

Abstract
NMR spectroscopy and ITC are powerful methods to investigate ligand-protein interactions. Here, we present a versatile and sensitive fluorine NMR approach that exploits the 19F nucleus of 19F labeled carbohydrates as a sensor to study glycan binding to lectins. Our approach is illustrated with the 11 kDa Cyanovirin-N, a mannose binding anti-HIV lectin. Two fluoro-deoxy sugar derivatives, methyl 2-deoxy-2-fluoro-α-D-mannopyranosyl-(1→2)-α-D-mannopyranoside and methyl 2-deoxy-2-fluoro-α-D-mannopyranosyl-(1→2)-α-D-mannopyranosyl-(1→2)-α-D-mannopyranoside were utilized. Binding was studied by 19F-NMR spectroscopy of the ligand and 1H-15N HSQC NMR spectroscopy of the protein. The NMR data agree well with those obtained from the equivalent reciprocal and direct ITC titrations. Our study shows that strategic design of fluorinated ligands and fluorine NMR spectroscopy for ligand screening holds great promise for easy and fast identification of glycan binding as well as for their use in reporting structural and/or electronic perturbations that ensue upon interaction with a cognate lectin.
Keywords: 19FNMR spectroscopy, fluoro-deoxy sugar synthesis, protein-ligand interaction, reverse and direct titration, lectin
Introduction
The growing realization of the enormous potential of glycans in information coding, embodied by the term “sugar code”,[1] has led to define oligosaccharides as functionally active units and provides the rational for identifying new targets in drug design. Aided by the advances in chemical synthesis of such determinants,[2] detection of glycan receptors (lectins) and the analyses of the specificity/mechanisms of protein-carbohydrate interactions are key steps in this endeavor.[3] With glycan-receptor recognition critically involved in viral and bacterial cell attachment, as well as in host defense, immune(dys)regulation and tumor progression,[4–8] versatile and sensitive analysis/screening tools in this area are urgently needed. NMR can satisfy this need, having proved its value by providing high-resolution molecular structures in solution and valuable information on ligand binding.[9–12]
Most biomolecular NMR applications employ proton/carbon/nitrogen experiments and 2D heteronuclear NMR is a powerful method to map ligand binding sites on proteins and to quantify protein-ligand interactions.[9–12] It has, however, some limitations. It is best suited for medium size proteins (< 50kDa) since amide resonance assignments are a prerequisite. In addition, for titration experiments with low affinity ligands, large amounts of ligand are needed to reach saturation, introducing uncertainty in the estimation of affinity parameters, especially for synthetic compounds of limited water-solubility. Thus, ligand-centered methods hold some promise to overcome these problems.
Among ligand-detected NMR approaches, only few exploit the favorable properties of the fluorine nucleus.[13,14] Mono- or trifluoro N-acetylglucosamines were used to measure affinity constants and map their binding environment.[15–18] Using fluorinated sugars,[19–21] saturation transfer difference (STD) experiments in which magnetization was relayed to the vicinal and geminal F-atoms in a monosaccharide were used in a competition screening assay in the search for lead compounds in inhibitor design.[22,23] Analogous to ITC, where in addition to titrating ligand into the protein solution, a second, reverse titration can be performed in which protein is titrated into the ligand solution, ligand-based NMR titrations provide the reciprocal data to information commonly extracted from 2D 1H,15N heteronuclear correlation spectra of the protein. If the correct model is used for data fitting, identical thermodynamic parameters should be obtained, regardless of the direction of the titration.[24] Here we present a NMR study in which the 19F-NMR signal of a singly fluorinated small molecule ligand is monitored upon protein addition. We compare these data to results obtained by recording changes in 1H,15N amide protein resonance frequencies upon ligand binding. For our study we selected the 11 kDa antiviral lectin cyanovirin-N (CV-N) as the sugar receptor, since its structure and glycan binding properties have been carefully analyzed by NMR spectroscopy, crystallography, and titration calorimetry.[25–28] CV-N harbors two carbohydrate-binding sites in its pseudo symmetric, bilobal structure, site 1 in domain B (residues 39–89) and site 2 in domain A (residues 1–38/90–101).[25–28] The minimal glycan recognition unit is a α1,2-linked dimannoside, either as the individual oligosaccharide or as part of the terminal arms of the branched Man-8 and Man-9 structures.[29,30] Domain A exhibits a slight preference for trimannoside and domain B for dimannoside.[28,31,32] The availability of detailed STD data for these ligands as well as results with specific deoxy sugars allowed us to identify the pivotal hydroxyl groups responsible for binding.[33,34] In particular, we determined that the 3′- and 4′-hydroxyl groups are essential for the interaction, whereas the 2′- and 6′-hydroxyl groups of the Manα(1–2)Manα (Man2) terminal unit are not directly involved in CV-N binding.[34] We therefore prepared methyl 2-deoxy-2-fluoro-α-D-mannopyranosyl-(1→2)-α-D-mannopyranoside (19F-Manα(1–2)Manα:19F-Man2) and methyl 2-deoxy-2-fluoro-α-D-mannopyranosyl-(1→2)-α-D-mannopyranosyl-(1→2)-α-D-mannopyranoside (19F-Manα(1–2)Manα(1–2)Manα:19F-Man3) (Supplementary Scheme S1) for use in our current work.
We show that fluorination at the 2′ or 2″ position of the sugar does not curtail its bioactivity in a cell-binding assay in which 19F-Man2 and 19F-Man3 were used as inhibitors for CV-N’s interaction with the cell surface. We determined affinity constants and exchange rates for 19F-Man2 and 19F-Man3 interaction with two of CV-N variants in which one of the binding sites had been impaired,[35–37] and compared the results to values derived from titration calorimetry.
In addition, due to the inherent high sensitivity of the fluorine chemical shift to its local environment, it was possible not only to distinguish between free and bound signals for 19F-Man2 and 19F-Man3, but also to identify two different modes of binding for 19F-Man3.
Results and Discussion
NMR is becoming a standard approach for studying protein-ligand interactions since it provides powerful means for mapping the structural details of binding sites on the protein as well as the ligand. In addition, affinity parameters can be determined. Here, we employed 19F-NMR and fluorinated ligands to evaluate the interactions between di- and trimannoside with CV-N and demonstrate that, based on the favorable properties of the 19F nucleus, novel features can be discerned.
