encephalopathy [1]. Patients exhibit developmental regression during early childhood and frequent pharmacoresistant seizures [1-3]. Up to 15% of DS subjects die during early childhood or adolescence, and most of these cases are thought to represent SUDEP [2], defined as Sudden, Unexpected, witnessed or unwitnessed, nontraumatic and nondrowning Death in patients with EPilepsy [4], excluding cases of documented status epilepticus. In the most widely used definition, death may occur with or without evidence of a seizure, and postmortem examination does not reveal a toxicological or anatomical cause of death. SUDEP accounts for 7.5-17% of all deaths in epilepsy [5,6]. Indirect evidence has linked SUDEP to seizure-induced apnea, pulmonary edema, dysregulation of cerebral circulation, and cardiac arrhythmias [5-7]. Arrhythmias may also occur secondary to hormonal or metabolic changes or autonomic discharges [6-8].
More than 80% of DS patients carry de novo mutations in SCN1A [9,10] that result in haploinsufficiency. SCN1A encodes the voltage-gated Na+ channel (VGSC) α subunit Nav1.1. The tetrodotoxin–resistant (TTX-R) Nav1.5 channel, encoded by SCN5A, is the predominant VGSC in the mammalian heart [11]. TTX-sensitive (TTX-S) VGSCs, including Nav1.1, Nav1.3, and Nav1.6, are also expressed in areas of the heart that include the ventricles and sino-atrial node, although their function is not well understood [11-16]. More recently, Kaufmann and colleagues [17] showed that, in addition to Nav1.5, human atrial myocytes express TTX-S VGSCs Nav1.1, Nav1.2, Nav1.4, and Nav1.6. VGSCs provide a pore for the movement of Na+ into the cell, resulting in a rapidly activating and inactivating transmembrane current (INa) responsible for the action potential (AP) upstroke and impulse propagation. The level of functional expression and biophysical properties of ion channels give the cardiac AP its characteristic shape. The balance between depolarizing currents (e.g., INa and ICaL) and repolarizing currents (e.g., Ito, IKr, IKs, and IK1) determines the level of excitability, AP morphology, AP duration (APD), and dynamics of impulse propagation [18]. Channelopathies disrupt this balance, leading to AP changes and increased susceptibility to arrhythmias and sudden death [10,19]. Cardiac arrhythmogenic diseases can result from gain-of-function (e.g., Long QT Syndrome-3, LQTS-3) or loss-of-function (e.g., Brugada Syndrome) mutations in SCN5A [20]. LQTS-3 mutations result in increased persistent INa during the AP plateau, leading to triggered activity in the form of early after-depolarizations (EADs), and providing a substrate for arrhythmogenesis [19]. Homozygous deletion of Scn1a, Scn2a, Scn5a, or Scn8a in mice is lethal, revealing their non-redundant functions [10,21-23]. Blockade of TTX-S VGSCs in the heart results in altered heart rate (HR) and cardiac contractility [11,12,14,17,24]. Despite many studies examining the effects of SCN1A mutations in the nervous system [3], the influence of SCN1A abnormalities on cardiac function remains unknown.
We propose that the strong association between epilepsy and SUDEP in DS subjects is a consequence of expression of mutant SCN1A in both brain and heart. Recent work, using a Scn1a +/- DS mouse model as well as an inhibitory neuron-specific Scn1a +/- line, suggested that SUDEP may be caused by parasympathetic hyperactivity immediately following seizures, leading to atrioventricular nodal block and lethal bradycardia [25]. While this study implicated cardiac dysfunction in DS-linked SUDEP, the excitability of individual cardiac myoyctes was not investigated. Further, instead of studying the effects of spontaneous seizures, as proposed to occur in SUDEP patients, this study utilized acute hyperthermia-induced seizures. Our objective here was to fill a critical gap in the literature by determining whether cardiac myocytes isolated from mice expressing a human SCN1A DS mutation [26] have altered excitability and whether DS mice exhibit cardiac dysfunction following spontaneous seizures. We propose that, in addition to neuronal dysfunction, Scn1a haploinsufficiency produces altered cardiac electrical function and arrhythmias, providing a cardiac contribution to the mechanism of SUDEP. We report that Scn1a-R1407X heterozygous mice have increased TTX-R, but not TTX-S, cardiac INa, as well as altered AP and ECG properties, EADs, and arrhythmias that produce SUDEP-like events. Our results provide novel insights into an ion channelopathy that provides critical conditions for arrhythmogenesis, and suggest a mechanism for SUDEP that includes changes in cardiac INa.
Materials and Methods
Animals
SCN1A R1407X/+ mice, previously maintained on the C57BL/6J background [26], were backcrossed to C3HFeB/HeJ (Jackson Laboratory, Bar Harbor, ME) to increase litter size. Heterozygous mutant mice of both genders from the N3 and N4 generations were studied at postnatal day (P)15-21. Whenever possible, all data analysis was conducted blinded to genotype. Heterozygous Scn1a R1407X/+ mice are designated DS throughout the manuscript.
Ethics Statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the University Committee on the Use and Care of Animals at the University of Michigan (Approval Numbers: 04695 and 09790). All efforts were made to minimize suffering.
Genotyping
The R1407X mutation abolishes a HpaII restriction site in the wildtype Scn1a sequence. DS mice were genotyped by PCR amplification of a 518 bp genomic fragment with the primers DS-F (5’ CAATGATTCCTAGGGGGATGTC 3’) and DS-R (5’ GTTCTGTGCACTTATCTGGATTCAC 3’). Digestion of the PCR product with HpaII generated 2 fragments, 295 and 223 bp, from the wildtype allele and an uncut 518 bp fragment from the mutant allele. Genomic DNA was amplified in a 25 µl reaction containing 1X GoTaq Buffer, 0.2 mM dNTPs, 0.5 µM each primer, 1 unit GoTaq DNA Polymerase (Promega). Incubation at 94°C for 3 min was followed by 31 cycles of 94°C for 30 sec, 62°C for 1 min, 72°C for 1 min followed by incubation at 72°C for 6 min. After digestion with HpaII, PCR products were separated on 2% agarose gels containing 0.15 µg/ml ethidium bromide.
Acutely Isolated Adult Mouse Ventricular Myocytes
WT and DS cardiac myocytes were acutely isolated from P15–21 mice using a protocol modified from Cerrone et al. [27]. In brief, hearts were isolated from WT and DS mice, and placed in ice-cold perfusion buffer. The Ca2+ free perfusion buffer consisted of (mM): 10 HEPES, 0.6 Na2HPO4, 113 NaCl, 4.7 KCl, 12 NaHCO3, 0.6 KH2PO4, 1.2 MgSO4, 10 KHCO3, 30 Taurine, 5.5 glucose, and 10 butaneodione monoxime. The hearts were cannulated and cleared with perfusion buffer (37°C, 3 ml/min). Next, type-II collagenase (0.87 mg/ml, Worthington Biochemical), trypsin (0.14 mg/ml), and 12 µM CaCl2 were added to the perfusion buffer for the enzymatic digestion. The lower two-thirds of the hearts were isolated and minced into small pieces in digestion buffer. The digestion reaction was stopped by resuspension in stopping buffer, which included perfusion buffer plus 10% fetal bovine serum and 12.5 µM CaCl2. The solution was then incrementally brought up to 1 mM CaCl2. Healthy ventricular cardiac myocytes were defined as those that were Ca2+ tolerant, rod shaped, striated, and quiescent, with a resting membrane potential less than or equal to -65 mV. All myocyte recordings were acquired within 8 h of the cell isolation.
