

Abstract
Phylogenetic analyses of gene expression have great potential for addressing a wide range of questions. These analyses will, for example, identify genes that have evolutionary shifts in expression that are correlated with evolutionary changes in morphological, physiological, and developmental characters of interest. This will provide entirely new opportunities to identify genes related to particular phenotypes. There are, however, 3 key challenges that must be addressed for such studies to realize their potential. First, data on gene expression must be measured from multiple species, some of which may be field-collected, and parameterized in such a way that they can be compared across species. Second, it will be necessary to develop comparative phylogenetic methods suitable for large multidimensional datasets. In most phylogenetic comparative studies to date, the number n of independent observations (independent contrasts) has been greater than the number p of variables (characters). The behavior of comparative methods for these classic 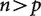 problems is now well understood under a wide variety of conditions. In studies of gene expression, and in studies based on other high-throughput tools, the number n of samples is dwarfed by the number p of variables. The estimated covariance matrices will be singular, complicating their analysis and interpretation, and prone to spurious results. Third, new approaches are needed to investigate the expression of the many genes whose phylogenies are not congruent with species phylogenies due to gene loss, gene duplication, and incomplete lineage sorting. Here we outline general considerations of project design for phylogenetic analyses of gene expression and suggest solutions to these three categories of challenges. These topics are relevant to high-throughput phenotypic data well beyond gene expression.
problems is now well understood under a wide variety of conditions. In studies of gene expression, and in studies based on other high-throughput tools, the number n of samples is dwarfed by the number p of variables. The estimated covariance matrices will be singular, complicating their analysis and interpretation, and prone to spurious results. Third, new approaches are needed to investigate the expression of the many genes whose phylogenies are not congruent with species phylogenies due to gene loss, gene duplication, and incomplete lineage sorting. Here we outline general considerations of project design for phylogenetic analyses of gene expression and suggest solutions to these three categories of challenges. These topics are relevant to high-throughput phenotypic data well beyond gene expression.
Introduction
RNA-seq now enables inexpensive studies of gene expression in a broad diversity of species (’t Hoen et al. 2008; Siebert et al. 2011). To date, most such studies have focused on making comparisons within species. They have, for example, examined differences in gene expression among different experimental conditions, disease status, cell types, tissue types, and genetic backgrounds.
Many questions of great interest, however, will require phylogenetic comparative analyses of gene expression across species. The phylogenetic analysis of gene expression, both by itself and in combination with other types of phenotypic data, will generate biological insight in a variety of ways.
– Sets of genes with correlated evolutionary changes in expression will reveal shared function, shared mechanisms regulating expression, or both.
– An investigator often knows of only one or two genes involved in a particular biological process, but wants to find others. These known genes will be used as “bait” to identify other genes with correlated evolutionary changes in expression. These are strong candidates for also being involved in the biological process of interest.
– Significant evolutionary changes in the covariance structure of expression data may indicate evolutionary changes in gene regulation, gene function, or both.
– Gene expression will be analyzed in combination with other data, such as physiological or morphological measurements, to identify genes with evolutionary changes in expression that are correlated with evolutionary changes in specific biological processes of interest.
– Phylogenetic analyses of gene expression will test alternative hypotheses about how selection acts on expression. It will allow for rigorous tests of the ortholog conjecture (Nehrt et al. 2011), as well as specific models of the evolution of gene function such as DDC (Force et al. 1999). This will also provide a much better understanding of expression neutral evolution.
– Comparative analyses of gene expression across species will be of great use even when an investigator is concerned with only a single species. This is because RNA-seq and other high-throughput tools are so powerful that they often detect hundreds, or even thousands, of genes with significant differential expression among treatments. It is still very difficult, though, to identify which differential expression is biologically meaningful. The most informative and cost-effective way to understand expression data from any particular species may be to collect similar data from closely related species and analyze them in a combined phylogenetic analysis.
It is not statistically valid to simply analyze the correlation of expression across species. This is because observations of any trait (including expression) made across multiple species are not independent, because some species are more closely related to each other than to others. If these evolutionary relationships between species are not taken into account, one can be severely misled by the strong similarity between closely related species and the many differences that are expected to arise by chance between distantly related species. The seminal article by Felsenstein (1985) demonstrated this problem and introduced an ingenious solution: phylogenetically independent contrasts. Based on the structure of the species phylogeny, it transforms the original dependent observations into a series of statistically independent contrasts. There is one independent contrast for each of the internal nodes on the phylogeny. Phylogenetically independent contrasts can then be analyzed to assess the correlation of the measured variables through the course of evolution. Since the introduction of phylogenetically independent contrasts, other comparative phylogenetic methods have also been developed. Phylogenetic generalized least-squares (Grafen 1989), for example, provide a more flexible framework for implementing comparative phylogenetic analyses.