Bioactivity of the 19F-containing glycans
In order to ascertain that substituting a fluorine at the 2′ or 2″ position for the hydroxyl group does not alter the properties of the sugars significantly and that the two 2′-19F-substituted dimannoside- and the 2″-19F-substituted trimannoside are capable to act as specific and potent ligands, we tested their activity in blocking lectin binding to cells. As shown in Figure 1, 2′-19F-substituted glycans strongly inhibited lectin binding to human carcinoma cells, when compared to the natural monosaccharide. Similar data were also obtained for a T lymphoma (Jurkat) line (not shown).
Figure 1.

Cell-binding of monofluorinated di- and trimannosides. Human colon adenocarcinoma cells (SW480) were stained with fluorescein-labelled CV-N. Controls representing staining with CV-N (100%-value at 1 μg/ml; green line) and without (0%-value; shaded area) are included in each panel (quantitative data for the percentage of positive cells/mean fluorescence intensity are listed). Top panel: control experiment without added glycan (green) and in the presence of 100 mM mannoside (left), and in the presence of 4 mM or 8 mM 19F-Man2 (right). Bottom panel: control experiment without added glycan (green) and in the presence of 100 mM mannoside (left), in the presence of 1 mM or 2 mM 19F-Man3 (right).
Therefore, incorporation of the 19F sensor did not impair bioactivity, confirming that the 2′ group5 are not critical for cell surface receptor binding on the two cell types tested, in line with expectations based on results with the 2′-deoxy derivative of the dimannoside.[34] These results prompted us to proceed to studying the interaction with the lectin by 19F-NMR spectroscopy.
Ligand-detected binding of fluorinated sugars to CV-N
For ligand-detected NMR experiments we recorded 1D-19F NMR spectra of 19F-Man2 (200 μM) and 19F-Man3 (50 μM) at 280 K. Spectra of 1:1 mixtures of sugar:protein are provided in Figure 2A, and Figure 2B for [CVNΔA]ssm, CVNmDB and CV-NP51G with 19F-Man2 and 19F-Man3, respectively.
Figure 2.

1D-19F NMR spectra of 19F-Man2/Man3 in the presence of [CVNΔA]ssm, a variant that contains a single glycan binding site on domain B, CVNmDB, a variant that contains a single glycan binding site on domain A, and CV-NP51G that is wild-type-like with both glycan binding sites present, at 280 K. CV-N domains are represented by two elongated crescents, representing the two binding sites. Site 1 on domain B is colored pink and site 2 on domain A in green. A) 19F-1D NMR spectra of 200 μM 19F-Man2 in the presence of [CVNΔA]ssm, CVNmDB, and CV-NP51G. B) 19F-1D NMR spectra of 50 μM 19F-Man3 in the presence of [CVNΔA]ssm, CVNmDB and CV-NP51G. Bound-state and free-state signals are in slow exchange for [CVNΔA]ssm in C) and CVNmDB in D. In the titration curves (bottom panels) the bound fraction is derived from the signal intensity (1-Ifree/I0) during the titration, with Ifree the intensity of the free ligand signal at each point in the titration, and I0 the intensity of the free ligand signal at the beginning of the titration. In C) [CVNΔA]ssm was titrated into 50 μM 19F-Man3 (top panel), with molar ratios of protein/glycan: 0:1, 0.5:1, 1:1, 1.5:1. In D) CVNmDB was titrated into 50 μM 19F-Man3 (top panel), with protein/glycan molar ratios: 0:1, 0.5:1, 1:1, 2:1, and 3:1.
Two distinct resonances, separated by a Δδ >1 ppm, correspond to the protein-free and protein-bound conformations. The resonance corresponding to the protein-bound conformation of 19F-Man2 when bound to domain A is downfield shifted from the free resonance while that corresponding to the domain B protein-bound is upfield shifted. The two resonances of 19F-Man3 when bound to domain A and B both experience a downfield shift, compared to the free sugar signal. Unambiguous assignments to the domain A-bound or domain B-bound state was achieved by using [CVNΔA]ssm and CVNmDB, variants of CV-N in which the binding site in domains A and B were obliterated.[35–37] Gratifyingly, the two resonances corresponding to bound 19F-Man2 in the CV-NP51G wild-type variant of CV-N that possesses both binding sites, exhibit identical chemical shifts to those observed in the single site mutants, confirming that the site that remains in the mutant is identical to its wild-type counterpart. The same holds for the 19F-Man3 resonances, although as pointed out above, both bound resonances are downfield of the free sugar signal.
Much of the power of fluorine NMR studies of biological systems derives from the high sensitivity of the fluorine shielding parameter to changes in local environment. Shielding is observed when the fluorine is in the close contact to H bond donors of the protein or solvent molecules.[38–40] On the contrary, a fluorine nucleus in the vicinity of an electronegative atom downfield shifts. Also, deshielded fluorines are found in the close contact with hydrophobic side chains.[38–40] Inspection of the crystal structure of a mutant CV-N protein, in which the carbohydrate binding site in domain A was abolished, complexed with a dimannoside ligand, P51G-m4-CVN: Man2 (PDB accession code 2RDK),[41] revealed that in the binding site on domain B, a 2′-19F substitution could act as a H-bond acceptor with a vicinal water molecule, causing an upfield shift. Unfortunately, structural data for dimannoside binding in domain A are not available, therefore we can only speculate that such H-bond is not present for 2′-19F-Man2 interacting with domain A. The small 19F downfield chemical shift observed in this case is most likely caused by either the anionic repulsion from an electronegative protein atom or the interaction of fluorine atom with the hydrophobic protein binding pocket. For Man3, the crystal structures of wild type CV-N bound to Man-9 (PDB accession code 3GXZ)[32] allows to delineate residues in domain A that interact with the trimannoside on the D1 arm of Man-9. The negative Glu101 side chain carboxylate causes electrostatic repulsion of the anionic 2″-19F, causing a downfield shift. A similar scenario or possibly contacts with hydrophobic protein side chains are likely for 19F-Man3 binding to domain B, although direct supporting structural data are not available.