Single Cell Electrophysiology
Standard voltage and current clamp techniques were used to assess the effects of DS mutatons on cardiac INa and AP properties, respectively [28,29]. Single cell cardiac electrophysiological properties were acquired from healthy cardiac myocytes. Experiments were performed using borosilicate glass pipettes with resistance of <3MΩ for INa and 4-5 MΩ for AP recordings. Data were acquired using an Axopatch 200B amplifier (Molecular Devices, USA). The data were acquired and analyzed using pCLAMP9-10 (Molecular Devices, USA) and custom AP analysis software (National Instruments LabView, USA)
Voltage Clamp Recordings
Voltage clamp INa recordings were performed at room temperature (21–22°C) with 5 mM [Na+]o. The extracellular solution contained (in mM): 5 NaCl, 1 MgCl2, 1 CaCl2, 0.1 CdCl2, 11 Glucose, 132.5 CsCl, and 20 HEPES. The filling solution contained (in mM): 5 NaCl, 135 CsF, 10 EGTA, 5 MgATP, and 5 HEPES. Upon gaining access to the cell, appropriate whole cell and series resistance compensation (<70%) and leak subtraction were applied. Whole cell INa, TTX-R INa, TTX-S INa density and biophysical properties were assessed. Assessment of transient and persistent INa density, INa inactivation time, and the voltage dependence of INa conductance were obtained by holding the cell at -120 mV, followed by stepping to voltages between -100 and +30 mV, in 5 mV steps, for 200 ms, with 2800 ms interpulse intervals. The voltage dependence of INa availability was determined by holding at various voltages (-160 mV to 0 mV, 5 mV increments, 200 ms duration) and stepping to -40 mV (30 ms), with 2770 ms interpulse intervals at -120 mV. The normalized voltage dependence of INa availability and conductance (based upon each cells’ reversal potential) were fit to a Boltzmann function, and differences in the V½ and slope factor were compared between groups. The time dependence of INa recovery was assessed by holding at -120 mV and stepping to -30 mV for 20 ms (P1), followed by a 1–40 ms (1 ms increments) interpulse interval at -120 mV, and a second step to -30 mV for 20 ms (P2). The time dependence of INa recovery was calculated by P2/P1 at each timepoint, and these results were fit to a single exponential function. The rate of INa inactivation was fit to a double exponential function. 100 nM TTX was added to block only the TTX-S INa, and therefore pharmacologically separate the TTX-R and TTX-S INa. TTX (30 µM) was then given to block all INa, and used for measuring the persistent INa (pre- minus post-30 µM TTX). The persistent current was measured 30-35 ms after the voltage step, which was a time when the current amplitude was stable. These persistent INa results were also confirmed using the P/4 method, yielding similar results.
Current Clamp Recordings
Current clamp AP recordings were acquired at 37°C in standard Tyrodes solution (in mM): 148 NaCl, 0.4 NaH2PO4, 1 MgCl2, 5.4 KCl, 1 CaCl2, 5.5 glucose, 15 HEPES. The internal solution included (in mM): 148 KCl, 1 MgCl2, 5 EGTA, 5 HEPES, 2 creatine, 5 K2-ATP, 5 phosphocreatine. Incremental amounts of current (0.1 nA steps, 0.3 ms) and pacing cycle lengths (2000 ms, 1000 ms, and up to the fastest pacing cycle length indicated, in 1 hertz (Hz) increments) were used to assess changes in excitability, AP morphology, and susceptibility to triggered activity (i.e. EADs). Only cells with a diastolic membrane potential more negative than -65 mV were used for analysis.
In Vivo ECG Recordings
P15-17 WT (N = 8) and DS (N = 13) mice were implanted with radiotelemetry ECG devices (DSI ETA-F10) at the University of Michigan Phenotyping Core. Animals were anesthetized (isoflurane) and the unit was implanted on the dorsal surface via a small 1 cm incision. Next, a small midline incision was made from the xiphoid process to the manubrium. The leads were passed over the shoulder subcutaneously and sutured onto the intercostal muscles of the rib cage, for ECG lead configuration II. The incisions were sutured closed, the animals were treated with prophylactic antibiotics, and all mice successfully recovered from the procedure without any signs of complications. The first WT mouse with a non-working test unit remained viable without any pathologies (>180 days). ECG (1 KHz sampling), temperature, activity, and running wheel activity were acquired continuously until P70 to provide mechanistic insights into the DS in vivo cardiac phenotype and the events precipitating SUDEP. Mice were housed in a temperature controlled room (21°C) in separate cages on a 12 h light-dark cycle (6 AM - 6 PM). Recordings were monitored and analyzed remotely. The alterations in the ECG waveform were documented during pentylenetetrazole-induced convulsive seizures (PTZ, 40 mg/kg loading and then 20 mg/kg repeated every 20 min intraperitoneally) and the period leading up to spontaneous death in non-PTZ treated mice. All surviving animals were euthanized at the end of the study.
Quantitative Reverse Transcriptase-Polymerase Chain Reactions (qRT-PCR)
Total RNA was isolated from individual hearts using Trizol Reagent (Abion/RNA). Aliquots (1.5 ug) of total RNA were treated with DNAse I (Invitrogen) and cDNA was prepared using the SuperScript II First Strand Synthesis System for RT-PCR (Invitrogen). The Scn5a transcript was quantified using TaqMan gene expression assays Mm01342518_m1 and Mm00451971_m1 (ABI) which span introns 7 and 12, respectively. As an internal control, the 18S transcript was quantified using TaqMan assay Mm03928990_g1. Fluorescence was measured on a Step 1 Real Time PCR System (ABI) at the Microarray Core at the University of Michigan. The mean CT value was determined from quadruplicate assays of each sample. The value of ΔCT was calculated by subtracting the CT for 18S from the CT for Scn5a.
Western Blot Analysis
Western blot analysis was performed as previously described [30]. Anti-Nav1.5 antibody was provided by Dr. Peter Mohler [31] and used at a dilution of 1:1000. Secondary antibody was goat anti-rabbit antibody conjugated to horseradish peroxidase used at a dilution of 1:800 (Thermo Scientific). Signals were visualized using a chemiluminescence system (Supersignal West Femto Maximum Sensitivity Substrate, Thermo Scientific), detected on a Li-Cor Odyssey using ImageStudio software.(Li-Cor).
Statistical Analyses
Results are expressed as mean ± standard error of the mean. Unpaired t test with Welch’s correction, χ2 Test and Log-rank, Mantel-Cox, Survival Test were used as appropriate to test for significance between genotypes, and significance was considered as p ≤ 0.05.
Results
DS mice have increased TTX-R transient and persistent INa density
To determine whether DS mice have altered cardiac excitability, we examined the INa properties of acutely isolated ventricular myocytes. We reported previously that Scn1a haploinsufficiency results in increased, rather than decreased, INa density and hyperexcitability in DS patient-specific induced pluripotent stem cell (iPSC)-derived neurons [32]. Consistent with this, we observed a 2-fold increase in the peak transient (p < 0.0001) and persistent (p ≤ 0.05) INa density in cardiac myocytes from DS vs. WT littermates (Figure 1 , A and B), with a hyperpolarizing shift in the voltage dependence of INa availability and conductance (Figure 1 C , p ≤ 0.05). Since the voltage dependence for pure or predominantly TTX-R Nav1.5 expressing cells is more negative compared to TTX-S (e.g. Nav1.1, Nav1.3, or Nav1.6) VGSC expressing cells, these results suggested a change in the proportion of total cellular INa carried by Nav1.5 [28,32-41]. We administered 100 nM TTX to test for potential changes in functional TTX-S INa density in the DS cardiac myocytes. We observed a similar reduction in DS and WT cells (Figure 1 D ), consistent with previous reports [42,43]. These data suggest that the observed increase in INa in DS cardiac myocytes was not due to increased functional TTX-S VGSC expression and was instead due to an increase in activity of the predominant cardiac TTX-R VGSC, Nav1.5.
Figure 1. DS Mice Have Altered Cardiac INa Properties.

A. Current-voltage (I-V) relationship of transient INa. Peak transient INa density is increased 2-fold in the DS (N = 6, n = 14) vs WT cardiac myocytes (N = 8, n = 20, p < 0.0001). Inset: Representative traces from each group. B. I-V relationship for persistent INa (pre- minus post-30 µM TTX) also shows a 2-fold increase in peak persistent INa in the DS vs. WT groups. To further confirm these results we employed the P/4 method to measure the persistent INa, yielding similar results (-60 mV, WT, -1.72 ± 0.50; DS, -3.88 ± 0.72, N = 2, n = 5-9, p = 0.02). C. Leftward shift (V½ of Boltzman fit, p = 0.04) in the voltage dependence of INa availability and conductance in the DS group. D. Similar percent change in peak transient INa density upon administration of 100 nM TTX in the WT and DS groups. Unpaired t-test with Welch’s correction.
We used 100 nM TTX to pharmacologically isolate the TTX-R and TTX-S INa. Regardless of genotype, and as expected, the V½ values for TTX-R INa (following blockade of TTX-S INa with 100 nM TTX) vs. TTX-S INa (defined as total INa minus TTX-R INa) were significantly different. Figure 2 illustrates that pharmacological isolation of each current led to the expected shifts between TTX-R vs. TTX-S INa voltage dependent properties (p ≤ 0.05), with V½ values for TTX-R INa being more negative than TTX-S INa availability and conductance. However, a comparison of V½ values for the pharmacologically separated currents between genotypes showed no differences, suggesting that the observed increases in total INa were due to increases in the level of functional channel expression rather than changes in voltage-dependence.
Figure 2. Isolation of TTX-R and TTX-S INa Biophysical Properties.