Ives et al. (2007) and Felsenstein (2008) have since expanded upon the original independent contrasts to account for variation within species. These updated methods consider a covariance matrix for within species variation, in addition to the covariance matrix that describes among species variation. There is now a rich set of methods for examining the evolution of quantitative characters, most of which have been developed to examine morphological and ecological data but have not yet been applied to functional genomic data.
To apply comparative phylogenetic tools to evolutionary analyses of gene expression, we must overcome three specific challenges. First, we must measure and parameterize expression data so that they can be compared across species. Second, we must confront the statistical challenges that arise when the number of variables under consideration far outnumbers the observations available. Third, new comparative methods must be developed which can accommodate gene-specific data, such as expression, when the phylogenies of genes are not congruent with those of species.
There has long been interest in comparing gene expression across species. Using microarray data, Rifkin et al. (2003) compared differential expression between two developmental time points across six lineages of Drosophila. More recently, Brawand et al. (2011) investigated the evolution of expression in six organs across 10 species of amniotes (one bird and nine mammals). Both studies included phylogenies that were inferred from the expression data. Neither of these studies, however, mapped the expression data onto phylogenies or performed independent contrasts. This potentially leaves some of their results in question, as the observations made in each species are not independent.
There have been other studies and reviews that have considered various aspects of the evolution of gene expression. Romero et al. (2012) made a survey of the potential for comparative studies of expression to reveal the evolution of regulatory mechanisms. Gilad et al. (2006) reviewed the evidence for different types of selection on expression and concluded that stabilizing selection dominates in most cases. Bedford and Hartl (2009) modeled the strength of stabilizing selection on expression in seven species of Drosophila and found it to be small though of major impact. Zheng et al. (2011) reviewed the current understanding of regulatory variation among species. Hodgins-Davis and Townsend (2009) highlighted the importance of taking environmental effects into account, which can greatly complicate expression studies that consider multiple species. These broad perspectives on the evolution of expression highlight the great potential for phylogenetic comparative analyses of gene expression, once key technical challenges are addressed.
Project design
Before describing the challenges that are specific to phylogenetic analyses of data on gene expression, it is first necessary to address project design (Fig. 1). This is the single most important aspect of a study on gene expression—if the data are not collected in such a way that they can answer the questions at hand, then they will be useless. There are many general considerations to take into account when designing an RNA-seq study (Auer and Doerge 2010), regardless of whether the data are to be analyzed in a phylogenetic perspective or not. Most are the same issues, such as replication and randomization, that have been identified and addressed in more than a decade of micro-array studies, although unfortunately many quantitative RNA-seq studies are recapitulating the problems that were identified in early micro-array studies. There are also additional project design issues that are specific to phylogenetic analysis, which are the focus of the present considerations.
Fig. 1.

A typical project design for phylogenetic analysis of gene expression. The tree depicts the phylogeny of the species, and each of the gray boxes represents a sample. A read count (i.e., an expression measurement) is available for each gene for each sample. In this example, there are 12 individuals sampled across 4 species. In each individual, data on expression are available for two treatments (e.g., tissue types). In some projects, data for different treatments may come from different individuals.
In an RNA-seq study, mRNA is isolated from each sample and shotgun sequenced. The resulting reads are then mapped to gene reference sequences, and the numbers of reads that map to each gene are counted to give quantitative indications of levels of expression. These counts are then normalized across samples to account for differences in sequencing efforts (Robinson and Oshlack 2010; Hansen et al. 2012). There is therefore one normalized read count for each gene for each sample.
The reference sequences to which the reads are mapped to derive counts can be derived from fully annotated genomes or from transcriptome assemblies if genome sequences are not available. Transcriptome assemblies can be based on the same data that are used to quantify assembly, usually by assembling across all samples prior to mapping each sample individually. In most cases, especially as the number of samples grows, it is more cost-effective to use long-read sequencing to assemble a high-quality reference and short-read sequencing across samples to quantify expression (Siebert et al. 2011).
Expression data from a single sample are not very interesting. It cannot be related to biological variation, and it is not even possible to compare expression across genes for technical reasons described below. Studies of gene expression therefore consider differences in expression across multiple samples. In general, the goal of an expression study is to identify which genes have a greater difference in expression between treatments than would be expected by chance. Multiple samples are therefore collected across multiple treatments. “Treatments” is used generically here for any biological variation, whether it is experimentally induced or not. Treatments could represent control and pharmacological treatments, different tissue types, different cell types, different developmental stages, different environmental conditions, or any number of other differences.
In order to have a sense of how much variation is expected by chance, it is critical to collect replicate samples for each treatment. Replication is not just an expensive technical nuisance, it is the investigator’s friend. By revealing how much variation there is when measuring expression across samples within a treatment, it provides a much better understanding of how to interpret variation across samples.