We also carried out 1D-19F NMR experiments at 298 K. However, except for the resonance of the free fluorinated glycan, no signal for the ligand-bound to protein conformation was detected in the case of 19F-Man2, complexed with all CV-N variants. This is probably due to severe line broadening in the intermediate exchange regime at room temperature. For 19F-Man3, on the other hand, both ligand-free and ligand-bound resonances were detected at 298 K when [CVNΔA]ssm, CVNmDB or CV-NP51G was added, although ligand-bound resonances were significantly broader than the ones at 280 K (data not shown). The 1D 19F-NMR titration data, monitoring the 19F-Man3 resonance intensities upon addition of [CVNΔA]ssm and CVNmDB at 280 K, are provided in Figure 2C, and Figure 2D respectively. The spectra for 19F-Man3 (50 μM) are displayed in the top panels and the resulting binding isotherm in the bottom panel. For both mannosides, free and bound resonances are in slow exchange. The KDF values were extracted from the binding isotherms, monitoring the intensity changes of the protein-bound ligand signal versus the protein:ligand molar ratio. The bound fraction (fb) was estimated from the intensity changes of the free ligand resonance during the titration using fb=1-Ifree/I0. The extracted KDF values were 10.8±0.3 μM and for [CVNΔA]ssm and 33.5±3.5 μM for CVNmutDB binding to 19F-Man3, respectively.
Exchange kinetics between free and protein-bound fluorinated glycans
The 19F-NMR resonances for free and bound states at 280 K are in slow exchange on the chemical shift scale, as evidenced by exchange peaks in the 2D 19F-19F NOESY spectra.
In order to extract kinetic parameters for the exchange process, a series of 2D 19F-19F homonuclear NOESY spectra with different mixing times were recorded. Representative spectra of 19F-Man2 (200 μM) in the presence of [CVNΔA]ssm and CVNmDB (1:1 molar ratio) at 280 K are provided in Figure 3A and Figure 3B. The exchange curves obtained from the intensity ratios of the bound, diagonal peak, to the exchange cross-peak (IBB/IBF) as a function of mixing time are shown next to the NOESY spectra. Analysis of the exchange curve for 19F-Man2 when bound to the site on domain B yields a free-to-bound exchange rate (kex) of 56.8±1.4 s−1, while that for binding to domain A yields a kex value of 99.2±2.1 s−1.
Figure 3.

2D 19F-19F NOESY exchange spectra of 19F-Man2/Man3 in the presence of [CVNΔA]ssm and CVNmDB protein. A) 19F-Man2 (200μM) bound to [CVNΔA]ssm(left panel; tmix=0.1 s), and the exchange curve of bound/bound over free/bound intensity ratio (IBB/IBF) versus mixing time (right panel) tmix: 0.05, 0.1, 0.3, 0.5 s. B) 19F-Man2 bound to CVNmDB (left panel; tmix=0.05 s) and the exchange curve of bound/bound over free/bound intensity ratio (IBB/IBF) versus mixing time (right panel) tmix: 0.025, 0.05, 0.1, 0.2 s. C) 19F-Man3 (50 μM) bound to [CVNΔA]ssm (left panel; tmix=0.1 s) and the exchange curve of bound/bound over free/bound intensity ratio (IBB/IBF) versus mixing time (right panel) tmix: 0.1, 0.3, 0.5, 0.7, 0.9 s. D) 19F-Man3 (50 μM) bound to CVNmDB (left panel; tmix=0.05 s) and the exchange curve of bound/bound over free/bound intensity ratio (IBB/IBF) versus mixing time (right panel) tmix: 0.05, 0.1, 0.15, 0.3 s. All spectra were recorded at 280 K for 1:1 molar ratios. The auto-correlation (diagonal) peaks IFF, and IBB arise from the free and bound states, while cross-correlation (off-diagonal) peaks IFB=IBF report on the exchange between free and bound states.
The equivalent data of 19F-Man3 (50 μM) in the presence of [CVNΔA]ssm and CVNmDB (1:1 molar ratio) are shown in Figure 3C and Figure 3D and resulted in kex values of 2.29±0.1 s−1 and 10.5±2.9 s−1 for the binding to domains B and A, respectively. Thus, the exchange between free and bound state of the di-and trimannoside is somewhat faster from domain A than domain B. Although easily observed at 280 K, no exchange peak was observed at 298 K for 19F-Man2 due to intermediate exchange that caused line broadening beyond detection. Those for 19F-Man3, on the other hand, are still present at 298 K.
Protein-detected binding between 19F-Man2 and 19F-Man3 with CV-N
Direct protein-observed NMR titration experiments were carried out with 19F-Man2 at 298 K by following chemical shift perturbations in the 1H-15N HSQC spectrum of 15N uniformly labeled [CVNΔA]ssm (75 μM, Figure 4) and CVNmDB (400 μM, Figure S1) as a function of added sugar. Spectra in the absence (black contours) and presence (green contours) of 25 molar equivalents of 19F-Man2 are provided in Figure 4A and Figure S1A, respectively. In the 1H-15N HSQC NMR spectra of [CVNΔA]ssm and CVNmDB titrations at 298 K, resonances are in fast exchange, in contrast to the situation in the 19F-NMR spectra, where resonances at 298 K were in intermediate exchange and broadened beyond detection. Following the chemical shift changes throughout the titration yielded the binding isotherm displayed in the right panels of Figure 4A and Figure S1A, respectively.
Figure 4.

NMR titration of [CVNΔA]ssm with 19F-Man2 recorded at 298 K and 19F-Man3 recorded at 280 K, respectively. A) Perturbed amide resonances in the [CVNΔA]ssm spectrum clearly reside in domain B and are labeled by amino acid name and number. The left panel display a superposition of 1H-15N HSQC NMR spectra of [CVNΔA]ssm in the absence (black) and presence of 25 molar equivalents of 19F-Man2 (green). The right panel displays the titration curve (chemical shift difference versus ligand/protein molar ratio), with the chemical shift difference defined as: Δδ = [(ΔδH)2 + (ΔδN × 0.17)2]1/2, where ΔδH and ΔδN are the observed chemical shift changes for 1H and 15N, respectively. The system is in fast exchange, as evidenced by a single signal corresponding to glycan free and glycan bound [CVNΔA]ssm. B) Superposition of 1H-15N HSQC spectra of 50 μM [CVNΔA]ssm in the absence (black) and presence (green) of 3 molar equivalents of 19F-Man3 (left panel). The right panel displays the titration curve at glycan/protein molar ratios: 0:1, 0.5:1, 1:1, 1.5:1, 2:1, and 3:1. The system is in slow exchange, as evidenced by separate signals for glycan free and glycan bound [CVNΔA]ssm. The binding isotherm is derived from the relative signal intensities (1-Ifree/I0) (right panel).