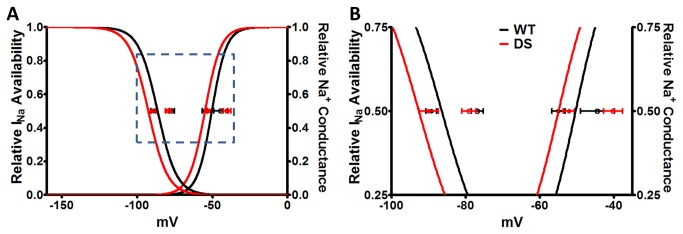
A. Boltzman curves for the voltage dependence of INa availability and conductance for the total cardiac INa (TTX-S + TTX-R INa; reproduction of the curve-fits from Figure 1C). In both WT and DS myocytes the V½ values of TTX-R INa (closed circles, following blockade of TTX-S INa with 100 nM TTX) and TTX-S INa (open circles, defined as total INa minus TTX-R INa) are plotted. Pharmacological separation of TTX-S and TTX-R INa was confirmed by the loss of difference in the V½ values between WT vs DS, and the development of a significant difference between the TTX-S vs. TTX-R V ½ values for INa availability and conductance. B. Zoom-in of the boxed region in A.
Table 1 shows a detailed biophysical characterization of INa properties. No changes were observed in the voltage of peak INa, INa reversal potential, or normalized INa-voltage relationships between genotypes. While the slope factors for the voltage dependence of the total INa availability and conductance did not differ between genotypes (Figure 1C , Table 1 ), when we pharmacologically separated the TTX-S and TTX-R INa (Figure 2 , Table 1 ), we observed slope factor differences. The slope factors for TTX-R Na+ conductance and TTX-S INa availability were significantly increased in DS, suggesting changes in the sensitivity of these channels to changes in voltage.
Table 1. INa biophysical Properties in Each Group.
|
Mouse
|
|||
|---|---|---|---|
| Parameter | WT | DS | |
| N=mice, n=cells | N=5-6, n=10-14 | N=6-8, n=14-20 | |
| Capacitance (pF) | 84.0 ± 6.4 | 61.0 ± 3.5 | p=0.005 |
| Peak INa (pA/pF) | -48.6 ± 4.0 | -94.8 ± 6.3 | p<0.0001 |
| V of Peak INa (mV) | -40.4 ± 1.0 | -42.5 ± 1.2 | p=0.17 |
| INa Rev. Pot. (mV) | 5.1 ± 1.9 | 9.2 ± 1.5 | p=0.09 |
| INa Availability | |||
| V ½ | -86.5 ± 1.8 | -92.6 ± 1.8 | p=0.02 |
| Slope Factor | 6.3 ± 0.2 | 6.4 ± 0.1 | p=0.81 |
| INa Conductance | |||
| V ½ | -50.4 ± 1.3 | -55.0 ± 1.4 | p=0.02 |
| Slope Factor | 4.9 ± 0.2 | 5.3 ± 0.3 | p=0.19 |
| INa Decay (τfast) | |||
| -50 | 2.4 ± 0.3 | 1.7 ± 0.1 | p=0.02 |
| -45 | 1.8 ± 0.1 | 1.4 ± 0.1 | p=0.04 |
| -40 | 1.3 ± 0.1 | 1.2 ± 0.1 | p=0.31 |
| -35 | 1.2 ± 0.1 | 1.0 ± 0.1 | p=0.03 |
| -30 | 1.1 ± 0.1 | 0.8 ± 0.1 | p=0.01 |
| INa Decay (τslow) | |||
| -50 | 5.2 ± 0.8 | 5.9 ± 0.7 | p=0.50 |
| -45 | 4.1 ± 0.5 | 4.5 ± 0.4 | p=0.59 |
| -40 | 4.1 ± 0.5 | 3.8 ± 0.4 | p=0.59 |
| -35 | 2.5 ± 0.3 | 3.0 ± 0.3 | p=0.32 |
| -30 | 2.1 ± 0.3 | 2.6 ± 0.3 | p=0.28 |
| INa Recovery (tau) | 4.0 ± 0.4 | 4.8 ± 0.4 | p=0.19 |
| Persistent INa @ ………. -60 millivolts (pA/pF) | -2.0 ± 0.7 | -4.3 ± 0.6 | p=0.04 |
| (N=2; n=7) | (N=1; n=5) | ||
| TTX Resistant INa (%) | 86.7 ± 3.1 | 87.0 ± 2.1 | p=0.94 |
| (N=2; n=7) | (N=1; n=5) | ||
| TTX-Resistant INa Availability | |||
| V ½ | -89.3 ± 1.4 | -90.0 ± 2.6 | p=0.81 |
| Slope Factor | 6.2 ± 0.3 | 7.2 ± 0.5 | p=0.11 |
| TTX Resistant INa Conductance | |||
| V ½ | -55.0 ± 1.7 | -52.1 ± 1.5 | p=0.24 |
| Slope Factor | 4.7 ± 0.4 | 6.1 ± 0.4 | p=0.04 |
| TTX-Sensitive INa Availability | |||
| V ½ | -76.9 ± 1.6 | -79.1 ± 1.9 | p=0.40 |
| Slope Factor | 4.1 ± 0.3 | 5.2 ± 0.2 | p=0.01 |
| TTX Sensitive INa Conductance | |||
| V ½ | -44.5 ± 4.6 | -40.2 ± 2.5 | p=0.43 |
| Slope Factor | 4.7 ± 1.0 | 6.5 ± 0.5 | p=0.15 |
Significant differences between the WT vs. DS myocytes are defined as p ≤ 0.05.
Differences in Scn5a transcription and translation are undetectable
DS cardiac myocytes exhibited a 2-fold increase in TTX-R transient and persistent INa density compared to WT myocytes. In an effort to understand the molecular mechanism for this difference, we performed quantitative RT-PCR (qRT-PCR) to ascertain whether we could detect differences in the level of Scn5a transcripts between genotypes. Using two different Scn5a primer pairs and two independent cDNAs per animal (n=4-5), we observed no change in the amount of Scn5a transcript (Figure 3A ). To assess differences in channel protein expression, we quantified Nav1.5 polypeptide levels in membrane enriched ventricular myocyte fractions from each genotype. As Figure 3B-C indicate, we found no measurable changes in Nav1.5 expression, suggesting a post-translational mechanism, e.g. altered channel trafficking, phosphorylation, or association with cytoskeletal proteins in specific subcellular domains.
Figure 3. mScn5a and Nav1.5 levels are unchanged in DS mutant hearts.

A. Heart RNA from biological replicates (DS mice, n = 4; WT mice, n = 5) were used to generate two independent cDNAs per animal. The cDNAs were assayed using qPCR in quadruplicate with two independent Scn5a TaqMan primer sets and normalized to 18s RNA. B. Western blots of membrane proteins isolated from DS and WT ventricular CMs. 50 µg of protein was loaded in each lane, and probed with anti-Nav1.5 (Mohler 1:1000), and anti-α-actin (Sigma 1:500), which served as the loading control. C. Quantification of Nav1.5 expression normalized to α-actin expression.
DS cardiac myocytes are hyperexcitable
To determine whether the observed changes in INa resulted in altered cardiac myocyte excitability, we recorded APs from isolated WT and DS ventricular cardiac myocytes. DS cardiac myocytes were hyperexcitable compared to WT. DS cells required significantly less current to initate AP firing (p ≤ 0.05, Figure 4A ). At all pacing cycle lengths, WT and DS mice had similar diastolic membrane potentials (ranging between -75 mV to -72 mV). At each pacing cycle length, we observed non-significant trends for increased AP upstroke velocity and APD in DS vs. WT cardiac myocytes (Figure 4B and C ). Despite these values not reaching statistical significance, we observed a significant increase in the incidence of EADs in DS (67%) vs. WT cardiac myocytes (18%, p ≤ 0.05, Figure 4D ), which provides a substrate for the initiation of cardiac arrhythmias [19].
Figure 4. DS myocytes exhibit increased excitability and incidence of early after depolarizations (EADs).

A. DS myocytes require significantly less injected current to fire APs. B. DS myocyte AP upstroke velocity is faster at all pacing cycle lengths (p = ns). C. Slight prolongation of the AP duration at 30%, 50%, and 75% repolarization at many pacing cycle lengths (p = ns). D. DS myocytes are significantly more susceptible to EADs, a substrate for arrhythmogenesis. Inset: Representative EADs from DS myocytes (red.) Panels A-C, unpaired t-test with Welch’s correction. Panel D χ2 Test (WT, N = 9, n = 11, DS, N = 8, n = 17).
DS leads to changes in cardiac excitability and sudden death
We observed that 21% of DS mice die by P150, with 38% of these deaths occurring before P25 (similar to [25]), and 69% of the deaths by P52 (n = 75 for each group, Figure 5A ) [26]. A similar incidence of SUDEP was observed in DS mice that were implanted with radiotelemetry devices (Figure 5B ). At the termination of the in vivo radiotelemetry ECG study a subset of WT and DS mice were tested for the susceptibility to PTZ-induced convulsive seizures (rated on the Racine Scale). Administration of PTZ led to marked bradycardia, which may be similar to previously observed seizure-induced bradycardia [44] and similar to that observed in Scn1a +/- mice following acute hyperthermia induced seizures [25]. Interestingly, the initial injection of PTZ resulted in a sudden 32 ± 5% decrease in the HR, which was further diminished with increasing doses of PTZ (58 ± 3% of pre-drug). Consistent with the increased susceptibility to seizures in DS [26,45-48], and the previously documented incidence of spontaneous seizures in DS mice [26], the minimum additive concentration of PTZ after which a convulsive seizure was first observed was less in DS than in WT mice (p = 0.02 at Racine Scale 5, Figure 6 ).