Replicate samples are usually collected across individuals (or, sometimes, pools of individuals, e.g., clutches of embryos that are each from a single spawning event). In some cases, each sample comes from a different individual. This is necessary when the treatment affects the individual as a whole, as would be the case for a drug treatment or environmental stress. In other cases, it is possible to collect samples for different treatments from the same individual, as when comparing expression across tissue types. It is usually desirable to collect samples for different treatments from the same individual when possible, as this provides an opportunity for considering individual effects as well as the effects of treatments.
The aspects of project design considered above apply to all studies of expression. In a phylogenetic study of gene expression, there is one more component to experimental design—species (Fig. 1). Each individual belongs to a particular species, and there can (and should) be multiple individuals per species. This makes it possible to look at treatment, individual, and species effects on levels of expression. Ideally, samples are available for each treatment for each species and, if possible, samples for different treatments are taken from the same individual (Fig. 1).
When collecting expression data from wild-caught specimens, as is likely to be the case in many studies that consider multiple species, there are many potential sources of variation that could complicate the interpretation of variation in expression among and within species (Hodgins-Davis and Townsend 2009). This is because wild-collected specimens usually have unknown environmental histories and genetic backgrounds. Expression from two samples from two individuals may differ because one ate last week and one ate an hour ago, not because the samples are drawn from two treatments. In some cases different species may live in different habitats, and differences in expression between these species may be due to differences in environment that the specimens experienced rather than to evolutionary changes in expression across species. The specifics of minimizing variation due to these extraneous factors will differ from study to study but are very important to consider. Common-garden approaches and thorough replication are two general strategies that should be used when possible.
Challenge I: measuring and parameterizing RNA-seq data so that it can be compared across species
The problem
The normalized read counts produced by a quantitative RNA-seq study are not direct measurements of expression. These counts are proportional to expression, but they are also impacted by other effects that can differ across genes and species. This is because the probability of sequencing a read for a gene is impacted by both the sequence of a gene (Hansen et al. 2010, 2012) and its length. These effects can be modeled with unknown species and gene-specific counting-efficiency coefficients. For gene g in species s, and treatment t, let kgs denote the gene and species-specific counting efficiency, the expectation of gene count Cgst is proportional to both kgs, and the gene expression level Egst.
| (1) |
Since the counting efficiency kgs is inconsistent across genes and species, the direct comparison using Cgst can be misleading. This is because differences in counts may simply be due to differences in kgs. Imagine the trivial example in which a given gene is twice as long in one species as in another, but the expression level (i.e., the number of transcripts per cell) is the same. The number of counts for this gene will differ by a factor of two across species, even though there has been no evolutionary change in levels of expression. If not taken into account, this could severely mislead comparative analyses. The same types of impacts can be realized if a given gene has a sequence that is sequenced more efficiently than the different sequence of the same gene in another species.
Addressing kgs is especially important when reference sequences are incomplete, as reads that map outside the reference will not be counted toward a gene. If the reference sequence for a gene is complete in one species but not in another, then the number of reads that map to the incomplete reference sequence will be an underestimate relative to the number of the species with the complete transcript prediction for the gene. As phylogenetic comparative studies of expression are likely to include species with reference sequences of varying quality, the ability to minimize the impacts of these differences on the evolutionary interpretation of expression is critical. If the reference sequences for all species are based on well-annotated genomes, it may be possible to approximate some components of kgs. A recent study of expression across mammals and a bird, for example, accounted for transcript length when normalizing RNA-seq count data (Brawand et al. 2011). It did not, however, account for differences in sequence composition or other factors that could contribute to kgs. This may have led, in some cases, to evolutionary differences in gene sequences being misinterpreted as evolutionary changes in expression, as both will impact RNA-seq counts.
Below we outline two distinct approaches to addressing the technical challenges imposed by kgs. Each has its own advantages and drawbacks, and the approach chosen for a particular project will depend on details of study design and further evaluation of these methods.
A solution: evolutionary analyses of expression ratios
In the first analysis approach, kgs is canceled out within species before any comparisons are made across species. Rather than compare counts across species, the investigator compares ratios of expected counts across species. Given Equation (1) above, consider the comparison of the ratio of gene expression in tissue type 1 with tissue type 2:
| (2) |
Because kgs is in both the numerator and the denominator, the ratio of counts is determined exclusively by the ratio of expression. These ratios can then be compared across species and genes without the confounding effects of kgs. This ratio is the Fold Change (FC), which is already widely used in visualizations of data on differential expression. FC has been compared across species in an analysis of the evolution of differential expression in Drosophila based on microarray data (Rifkin et al. 2003).
The advantage of this approach is that it greatly simplifies downstream analyses since kgs is removed prior to any comparative phylogenetic analyses. The drawback is that, by transforming all observations to ratios, downstream tests of significance cannot take into consideration the absolute magnitude of counts. The observed variance across replicates could, however, still be used to assess significance. If the treatment samples are taken from the same individuals, as in Fig. 1, the independent contrasts can be computed from the transformed data according to Felsenstein (2008), which accommodates multiple individuals per species.