The equilibrium dissociation constant (KDF) for 19F-Man2 binding was determined from the titration shifts (chemical shift Δδ (ppm) versus the ratio of ligand to protein concentration) using several resonances. 19F-Man2 binding to the single sites on domains B and A yielded KDF values of 960±91 μM and 1.37±0.075 mM, respectively. The latter value is about a factor of two larger than the one determined for Man2 binding to site A on CVNmutDB (757±80 μM),[36] suggesting that the substitution of fluorine at the 2′ position reduces the binding affinity only by a very small amount. As noted previously, Man2 exhibits a slight preference for Domain B over Domain A, respectively.[28,31,32]
Equivalent NMR titration experiments, monitoring chemical shift perturbations in the 1H-15N HSQC spectra of [CVNΔA]ssm and CVNmDB were carried out at 280 K with 19F-Man3. A superposition of [CVNΔA]ssm (50 μM) spectra in the absence (black contours) and presence (green contours) of 3 molar equivalents of 19F-Man3 is provided in Figure 4B. The binding isotherm derived from the intensity changes of free resonances is depicted in the right panel. The equivalent titration experiment was performed for CVNmDB (50 μM) and the data are provided in Figure S1B. The dissociation constants were extracted as described for 19F-Man2 (above) and binding to a single site on domain B (in [CVNΔA]ssm) yielded a KDF value of 63.5±5 μM, while the one for binding to a single site on domain A (in CVNmDB) was 150.3±7 μM.
ITC binding
A direct comparison of binding parameters that were extracted from the different NMR titrations with those from ITC measurements was performed.
In particular, reverse titrations in which 19F-Man3 (50 μM) was placed into the cell and protein into the injector were carried out. In this manner, an equivalent situation to the one in 19F-glycan-observed NMR titrations at 280 K is created. The measured titration heat as well as the derived binding isotherms are provided in Figure 5A and Figure S2A for [CVNΔA]ssm and CVNmDB, respectively. The extracted KDF values were 12.6±1.2 μM for [CVNΔA]ssm and 28.5±3.5 μM for CVNmutDB, in excellent agreement with the values extracted from the 19F-glycan-observed NMR titrations (see above).
Figure 5.

ITC titration experiments for the interaction between 19F-Man3 and [CVNΔA]ssm at 280 K. In A) the trace for titrating [CVNΔA]ssm into 50 μM 19F-Man3 (reverse titrations) for 28 automated injections is shown (top panel), and the derived binding isotherm is displayed in the bottom panel. The nonlinear least-squares best fit to the experimental data using a one-site model is shown by the line connecting the data points. In B) the ITC trace of titrating 19F-Man3 into 30 μM [CVNΔA]ssm (direct titrations) for 28 automated injections (top panel) with the derived binding isotherm (bottom panel) is displayed. The nonlinear least-squares best fit to the experimental data using a one-site model is shown by the line connecting the data points.
We also carried out direct ITC titrations at 280 K with 19F-Man3 in the injector and [CVNΔA]ssm(30 μM) and CVNmDB(50μM) in the cell. The observed binding isotherms are provided in Figure 5B and Figure S2B yielding KDF values of 62.5±2 μM and KDF of 153.3±5 μM for [CVNΔA]ssm and CVNmDB, respectively. These values are in good agreement with those obtained from the NMR direct titration experiment for both [CVNΔA]ssm and CVNmDB variants, that were 63.5±5 μM and 150.3±7 μM, respectively. Interestingly, it appears that 19F-Man3 no longer preferentially binds to domain A, as was observed for the non-fluorinated Man3,[28,31,32] possibly due to the 19F substitution. The extracted dissociation constants for 19F-Man3 binding are provided in Table 1.
Table 1.
Dissociation constants for the interaction of 19F-Man3 with single binding site CV-N variants determined by ligand-observed 19F-NMR, reverse ITC, protein-observed NMR, and direct ITC experiments.
| Protein | KDF (μM) 19F-NMR | KDF (μM) reverse ITC | KDF (μM) 1H-15N HSQC | KDF (μM) direct ITC |
|---|---|---|---|---|
| [CVNΔA]ssm | 10.8 ± 0.3 | 12.6 ± 1.2 | 63.5 ± 5 | 62.5 ± 2 |
| CVNmDB | 33.5± 3.5 | 28.5 ± 3.5 | 150.3 ± 7 | 153.3 ± 5 |
Calorimetry is the only technique that directly permits to evaluate the basic physical forces in sufficient detail between and within molecules by measuring heat quantities or heat effects.42 Therefore, a deeper understanding of the molecular basis of protein–ligand interactions can be gained by thoroughly characterizating and quantifying the energetics that govern complex formation.24,42 Analysis of the binding isotherm for 19F-Man3 with [CVNΔA]ssm reverse ITC titrations yielded a ΔG value of −5.1 kcal mol−1, ΔH= −2.9 kcal mol−1, and a binding entropy TΔS of 2.2 kcal mol−1, while for the direct ITC titration a ΔG value of −5.3 kcal mol−1, ΔH= −5.1 kcal mol−1, and a binding entropy TΔS of 0.2 kcal mol−1 was obtained. Similarly, a ΔG value of −5.8 kcal mol−1, ΔH= −1.5 kcal mol−1 and TΔS of 4.3 kcal mol−1 was measured in the reverse ITC titration of 19F-Man3 with CVNmDB, while a ΔG value of −4.88 kcal mol−1, ΔH= −4.08 kcal mol−1 and TΔS of 0.8 kcal mol−1 was obtained for the direct titration. A summary of all thermodynamic data determined for 19F-Man3 binding to [CVNΔA]ssm and CVNmDB in reverse and direct ITC titrations is provided in Table 2. It should be mentioned that standard deviation values arise from the fitting procedure.
Table 2.
Thermodynamic parameters extracted from ITC binding data
| Protein | Enthalpy ΔH (kcal mol−1) | Entropy TΔS (kcal mol−1) | Free energy ΔG (kcal mol−1) | Stoichiometry n |
|---|---|---|---|---|
| [CVNΔA]ssm reverse ITC | −2.9 ± 0.05 | 2.2 ± 0.04 | −5.1 ± 0.06 | 1.01 ± 0.03 |
| [CVNΔA]ssm direct ITC | −5.1 ± 0.1 | 0.2 ± 0.08 | −5.3 ± 0.1 | 1.0 ± 0.0 |
| CVNmDB reverse ITC | −1.5 ± 0.03 | 4.3 ± 0.02 | −5.8 ± 0.04 | 1.02 ± 0.04 |
| CVNmDB direct ITC | −4.08 ± 0.06 | 0.8 ± 0.03 | −4.88 ± 0.05 | 1.0 ± 0.0 |
For both mutants, binding to 19F-Man3 was driven by enthalpic contributions (with negative ΔH values of −2.9 kcal mol−1 and −5.1 kcal mol−1 in the case of [CVNΔA]ssm, and −1.5 kcal mol−1 and −4.08 kcal mol−1 for CVNmDB, for the reverse and direct ITC titrations, respectively), with favorable entropic contributions. Somewhat larger positive TΔS values (2.2 kcal mol−1 for [CVNΔA]ssm and 4.3 kcal mol−1 for CVNmDB) were obtained for the reverse ITC titrations, compared to the direct ones (0.2 kcal mol−1 for [CVNΔA]ssm and 0.8 kcal mol−1 for CVNmDB), suggesting that water molecules are more eficiently released from the complex surface in the case of reverse titrations.42 The overall ΔG values, however, are not very different.
