

Abstract
MicroRNAs (miRNAs) are endogenous small RNAs playing a crucial role in plant growth and development, as well as stress responses. Among them, some are highly evolutionally conserved in the plant kingdom, this provide a powerful strategy for identifying miRNAs in a new species. Tea (Camellia sinensis) is one of the most important commercial beverage crops in the world, but only a limited number of miRNAs have been identified. In the present study, a total of 14 new C. sinensis miRNAs were identified by expressed sequence tag (EST) analysis from 47 452 available C. sinensis ESTs. These miRNAs potentially target 51 mRNAs, which can act as transcription factors, and participate in stress response, transmembrane transport, and signal transduction. Analysis of gene ontology (GO), based on these targets, suggested that 37 biological processes were involved, such as oxidation-reduction process, stress response, and transport. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis inferred that the identified miRNAs took part in 13 metabolic networks. Our study will help further understanding of the essential roles of miRNAs in C. sinensis growth and development, and stress response.
Keywords: MicroRNA (miRNA), Camellia sinensis, Tea, Gene ontology, Pathway
1. Introduction
MicroRNAs (miRNAs) are a class of endogenous non-coding small RNAs (about 21 nucleotides (nt)), which negatively regulate the expression of genes by targeting mRNA for cleavage or translational repression in a sequence-complementary dependent manner (Bartel, 2004; He and Hannon, 2004). They play crucial roles in plant growth and development, including flower development (Chen, 2004), leaf organ morphogenesis and polarity (Palatnik et al., 2003; Juarez et al., 2004; Mallory et al., 2004), root development (Guo et al., 2005; Williams et al., 2005), and fruit ripening (Moxon et al., 2008; Carra et al., 2009). Additionally, plant miRNAs also respond to drought, cold, salt, and other abiotic stress, as well as biotic stress (Sunkar et al., 2012).
The first plant miRNA was discovered in Arabidopsis in 2002 by small RNAs cloning (Reinhart et al., 2002). Subsequently, a large number of plant miRNAs were identified in a wide range of plant species. Currently, a total of 5 940 plant miRNAs from 67 species are published in the miRBase database (http://www.mirbase.org/, Release 19: August 2012) (Kozomara and Griffiths-Jones, 2011). miRNA-related research is steadily growing, with researchers identifying miRNAs and studying their functions using a series of computational tools and/or experimental methods including small RNAs cloning, high-throughput sequencing, and degradome sequencing. Comparison of miRNAs in different plant species by expressed sequence tag (EST) analysis had shown that some miRNAs were highly evolutionary conserved among species (Zhang et al., 2006a); this provided a powerful strategy for identifying miRNAs in a new species. Identification of miRNAs using EST analysis has two significant advantages (Frazier and Zhang, 2011): (1) There is no specialized software required and it can be used to identify miRNAs in any species if they are previously registered EST sequences; (2) Since EST are derived from transcribed sequences, EST analysis also provided direct evidence for miRNA expression. In view of these advantages, EST analysis had been used to identify conserved miRNAs in Brassica napus (Xie et al., 2007), Medicago truncatula (Zhou et al., 2008), Lycopersicon esculentum (Yin et al., 2008), Glycine max (Zhang et al., 2008), citrus (Song et al., 2010), Nicotiana tabacum (Frazier et al., 2010), Panicum virgatum (Xie et al., 2010), Solanum tuberosum (Xie et al., 2011), Malus domestica (Yu et al., 2011), strawberry (Dong et al., 2012), etc.
Tea (Camellia sinensis) is an important commercial beverage crop grown in different agro-climatic zones in the world. Because of its extensive secondary metabolites in leaves, including theanine, polyphenols, caffeine, and volatile oils, the tea beverage possesses many health benefits to humans (Rogers et al., 2008; Prabu and Mandal, 2010; Shi et al., 2011). In addition to its health benefits and economic value, C. sinensis is also a wonderful source of experimental material to expound gene expression and regulation because of the availability of a mass of ESTs. Though numbers of miRNAs were identified from a wide range of species, there was no registered C. sinensis miRNA in miRBase (http://www.mirbase.org, Release 19: August 2012). Recently, Das and Mondal (2010) and Prabu and Mandal (2010) identified several miRNAs from C. sinensis using computational methods, and Mohanpuria and Yadav (2012) discovered six tea-specific miRNAs using a direct cloning approach. However, compared to Arabidopsis (703) or rice (708) (http://www.mirbase.org/, Release 19: August 2012), more miRNA genes still remain to be discovered in C. sinensis. Furthermore, little attention has been focused on the function of C. sinensis miRNAs. In this study, we aim to identify miRNAs and their potential targets in C. sinensis and study their functions. To achieve this goal, EST analysis was performed to discover miRNAs and potential targets in C. sinensis, and Blast2GO (Conesa et al., 2005; Conesa and Götz, 2008) was employed to further understand their functions.