A solution: consider measurements from different treatments as different characters
In the second approach, gene-specific counting efficiencies are not addressed prior to comparative phylogenetic analyses. Rather than considering the ratio of counts for the same gene across treatments, the counts for each gene for each treatment are initially considered as if they are different characters. If, for example, there are measurements for 5000 genes in two treatments, these measurements are treated as if they are 10,000 different characters. The phylogenetically independent contrasts of these characters are then calculated as usual, and the covariance matrix estimated from these contrasts. Particular cells in the resulting covariance matrix will correspond to covariances between the same treatment for the same gene (i.e., the variances), different treatments for the same gene, the same treatment for different genes, or different treatments for different genes. The covariance matrix can then be decomposed into these various categories, and those that are relevant to the question at hand considered further. Since kgs embodies sequencing efficiency, which is a technical factor, it would be possible to borrow information across categories that are measured under the same technical condition to estimate and correct for kgs.
There are several advantages to this approach. It preserves the magnitudes of the normalized counts, which improves the ability to assess the significance of differences in expression. It is also a very general framework that allows for more complex experimental designs. The primary disadvantage is that it greatly increases the dimensionality of the problem, which in turn exacerbates the challenges described in the next section.
Challenge II: a large number of genes and a small number of species
The problem
In most studies that make use of phylogenetically independent contrasts, the number n of phylogenetically independent contrasts has been far greater than the number p of variables. The behavior of these classic 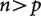 evolutionary problems is now well understood under a wide variety of conditions. In particular, it is possible to accurately infer all cells of the covariance matrix, which describes the evolutionary relationship between the variables, from the contrasts. In phylogenetic analyses of expression based on high-throughput data on expression, though, the number n of independent contrasts (i.e., the number of species minus one) is dwarfed by the number p of variables (the number of genes considered, if differential expression ratios are plotted onto the trees, or the number of treatments times the number of genes, if the counts for each gene for each treatment are considered as separate characters). These new analyses with
evolutionary problems is now well understood under a wide variety of conditions. In particular, it is possible to accurately infer all cells of the covariance matrix, which describes the evolutionary relationship between the variables, from the contrasts. In phylogenetic analyses of expression based on high-throughput data on expression, though, the number n of independent contrasts (i.e., the number of species minus one) is dwarfed by the number p of variables (the number of genes considered, if differential expression ratios are plotted onto the trees, or the number of treatments times the number of genes, if the counts for each gene for each treatment are considered as separate characters). These new analyses with 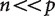 raise a variety of challenges.
raise a variety of challenges.
The crux of the problem is that when 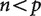 , the information provided by the contrasts is not sufficient to uniquely construct the covariance matrix from observed data (Fig. 2). The covariance matrix is always of size
, the information provided by the contrasts is not sufficient to uniquely construct the covariance matrix from observed data (Fig. 2). The covariance matrix is always of size 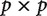 . This applies to the true, but unknown, covariance matrix, as well as to the covariance matrix that is estimated from the observed data. The matrix of observations (i.e., independent contrasts), however, has size
. This applies to the true, but unknown, covariance matrix, as well as to the covariance matrix that is estimated from the observed data. The matrix of observations (i.e., independent contrasts), however, has size 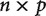 . When
. When 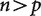 , this matrix is larger than the covariance matrix and can contain enough information to uniquely infer each element of the covariance matrix. When
, this matrix is larger than the covariance matrix and can contain enough information to uniquely infer each element of the covariance matrix. When 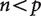 , this matrix is smaller than the covariance matrix and the elements of the inferred covariance matrix cannot be uniquely determined.
, this matrix is smaller than the covariance matrix and the elements of the inferred covariance matrix cannot be uniquely determined.
Fig. 2.

An illustration of relative matrix dimensions in comparative analyses. n is the number of observations (independent contrasts) and p is the number of variables (characters) measured in each observation. (a) 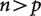 , so the independent contrasts matrix is larger than the covariance matrices and it is possible to uniquely estimate the true covariance matrix. This is the case for most comparative studies to date, which consider many more species than variables. (b)
, so the independent contrasts matrix is larger than the covariance matrices and it is possible to uniquely estimate the true covariance matrix. This is the case for most comparative studies to date, which consider many more species than variables. (b) 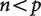 , so the independent contrasts matrix is smaller than the covariance matrices and it is not possible to uniquely estimate the true covariance matrix. This is the situation for phylogenetic comparative analyses of RNA-seq expression data, as well as other high-throughput phenotype data.
, so the independent contrasts matrix is smaller than the covariance matrices and it is not possible to uniquely estimate the true covariance matrix. This is the situation for phylogenetic comparative analyses of RNA-seq expression data, as well as other high-throughput phenotype data.