Figure 5. DS Mice Undergo SUDEP.

A. Kaplan-Meier survival curves for WT and DS mice (N = 75 for each group, p < 0.0001, Log-rank, Mantel-Cox, Survival Test). B. Percent survival in WT (N = 8) and DS (N = 13) mice implanted with radiotelemetry units. SUDEP or near-SUDEP in 3 DS mice (at P41, P45, and P51, respectively).
Figure 6. Decreased Threshold for PTZ Induced Seizures in DS Mice.

WT and DS mice were administered incremental doses of pentylenetetrazole (PTZ), monitored for observable seizures, and classified on the Racine Scale.
In vivo ECG recordings suggest a cardiac mechanism for SUDEP in DS
Consistent with the increased persistent INa and AP changes reported above, DS mice exhibited significant alterations in the ECG that may provide insights into a potential human DS cardiac phenotype. While there were no differences in HR, PQ interval, PR interval, or QRS duration, we observed a significant prolongation of the QT interval (HR corrected and uncorrected QT50-90%, Table 2 and Figure 7A ). We were able to record the events preceding spontaneous SUDEP (or near-SUDEP) in 3 of the 13 DS mice at P41, P45, and P51 (refered to as DS-1, DS-2, and DS-3), respectively. All 8 WT mice were alive at the end of the study (Figure 5B ). DS-1 and DS-2 died suddenly subsequent to ventricular fibrillation (VF), while DS-3 became moribund, with lack of movement, severe bradycardia, and hypothermia, and was euthanized for ethical reasons. All 3 DS mice that died exhibited R-R variability. Focal and idioventricular rhythms, conduction abnormalities, and ultimately a catastrophic cardiac arrhythmia preceded death in DS-1 and DS-2. One hundred minutes prior to putative SUDEP, which occurred at 7:46 PM and 10:16 PM in DS-1 and DS-2, respectively, the animals became bradycardic, followed by a sharp increase in the HR just prior to the terminal event (Figure 7B ). In contrast, the HR was steady in time matched WT-1 and WT-2 mice. DS-3 exhibited sudden non-lethal bradycardic events, resulting in large HR fluctations and progressive HR slowing (Figure 7C ). To further investigate the DS ECG phenotype preceding putative SUDEP, we analyzed the R-R interval. One day prior to death in DS-1 and DS-2 the R-R interval was regular. In contrast, 1 hour preceding death in both animals the R-R interval became irregular, with more frequent episodes of R-R variability preceding the fatal cardiac arrhythmia and sudden death (Figure 7D and E ). DS-3 developed increased R-R variability in the 48 hours preceding euthanasia, with the most pronounced R-R variability in the hour preceding death (Figure 7F ).
Table 2. Telemetry ECG Measurements of various parameters illustrating a significant prolongation of the QT interval and the corrected QT interval (QTc). QTc calculated by the Bazett formula.
|
Mouse
|
|||
|---|---|---|---|
| Parameter | WT | DS | Significance |
| N = # mice | N = 5 | N = 13 | |
| RR | 87.3 ± 2.9 | 86.9 ± 2.2 | p = 0.93 |
| PQ | 29.1 ± 1.3 | 28.0 ± 0.6 | p = 0.46 |
| PR | 33.2 ± 0.9 | 32.4 ± 0.4 | p = 0.41 |
| QRS | 7.1 ± 0.5 | 7.5 ± 0.3 | p = 0.53 |
| QT50 | 30.6 ± 1.4 | 35.6 ± 1.9 | p = 0.05 |
| QT75 | 33.6 ± 1.2 | 39.3 ± 2.2 | p = 0.04 |
| QT90 | 35.6 ± 1.0 | 42.2± 2.5 | p = 0.03 |
| QT50c | 3.6 ± 0.2 | 4.2 ± 0.2 | p = 0.04 |
| QT75c | 3.3 ± 0.2 | 3.8 ± 0.2 | p = 0.06 |
| QT90c | 3.8 ± 0.2 | 4.5 ± 0.2 | p = 0.03 |
Significant differences between the WT vs. DS myocytes are defined as p ≤ 0.05.
Figure 7. Altered Heart Rates Precede Death.

A. DS mice exhibit significant QT prolongation (50 - 90%). B. Heart rates in DS mice decrease 100 min before death, followed by a sharp increase just prior to the terminal event, while the WT heart rates remains high and constant. (100 minutes = 10:16 PM in WT-1 and DS-1; 7:46 PM in WT-2 and DS-2). C. WT-3 and DS-3 HR cycling, followed by DS exhibiting sudden drops in heart rate in the 72 h preceding death. D and E. Increased R-R variability 60 min prior to SUDEP in DS-1 (blue) and DS-2 (red), respectively, with further increased variability immediately preceding the lethal arrhythmia, while 1 day prior at the same time the R-R interval was constant (black). F. Progressive bradycardia and increased R-R variability in DS-3 at several time points preceding an agonal state and euthanasia (denoted by colored arrows in C).
Cardiac arrhythmias in DS mice often preceded apparent spontaneous convulsive seizures observed as high frequency muscle artifacts (Figure 8 ). These artifacts were not observed in untreated WT mice, but arose during PTZ-induced convulsive seizures in both WT and DS mice (Figures 9 D ). DS-1 and DS-2 exhibited periods of premature ventricular complexes (PVCs) and bundle branch block (BBB) that often preceded or occurred during apparent convulsive seizures (Figures 8 , A and B). In contrast, the incidence of PVCs and BBB during the PTZ study was low (3 of 8 mice with 1-3 PVCs per mouse over a 3 hour period), was not correlated with PTZ induced convulsive seizures, and was not closely coupled to the catastrophic event (occuring more than 30-60 minutes prior to death from status epilepticus).
Figure 8. Cardiac Arrhythmias Precede SUDEP in DS.

Lead II ECG traces illustrating cardiac arrhythmias preceding death. A-C. In mouse DS-2, muscle artifact consistent with convulsive seizures was preceded by idioventricular rhythms, including premature ventricular complexes (PVCs), bundle branch block (BBB), altered QRS morphology, and R-R variability. D and E. Initiation of high frequency electrical activity without any discernible sinus activity, consistent with VF. F. Low amplitude wide complex focal bradycardia with a BBB morphology, and eventual asystole.
Figure 9. Dominant Frequency Analysis.

A. Sinus rhythm 1 day prior to SUDEP, which is consistent with heart rate (728 bpm) analysis. B. Muscle artifact embedded in the sinus ECG (same as Figure 9 C) without any clear frequency peaks. C. High frequency electrical activity without any discernible sinus activity, consistent with VF (~25 Hz, same as Figure 9, D and E). D. PTZ induced seizures lead to a lower frequency electrical signal (~10 - 20 Hz). Inset: Representative snapshots of the ECG signal included in the fast-fourier transformation.
All 3 DS mice developed large R-R variability and idioventricular arrhythmias with changing QRS morphology, which were closely coupled with high frequency electrical activity, consistent with the signal during an apparent convulsive seizure (Figure 8 , A-C). Other than a few PVCs in WT-2, time matched ECG recordings from WT-1, WT-2, and WT-3 did not exhibit any of these ECG manifestations and arrhythmias that preceded sudden death in DS mice. In contrast to the ECG changes preceding death in DS-1 and DS-2, the R-R and QRS changes recorded after PTZ administration did not directly precede the convulsive seizures. Ultimately, DS-1 and DS-2 underwent a catastrophic event with high frequency electrical activity without any discernible QRS complexes. A contribution from muscle artifact cannot be entirely excluded, but the ECGs from both mice are consistent with VF (Figure 8, D and E ), as discussed below.