2. Methods
2.1. Sequence sources
The test sequences were obtained from the miRBase database and the National Center for Biotechnology Information (NCBI). Currently, a total of 5 940 known plant miRNAs were available in the miRBase database, and 47 452 ESTs and 154 468 mRNAs sequences were available for C. sinensis in the NCBI by October 2012. To identify more potential C. sinensis miRNAs, all of these sequences were downloaded for identifying miRNAs.
2.2. Identification of potential miRNAs in C. sinensis using EST analysis
The prediction of potential miRNA adopted a previously reported method (Zhang et al., 2005; Frazier et al., 2010). There were two crucial filter conditions in EST analysis: one is the conservation of mature miRNA sequences, another is the secondary structure of the pre-miRNAs (Zhang et al., 2008). Briefly, the mature sequences of all known plant miRNAs were used as a query for homologous search against C. sinensis EST database using BLAST+2.2.25 program (Altschul et al., 1997). The parameters used in the BLASTn were adjusted as follows: E value cutoff of 0.01; the word size was set at seven; and all other parameters used default settings. After removing the repeated ones, the rest of the ESTs with no more than 3 nt mismatches were used for additional analysis of secondary structure, based on the following criteria (Frazier et al., 2010) using MFOLD V3.2 (Zuker, 2003) (http://mfold.rit.albany.edu/?q=mfold): (1) pre-miRNA could fold into a typical hairpin secondary structure and the mature miRNA was located in one stem; (2) the length of the pre-miRNA was no less than 50 nt; (3) pre-miRNA had a high minimal folding free energy (MFE) and MFE index (MFEI), which was calculated by
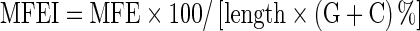 , ,
|
where length is the length of RNA sequence and MFE is the negative folding free energy (−ΔG) (Zhang et al., 2006b); (4) the maximum number of nucleotides mismatches between the mature miRNA and its opposite miRNA* sequnence was six; and, (5) no loops or breaks in the miRNA/miRNA* duplex was allowed.
2.3. Prediction of miRNA targets in C. sinensis
In brief, we used the potential C. sinensis miRNAs blast against the C. sinensis mRNA database to search sequences conforming to the following standards as the C. sinensis candidate target gene: (1) the maximum number of mismatched nucleotides between the mature miRNA and its potential target genes was four; (2) the maximum number of mismatched nucleotides at positions 1–9 was one; (3) no mismatches were allowed at positions 10–11; (4) more than two continuous mismatches at any position were not allowed (Xie et al., 2010).
2.4. Analysis of gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway
To better understand the function of C. sinensis miRNAs, Blast2GO (Conesa et al., 2005; Conesa and Götz, 2008) was employed to investigate the predicted target genes. First, the identified miRNA targeted mRNAs were used to BLASTX against NR database with an E value of 10−25. Second, the best hits identified by BLASTX were further searched against the GO and KEGG databases using default settings.
3. Results and discussion
3.1. Potential miRNAs in C. sinensis
In this study, we identified 14 potential miRNAs from a total of 47 452 available C. sinensis ESTs by homologous search. This indicates that about 0.0295% of C. sinensis ESTs contain potential miRNAs. The ratio is as high as the previously reported about 0.0277% for switchgrass (Xie et al., 2010).
The 14 new C. sinensis miRNAs belong to nine families, of which miR171, miR2911, miR5021, miR5368, and miR6483 have two members, while miR156, miR397, miR399, and miR2863 have only one member. Mature miRNAs have been observed to be located on the arm of pre-miRNA, but the located positions were found either on the 5′ arm of the stem (50%) or on the 3′ arm (50%). The length of mature miRNAs varies from 18 to 22 nt and 42.86% (6 out of 14) of them are 21 nt in length. The length of C. sinensis pre-miRNAs also varies from 85 to 201 nt with an average of (122.79±38.13) nt (Table 1). Fig. 1 showed predicted pre-miRNAs of miR171a in C. sinensis, and others were given in Data S1.
