

Abstract
Beta-glucosidase (3.2.1.21) plays an essential role in the removal of non-reducing terminal glucosyl residues from glycosides. Recently, beta-glucosidase has been of interest for biomass conversion that acts in synergy with two other enzymes, endoglucanase and exo-glucanase. However, there is not much information available on the catalytic interactions of beta-glucosidase with its substrates. Thus, this study reports on the binding modes between beta-glucosidase from glycoside hydrolase family 1 namely BglB with cellobiose, cellotetraose and cellotetriose via molecular docking simulation. From the results, the binding affinities of BglB-cellobiose, BglB-cellotetraose, and BglB-cellotetriose complexes were reported to be -6.2kJ/mol , -5.68 kJ/mol and -5.63 kJ/mol, respectively. The detail interactions were also been investigated that revealed the key residues involved in forming hydrogen bonds (h-bond) with the substrates. These findings may provide valuable insigths in designing beta-glucosidase with higher cellobiose-hydrolyzing efficiency.
Keywords: Beta-glucosidase, binding mode, molecular docking, cellobiose, cellotetraose, cellotetriose
Background
Cellulose is a highly unbranched polymer consisting of glucose residues linked together by β-1, 4-glycosidic bonds with cellobiose as its structural unit. The glucose chains in cellulose are tightly packed to form insoluble crystallite which is impenetrable to any molecules including water.The structure of lignocelluloses is very complex where cellulose forms a skeleton which surrounded by hemicelluloses and lignin like matrix compose. Cellulose is one of the most abundant polysaccharide compound in nature and is thought to be a promisingly renewable biomass resource for alternative fuels [1]. The cellobiose unit in cellulose structure can be further degraded into glucose monomer unit by cellulases through enzymatic hydrolysis. There are three major enzymes in cellulase system that work in synergy to hydrolyze cellulose into glucose monomer which are endocellulase (endo-1,4-β- glucanohydrolase, EC 3.2.1.4), exocellulase (1,4-β-D-glucan cellobiohydrolase, EC 3.2.1.91) and β-glucosidase (β-1,4- glucosidase, EC 3.2.1.21). The biodegradation process of cellulose starts by endocellulase by cutting randomly at β-1,4- glucosidic linkages in the cellulose by endocellulases producing various lengths of oligosaccharides with the new chain ends. This action eventually breaks down the crystalline structure of the cellulose. Then, exocellulase cleaves the reducing and nonreducing ends of this new oligosacarides chain generating either glucose (glucanohydrolases) or cellobiose (cellobiohydrolase) as the major products. Finally, β-glucosidase completes this process by hydrolyzing the remaining cellobiose or cellotetraose into glucose monomer unit. Cellobiose which is an intermediate product is also a strong inhibitor for endoglucanase and exoglucanase and it becomes one of the key bottlenecks in enzymatic hydrolysis. In order to prevent this inhibition process, cellobiose unit must be immediately removed. Thus, it is important to understand the catalytic activity of β-glucosidase in order to improve the efficiency of this enzyme. This will help in designing an enhanced β-glucosidases. However, little is known about the catalaytic interactions between β-glucosidase and cellobiose. By employing molecular docking simulation, the binding modes between enzyme and substrate can be further explored.
Methodology
Preparation of Protein structure:
β-glucosidase B (BglB) from Paenibacillus polymyxa from Family 1 glycosidase hydrolases (GH-1) was chosen and its crystal structure was obtained from Protein Data Bank (http://www.rscb.org) with PDB IDs 2O9R [2]. The protein consists of one single chain and 452 residues length with resolution 2.30Å. The quality of structure was evaluated using a global QMEAN scoring function [3] and PROCHECK [4] was used to assess the stereo-chemical properties of the polypeptide structure. Waters, cofactors and originally bounded ligand in BglB was removed from the original PDB file. Polar hydrogen atoms were added and Kollman charges, atomic solvation parameters and fragmental volumes were assigned to the protein using AutoDock Tools [5].
Preparation of ligand structure:
The structure of cellobiose (CBI), cellotetraose (CTT) and cellotetriose (CTR) were sketched and cleaned using gradient optimization after adding explicit H-atoms using Marvin Sketch 5.10.2 (ChemAxon) software. Obminimize utility script of OpenBabel 2.3.1 [6] was used to minimize ligands molecule using MMFF94 forcefield. The prepared ligands were used as input files for AutoDock 4.2 5.The ligand rigid roots were automatically set and all possible rotatable bonds and torsions were defined as active.