Figure 9A illustrates sinus activity with the corresponding Fast Fourier Transform (FFT) spectrum showing a dominant frequency peak at ~12 Hz that is consistent with the measured HR of 728 bpm. Figure 9B is the FFT spectrum of an ECG pattern consistent with a convulsive seizure with the maintenance of sinus activity. There were not any clear dominant frequency peaks (ECG signal from Figure 8C ). Figure 9C shows the FFT spectrum during the apparent VF ECG pattern. The dominant frequency of the complex electrical activity was ~25 Hz, which is consistent with previously reported frequencies of mouse VF [49,50]. To confirm that the muscle artifact from a spontaneous lethal convulsive seizure would not yield a similar FFT spectrum as in Figure 9C , we assessed the FFT spectrum during a PTZ-induced convulsive seizure (Racine Scale 5, Figure 9D ). Unlike the dominant frequency peak at ~25 Hz in Figure 9C , the ECG signal during a PTZ induced seizure was ~10 Hz (Figure 9D ). In summary, neither putative spontaneous seizures nor PTZ-induced seizures in DS mice, with and without identifiable QRS complexes embedded within the high frequency muscle artifact, respectively, yielded a peak at 25 Hz on the ECG FFT spectrum (Figure 9B and D ). Therefore, PTZ-induced seizures and death did not phenocopy the cardiac ECG phenotype of putative SUDEP in DS-1 and DS-2. Figure 9 demonstrates the sensitivity of FFT analysis to isolate the multipe frequency components and provides further evidence that one mechanism for SUDEP in DS may be cardiac arrhythmia.
Ultimately, DS-1 and DS-2 developed wide complex, low amplitude, and bradycardic focal discharges with a BBB morphology that progressively decreased in rate to eventual asystole (Figure 8F ). In contrast to DS-1 and DS-2, DS-3 did not have the opportunity to undergo a lethal cardiac arrhythmia, as it was euthanized for ethical reasons. Yet, during the final hour of ECG recordings DS-3, but not WT-3, became bradycardic (<180 bpm) and exhibited abrupt changes in the QRS morphology and amplitude. Some aspects of the spontaneous bradycardia and periods of bradycardia followed by tachycardia, observed here, are similar to effects reported in the Scn1a +/- DS mouse model following acute hyperthermic seizures [25], although there are key differences, for example, the absence of atrioventricular nodal block and the initiation of VF in our model. Nevertheless, taken together, these data support the hypothesis that significant cardiac pathophysiological changes occur in DS mice.
Discussion
SUDEP is a catastrophic, multi-system failure that involves seizures, changes in autonomic tone, respiratory dysregulation, and cardiac arrhythmias [5,7,51]. Recent work suggested that parasympathetic hyperactivity following hyperthermia induced tonic-clonic seizures resulted in severe bradycardia and death in the Scn1a +/- mouse model of DS [25]. This study, however, left open important gaps in our knowledge: first, whether cardiac myocytes isolated from a mouse model of DS have altered excitability and second, whether DS mice exhibit cardiac dysfunction following spontaneous seizures. Here, we used a multilevel approach to investigate the DS cardiac phenotype in a global knockin mouse model expressing a human DS mutation. Electrophysiological recordings of acutely dissociated ventricular myocytes demonstrated that Scn1a haploinsufficiency leads to a 2-fold increase in transient and persistent INa density. The pharmacological and biophysical properties of INa in the DS myocytes suggest that the observed increase in INa is the result of an increased number of functional Nav1.5 channels at the plasma membrane. DS ventricular myocytes exhibited alterations in AP morphology and incidences of triggered activity. In vivo, DS mice developed spontaneous seizures and pathological ECG manifestations, including bradycardia, idioventricular rhythms, RR variability, PVCs, BBB, and VF. Ultimately, these results provide mechanistic insights into how alterations in cardiac electrical function establish ideal conditions for arrhythmogenesis and SUDEP. Thus, DS mutations in Scn1a lead not only to alterations in neuronal excitability, but also to cardiac electrophysiological abnormalities in isolated ventricular myocytes, contributing to the mechanism underlying SUDEP.
DS venticular myocytes have increased INa density with AP changes
Neuronal hyperexcitability in DS mouse models has been proposed to occur through a selective decrease in INa in inhibitory neurons [25,26,34,52]. Interestingly, more recent results demonstrate that at P21-24, an age older than previously investigated, INa is increased in excitatory pyramidal neurons in Scn1a+/- mice [53]. Further, DS patient-specific forebrain-like neurons generated from iPSCs have increased INa in both excitatory and inhibitory neuronal cell types [32]. Here, we observed a significant increase in TTX-R INa density and hyperpolarizing shifts in the voltage dependence of INa conductance and availability in DS cardiac myocytes. Our data are consistent with previous reports showing that increased Nav1.5 expression leads to hyperpolarizing shifts in the voltage dependence of INa conductance and availability [28,29]. These results suggest that the increased INa in DS can be explained by increased functional TTX-R Nav1.5 activity. In another mouse model of DS (Scn1b null mice [54]) there was a 2-fold increase in Nav1.5-mediated transient and persistent INa. Furthermore, in Scn1b null mice and in ventricular myocytes over-expressing human SCN5A [28] there was action potential prolongation, with prolonged QT and RR intervals in the Scn1b mice [54]. Increased transient INa is predicted to provide more depolarizing current during the AP upstroke, resulting in a lower threshold of current injection to elicit an AP, and increased AP upstroke velocity. Increased transient INa alone would increase the safety factor and preserve the stability of impulse propagation [18]. However, as observed in models of LQTS-3 and Nav1.5 overexpression, increased persistent INa disrupts the balance between depolarizing and repolarizing currents, leading to APD/QT prolongation, EADs, arrhythmias, and sudden cardiac death [28,55].
DS Mice Exhibit LQTS Phenotype
Similar to the INa recordings from DS mice presented here, our previous work using the Scn1b null mouse model of DS demonstrated proportional increases in transient and persistent INa [54]. In contrast, genetic and pharmacological models of increased persistent INa leading to LQTS3 exhibit a disproportionate increase in persistent INa, with little change in transient INa [55-58]. As we previously demonstrated however [28], regardless of whether the increase in persistent INa scales with the transient current, an absolute increase in persistent INa disturbs the balance of inward and outward currents during the AP plateau (a time of high membrane resistance). Subsequently, this leads to APD prolongation and EAD formation, which is a known trigger for the initiation of arrhythmias and sudden cardiac death [19,59].
It remains incompletely understood whether the QT interval is prolonged in DS patients. QT prolongation has been shown to be a predisposing interictal and peri-ictal factor that ultimately leads to SUDEP in some (non-DS) epilepsy patients [7]. QT prolongation has been observed in children and adult patients with chronic epilepsy, during epileptic discharges, and the QT prolongation is even more prevalent in SUDEP vs. without SUDEP epileptic patients [4,60-64]. Interestingly, 33-44% of epilepsy patients and mouse models of epilepsy are prone to cardiac arrhythmias and LQTS, and conversely, one-third of LQTS patients have a history of seizures [8,65,66]. In two studies that examined ECG parameters in patients with DS, increased QT dispersion [67] and decreased heart rate variability [67,68] were observed, with no changes in the QT interval [67,68]. Studies that have examined the QT interval in DS mouse models have yielded contrasting results, with QT prolongation observed in the Scn1b null model and no change in the Scn1a+/- model [25,54]. The present study, using a human SCN1A knockin mutant mouse model, is the first to record ECG properties in conscious, unrestrained, and telemetered DS mice that have regained circadian heart rate, temperature, and activity cycling (e.g., Figure 7C).
DS mutations lead to alterations in the expression of other ion channels
Changes in the expression of a single ion channel gene often result in changes in the expression levels of other ion channel genes [29,69,70]. Scn1a null mice have increased Nav1.3 expression in the hippocampus [34]. Scn1b null mice have decreased Nav1.1 and increased Nav1.3 expression in the hippocampus, increased Scn5a/Nav1.5 expression and 3H-saxitoxin binding (suggesting increased TTX-S VGSC expression) in the heart [54], as well as altered TTX-R and TTX-S INa biophysical properties and decreased Nav1.9 expression in dorsal-root-ganglia (DRG) [48,69]. DS patient-specific iPSC excitatory and inhibitory neurons have increased INa in spite of SCN1A haploinsufficiency [32]. These compensatory changes in ion channel expression are not limited to VGSCs, as there is also a reduction in K+ current in Scn1b null DRGs and cortical pyramidal neurons, contributing to hyperexcitability [69,71]. Dissociated DRG neurons from Scn1b null mice have a reduction in not only INa density, but also in Kv4.2 expression and IA density [74]. Furthermore, Navβ1 has been shown to interact with Kv4.2 [71]. Cortical neurons from Scn1b null mice exhibited reduced IA current, and cortical pyramidal neurons exhibited APD prolongation with repetitive firing [71]. Additionally, Nav1.5 and Kir2.1 have been shown to be part of a macromolecular complex, with reciprocal interactions between Nav1.5 and Kir2.1 expression, and subsequent changes in INa and IK1 density [29]. Here, we show that Scn1a haploinsufficiency leads to increased functional expression of TTX-R INa in the heart. Taken together, these data suggest a homeostatic-like mechanism in response to VGSC subunit gene mutation that overcompensates by increased activity of a different VGSC subunit.