Table 1.
C. sinensis miRNA identification by homolog search
| New miRNA | Query miRNA | GenBank acc. No. | Tissue type | Arm | SP | EP | ME | Mature sequence* | E value | PL | A+U (%) | MFE (kcal/mol) | MFEI |
| miR156 | aly-miR157a-5p | HS396956.1 | Leaf | 5′ | 212 | 232 | 20/21 | UUGACAGAAGAUAGAGAGCAu | 1.00E−04 | 100 | 54.00 | 45.80 | 1.00 |
| miR171a | ptc-miR171k | FS948108.1 | Roots | 3′ | 273 | 293 | 20/21 | GGAUUGAGCCGCGCCAAUAUu | 1.00E−04 | 100 | 65.00 | 39.20 | 1.12 |
| miR171b | ptc-miR171k | FS948109.1 | Roots | 5′ | 163 | 143 | 20/21 | GGAUUGAGCCGCGCCAAUAUu | 1.00E−04 | 97 | 63.92 | 40.40 | 1.15 |
| miR397 | osa-miR397b | CV699725.1 | Leaf | 5′ | 99 | 79 | 21/21 | UUAUUGAGUGCAGCGUUGAUG | 3.00E−05 | 126 | 59.52 | 39.20 | 0.77 |
| miR399 | osa-miR399j | FS958856.1 | Young leaves | 3′ | 109 | 129 | 20/21 | UGCCAAaGGAGAGUUGCCCUA | 8.00E−03 | 103 | 51.46 | 52.80 | 1.06 |
| miR2863 | osa-miR2863a | FS950435.1 | Roots | 5’ | 37 | 57 | 18/21 | UauaUAUUGUUGAAAUGGCUU | 8.00E−03 | 85 | 64.71 | 21.50 | 0.72 |
| miR2911a | nta-miR2911 | JK476023.1 | Leaf | 3′ | 364 | 383 | 20/20 | GGCCGGGGGACGGACUGGGA | 1.00E−04 | 97 | 22.68 | 65.40 | 0.87 |
| miR2911b | nta-miR2911 | FS953337.1 | Shoot stems | 3′ | 177 | 196 | 20/20 | GGCCGGGGGACGGACUGGGA | 1.00E−04 | 97 | 23.71 | 69.20 | 0.94 |
| miR5021a | ath-miR5021 | GW690847.1 | Bud | 3′ | 256 | 237 | 18/20 | aGAGAAGAAGAAGAAGAAAg | 2.00E−03 | 195 | 55.90 | 71.90 | 0.84 |
| miR5021b | ath-miR5021 | GE651759.1 | Tender root | 5′ | 73 | 92 | 18/20 | aGAGAAGAAGAAGAAGAAAg | 2.00E−03 | 201 | 56.22 | 72.20 | 0.82 |
| miR5368a | gma-miR5368 | GE653011.1 | Tender root | 3' | 505 | 523 | 19/19 | GGACAGUCUCAGGUAGACA | 4.00E-04 | 163 | 42.33 | 76.10 | 0.81 |
| miR5368b | gma-miR5368 | FS945766.1 | Mature leaves | 3′ | 196 | 214 | 19/19 | GGACAGUCUCAGGUAGACA | 4.00E-04 | 144 | 42.36 | 68.50 | 0.83 |
| miR6483a | hbr-miR6483 | HS398296.1 | Leaf | 5′ | 253 | 232 | 21/22 | UAUUGUAGAAAUUUUCgGGAUC | 2.00E−03 | 101 | 62.38 | 29.80 | 0.78 |
| miR6483b | hbr-miR6483 | JK714410.1 | Leaf | 5′ | 303 | 282 | 21/22 | UAUUGUAGAAAUUUUCgGGAUC | 2.00E−03 | 110 | 63.64 | 30.00 | 0.75 |
Lowercase letters in mature sequence mean mismatch
SP: start point; EP: end point; ME: match extent; PL: pre-miRNA length; MFE: minimal folding free energy; MFEI: MFE index
Fig. 1.