ITC also allows determining the stoichiometry of binding, independent of the binding affinity.24 Since a 1:1 stoichiometry was obtained from the experimental (sigmoid) curves in the cases of reciprocal ITC titrations, a one binding site model was used to fit the data and binding affinity parameters were extracted. In the case of direct ITC titrations, since ~1:1 stoichiometry was observed, a stoichiometry value of n=1 was fixed in order to fit the experimental (parabolic) curves, based on the fact that a single binding site was detected for the equivalent, protein-observed 1H-15N HSQC titrations (Figure 4B and Figure S1B). Furthermore, the fully saturated samples resulted either at the end of protein-observed 1H-15N HSQC or direct ITC titrations, were tested on 1D-19F NMR as well, and indeed, only one protein-bound peak corresponding to a single binding site was observed. Therefore, the reverse titration allowed us not only to check the stoichiometry, but also to ensure that a suitable binding model was used to fit the ITC data. For a 1:1 binding stoichiometry, the measured thermodynamic parameters are expected to be invariant when changing the orientation of the experiment.24 However, this is rarely the case since one of the two species may display greater aggregation or less solubility when concentrated.24b Indeed, a comparison of the data in Table 1 revealed that titrating 19F-Man3 into solutions of [CVNΔA]ssm and CVNmDB using protein-observed 1H-15N HSQC spectra or direct ITC experiments yielded KDF values approximately five times higher than the ones obtained from ligand-observed 19F-NMR and reciprocal ITC titrations. Naturally, if aggregation occurs, this will happen at high concentrations of the ligand. Therefore, it is advantageous to measure binding on the ligand, either by NMR or reverse ITC, since lower ligand concentrations are needed.
Thus, ligand-detected binding methods are preferable compared to methods that measure protein signals, since any errors associated with the solubility issue, which therefore might introduce incorrect ligand concentrations, are avoided.
Detection of a second binding mode in the interaction of 19F-Man3 with CV-N
The concentration of 19F-Man3 in the ligand-detected 19F-NMR experiments was 50 μM, while for those with 19F-Man2 was 200 μM. To directly compare the results of [CVNΔA]ssm, CVNmDB and CV-NP51G binding to the two fluorinated glycans, we also recorded a ligand-detected 19F-spectrum of 19F-Man3 (200 μM) with CV-N variants (1:1 molar ratio). As can be observed from the comparison of the spectra illustrated in Figure 6, at higher 19F-Man3 concentrations ( >150 μM) additional resonances emerge. These new resonances exhibit exactly the same chemical shifts (Figure 6B) that were seen for the signals of 19F-Man2 when bound to domains A and B of CV-N, respectively (Figure 6A).
Figure 6.

1D-19F spectra of 200 μM 19F-Man2/Man3 in the presence of different CV-N variants, illustrating the power 19F NMR for discriminating between different binding sites and modes. The presence of a second binding mode for CV-N’s interaction with 19F-Man3 is clearly observed. All spectra were recorded at 280 K. A) 1D-19F spectra of 200 μM 19F-Man2 in the presence of [CVNΔA]ssm, CVNmDB and CV-NP51G. B) 1D-19F spectra of 200 μM 19F-Man3 in the presence of [CVNΔA]ssm, CVNmDB and CV-NP51G. Schematic depiction of 19F-Man2 and the two binding modes of 19F-Man3 to CV-N variants are represented on the right panels of A) and B). The individual sugar units on the oligosaccharide are color coded according to their linkage pattern. C) 2D 19F-19F NOESY spectra 19F-Man3 in the presence of 200 μM [CVNΔA]ssm and CVNmDB (1:1 molar ratio of glycan:protein). The auto-correlation (diagonal) peak intensities IFF, IBB in the 2D 19F-19F NOESY spectra arise from the free and bound states, while cross-correlation (off-diagonal) resonances IFB=IBF and IFB’=IBF’ arise from exchange between the two binding modes for CV-N and 19F-Man3. D) Competitive binding of Man2 to the complex of 19F-Man3:[CVNΔA]ssm (200 μM;19F-Man3:[CVNΔA]ssm:Man2 molar ratios: 1:1:0; 1:1:1; 1:1:5; 1:1:10; and 1:1:20). Schematic representation of Man2 competing with 19F-Man3 binding to [CVNΔA]ssm is represented on the bottom panel. Man2 competitor displace first 19F-Man3 bound to protein with non-reducing 19F-Manα(1–2)Manα unit (the low affinity binding mode: solid arrow), and later the one bound with the unit closer to O-methyl group (high affinity binding mode: dashed arrow).
The presence of the second binding mode can also be observed in the 2D 19F-19F exchange NOESY experiment recorded with a 50 ms mixing time on the sample of 200 μM 19F-Man3 with [CVNΔA]ssm and CVNmDB (Figure 6C). Given that the additional peaks exactly match the chemical shifts of the bound conformations of 19F-Man2 when sitting in domains A and B of CV-N and are only observed at relatively high concentrations, it appears that each domain of CV-N binds first to the Manα(1–2)Manα unit that is closer to O-methyl group in 19F-Man3 (high affinity binding mode: 1) and subsequently to next non-reducing end 19F-Manα (1–2)Manα unit (low affinity binding mode: 1′).
To further substantiate our findings we also carried out a competition binding experiment, adding non-fluorinated Man2 to a solution of 19F-Man3 (200 μM) and [CVNΔA]ssm (1:1 molar ratio) and monitored the change in 1D-19F NMR ligand signal intensity as a function of Man2 addition. The resulting data up to a 20 fold molar excess of Man2, are provided in Figure 6D. As can be noted, the resonance that corresponds to the bound conformation of the non-reducing end 19F-Manα(1–2)Manα unit is the one that is affected first by the addition of non-fluorinated Man2, while the resonance corresponding to the bound conformation of the second Manα(1–2)Manα unit, the one closer to O-methyl group, is affected only at higher Man2 addition. Such differentiation was not observed when 19F-Man3 or non-fluorinated Man3 was used in protein-detected titration experiments.