In essence, when 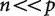 the true covariance matrix is being squeezed through the much smaller data matrix and then expanded back out to the observed covariance matrix (Fig. 2). It is analogous to compressing and then expanding a digital photograph. If the original photograph is a thousand by a thousand pixels and you compress it to a format that is a thousand elements by ten elements, there is no way to uniquely reconstruct each pixel of all possible original photographs. The missing information must be interpolated, and the resulting image may differ substantially from the original image.
the true covariance matrix is being squeezed through the much smaller data matrix and then expanded back out to the observed covariance matrix (Fig. 2). It is analogous to compressing and then expanding a digital photograph. If the original photograph is a thousand by a thousand pixels and you compress it to a format that is a thousand elements by ten elements, there is no way to uniquely reconstruct each pixel of all possible original photographs. The missing information must be interpolated, and the resulting image may differ substantially from the original image.
More formally, when 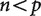 the estimated covariance matrix will be singular. This means that the rows of the matrix will not be independent of each other, and that some cells may not be accurately inferred. Singular matrices are not invertible, which means that many basic linear algebra manipulations cannot be carried out exactly. There are several important implications of this. Some common statistical procedures cannot be performed on singular covariance matrices. It also means that the number of principal components that could be derived from the estimated covariance matrix will be determined by the number of independent observations, not the number of variables. A covariance matrix of size
the estimated covariance matrix will be singular. This means that the rows of the matrix will not be independent of each other, and that some cells may not be accurately inferred. Singular matrices are not invertible, which means that many basic linear algebra manipulations cannot be carried out exactly. There are several important implications of this. Some common statistical procedures cannot be performed on singular covariance matrices. It also means that the number of principal components that could be derived from the estimated covariance matrix will be determined by the number of independent observations, not the number of variables. A covariance matrix of size 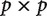 could have up to p principal components (one corresponding to each eigen vector). In the singular matrices described above, however, only n of these will correspond to non-zero eigen values.
could have up to p principal components (one corresponding to each eigen vector). In the singular matrices described above, however, only n of these will correspond to non-zero eigen values.
A solution
To address the 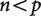 challenge in the context of phylogenetic analyses of data on gene expression, we propose to adapt a recently developed and powerful statistical framework, matrix regularization. Compared with alternative approaches, such as principal component analysis, regularization has advantages in computational speed, interpretability, robustness, and good power in small sample sizes (Hastie et al. 2009). These regularization approaches try to match the observed data with a set of most likely patterns arising from a model. They remain data-driven because the target pattern is general enough that only significant correlations will be retained by the regularization methods.
challenge in the context of phylogenetic analyses of data on gene expression, we propose to adapt a recently developed and powerful statistical framework, matrix regularization. Compared with alternative approaches, such as principal component analysis, regularization has advantages in computational speed, interpretability, robustness, and good power in small sample sizes (Hastie et al. 2009). These regularization approaches try to match the observed data with a set of most likely patterns arising from a model. They remain data-driven because the target pattern is general enough that only significant correlations will be retained by the regularization methods.
Fig. 3 presents the result of simulation analyses, comparing the true covariance matrix (Fig. 3a) with inferred covariance matrices (Fig. 3b–d). We considered 100 variables (which could each be the ratio of expression of 100 genes in two treatments) on an eight-taxon tree. The source code for these analyses is available at https://bitbucket.org/caseywdunn/sicb2013.
Fig. 3.

Simulation analyses of correlation matrix reconstruction. The evolution of expression of 100 genes was simulated on an eight-taxon phylogeny. The legend indicates the magnitude of correlation. The true covariance matrix is block-diagonal (a). The other three matrices (b–d) show the results of alternative approaches to reconstructing the correlation matrix. The correlation matrix inferred directly from the independent contrasts has spurious high and low correlations for many genes that do not have covariance (b). Regularization of matrix b reduces the number of these false positives (c and d). Convex minimization (c; Luo 2011) is less conservative than thresholding (d; Bickel and Levina 2008).
It is clear that the covariance matrix derived directly from the independent contrasts (Fig. 3b) is extremely noisy in comparison to the true covariance matrix that was used to simulate the data. Strong covariance (both negative and positive) is spuriously detected between genes that have no true covariance. Although strong covariance is recovered for genes that do indeed have strong covariance (see the upper left of Fig. 3b), some genes with moderate covariance are found to have nearly zero covariance. This noise is expected since the 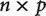 independent contrast matrix does not have enough information to uniquely estimate the
independent contrast matrix does not have enough information to uniquely estimate the 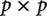 covariance matrix (Fig. 2).
covariance matrix (Fig. 2).