Predicted pre-miRNAs of miR171a in C. sinensis
MFE is an important parameter for RNA folding into their secondary structures. Usually, the stability of the secondary structure of an RNA sequence increases with the reduction of the MFE. The MFEI was a sufficient criterion for distinguishing miRNAs from other RNAs. Previous research also suggested that it is more likely to be a potential miRNA if the pre-miRNA met the following criteria: MFEI>0.85 (Zhang et al., 2006b). All predicted C. sinensis pre-miRNAs have a typical stem-loop secondary structure, pairing diversity depends on the length of precursor, and we only select the most stable one as the candidate pre-miRNA. Namely, they have a higher MFE, as well as MFEI. The MFE of new identification C. sinensis miRNAs ranges from 21.50 to 76.10 kcal/mol (1 kcal=4.184 kJ) with an average of (51.57±18.68) kcal/mol and the MFEI ranges from 0.72 to 1.15 with an average of 0.89±0.14 (Table 1).
Furthermore, the expression of C. sinensis miRNAs, according to the tissue type reported for each EST in the NCBI database, may be observed in leaf, root, stem, and bud (Table 1).
3.2. C. sinensis miRNA targets and their functions
Increasing evidences have demonstrated that most plant miRNAs bind to their target mRNA sequences with perfect or near-perfect sequence complementarity (Wang et al., 2004; Schwab et al., 2005). This provides a powerful strategy for discovering potential miRNA targets by comparing and aligning miRNAs with mRNAs sequences. Here, we performed more stringent criterion (Schwab et al., 2005) to identify potential C. sinensis targets. After a set of screening criteria as described in the method, we achieved 51 target genes. Among the 51 predicted targets, 17 mRNAs encoded transcriptional factors, 14 mRNAs were stress responsive genes, and others were involved in transmembrane transport, signal transduction and transcription regulation. Unfortunately, 15 out of 51 targets’ function remain unknown (Table 2). The results imply that miRNAs may play an important role in C. sinensis growth and development, as well as environmental stress.
Table 2.
Potential targets of the identified miRNAs in C. sinensis
| miRNA family | Accession ID for targets | Target description | Function |
| miR156 | KA284177, KA295488, HP757423, KA282627, KA285159, HP745756, HP751450, KA284930, KA293068, GAAC01043871, GAAC01052380 | Squamosa promoter-binding-like protein | TF |
| miR171 | HP735040, HP713619, GAAC01007557 | Gras family transcription factor | TF |
| HP757272, KA297400, GAAC01010861 | Scarecrow-like protein | TF | |
| miR397 | HP737460 | Laccase precursor | SR |
| HP763272, GAAC01026665, GAAC01009301, KA285173 | Laccase | SR | |
| miR399 | HP729908 | Probable ubiquitin-conjugating enzyme e2 24-like | SR |
| miR2911 | KA283566 | Cytochrome p450 like_tbp | SR |
| KA280075, KA285244, KA300874, KA279444, KA281442, KA287941, KA296981, KA300579, KA298382 | Hypothetical protein MTR | Unknown | |
| miR5021 | KA279939, KA291019, KA303064 | 60s ribosomal protein | Unknown |
| HP701326 | Transcription activator glk1-like | TR | |
| KA281241 | Conserved hypothetical protein | Unknown | |
| GAAC01052403 | Probable LRR receptor-like serine threonine-protein kinase at1g14390 | ST | |
| GAAC01011182 | PREDICTED: uncharacterized protein LOC100266927 | Unknown | |
| HP701293 | Uncharacterized F-box/LRR-repeat protein C02F5.7-like | Unknown | |
| HP748043 | Monocopper oxidase-like protein sku5-like | SR | |
| KA281010 | Serine/threonine-protein phosphatase PP2A catalytic subunit | ST | |
| GAAC01045495 | Erd6-like transporter | TMT | |
| miR5368 | KA283870 | Metallocarboxypeptidase inhibitor | SR |
| KA279481, KA283770, KA303168, KA303031, GAAC01046756 | Cell wall-associated hydrolase, partial | SR | |
| miR6483 | HP736555 | Envelope membrane protein | TMT |
TF: transcription factor; SR: stress response; ST: signal transduction;TMT: transmembrane transport; TR: transcriptional regulation
Many miRNA targets identified by bioinformatics and/or experimental methods were transcription factors that help control plant growth and development. Here, we also found this type of targets. SQUAMOSA promoter binding protein-like (SPL) transcription factors, a class targets of miR156, play an important role in controlling flowering time, regulating plant transition from vegetative phase to reproductive phase, while overexpression of miR156 delays flowering and extends the vegetative stage (Wang et al., 2009; Wu et al., 2009). Furthermore, SPL is also involved in leaf development (Chen et al., 2010) and anthocyanin biosynthesis (Gou et al., 2011). SCARECROW-LIKE (SCL) transcription factor, miR171 target gene, was reported to act as a positive regulator in root development by integrating and maintaining a functional gibberellic acid (GA) pathway (Heo et al., 2011).