19F-NMR competition assay
To further highlight the advantages of using 19F-NMR, we also carried out a quantitative competition binding experiment, titrating non-fluorinated Man3 into a complex of 19F-Man3 (250 μM) with relatively low (25 μM) of CVNmDB (10:1 molar ratio), that ensure that only the high affinity binding mode is involved. Indeed the inspection of 1D-19F NMR spectra show only the presence of the peak corresponding to high affinity protein-ligand binding mode, while no additional low affinity binding peak is observed.
The intensity change in the 1D-19F NMR free ligand signal was monitored as a function of Man3 addition. A schematic representation of competitive binding scenario is illustrated in Figure 7A. The resulting data are provided in Figure 7B. The intensity increase in the 1D-19F NMR spectrum of the free 19F-Man3 signal in complex with CVNmDB, upon addition of the non-fluorinated competitor (Man3), corresponds to the amount of non-labeled ligand bound to CVNmDB. The dissociation constant of Man3 binding to CVNmDB was obtained from the relevant competition titration data for the 19F-Man3:CVNmDB:Man3 complex (Figure 7B). In order to extract the binding constant kd of Man3 interacting with CVNmDB, it is necessary to know the KDF value of 19F-Man3 binding to CVNmDB (Figure 7A). We determined the value by 19F-NMR and reciprocal ITC titrations (see above). The extracted kd value of 4.65 ± 0.6 μM is consistent with our previous affinity data of the non-labeled Man3 with CVNmDB (3.4±0.05 μM), obtained from ITC titration experiment.[36]
Figure 7.
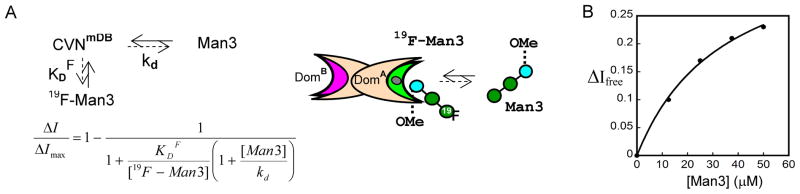
Competition of Man3 and 19F-Man3 for 250 μM 19F-Man3 in complex with 25 μM CVNmDB. A) Schematic representation of the competitive binding scenario. Cheng–Prusoff equation based on a competitive site binding model, where [19F-Man3] and [Man3] are the concentrations of the fluorinated and non-fluorinated ligand, and KDF the dissociation constants of 19F-Man3 for the glycan-binding site on domain A of CVNmDB. The data were fitted using the maximum increase in free 19F-Man3 resonance intensity upon Man3 addition, ΔImax, and kd the dissociation constant for Man3 binding as adjustable parameters. B) 19F-NMR spectra were recorded for 19F-Man3:CVNmDB:Man3 molar ratios of 10:1:0, 10:1:0.25, 10:1:0.5, 10:1:0.75, and 10:1:1. In the titration curve the bound fraction of Man3 is derived from the difference in signal intensity of the 19F-Man3 free signal ΔI=Ifree-I0 during the titration, with Ifree the intensity of the 19F-Man3 free signal at each point in the titration, and I0 the intensity of 19F-Man3 free signal at the beginning of the titration.
Conclusion
We presented an approach to quantitatively assess glycan-lectin interactions in which the changes in the 19F-NMR resonances of fluorinated glycans are monitored upon protein addition. Our method exploits the relatively large 19F-NMR chemical shift range that results in well-resolved resonances in 1D spectrum. Employing fluorinated ligands permits studying protein-ligand interaction by NMR even for high molecular mass proteins (> 100kDa) and requires only minimal amounts of ligand. Therefore, novel avenues for screening in a facile fashion and relatively short time can be envisaged.
In particular we show that:
Quantitative binding affinity data can be obtained using 19F-NMR experiments with fluorinated glycans.
Information about protein-ligand binding exchange can be extracted from 19F-19F NOESY experiments and we determined chemical exchange rate constant, kex, for each of the two binding sites of CV-N.
Different binding modes can be discerned for the two neighboring Manα(1–2)Manα units in 19F-Man3 when interacting with CV-N.
19F-NMR competition experiments can be employed for quantitatively extracting affinity constants of non-fluorinated glycans with target lectins.
In summary, utilizing 19F as sensor for studying protein-ligand interactions represents a promising perspective for 19F-NMR spectroscopy as a universal tool in drug discovery for any target receptors for which fluorinated ligands are available or can be synthesized.
Experimental Section
Lectins
The Cyanovirin-N wild-type stabilized variant CV-NP51G and the two mutants [CVNΔA]ssm and CVNmDB that possess only one sugar-binding site, either in domain B ([CVNΔA]ssm) or domain A (CVNmDB) were prepared as described previously.[35–37] The two fluoro-deoxy sugar derivatives, methyl 2-deoxy-2-fluoro-α-D-mannopyranosyl-(1→2)-α-D-mannopyranoside and methyl 2-deoxy-2-fluoro-α-D-mannopyranosyl-(1→2)-α-D-mannopyranosyl-(1→2)-α-D-mannopyranoside were obtained using synthetic procedures outlined in Supplementary Scheme S1.
Cell assays
CV-NP51G was fluorescently labeled by fluorescein isothiocyanate under activity-preserving conditions, separated from reagents by gel filtration and applied in cytofluorometry as described for other lectins previously.[43]
NMR Spectroscopy
19F-NMR spectra were recorded on a Bruker 600 MHz AVANCE spectrometer, equipped with a Bruker CP TXO triple-resonance, X-nuclei observe, z-axis gradient cryoprobe (Bruker Biospin, Billerica, MA). 1H-15N HSQC experiments were recorded on Bruker 600 MHz AVANCE spectrometer equipped with CP TCI triple-resonance, z-axis gradient 1H detect cryoprobe (Bruker Biospin, Billerica, MA). Spectra were processed with NMRPipe[44] and analyzed with NMRview.[45]
NMR Titrations –monitoring resonances of the fluorinated ligand
Ligand-observed 19F-NMR experiments were performed using fluorinated deoxy sugars, 19F-Man2 (200 μM) and 19F-Man3 (50 μM), and single binding site variants, [CVNΔA]ssm, CVNmDB as well as the stabilized CV-NP51G wild-type protein at 1:1 protein:ligand molar ratios, in 20 mM sodium phosphate buffer, pH 6.0, 0.01% NaN3, 90% H2O/10% D2O.