Docking Procedure:
The computational docking was performed in machine running Intel Core Duo processor with 4GB RAM and 500GB hardick. The ligand centered maps were generated by AutoGrid program with a spacing of 0.375 Å and dimensions of 60 × 60 × 60 points. The gridbox centre was set to coordinate 66.14, 28.0619 and 38.081 in x y, and z respectively. The default settings were used for all other parameters. Lamarckian genetic algorithm (LGA) method was employed in this docking simulation [7]. For each docking simulation 100 different conformers were generated and were clustered into group with RMSD lower than 2.0Å. The clusters were ranked by the lowest energy representative for each cluster. The conformer with lowest binding energy was chosen and been analyzed for hydrogen bonding and hydrophobic interaction using LigPlot [8]. For the estimated binding energy (kcal/mol), the inhibition constant (Ki) for each ligand was calculated.
Result & discussions
From the docking simulation, the best conformation complexes were selected from 100 docking poses namely BglB-CBI, BglBCTT, and BglB-CTR. Table 1 (see supplementary material) shows the AutoDock binding free energies (ΔGb, kcal/mol) and inhibition constant (Ki , µm) for the three complexes. Cellobiose gives the highest score followed by cellotetraose and cellotetriose. The view of binding sites of each complex is shown in Figure 1. The detailed interactions between betaglucosidase and each ligands was further analyzed using LigPlot in order to reveal the key residues involved in the binding process. The results indicated that each ligands interacted with beta–glucosidase by forming hydrogen bonds and hydrophobic interactions as shown in Figure 2.
Figure 1.
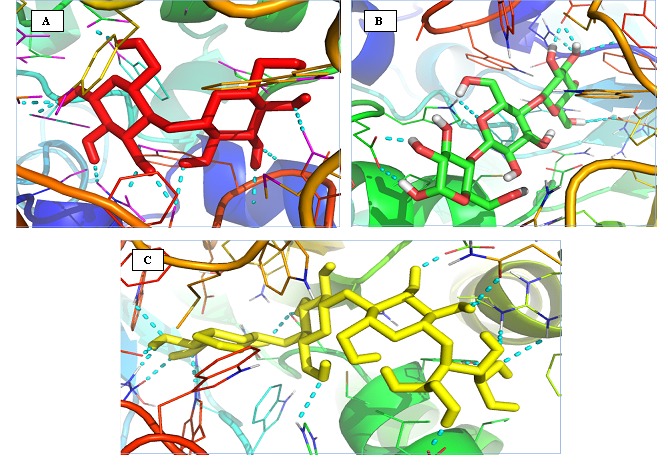
Image showing docked ligand with stick representation of cellobiose (red) , cellotetraose (green) and cellotriose (yellow) into the binding site of BglB (PDB ID : 2O9R) with cartoon representation . The cyan dashed lines represent hydrogen bond interaction with the enzymes. Images are generated by PyMol Version 1.4.1
Figure 2.
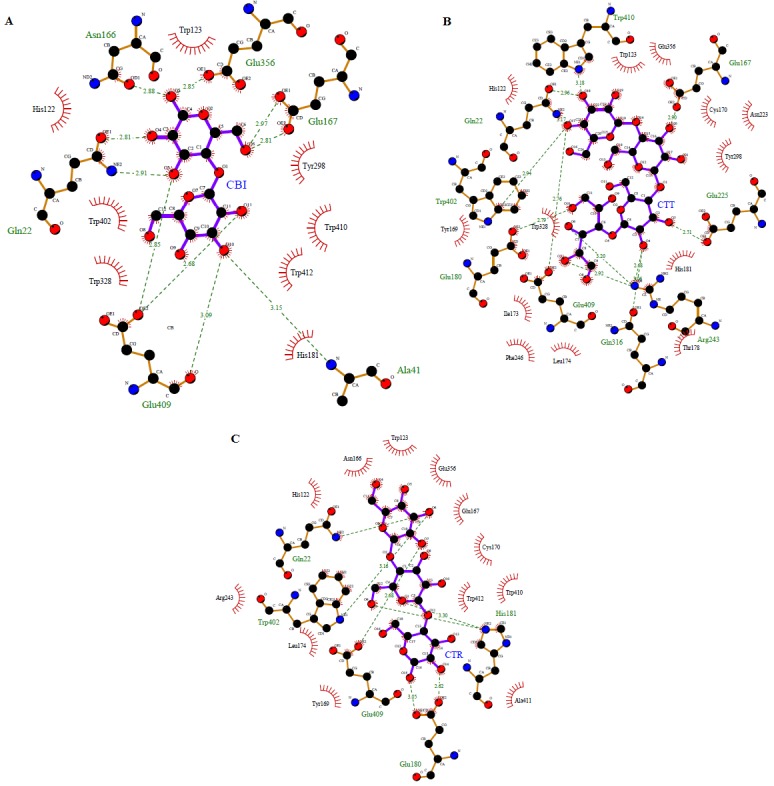
Image showing the residues within the active site region of beta-glucosidase B– BglB-CBI (Panel A) BglB-CTT (Panel B) and BgIB-CTR (Panel C). The models with the ball and stick belongs to cellobiose, cellotetraose and cellotetriose are shown in the ball and stick models respectively. The green dashed lines represent hydrogen bond formed between BglB and the respective ligands. The red spoke wise arcs pointing towards the ligands indicate the hydrophobic interactions while the ligand atoms with spokes indicate atoms that involved in hydrophobic contacts. Images were generated by LigPlot.