Recent studies have shown that miRNAs are also involved in plant adaptation to environmental stresses, such as cold (Zhang et al., 2009; Thiebaut et al., 2012), salt (Ding et al., 2009), drought (Li et al., 2011), and nutrient deficiency (Sunkar et al., 2007; Zhao et al., 2012). Interestingly, we identified 14 potential targets of miR397, miR399, miR2911, miR5021, and miR5368 that were responses to stress. Further analysis of GO suggested that miR397 and miR399 play essential roles in copper ion and phosphate starvation.
To further understand the function of C. sinensis miRNAs, the predicted target mRNAs were subjected to analysis by GO and KEGG, a database for analyzing gene functions systematically (Kanehisa and Goto, 2000), using Blast2GO. The result suggested that C. sinensis miRNAs were involved in 37 biological processes. Among them, 9 targets of miR397, miR2911, and miR5021 took part in oxidation-reduction process, 3 targets of miR397 and miR399 responded to stress, and others were related to regulation of transcription, transport, growth and development, metabolism and translation (Table 3). Pathway enrichment analysis, based on the KEGG database, demonstrates that the identified miRNAs participated in 13 metabolism networks. These networks were involved in caffeine metabolism, ascorbate and aldarate metabolism, fatty acid metabolism, T cell receptor signaling pathway, and other secondary metabolites process (Table 4). Interestingly, miR2911 was demonstrated to participate in the caffeine metabolism. Obviously, our study will help further understanding of the important regulation roles of miRNAs in C. sinensis growth and development, stress response, and likewise in research and development of low-caffeine tea.
Table 3.
GO analysis of miRNA targets in C. sinensis
| miRNA | Biological process | Accession ID for targets | GO |
| 397, 2911, 5021 | Oxidation-reduction process | HP748043; GAAC01045495; KA287941; KA283566; KA285173; GAAC01009301; HP763272; HP737460; GAAC01026665 | GO:0055114 |
| 156, 171 | Regulation of transcription | KA284177; GAAC01007557; KA295488; KA282627; KA285159; HP735040; HP713619; HP757272; KA297400 | GO:0006351; GO:0006355 |
| 399, 5021, 6483 | Transport | GAAC01045495; HP736555; HP729908 | GO:0006817; GO:0015992; GO:0055085; GO:0008643 |
| 397, 399 | Stress response | HP729908; HP737460; HP729908 | GO:0016036; GO:0046688; GO:0055062 |
| 397 | Metabolic | GAAC01009301; GAAC01026665; KA285173; HP763272; HP737460 | GO:0046274; GO:0010413; GO:0009809; GO:0045492 |
| 397 | Growth and development | HP737460; HP763272; GAAC01026665 | GO:0010228; GO:0009834; GO:0009832 |
| 5021 | Translation | KA279939; KA291019 | GO:0006412 |
Table 4.
KEGG analysis of miRNA targets in C. sinensis
| miRNA | Accession ID for targets | Target description | Enzyme | Pathway |
| 397 | HP737460 | Laccase precursor | EC:1.10.3.3 | Ascorbate and aldarate metabolism |
| GAAC01009301 | Laccase | EC:1.10.3.3 | ||
| 2911 | KA283566 | Cytochrome p450 like_tbp | EC:1.14.14.1 | Fatty acid metabolism, caffeine metabolism, aminobenzoate de-gradation, metabolism of xeno-biotics by cytochrome P450, drug metabolism-cytochrome P450, drug metabolism-other enzymes, arachidonic acid metabolism, linoleic acid metabolism, tryptophan metabolism, steroid hormone biosynthesis, retinol metabolism |
| KA287941 | ||||
| 5021 | KA281010 | Serine/threonine-protein phosphatase PP2A catalytic subunit | EC:3.1.3.16 | T cell receptor signaling pathway |
4. Conclusions
In this study, we identified 14 new C. sinensis miRNAs by EST analysis, which belong to 9 families. These C. sinensis miRNAs potentially target 51 mRNAs, which can act as transcription factors, and participate in stress response, transmembrane transport, and signal transduction. GO analysis suggested that 37 biological processes were involved, such as oxidation-reduction process, stress response, and transport. KEGG pathway enrichment analysis inferred that the identified miRNAs took part in 13 metabolism networks. Interestingly, miR2911 was demonstrated to participate in caffeine metabolism. Our study will help further understanding of the essential roles of miRNAs in C. sinensis growth and development, stress response, as well as in research and development of low-caffeine tea.