For titration experiments with 19F-Man3 sugars (50μM), 19F-spectra were monitored upon addition of increasing amounts of protein, [CVNΔA]ssm and CVNmDB, (5 mM stock solution) in 20 mM sodium phosphate buffer, pH 6.0, 0.01% NaN3, 90% H2O/10% D2O. A series of 1D-19F NMR spectra were recorded at 280 K at protein:sugar molar ratios of 0:1, 0.5:1, 1:1, and 1.5:1 for [CVNΔA]ssm and 0:1, 0.5:1, 1:1, 2:1 and 3:1 for CVNmDB. Free and bound 19F ligand resonances throughout the titration are in slow exchange on the chemical shift scale. The observed signal intensity change during the titrations is directly proportional to the fraction bound (fb), given by: fb=1-ff=1-Ifree/I0= [PL]/[L], where [L] is the total concentration of ligand and [PL] the concentration of the resulting ligand-protein complex. Ifree is NMR peak intensity of the free ligand signal at each point in the titration, and I0 is the peak intensity of the free ligand signal at the beginning of titration. Binding curves were derived from the relative ratios of ligand resonance intensities (1-Ifree/I0) versus molar ratio M of protein:sugar, and apparent KDF values were obtained by non-linear best fitting of the titration curves using KaleidaGraph (Synergy Software, Reading, PA), using the following equation:
NMR Titrations – monitoring the protein resonances
Protein-observed NMR titration experiments were performed on Bruker 600 MHz AVANCE spectrometers, equipped with 5-mm, triple resonance, three-axis gradient probes or z-axis gradient cryoprobes, using uniformly 15N-labeled [CVNΔA]ssm(75 μM) or CVNmDB(400 μM) with increasing amounts of 19F-Man2, in 20 mM sodium phosphate buffer, pH 6.0, 0.01% NaN3, 90% H2O/10% D2O. A series of 1H-15N HSQC spectra at 298 K were recorded after the addition of sugar aliquots from a stock solution (10 mM) at sugar:protein molar ratios of: 1:0, 2:1, 4:1, 6:1, 8:1, 10:1, 14:1, 18:1, 22:1, and 25:1 for [CVNΔA]ssm and 0:1, 0.5:1, 1:1, 1.5:1, 2:1, 2.5:1, 3:1, 3.5:1, 5:1, 6:1, 7:1, 8:1, 9:1, 11:1, 12:1, 15:1, 16:1, 21:1, and 25:1 for CVNmDB.
Free and sugar-bound protein resonances throughout the titration are in fast exchange on the chemical shift scale. The observed chemical shift changes during the titration is given by: Δδ= [PL]/[P] (δb − δf), with [P] and [PL] the concentrations of protein and ligand-protein complex and δb and δf the chemical shifts of protein resonances in the free and fully bound state. The chemical shift difference was calculated as: Δδ= [(ΔδH)2 + (ΔδN × 0.17)2]1/2, with ΔδH and ΔδN the observed chemical shift changes for 1H and 15N, respectively. The dissociation constant KDF, was obtained by best-fitting the titration curve (chemical shift change Δδ vs. molar ratio M of sugar:protein) using KaleidaGraph software and the following equation:
Analogous NMR titration experiments were performed using uniformly 15N-labeled [CVNΔA]ssm(50 μM) and CVNmDB(50 μM) with 19F-Man3, in 20 mM sodium phosphate buffer, pH 6.0, 0.01% NaN3, 90% H2O/10% D2O. A series of 1H-15N HSQC spectra were recorded at 280 K after addition of sugar aliquots from a stock solution of 10 mM at sugar:protein molar ratios of: 0:1, 0.5:1, 1:1, 1.5:1, 2:1, and 3:1 for [CVNΔA]ssm and 0:1, 0.5:1, 1:1, 1.5:1, 2:1, 2.5:1, 3.5:1, and 4:1 for CVNmDB. In this case, free and sugar-bound protein resonances throughout the titration were in slow exchange on the chemical shift scale. The observed signal intensity change during the titration is direct proportional to the fraction bound (fb), given by: fb=1-ff=1-Ifree/I0= [PL]/[P], with [P] the total concentration of protein and [PL] the concentrations of the protein-ligand complex. Ifree is the resonance intensity of the ligand-free protein signal at each point in the titration, and I0 is the resonance intensity of the ligand-free protein signal at the beginning of titration. Binding curves were derived from the relative ratios of ligand resonance intensities (1-Ifree/I0) versus molar ratio M of protein:sugar, and apparent KDF values were obtained by nonlinear best fitting of the titration curves using KaleidaGraph (Synergy Software, Reading, PA), averaging over the eight titration curves using the following equation:
2D 19F-19F NOESY exchange experiment
Two-dimensional exchange spectroscopy (NOESY) represents a powerful tool for probing ligand-receptor exchange.[46] NOESY experiments are particularly suitable to evaluate chemical exchange processes that occur on a slow time scale, where the exchange rate has little effect on the lineshape. A series of 2D homonuclear exchange 19F-19F NOESY experiments were recorded at 280 K on a sample containing 200 μM 19F-Man2, complexed with [CVNΔA]ssm and CVNmDB at 1:1 molar ratios and mixing times (tmix) of 0.05, 0.1, 0.3, 0.5 s and 0.025 0.05, 0.1, 0.2 s, respectively. An equivalent series was recorded for 50 μM 19F-Man3, complexed with [CVNΔA]ssm and CVNmDB at mixing times (tmix) of 0.1, 0.3, 0.5, 0.7, 0.9 s and 0.05, 0.1, 0.15, 0.3 s, respectively. The exchange rate constant was extracted by fitting the experimental intensity ratios (diagonal peak (IBB) of bound conformation, to the bound to free exchange cross-peak (IBF)) versus mixing time (tmix), using the following equation:
where keq is the equilibrium constant, tmix the mixing time, and kex the free-bound exchange rate.[47]
Isothermal Titration Calorimetry
Calorimetric titrations were performed using a VP-ITC isothermal titration calorimeter (MicroCal, LLC; Northampton, MA). Titrations were carried out at 280 K and all solutions contained 50 mM sodium phosphate buffer, pH 7.4, 0.2 M NaCl, 0.02 % NaN3. ITC measurements were carried out with [CVNΔA]ssm and CVNmDB solution in the cell (~1.44 ml active volume) at 30 μM and 50 μM concentrations, respectively, stirred at 310 rpm. 28 injections of 10 μl aliquots of 2.5 mM 19F-Man3 in the case of [CVNΔA]ssm, and 35 injections of 8 μl aliquots of 4 mM 19F-Man3 in the case of CVNmDB, were performed at 2 min intervals from a 285 μl stirring syringe. Reverse ITC measurements were performed as follows: 50 μM 19F-Man3 solution was placed in the calorimeter cell (~1.44 ml active volume), stirred at 310 rpm, while [CVNΔA]ssm or CVNmDB solution were placed in the injector. 10 μl aliquots of 0.6 mM [CVNΔA]ssm and 11 μl aliquots of 1 mM CVNmDB were added at 2 min intervals from the 285 μl stirring syringe. A total of 28 injections were performed for [CVNΔA]ssm and 25 injections for CVNmDB. Binding isotherms were fit using the Origin 7.0 software using a standard one-site model, to extract the apparent number of binding sites and affinity parameters. Values for the binding enthalpy, the apparent number of binding sites and affinities were obtained from the fit to the experimental data. Other thermodynamic parameters were calculated using the standard expressions: ΔG = −RT ln Ka; ΔG = ΔH −TΔS.