It is not surprising that we were unable to detect a measureable decrease in TTX-S INa in our experiments. Nav1.1 is one of at least three known TTX-S VGSCs expressed in cardiac myocytes, and TTX-S INa contributes only ~10% of the global INa [11,14,17,42,43]. Thus, it is unlikely that small decreases in TTX-S INa, due to Scn1a haploinsufficency, would be resolvable using the whole cell voltage clamp technique. In future studies, the use of super-resolution imaging, as in [72], will allow positioning of the recording electrode directly at the T-tubules and in the triad, where TTX-S VGSCs are localized, thus enabling more precise measurement of small, localized changes in cardiac TTX-S INa. In spite of this, it is possible that decreases in Nav1.1 expression in the T-tubules may disturb the macromolecular complex that includes the Na+-Ca2+ exchanger, Na+/K+-ATPase, inositol triphosphate receptor, Ca2+-calmodulin dependent protein kinase-II, and ankyrin-B [73,74]. Mutations in ankyrin-B are known to lead to altered Ca2+ handling and pathological ECG changes, including QT prolongation, bradycardia, sinus arrhythmia, idiopathic ventricular fibrillation, catecholaminergic polymorphic ventricular tachycardia, and risk of sudden death [75,76]. These results show that microdomain changes at the T-tubules can lead to whole cell implications. Here, in our DS model, altered Ca2+ handling downstream of reductions in Nav1.1 expression at the T-tubules may indirectly affect Nav1.5 function via changes in the binding of calmodulin to the channel or altered Ca2+-calmodulin dependent protein kinase-II-mediated channel phosphorylation, ultimately resulting in increased INa and arrhythmia [77,78]. Testing this hypothesis will be the focus of future studies.
Cardiac arrhythmias provide a mechanism for SUDEP in DS
Ion channelopathies are multi-organ diseases. Simultaneous electroencephalogram (EEG) and ECG studies confirm the high prevalence (33-44%) of arrhythmias in epileptic patients [8]. Conversely, approximatly one-third of LQTS patients have a history of seizures [65]. We propose that SUDEP may be caused by mutations in ion channel genes that are expressed in both the brain and the heart (e.g., KCNQ1 [8], KCNH2 [65], SCN1B [54], SCN5A [79], SCN8A [15], and SCN1A [this study and [25]). Just prior to death, some SUDEP patients [5] and LQTS mutant mice [8] exhibit loss of EEG activity, cardiorespiratory changes, and ultimately fatal cardiac arrhythmias. Sudden death due to cardiac arrhythmias in the mice shown here is consistent with published factors that precipitated and accompanied SUDEP [7]. Our mouse data mirror a human case of near-SUDEP in which the patient developed VF following a generalized seizure and ventricular tachycardia [66]. This study is the first to reveal the implications of DS mutations in Scn1a on the incidence and mechanisms of arrhythmias and SUDEP due to changes in cardiac myocyte excitability, and suggest targets for risk assessment and intervention to prevent SUDEP in DS and perhaps other epileptic channelopathies.
Acknowledgments
The authors thank Louis D’Alecy, DMD PhD and Steven Whiteshall from the University of Michigan Phenotyping Core, Krzysztof R. Grzeda, MD PhD for providing custom software for AP analysis, Larisa Kruger and Gustavo Patino, MD PhD for assistance with the PTZ-induced seizure experiments, and Peter Mohler, PhD, Ohio State University, for the anti-Nav1.5 antibody.
Funding Statement
This work was supported by grants NIH-NS076752 (to LLI), NIH-NS076916 (to JMP), the University of Michigan Center for Organogenesis (to LLI), Citizens United for Research in Epilepsy Foundation (to JMP) and the Dravet Syndrome Foundation(to JMP and MHM). DSA was supported by NIH T32HL007853 to the University of Michigan Cardiovascular Center and a Postdoctoral Fellowship from the Epilepsy Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Escayg A, Goldin AL (2010) Sodium channel SCN1A and epilepsy: mutations and mechanisms. Epilepsia 51: 1650-1658. doi: 10.1111/j.1528-1167.2010.02640.x. PubMed: 20831750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dravet C, Bureau M, Oguni H, Fukuyama Y, Cokar O (2005) Severe myoclonic epilepsy in infancy: Dravet syndrome. Adv_Neurol 95: 71-102. PubMed: 15508915. [PubMed] [Google Scholar]
- 3. Oakley JC, Kalume F, Catterall WA (2011) Insights into pathophysiology and therapy from a mouse model of Dravet syndrome. Epilepsia 52 Suppl 2: 59-61. doi: 10.1111/j.1528-1167.2011.03004.x. PubMed: 21463282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Surges R, Adjei P, Kallis C, Erhuero J, Scott CA et al. (2010) Pathologic cardiac repolarization in pharmacoresistant epilepsy and its potential role in sudden unexpected death in epilepsy: a case-control study. Epilepsia 51: 233-242. doi: 10.1111/j.1528-1167.2009.02330.x. PubMed: 19817816. [DOI] [PubMed] [Google Scholar]
- 5. Shorvon S, Tomson T (2011) Sudden unexpected death in epilepsy. Lancet, 378: 2028–38. PubMed: 21737136. [DOI] [PubMed] [Google Scholar]
- 6. Schuele SU, Widdess-Walsh P, Bermeo A, Lüders HO (2007) Sudden unexplained death in epilepsy: the role of the heart. Cleve_Clin_J_Med 74 Suppl 1: S121-S127. PubMed: 17455560. [DOI] [PubMed] [Google Scholar]
- 7. Surges R, Thijs RD, Tan HL, Sander JW (2009) Sudden unexpected death in epilepsy: risk factors and potential pathomechanisms. NatRev. Neurologist 5: 492-504. [DOI] [PubMed] [Google Scholar]
- 8. Goldman AM, Glasscock E, Yoo J, Chen TT, Klassen TL et al. (2009) Arrhythmia in heart and brain: KCNQ1 mutations link epilepsy and sudden unexplained death. Sci Transl_Med 1: 2ra6 PubMed: 20368164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C et al. (2001) De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am_J_Hum_Genet 68: 1327-1332. PubMed: 11359211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Meisler MH, Kearney JA (2005) Sodium channel mutations in epilepsy and other neurological disorders. J_Clin_Invest 115: 2010-2017. PubMed: 16075041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maier SK, Westenbroek RE, McCormick KA, Curtis R, Scheuer T et al. (2004) Distinct subcellular localization of different sodium channel alpha and beta subunits in single ventricular myocytes from mouse heart. Circulation 109: 1421-1427. doi: 10.1161/01.CIR.0000121421.61896.24. PubMed: 15007009. [DOI] [PubMed] [Google Scholar]
- 12. Maier SK, Westenbroek RE, Yamanushi TT, Dobrzynski H, Boyett MR et al. (2003) An unexpected requirement for brain-type sodium channels for control of heart rate in the mouse sinoatrial node. Proc_Natl_Acad Sci U_S_A 100: 3507-3512. PubMed: 12631690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Inoue N, Ohkusa T, Nao T, Lee JK, Matsumoto T et al. (2004) Rapid electrical stimulation of contraction modulates gap junction protein in neonatal rat cultured cardiomyocytes: involvement of mitogen-activated protein kinases and effects of angiotensin II-receptor antagonist. Jamcollcardiol 44: 914-922. PubMed: 15312880. [DOI] [PubMed] [Google Scholar]
- 14. Dhar Malhotra J, Chen C, Rivolta I, Abriel H, Malhotra R et al. (2001) Characterization of sodium channel alpha- and beta-subunits in rat and mouse cardiac myocytes. Circulation 103: 1303-1310. doi: 10.1161/01.CIR.103.9.1303. PubMed: 11238277. [DOI] [PubMed] [Google Scholar]
- 15. Noujaim SF, Kaur K, Milstein M, Jones JM, Furspan P et al. (2012) A null mutation of the neuronal sodium channel NaV1.6 disrupts action potential propagation and excitation-contraction coupling in the mouse heart. FASEB J 26: 63-72. doi: 10.1096/fj.10-179770. PubMed: 21948246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baruscotti M, Westenbroek R, Catterall WA, DiFrancesco D, Robinson RB (1997) The newborn rabbit sino-atrial node expresses a neuronal type I-like Na+ channel. J Physiol 498 ( 3): 641-648. PubMed: 9051576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kaufmann SG, Westenbroek RE, Maass AH, Lange V, Renner A et al. (2013) Distribution and function of sodium channel subtypes in human atrial myocardium. J Mol Cell Cardiol, 61: 133–41. PubMed: 23702286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kléber AG, Rudy Y (2004) Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol Rev 84: 431-488. doi: 10.1152/physrev.00025.2003. PubMed: 15044680. [DOI] [PubMed] [Google Scholar]