Acknowledgments
We are grateful to Danielle (Han GAO) (Institute of Tea Science, Zhejiang University, China) for her modification of this paper. We also appreciate Qing-feng NIU (Department of Horticulture, the State Agricultural Ministry Key Laboratory of Horticultural Plant Growth, Zhejiang University, China) for his suggestions on the revised manuscript.
List of electronic supplementary materials
The pre-miRNAs of potential miRNAs in C. sinensis
Footnotes
Electronic supplementary materials: The online version of this article (doi:10.1631/jzus.B1300006) contains supplementary materials, which are available to authorized users
Compliance with ethics guidelines: Quan-wu ZHU and Yao-ping LUO declare that they have no conflict of interest.
This article does not contain any studies with human or animal subjects performed by any of the authors.
References
- 1.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucl Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bartel DP. microRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 3.Carra A, Mica E, Gambino G, Pindo M, Moser C, Pe ME, Schubert A. Cloning and characterization of small non-coding RNAs from grape. Plant J. 2009;59(5):750–763. doi: 10.1111/j.1365-313X.2009.03906.x. [DOI] [PubMed] [Google Scholar]
- 4.Chen XM. A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science. 2004;303(5666):2022–2025. doi: 10.1126/science.1088060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen X, Zhang Z, Liu D, Zhang K, Li A, Mao L. Squamosa promoter-binding protein-like transcription factors: star players for plant growth and development. J Integr Plant Biol. 2010;52(11):946–951. doi: 10.1111/j.1744-7909.2010.00987.x. [DOI] [PubMed] [Google Scholar]
- 6.Conesa A, Götz S. Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int J Plant Genom. 2008;2008:619832. doi: 10.1155/2008/619832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Conesa A, Götz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 8.Das A, Mondal TK. Computational identification of conserved microRNAs and their targets in tea (Camellia sinensis) Am J Plant Sci. 2010;01(02):77–86. doi: 10.4236/ajps.2010.12010. [DOI] [Google Scholar]
- 9.Ding D, Zhang L, Wang H, Liu Z, Zhang Z, Zheng Y. Differential expression of miRNAs in response to salt stress in maize roots. Ann Bot. 2009;103(1):29–38. doi: 10.1093/aob/mcn205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dong QH, Han J, Yu HP, Wang C, Zhao MZ, Liu H, Ge AJ, Fang JG. Computational identification of microRNAs in strawberry expressed sequence tags and validation of their precise sequences by miR-RACE. J Heredity. 2012;103(2):268–277. doi: 10.1093/jhered/esr127. [DOI] [PubMed] [Google Scholar]
- 11.Frazier TP, Zhang B. Identification of Plant MicroRNAs Using Expressed Sequence Tag Analysis. In: Pereira A, editor. Plant Reverse Genetics. New York: Humana Press; 2011. pp. 13–25. [DOI] [PubMed] [Google Scholar]
- 12.Frazier TP, Xie F, Freistaedter A, Burklew CE, Zhang B. Identification and characterization of microRNAs and their target genes in tobacco (Nicotiana tabacum) Planta. 2010;232(6):1289–1308. doi: 10.1007/s00425-010-1255-1. [DOI] [PubMed] [Google Scholar]
- 13.Gou JY, Felippes FF, Liu CJ, Weigel D, Wang JW. Negative regulation of anthocyanin biosynthesis in Arabidopsis by a miR156-targeted SPL transcription factor. Plant Cell. 2011;23(4):1512–1522. doi: 10.1105/tpc.111.084525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo HS, Xie Q, Fei JF, Chua NH. MicroRNA directs mRNA cleavage of the transcription factor NAC1 to downregulate auxin signals for Arabidopsis lateral root development. Plant Cell. 2005;17(5):1376–1386. doi: 10.1105/tpc.105.030841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5(7):522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 16.Heo JO, Chang KS, Kim IA, Lee MH, Lee SA, Song SK, Lee MM, Lim J. Funneling of gibberellin signaling by the GRAS transcription regulator scarecrow-like 3 in the Arabidopsis root. PNAS. 2011;108(5):2166–2171. doi: 10.1073/pnas.1012215108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Juarez MT, Kui JS, Thomas J, Heller BA, Timmermans MCP. MicroRNA-mediated repression of rolled leaf1 specifies maize leaf polarity. Nature. 2004;428(6978):84–88. doi: 10.1038/nature02363. [DOI] [PubMed] [Google Scholar]