Here we consider two methods of regularization, convex minimization (Luo 2011) and thresholding (Bickel and Levina 2008). Thresholding is more conservative than convex minimization, with fewer false positives but many more false negatives. In convex minimization, covariance matrix is decomposed into two components. The first component is attributed to the effects of unmeasured factors (e.g., environmental effects and regulation pathways) and the second component is attributed to strong pairwise gene-expression correlations after accounting for the effects of unmeasured factors. The statistical model behind this decomposition is related to other popular models, including principal component analysis and surrogate variable analysis (Leek and Storey 2007), but generalizes to exploit the covariance structures in terms of both eigen values/eigen vectors and matrix entries. In thresholding, only strong (both positive and negative) pairwise gene correlations are retained, which parallels the second component of convex minimization but without accounting for unmeasured factors. Both regularization methods require input parameters on the regularization strength trading off false positives and false negatives, and we here only consider the theoretical choices as illustrations. Additional statistical research is needed for designing robust, interpretable, and data-driven ways for choosing the regularization strength in this context.
These results indicate that regularization can, at least in part, overcome some of the challenges that arise in the comparative phylogenetic analysis of high-dimensional functional genomic data. The exact approach that is taken for each study will depend on the goals. If the investigator would like to identify a small number of genes with high effect and avoid false positives, then a conservative regularization approach such as thresholding (Bickel and Levina 2008) would be appropriate. If the goal is to identify the greatest number of genes that covary with a particular phenotypic character, then a less conservative approach such as convex minimization (Luo 2011) would be appropriate. If no regularizations are applied, great care must be taken in interpreting covariances, even when they are inferred to be quite strong.
The limitations of these regularization approaches include that they presume many elements of the covariance matrix to be zero or that the covariance matrix is in a lower dimensional space (e.g., that the matrix is sparse or low rank) in general. Regularization may result in an inferred covariance matrix that is not in the same subspace as the true matrix. The extent of regularization can be chosen based on data, for example by cross-validation (Hastie et al. 2009). However, the optimal way of selecting the regularization is still an open and challenging problem in statistics, though there were many successful examples (Hastie et al. 2009). The extent of these limitations for the present application will only be apparent as these approaches are applied to real data.
Challenge III: accommodating duplication and loss of genes
The problem
In the analyses above, we assumed that each species had exactly the same set of genes. In reality, genes are duplicated and lost through the course of evolution. This leads to the expansion, refinement, and even complete loss of gene families in different species through time. As a result, genes have phylogenetic histories that are not always the same as the phylogenetic histories of the species under consideration (Fig. 4).
Fig. 4.
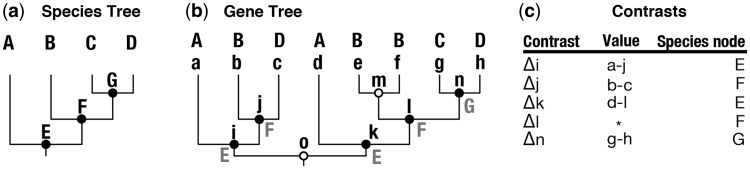
Calculating contrasts associated with speciation events on a gene tree that includes paralogs. (a) The species phylogeny. Nodes representing species and speciation events are labeled with capital letters. (b) The gene phylogeny, which includes paralogs and orthologs. Black nodes indicate speciation events; white nodes indicate gene duplications. The tips and internal nodes of the gene phylogeny are labeled with lowercase letters. The species for which each gene is drawn is indicated by a capital letter, and internal speciation nodes are labeled with a capital letter for the corresponding node in the species tree. (c) Table of contrasts that correspond to speciation events. There are five contrasts in the gene tree that together represent all three speciation events in the species tree. These contrasts can then be combined across gene trees to form an independent-contrasts matrix, in which the values for each gene tree correspond to a column and the rows correspond to species nodes (when there are multiple values from a given species node in a gene tree, they could be averaged before being added to the contrasts table). Calculating the contrast for node l (indicated by *) is complicated by the fact that one of the descendent nodes is a duplication. Such contrasts could be skipped or accommodated by more complicated calculations.
Previous high-throughput studies of gene expression across species have focused on the subset of genes that have only strict orthologs (Rifkin et al. 2003; Brawand et al. 2011). This approach greatly simplifies analyses but discards a large fraction of the data and precludes the investigation of many phenomena of broad interest, such as the evolution of gene expression following gene duplication.
A solution
Several methods that reconcile gene phylogenies with species phylogenies have recently been developed (Akerborg et al. 2009; Arvestad et al. 2009; Sennblad and Lagergren 2009; Rasmussen and Kellis 2010; Wu et al. 2012). These tools label each node in a gene phylogeny as either a speciation event or a gene duplication event. By definition, the speciation nodes of the gene trees each correspond to particular nodes in the species tree. This means that a common ontology can be used for these speciation nodes across all gene trees and the species tree. Each duplication node may be unique to a particular gene tree, as when a new copy of a particular gene arises by tandem duplication. Such unique duplication events must be named individually. It may also be that there are duplication events that are shared by multiple genes, such as the duplication of a whole genome. If there is additional external information that allows these types of duplications to be labeled in each gene tree, then a common name can be used for the shared duplication node across all gene trees. In any particular gene tree, the same speciation node, or shared duplication node, may be present multiple times.