- 19. Zareba W, Cygankiewicz I (2008) Long QT syndrome and short QT syndrome. Prog_Cardiovasc_Dis 51: 264-278. PubMed: 19026859. [DOI] [PubMed] [Google Scholar]
- 20. George AL Jr. (2005) Inherited disorders of voltage-gated sodium channels. J_Clin_Invest 115: 1990-1999. PubMed: 16075039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Catterall WA, Kalume F, Oakley JC (2010) NaV1.1 channels and epilepsy. JPhysiol J Physiol 588: 1849-1859. PubMed: 20194124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Trudeau MM, Dalton JC, Day JW, Ranum LP, Meisler MH (2006) Heterozygosity for a protein truncation mutation of sodium channel SCN8A in a patient with cerebellar atrophy, ataxia, and mental retardation. Med_Genet 43: 527-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Papadatos GA, Wallerstein PM, Head CE, Ratcliff R, Brady PA et al. (2002) Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proc Natl_Acad_Sci_U_S_A 99: 6210-6215. PubMed: 11972032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lei M, Jones SA, Liu J, Lancaster MK, Fung SS et al. (2004) Requirement of neuronal- and cardiac-type sodium channels for murine sinoatrial node pacemaking. JPhysiol 559: 835-848 [DOI] [PMC free article] [PubMed]
- 25. Kalume F, Westenbroek RE, Cheah CS, Yu FH, Oakley JC et al. (2013) Sudden unexpected death in a mouse model of Dravet syndrome. J Clin Invest 123: 1798-1808. doi: 10.1172/JCI66220. PubMed: 23524966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E et al. (2007) Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. JNeurosci J Neurosci 27: 5903-5914. PubMed: 17537961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cerrone M, Noujaim SF, Tolkacheva EG, Talkachou A, O'Connell R et al. (2007) Arrhythmogenic mechanisms in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ_Res 101: 1039-1048. PubMed: 17872467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Auerbach DS, Grzeda KR, Furspan PB, Sato PY, Mironov S et al. (2011) Structural heterogeneity promotes triggered activity, reflection and arrhythmogenesis in cardiomyocyte monolayers. JPhysiol 589: 2363-2381 [DOI] [PMC free article] [PubMed]
- 29. Milstein ML, Musa H, Balbuena DP, Anumonwo JM, Auerbach DS et al. (2012) Dynamic reciprocity of sodium and potassium channel expression in a macromolecular complex controls cardiac excitability and arrhythmia. Proc Natl Acad Sci U S A 109: E2134-E2143. doi: 10.1073/pnas.1109370109. PubMed: 22509027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brackenbury WJ, Davis TH, Chen C, Slat EA, Detrow MJ et al. (2008) Voltage-gated Na+ channel beta1 subunit-mediated neurite outgrowth requires Fyn kinase and contributes to postnatal CNS development in vivo. J Neurosci 28: 3246-3256. doi: 10.1523/JNEUROSCI.5446-07.2008. PubMed: 18354028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lowe JS, Palygin O, Bhasin N, Hund TJ, Boyden PA et al. (2008) Voltage-gated Nav channel targeting in the heart requires an ankyrin-G dependent cellular pathway. J Cell Biol 180: 173-186. doi: 10.1083/jcb.200710107. PubMed: 18180363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu Y, Lopez-Santiago LF, Yuan Y, Jones JM, Zhang H et al. (2013) Dravet syndrome patient-derived neurons suggest a novel epilepsy mechanism. Ann Neurol, 74: 128–39. PubMed: 23821540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen Y, Yu FH, Sharp EM, Beacham D, Scheuer T et al. (2008) Functional properties and differential neuromodulation of Na(v)1.6 channels. Mol_Cell Neurosci 38: 607-615. doi: 10.1016/j.mcn.2008.05.009. PubMed: 18599309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F et al. (2006) Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat_Neurosci 9: 1142-1149. PubMed: 16921370. [DOI] [PubMed] [Google Scholar]
- 35. Yamaji N, Little MJ, Nishio H, Billen B, Villegas E et al. (2009) Synthesis, solution structure, and phylum selectivity of a spider delta-toxin that slows inactivation of specific voltage-gated sodium channel subtypes. Biol_Chem 284: 24568-24582. PubMed: 19592486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Petitprez S, Zmoos AF, Ogrodnik J, Balse E, Raad N et al. (2011) SAP97 and dystrophin macromolecular complexes determine two pools of cardiac sodium channels Nav1.5 in cardiomyocytes. Circ_Res 108: 294-304. PubMed: 21164104. [DOI] [PubMed] [Google Scholar]
- 37. Fletcher EV, Kullmann DM, Schorge S (2011) Alternative splicing modulates inactivation of type 1 voltage-gated sodium channels by toggling an amino acid in the first S3-S4 linker. Biol_Chem 286: 36700-36708. PubMed: 21890636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Patino GA, Brackenbury WJ, Bao Y, Lopez-Santiago LF, O'Malley HA et al. (2011) Voltage-Gated Na+ Channel {beta}1B: A Secreted Cell Adhesion Molecule Involved in Human Epilepsy. JNeurosci 31: 14577-14591 [DOI] [PMC free article] [PubMed]
- 39. Estrada G, Restano-Cassulini R, Ortiz E, Possani LD, Corzo G (2011) Addition of positive charges at the C-terminal peptide region of CssII, a mammalian scorpion peptide toxin, improves its affinity for sodium channels Nav1.6. Peptides 32: 75-79. doi: 10.1016/j.peptides.2010.11.001. PubMed: 21078353. [DOI] [PubMed] [Google Scholar]
- 40. Chatelier A, Zhao J, Bois P, Chahine M (2010) Biophysical characterisation of the persistent sodium current of the Nav1.6 neuronal sodium channel: a single-channel analysis. Pflugers Arch 460: 77-86. doi: 10.1007/s00424-010-0801-9. PubMed: 20204400. [DOI] [PubMed] [Google Scholar]
- 41. Mercier A, Clément R, Harnois T, Bourmeyster N, Faivre JF et al. (2012) The beta1-subunit of Na(v)1.5 cardiac sodium channel is required for a dominant negative effect through alpha-alpha interaction. PLOS ONE 7: e48690. doi: 10.1371/journal.pone.0048690. PubMed: 23133651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brette F, Orchard CH (2006) Density and sub-cellular distribution of cardiac and neuronal sodium channel isoforms in rat ventricular myocytes. Biochem_Biophys_Res_Commun 348: 1163-1166. PubMed: 16904633. [DOI] [PubMed] [Google Scholar]
- 43. Haufe V, Cordeiro JM, Zimmer T, Wu YS, Schiccitano S et al. (2005) Contribution of neuronal sodium channels to the cardiac fast sodium current INa is greater in dog heart Purkinje fibers than in ventricles. Cardiovasc_Res 65: 117-127. PubMed: 15621039. [DOI] [PubMed] [Google Scholar]
- 44. Nashef L, Hindocha N, Makoff A (2007) Risk factors in sudden death in epilepsy (SUDEP): the quest for mechanisms. Epilepsia 48: 859-871. doi: 10.1111/j.1528-1167.2007.01082.x. PubMed: 17433051. [DOI] [PubMed] [Google Scholar]
- 45. Dravet C, Bureau M, Dalla BB, Guerrini R (2011) Severe myoclonic epilepsy in infancy (Dravet syndrome) 30 years later. Epilepsia 52 Suppl 2: 1-2. doi: 10.1111/j.1528-1167.2010.02921.x. PubMed: 21463271. [DOI] [PubMed] [Google Scholar]
- 46. Guerrini R, Aicardi J (2003) Epileptic encephalopathies with myoclonic seizures in infants and children (severe myoclonic epilepsy and myoclonic-astatic epilepsy). J_Clin_Neurophysiol 20: 449-461. PubMed: 14734934. [DOI] [PubMed] [Google Scholar]
- 47. Oakley JC, Kalume F, Yu FH, Scheuer T, Catterall WA (2009) Temperature- and age-dependent seizures in a mouse model of severe myoclonic epilepsy in infancy. Proc_Natl_Acad Sci U_S_A 106: 3994-3999. PubMed: 19234123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen C, Westenbroek RE, Xu X, Edwards CA, Sorenson DR et al. (2004) Mice lacking sodium channel beta1 subunits display defects in neuronal excitability, sodium channel expression, and nodal architecture. JNeurosci 24: 4030-4042 [DOI] [PMC free article] [PubMed]
- 49. Noujaim SF, Pandit SV, Berenfeld O, Vikstrom K, Cerrone M et al. (2007) Up-regulation of the inward rectifier K+ current (I K1) in the mouse heart accelerates and stabilizes rotors. JPhysiol 578: 315-326 [DOI] [PMC free article] [PubMed]