- 18.Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucl Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucl Acids Res. 2011;39:D152–D157. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li B, Qin Y, Duan H, Yin W, Xia X. Genome-wide characterization of new and drought stress responsive microRNAs in populus euphratica. J Exp Bot. 2011;62(11):3765–3779. doi: 10.1093/jxb/err051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mallory AC, Reinhart BJ, Jones-Rhoades MW, Tang GL, Zamore PD, Barton MK, Bartel DP. MicroRNA control of phabulosa in leaf development: importance of pairing to the microRNA 5′ region. EMBO J. 2004;23(16):3356–3364. doi: 10.1038/sj.emboj.7600340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mohanpuria P, Yadav SK. Characterization of novel small RNAs from tea (Camellia sinensis L.) Mol Biol Rep. 2012;39(4):3977–3986. doi: 10.1007/s11033-011-1178-3. [DOI] [PubMed] [Google Scholar]
- 23.Moxon S, Jing RC, Szittya G, Schwach F, Pilcher RLR, Moulton V, Dalmay T. Deep sequencing of tomato short RNAs identifies microRNAs targeting genes involved in fruit ripening. Genome Res. 2008;18(10):1602–1609. doi: 10.1101/gr.080127.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palatnik JF, Allen E, Wu XL, Schommer C, Schwab R, Carrington JC, Weigel D. Control of leaf morphogenesis by microRNAs. Nature. 2003;425(6955):257–263. doi: 10.1038/nature01958. [DOI] [PubMed] [Google Scholar]
- 25.Prabu GR, Mandal AK. Computational identification of miRNAs and their target genes from expressed sequence tags of tea (Camellia sinensis) Genom Prot Bioinf. 2010;8(2):113–121. doi: 10.1016/s1672-0229(10)60012-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reinhart BJ, Weinstein EG, Rhoades MW, Bartel B, Bartel DP. MicroRNAs in plants. Genes Dev. 2002;16(13):1616–1626. doi: 10.1101/gad.1004402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rogers PJ, Smith JE, Heatherley SV, Pleydell-Pearce CW. Time for tea: mood, blood pressure and cognitive performance effects of caffeine and theanine administered alone and together. Psychopharmacology. 2008;195(4):569–577. doi: 10.1007/s00213-007-0938-1. [DOI] [PubMed] [Google Scholar]
- 28.Schwab R, Palatnik JF, Riester M, Schommer C, Schmid M, Weigel D. Specific effects of microRNAs on the plant transcriptome. Dev Cell. 2005;8(4):517–527. doi: 10.1016/j.devcel.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 29.Shi CY, Yang H, Wei CL, Yu O, Zhang ZZ, Jiang CJ, Sun J, Li YY, Chen Q, Xia T, et al. Deep sequencing of the Camellia sinensis transcriptome revealed candidate genes for major metabolic pathways of tea-specific compounds. BMC Genom. 2011;12:131. doi: 10.1186/1471-2164-12-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song C, Jia Q, Fang J, Li F, Wang C, Zhang Z. Computational identification of citrus microRNAs and target analysis in citrus expressed sequence tags. Plant Biol. 2010;12(6):927–934. doi: 10.1111/j.1438-8677.2009.00300.x. [DOI] [PubMed] [Google Scholar]
- 31.Sunkar R, Chinnusamy V, Zhu J, Zhu JK. Small RNAs as big players in plant abiotic stress responses and nutrient deprivation. Trends Plant Sci. 2007;12(7):301–309. doi: 10.1016/j.tplants.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 32.Sunkar R, Li YF, Jagadeeswaran G. Functions of microRNAs in plant stress responses. Trends Plant Sci. 2012;17(4):196–203. doi: 10.1016/j.tplants.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 33.Thiebaut F, Rojas CA, Almeida KL, Grativol C, Domiciano GC, Lamb CRC, de Almeida Engler J, Hemerly AS, Ferreira PCG. Regulation of miR319 during cold stress in sugarcane. Plant Cell Env. 2012;35(3):502–512. doi: 10.1111/j.1365-3040.2011.02430.x. [DOI] [PubMed] [Google Scholar]