Results showed that the binding pocket of BglB mainly formed by residues Asn166 , Glu356 , Glu167 , Gln22 , Glu409 , Ala411 , Trp123 , Tyr123 , Tyr298 , Trp410 , Trp410 , Trp412 , His181 , Trp412 , His181, Trp402, and His122. Cellobiose is stabilized by BglB residues via both hydrogen bond and hydrophobic interactions. Ten hydrogen bonds were observed in the BglBCBI complexes as shown in Table 2 (see supplementary material), involving residues Glu356, Asn166, Glu167, Gln22, Glu409, and His181. While, another seven hydrogen bonds reported in BglB-CTT complex interacting with residues Gln22, Trp 402 , Glu409 , His181 and Glu180 , and hydrophobic interactions were formed by Trp413 , Glu356 , Cys170 , Asn223, Tyr298 , His181 , Thr178 , Leu174 , Phe246, Ile173 , Tyr169, Tyr328 and His122. As for BglB-CTR complex, the residues involve in forming hydrogen bonds cellotetriose were Trp402, His181, Glu409, and Glu180. While, His122, Asn166,Trp123, Glu356 , Glu167 ,Cys170 , Trp412 , Cys170 , Trp412 , Trp410 , Ala418 , Tyr169 , Leu174 , and Arg243 were found to formed hydrophobic interaction with the complex.
It has been reported that Glu167 act as a protonated agent of interglycosidic oxygen atom in hydrolysis mechanism, Glu356 act as nucleophilic in stabilizingthe transition state and Glu409 as catalytic residue [2]. In agreement with Isorna et al [2], it is observed in BglB complexes through hydrogen bonding to this three glutamate residue in cellobiose binding. Interestingly, Glu409 was found to form hydrogen bond involve in hydrogen bond in all three complexes suggesting that this residue might play as essential role in pathway of enzymatic hydrolysis. This glutamate residue is highly conserved among family glycoside hydrolase 1 [9]. The glutamate side-chain can adapt its position to an axial O4 and have ability to recognize galacto-configured substrates due to its conformational freedom characteristic [10]. In other study, Tiwari et al [11] reported that glutamate is important in reducing the energy barrier of the glycosylation step.
Conclusion
In this study the binding modes between beta-glucosidase with cellobiose, cellotetraose, cellotetriose and thio-cellobiose complexes was explored. From the results, there is nonbonded interaction exist between the complexes namely hydrogen bond and hydrophobic contacts. The binding affinities of the complexes BglB-cellobiose, BglB-cellotetraose, and BglBcellotetriose are reported to be -6.2kJ/mol, -5.68kJ/mol, - 5.63kJ/mol respectively. The common residues were found to be Trp123, Glu356, Glu167, Trp410, His181, Glu409, Trp402, Gln22, and His122.The result from this study will eventually give some idea to experimentalist to design better enzyme for more efficient enzymatic hydrolysis process with higher yields and also maintain the cost of production as low as possible.
Supplementary material
Acknowledgments
The authors would like to thank Malaysia-Japan International Institute of Technology (MJIIT), Universiti Teknologi Malaysia Kuala Lumpur for providing the financial support for this project(Institutional grant Vot 4J025).
Footnotes
Citation:Khairudin & Mazlan, Bioinformation 9(16): 813-817 (2013)
References
- 1.Maclellan J, et al. Strategies to Enhance Enzymatic Hydrolysis of Cellulose in Lignocellulosic Biomass. 2010;31:35. [Google Scholar]
- 2.Isorna P, et al. J Mol Biol. 2007;371:1204. doi: 10.1016/j.jmb.2007.05.082. [DOI] [PubMed] [Google Scholar]
- 3.Benkert P, et al. Proteins. 2008;71:261. doi: 10.1002/prot.21715. [DOI] [PubMed] [Google Scholar]
- 4.Unit M, Street G. J Appl Cryst. 1993;26:283. [Google Scholar]
- 5.Docking AJA, Autodock AJ. Energy Function. 2009;19:1639. [Google Scholar]
- 6.O'Boyle NM, et al. J cheminform. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trott O, Olson AJ. J Comput Chem. 2010;31:455. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Laskowski RA, Swindells MB. J Chem inf Model. 2011;51:2778. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- 9.Opassiri R. BMC Plant Biol. 2006;6:33. doi: 10.1186/1471-2229-6-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanz-Aparicio J, et al. J Mol Biol. 1998;275:491. doi: 10.1006/jmbi.1997.1467. [DOI] [PubMed] [Google Scholar]
- 11.Tiwari M, et al. Process Biochemistry. 2012 doi:10.1016/j.procbio.2012.09.015. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.