- 50. Noujaim SF, Berenfeld O, Kalifa J, Cerrone M, Nanthakumar K et al. (2007) Universal scaling law of electrical turbulence in the mammalian heart. Proc Natl Acad Sci U S A 104: 20985-20989. doi: 10.1073/pnas.0709758104. PubMed: 18093948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nei M, Hays R (2010) Sudden unexpected death in epilepsy. Curr Neurol Neurosci Rep 10: 319-326. doi: 10.1007/s11910-010-0116-4. PubMed: 20446062. [DOI] [PubMed] [Google Scholar]
- 52. Cheah CS, Yu FH, Westenbroek RE, Kalume FK, Oakley JC et al. (2012) Specific deletion of NaV1.1 sodium channels in inhibitory interneurons causes seizures and premature death in a mouse model of Dravet syndrome. Proc Natl Acad Sci U S A 109: 14646-14651. doi: 10.1073/pnas.1211591109. PubMed: 22908258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mistry A, Miller A, Thompson C, Kearney J, George AL (2012) Strain and age-dependent differences in hippocampal neuron sodium current densities in a mouse model of Dravet syndrome. New Orleans, LA.: Society for Neuroscience. [Google Scholar]
- 54. Lopez-Santiago LF, Meadows LS, Ernst SJ, Chen C, Malhotra JD et al. (2007) Sodium channel Scn1b null mice exhibit prolonged QT and RR intervals. Jmolcell Cardiol 43: 636-647. PubMed: 17884088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nuyens D, Stengl M, Dugarmaa S, Rossenbacker T, Compernolle V et al. (2001) Abrupt rate accelerations or premature beats cause life-threatening arrhythmias in mice with long-QT3 syndrome. Nat_Med 7: 1021-1027. PubMed: 11533705. [DOI] [PubMed] [Google Scholar]
- 56. Terrenoire C, Wang K, Tung KW, Chung WK, Pass RH et al. (2013) Induced pluripotent stem cells used to reveal drug actions in a long QT syndrome family with complex genetics. J Gen Physiol 141: 61-72. PubMed: 23277474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pignier C, Revenaz C, Rauly-Lestienne I, Cussac D, Delhon A et al. (2007) Direct protective effects of poly-unsaturated fatty acids, DHA and EPA, against activation of cardiac late sodium current: a mechanism for ischemia selectivity. Basic Res_Cardiol 102: 553-564. doi: 10.1007/s00395-007-0676-x. PubMed: 17891522. [DOI] [PubMed] [Google Scholar]
- 58. Malan D, Friedrichs S, Fleischmann BK, Sasse P (2011) Cardiomyocytes obtained from induced pluripotent stem cells with long-QT syndrome 3 recapitulate typical disease-specific features in vitro. Circ Res 109: 841-847. doi: 10.1161/CIRCRESAHA.111.243139. PubMed: 21799153. [DOI] [PubMed] [Google Scholar]
- 59. Moss AJ, Kass RS (2005) Long QT syndrome: from channels to cardiac arrhythmias. J_Clin_Invest 115: 2018-2024. PubMed: 16075042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tavernor SJ, Brown SW, Tavernor RM, Gifford C (1996) Electrocardiograph QT lengthening associated with epileptiform EEG discharges--a role in sudden unexplained death in epilepsy? Seizure 5: 79-83. doi: 10.1016/S1059-1311(96)80067-7. PubMed: 8777558. [DOI] [PubMed] [Google Scholar]
- 61. Oppenheimer S (1990) Cardiac dysfunction during seizures and the sudden epileptic death syndrome. Jrsocmed 83: 134-136. PubMed: 2182856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Surges R, Taggart P, Sander JW, Walker MC (2010) Too long or too short? New insights into abnormal cardiac repolarization in people with chronic epilepsy and its potential role in sudden unexpected death. Epilepsia 51: 738-744. doi: 10.1111/j.1528-1167.2010.02571.x. PubMed: 20384763. [DOI] [PubMed] [Google Scholar]
- 63. Kändler L, Fiedler A, Scheer K, Wild F, Frick U et al. (2005) Early post-convulsive prolongation of QT time in children. Acta Paediatr 94: 1243-1247. PubMed: 16278992. [DOI] [PubMed] [Google Scholar]
- 64. Brotherstone R, Blackhall B, McLellan A (2010) Lengthening of corrected QT during epileptic seizures. Epilepsia 51: 221-232. doi: 10.1111/j.1528-1167.2009.02281.x. PubMed: 19732135. [DOI] [PubMed] [Google Scholar]
- 65. Johnson JN, Hofman N, Haglund CM, Cascino GD, Wilde AA et al. (2009) Identification of a possible pathogenic link between congenital long QT syndrome and epilepsy. Neurology 72: 224-231. doi:10.1212/01.wnl.0000335760.02995.ca. PubMed: 19038855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Espinosa PS, Lee JW, Tedrow UB, Bromfield EB, Dworetzky BA (2009) Sudden unexpected near death in epilepsy: malignant arrhythmia from a partial seizure. Neurology 72: 1702-1703. doi: 10.1212/WNL.0b013e3181a55f90. PubMed: 19433745. [DOI] [PubMed] [Google Scholar]
- 67. Ergul Y, Ekici B, Tatli B, Nisli K, Ozmen M (2013) QT and P wave dispersion and heart rate variability in patients with Dravet syndrome. Acta Neurol Belg 113: 161-166. doi: 10.1007/s13760-012-0140-z. PubMed: 23065439. [DOI] [PubMed] [Google Scholar]
- 68. Delogu AB, Spinelli A, Battaglia D, Dravet C, De Nisco A et al. (2011) Electrical and autonomic cardiac function in patients with Dravet syndrome. Epilepsia 52 Suppl 2: 55-58. doi: 10.1111/j.1528-1167.2011.03003.x. PubMed: 21463281. [DOI] [PubMed] [Google Scholar]
- 69. Lopez-Santiago LF, Brackenbury WJ, Chen C, Isom LL (2011) Na+ channel Scn1b gene regulates dorsal root ganglion nociceptor excitability in vivo. Biol_Chem 286: 22913-22923. PubMed: 21555511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sato PY, Musa H, Coombs W, Guerrero-Serna G, Patiño GA et al. (2009) Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ_Res 105: 523-526. PubMed: 19661460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Marionneau C, Carrasquillo Y, Norris AJ, Townsend RR, Isom LL et al. (2012) The sodium channel accessory subunit Navbeta1 regulates neuronal excitability through modulation of repolarizing voltage-gated K(+) channels. J Neurosci 32: 5716-5727. doi: 10.1523/JNEUROSCI.6450-11.2012. PubMed: 22539834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Agullo-Pascual E, Reid DA, Keegan S, Sidhu M, Fenyö D et al. (2013) Super-resolution fluorescence microscopy of the cardiac connexome reveals plakophilin-2 inside the connexin43 plaque. Cardiovasc Res. PubMed: 23929525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mohler PJ, Davis JQ, Bennett V (2005) Ankyrin-B coordinates the Na/K ATPase, Na/Ca exchanger, and InsP3 receptor in a cardiac T-tubule/SR microdomain. PLOS Biol 3: e423. doi: 10.1371/journal.pbio.0030423. PubMed: 16292983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. J O-Uchi, K Komukai, Y Kusakari, T Obata, K Hongo, et al. (2005) alpha1-adrenoceptor stimulation potentiates L-type Ca2+ current through Ca2+/calmodulin-dependent PK II (CaMKII) activation in rat ventricular myocytes. Proc Natl Acad Sci U S A 102: 9400-9405. doi: 10.1073/pnas.0503569102. PubMed: 15964981. 10.1073/pnas.0503569102 PubMed: 15964981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim S et al. (2003) Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature 421: 634-639. doi: 10.1038/nature01335. PubMed: 12571597. [DOI] [PubMed] [Google Scholar]
- 76. Mohler PJ, Splawski I, Napolitano C, Bottelli G, Sharpe L et al. (2004) A cardiac arrhythmia syndrome caused by loss of ankyrin-B function. Proc Natl Acad Sci U S A 101: 9137-9142. doi: 10.1073/pnas.0402546101. PubMed: 15178757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Adsit GS, Vaidyanathan R, Galler CM, Kyle JW, Makielski JC (2013) Channelopathies from mutations in the cardiac sodium channel protein complex. J Mol Cell Cardiol 61: 34-43. doi: 10.1016/S0735-1097(13)60035-2. PubMed: 23557754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Herren AW, Bers DM, Grandi E (2013) Post-translational modifications of the cardiac Na channel: contribution of CaMKII-dependent phosphorylation to acquired arrhythmias. Am J Physiol Heart Circ Physiol 305: H431-H445. doi: 10.1152/ajpheart.00306.2013. PubMed: 23771687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Aurlien D, Leren TP, Taubøll E, Gjerstad L (2009) New SCN5A mutation in a SUDEP victim with idiopathic epilepsy. Seizure 18: 158-160. doi: 10.1016/j.seizure.2008.07.008. PubMed: 18752973. [DOI] [PubMed] [Google Scholar]