- 34.Wang JW, Czech B, Weigel D. miR156-regulated spl transcription factors define an endogenous flowering pathway in Arabidopsis thaliana . Cell. 2009;138(4):738–749. doi: 10.1016/j.cell.2009.06.014. [DOI] [PubMed] [Google Scholar]
- 35.Wang XJ, Reyes JL, Chua NH, Gaasterland T. Prediction and identification of Arabidopsis thaliana microRNAs and their mRNA targets. Genome Biol. 2004;5(9):R65. doi: 10.1186/gb-2004-5-9-r65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Williams L, Grigg SP, Xie MT, Christensen S, Fletcher JC. Regulation of Arabidopsis shoot apical meristem and lateral organ formation by microRNA miR166g and its AtHD-ZIP target genes. Development. 2005;132(16):3657–3668. doi: 10.1242/dev.01942. [DOI] [PubMed] [Google Scholar]
- 37.Wu G, Park MY, Conway SR, Wang JW, Weigel D, Poethig RS. The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis . Cell. 2009;138(4):750–759. doi: 10.1016/j.cell.2009.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xie F, Frazier TP, Zhang B. Identification and characterization of microRNAs and their targets in the bioenergy plant switchgrass (Panicum virgatum) Planta. 2010;232(2):417–434. doi: 10.1007/s00425-010-1182-1. [DOI] [PubMed] [Google Scholar]
- 39.Xie F, Frazier TP, Zhang B. Identification, characterization and expression analysis of microRNAs and their targets in the potato (Solanum tuberosum) Gene. 2011;473(1):8–22. doi: 10.1016/j.gene.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 40.Xie FL, Huang SQ, Guo K, Xiang AL, Zhu YY, Nie L, Yang ZM. Computational identification of novel microRNAs and targets in Brassica napus . FEBS Lett. 2007;581(7):1464–1474. doi: 10.1016/j.febslet.2007.02.074. [DOI] [PubMed] [Google Scholar]
- 41.Yin Z, Li C, Han X, Shen F. Identification of conserved microRNAs and their target genes in tomato (Lycopersicon esculentum) Gene. 2008;414(1-2):60–66. doi: 10.1016/j.gene.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 42.Yu H, Song C, Jia Q, Wang C, Li F, Nicholas KK, Zhang X, Fang J. Computational identification of microRNAs in apple expressed sequence tags and validation of their precise sequences by miR-RACE. Physiol Plant. 2011;141(1):56–70. doi: 10.1111/j.1399-3054.2010.01411.x. [DOI] [PubMed] [Google Scholar]
- 43.Zhang BH, Pan XP, Wang QL, Cobb GP, Anderson TA. Identification and characterization of new plant microRNAs using EST analysis. Cell Res. 2005;15(5):336–360. doi: 10.1038/sj.cr.7290302. [DOI] [PubMed] [Google Scholar]
- 44.Zhang B, Pan X, Cannon CH, Cobb GP, Anderson TA. Conservation and divergence of plant microRNA genes. Plant J. 2006;46(2):243–259. doi: 10.1111/j.1365-313X.2006.02697.x. [DOI] [PubMed] [Google Scholar]
- 45.Zhang BH, Pan XP, Cox SB, Cobb GP, Anderson TA. Evidence that miRNAs are different from other RNAs. Cell Mol Life Sci. 2006;63(2):246–254. doi: 10.1007/s00018-005-5467-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang B, Pan X, Stellwag EJ. Identification of soybean microRNAs and their targets. Planta. 2008;229(1):161–182. doi: 10.1007/s00425-008-0818-x. [DOI] [PubMed] [Google Scholar]
- 47.Zhang J, Xu Y, Huan Q, Chong K. Deep sequencing of Brachypodium small RNAs at the global genome level identifies microRNAs involved in cold stress response. BMC Genom. 2009;10:449. doi: 10.1186/1471-2164-10-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao M, Tai H, Sun S, Zhang F, Xu Y, Li WX. Cloning and characterization of maize miRNAs involved in responses to nitrogen deficiency. PLoS One. 2012;7(1):e29669. doi: 10.1371/journal.pone.0029669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou ZS, Huang SQ, Yang ZM. Bioinformatic identification and expression analysis of new microRNAs from Medicago truncatula . Biochem Biophys Res Commun. 2008;374(3):538–542. doi: 10.1016/j.bbrc.2008.07.083. [DOI] [PubMed] [Google Scholar]
- 50.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucl Acids Res. 2003;31(13):3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The pre-miRNAs of potential miRNAs in C. sinensis