Once each node in each gene tree is labeled as a speciation, shared duplication, or unique duplication, it is possible to proceed with phylogenetically independent contrasts across all gene trees. The calculation of contrasts associated with speciation events is the most straightforward (Fig. 4). For each internal node in the species tree, identify the two descendent nodes as in a typical contrast analysis (e.g., the descendants of F are B and G in Fig. 4a). Then, find the corresponding internal node (in Fig. 4b, nodes j and l correspond to speciation event F) and descendent nodes (in Fig. 4b, nodes b, e, and f correspond to node B in the species tree, and node n corresponds to speciation node G) in each gene tree and calculate the contrasts based on the difference in expression values as reconstructed on the gene tree. One complication is that the number of nodes for a given speciation event will not be consistent across gene trees. There are various ways to address this, the most simplistic being to take the average contrast value across the different nodes of a gene tree that correspond to the same speciation event.
In this way, the investigator builds up the contrasts that correspond to each internal node on the species tree across all gene trees. This results in a 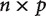 contrast matrix, where n is the number of internal nodes on the species tree and p is the number of gene trees. The
contrast matrix, where n is the number of internal nodes on the species tree and p is the number of gene trees. The 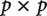 covariance matrix can then be estimated from this matrix of contrasts. A similar approach could be taken for calculating contrasts across shared duplication events. Covariance cannot be computed across unique duplication events since they are gene-specific. Variances (after normalizing by branch length) could be compared across the different categories of nodes to see whether significantly larger changes are realized for one category of nodes relative to the others.
covariance matrix can then be estimated from this matrix of contrasts. A similar approach could be taken for calculating contrasts across shared duplication events. Covariance cannot be computed across unique duplication events since they are gene-specific. Variances (after normalizing by branch length) could be compared across the different categories of nodes to see whether significantly larger changes are realized for one category of nodes relative to the others.
Depending on the objectives of a study, it may be desirable to consider only a subset of the contrasts that are made on a gene tree. This is because a given contrast on the species tree may also span one or more duplication events on the gene tree. This is the case for contrast l in Fig. 4b and c. This means that the observed differences may be due to the effects of duplication as well as evolution between species. To avoid these potentially confounding complications, the investigator could consider only the contrasts in which the descendent node is connected to the internal node by unbroken branches that do not include duplication nodes.
This general approach could be expanded to accommodate other sources of incongruence between gene trees and species trees, such as incomplete lineage sorting.
Conclusion
Although there are multiple challenges that must be addressed to enable comparative phylogenetic analyses of high-throughput data on gene expression, they are not insurmountable. Some solutions, such as the use of count ratios rather than counts, can be immediately implemented with off-the-shelf tools. Others will require further refinement and implementation with new tools.
Several of the approaches presented here are applicable to any high-dimensional quantitative phenotypic data, not just gene expression. They will therefore be useful for interpreting other categories of functional genomic and proteomic data, as well as the high-throughput approaches just now being applied to morphology, development, and physiology. It is likely that the greatest biological insight will come from combining these different types of data into single analyses, allowing for the examination of functional genomic and other phenotypic data in a single evolutionary framework.
Acknowledgments
We thank Joe Felsenstein for insightful conversations that helped to frame and approach the problem. Thanks to Rebecca Helm, Stefan Siebert, and Felipe Zapata for providing comments on earlier drafts. C.W.D. conceived and initiated the study and wrote most of the manuscript. X.L. and Z.W. developed and applied the regularization approaches. All three authors wrote the code together for the simulations and analyses.
Funding
C.W.D. was supported by the National Science Foundation Waterman Award. X.L. was partially supported by National Institutes of Health grants P01-AA019072 and P30-AI042853, and a Brown University faculty startup fund.
Supplementary Data
The R code that produced the simulations and generated Fig. 3 can be found at https://bitbucket.org/caseywdunn/sicb2013.
References
- Akerborg O, Sennblad B, Arvestad L, Lagergren J. Simultaneous Bayesian gene tree reconstruction and reconciliation analysis. Proc Natl Acad Sci USA. 2009;106:5714–9. doi: 10.1073/pnas.0806251106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvestad L, Lagergren J, Sennblad B. The gene evolution model and computing its associated probabilities. J ACM. 2009;56:1–44. [Google Scholar]
- Auer PL, Doerge RW. Statistical design and analysis of RNA sequencing data. Genetics. 2010;185:405–16. doi: 10.1534/genetics.110.114983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford T, Hartl DL. Optimization of gene expression by natural selection. Proc Natl Acad Sci USA. 2009;106:1133–8. doi: 10.1073/pnas.0812009106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickel PJ, Levina E. Covariance regularization by thresholding. The Annals of Statistics. 2008;36:2577–604. [Google Scholar]
- Brawand D, Soumillon M, Necsulea A, Julien P, Csárdi G, Harrigan P, Weier M, Liechti A, Aximu-Petri A, Kircher M, et al. The evolution of gene expression levels in mammalian organs. Nature. 2011;478:343–8. doi: 10.1038/nature10532. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Phylogenies and the comparative method. Am Nat. 1985;125:1–15. [Google Scholar]
- Felsenstein J. Comparative methods with sampling error and within-species variation: contrasts revisited and revised. Am Nat. 2008;171:713–25. doi: 10.1086/587525. [DOI] [PubMed] [Google Scholar]
- Force A, Lynch M, Pickett FB, Amores A, Yan YL, Postlethwait J. Preservation of duplicate genes by complementary, degenerative mutations. Genetics. 1999;151:1531–45. doi: 10.1093/genetics/151.4.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilad Y, Oshlack A, Rifkin SA. Natural selection on gene expression. Trends Genet. 2006;22:456–61. doi: 10.1016/j.tig.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Grafen A. The phylogenetic regression. Philos Trans R Soc B Biol Sci. 1989;326:119–57. doi: 10.1098/rstb.1989.0106. [DOI] [PubMed] [Google Scholar]
- Hansen KD, Brenner SE, Dudoit S. Biases in Illumina transcriptome sequencing caused by random hexamer priming. Nucleic Acids Res. 2010;38:e131. doi: 10.1093/nar/gkq224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KD, Irizarry RA, Wu Z. Removing technical variability in RNA-seq data using conditional quantile normalization. Biostatistics. 2012;13:204–16. doi: 10.1093/biostatistics/kxr054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastie T, Tibshirani R, Friedman J. The elements of statistical learning. Data mining, inference, and prediction. 2nd ed. 2009. New York: Springer-Verlag. [Google Scholar]
- Hodgins-Davis A, Townsend JP. Evolving gene expression: from G to E to G x E. Trends Ecol Evol. 2009;24:649–58. doi: 10.1016/j.tree.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ives AR, Midford PE, Garland TJ. Within-species variation and measurement error in phylogenetic comparative methods. Syst Biol. 2007;56:252–70. doi: 10.1080/10635150701313830. [DOI] [PubMed] [Google Scholar]
- Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007;3:1724–35. doi: 10.1371/journal.pgen.0030161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X. High dimensional low rank and sparse covariance matrix estimation via convex minimization (arXiv.org) 2011 [Google Scholar]
- Nehrt NL, Clark WT, Radivojac P, Hahn MW. Testing the ortholog conjecture with comparative functional genomic data from mammals. PLoS Comput Biol. 2011;7:e1002073. doi: 10.1371/journal.pcbi.1002073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen MD, Kellis M. A Bayesian approach for fast and accurate gene tree reconstruction. Mol Biol Evol. 2010;28:273–90. doi: 10.1093/molbev/msq189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rifkin SA, Kim J, White KP. Evolution of gene expression in the Drosophila melanogaster subgroup. Nat Genet. 2003;33:138–44. doi: 10.1038/ng1086. [DOI] [PubMed] [Google Scholar]
- Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero IG, Ruvinsky I, Gilad Y. Comparative studies of gene expression and the evolution of gene regulation. Nat Rev Genet. 2012;13:505–16. doi: 10.1038/nrg3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sennblad B, Lagergren J. Probabilistic orthology analysis. Syst Biol. 2009;58:411–24. doi: 10.1093/sysbio/syp046. [DOI] [PubMed] [Google Scholar]
- Siebert S, Robinson MD, Tintori SC, Goetz F, Helm RR, Smith SA, Shaner N, Haddock SHD, Dunn CW. Differential gene expression in the siphonophore Nanomia bijuga (Cnidaria) assessed with multiple next-generation sequencing workflows. PLoS One. 2011;6:e22953. doi: 10.1371/journal.pone.0022953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ’t Hoen PAC, Ariyurek Y, Thygesen HH, Vreugdenhil E, Vossen RHAM, de Menezes RX, Boer JM, van Ommen GJB, den Dunnen JT. Deep sequencing-based expression analysis shows major advances in robustness, resolution and inter-lab portability over five microarray platforms. Nucleic Acids Res. 2008;36:e141. doi: 10.1093/nar/gkn705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YC, Rasmussen MD, Bansal MS, Kellis M. TreeFix: statistically informed gene tree error correction using species trees. Syst Biol. 2012;62:110–20. doi: 10.1093/sysbio/sys076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Gianoulis TA, Karczewski KJ, Zhao H, Snyder M. Regulatory variation within and between species. Ann Rev Genomics Hum Genet. 2011;12:327–46. doi: 10.1146/annurev-genom-082908-150139. [DOI] [PubMed] [Google Scholar]