19F-NMR competition binding assay
The binding affinities of Man3 for the CVNmDB variant were quantified using a 19F-NMR-based competition assay. In this experiment, the non-labeled competitor ligand Man3 was titrated into a solution of 250 μM 19F-Man3 and 25 μM CVNmDB in 20 mM sodium phosphate buffer, pH 6.0, 0.01% NaN3, 90% H2O/10% D2O. A series of 1D-19F NMR spectra were recorded at 280 K with 19F-Man3:CVNmDB:Man3 molar ratios of: 10:1:0; 10:1:0.25; 10:1:0.5; 10:1:0.75; and 10:1:1. The intensity increase of the free 1D NMR signal of 19F-Man3 complexed with CVNmDB, upon addition of the inhibitor (Man3), is proportional with the amount of non-labeled ligand (Man3) bound to CVNmDB. The apparent dissociation constants kd for Man3 binding to the Domain A of CVNmDB, was obtained from best fitting the response curves with KaleidaGraph (Synergy Software, Reading, PA), using a corresponding competitive binding Cheng–Prusoff equation,[48] where KDF is the dissociation constant of 19F-Man3 binding to the Domain A of CVNmDB (see above).
Supplementary Material
Acknowledgments
We thank Mike Delk for NMR technical support. This work was supported by a Science Foundation of Ireland Grant 08/IN.1/B2067 (to S. O.), funding from the EC (contract no. 26060, GlycoHIT; to H.J.G) and a National Institutes of Health Grant RO1GM080642 (to A.M.G.).
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/chem.201204070
References
- 1.Gabius H-J, editor. The Sugar Code. Fundamentals of glycosciences. Wiley-VCH; Weinheim, Gemany: 2009. [Google Scholar]
- 2.Oscarson S. In: The Sugar Code. Fundamentals of glycosciences. Gabius H-J, editor. Wiley-VCH; Weinheim, Gemany: 2009. pp. 31–51. [Google Scholar]
- 3.Gabius HJ, André S, Jiménez-Barbero J, Romero A, Solís D. Trends Biochem Sci. 2011;36:298–313. doi: 10.1016/j.tibs.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 4.Imberty A, Chabre YM, Roy R. Chemistry. 2008;14:7490–7499. doi: 10.1002/chem.200800700. [DOI] [PubMed] [Google Scholar]
- 5.Holgersson J, Gustafsson A, Gaunitz S. In: The Sugar Code. Fundamentals of glycosciences. Gabius H-J, editor. Wiley-VCH; Weinheim, Gemany: 2009. pp. 31–51. [Google Scholar]
- 6.Sperandio M, Gleissner CA, Ley K. Immunol Rev. 2009;230:97–113. doi: 10.1111/j.1600-065X.2009.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chabre YM, Giguere D, Blanchard B, Rodrigue J, Rocheleau S, Neault M, Rauthu S, Papadopoulos A, Arnold AA, Imberty A, Roy R. Chemistry. 2011;17:6545–6562. doi: 10.1002/chem.201003402. [DOI] [PubMed] [Google Scholar]
- 8.Kaltner H, Gabius HJ. Histol Histopathol. 2012;27:397–416. doi: 10.14670/HH-27.397. [DOI] [PubMed] [Google Scholar]
- 9.Clore GM, Gronenborn AM. J Magn Reson. 1982;48:402–417. [Google Scholar]
- 10.Clore GM, Gronenborn AM. Protein Sci. 1994;3:372–390. doi: 10.1002/pro.5560030302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ni F, Zhu Y. J Magn Reson B. 1994;103:180–184. doi: 10.1006/jmrb.1994.1027. [DOI] [PubMed] [Google Scholar]
- 12.Roldos V, Canada FJ, Jiménez-Barbero J. Chembiochem. 2011;12:990–1005. doi: 10.1002/cbic.201000705. [DOI] [PubMed] [Google Scholar]
- 13.Dalvit C. Prog NMR Spectr. 2007;51:243–271. [Google Scholar]
- 14.Vulpetti A, Hommel U, Landrum G, Lewis R, Dalvit C. J Am Chem Soc. 2009;131:12949–12959. doi: 10.1021/ja905207t. [DOI] [PubMed] [Google Scholar]
- 15.Millett F, Raftery MA. Biochem Biophys Res Commun. 1972;47:625–632. doi: 10.1016/0006-291x(72)90924-2. [DOI] [PubMed] [Google Scholar]
- 16.Alter GM, Magnuson JA. Biochemistry. 1974;13:4038–4045. doi: 10.1021/bi00716a035. [DOI] [PubMed] [Google Scholar]
- 17.Midoux P, Grivet JP, Monsigny M. FEBS Lett. 1980;120:29–32. doi: 10.1016/0014-5793(80)81038-6. [DOI] [PubMed] [Google Scholar]
- 18.Jordan F, Bahr H, Patrick J, Woo PW. Arch Biochem Biophys. 1981;207:81–86. doi: 10.1016/0003-9861(81)90011-4. [DOI] [PubMed] [Google Scholar]
- 19.Lemieux RU. Chem Soc Rev. 1989;18:347–374. [Google Scholar]
- 20.Solis D, Romero A, Kaltner H, Gabius HJ, Diaz-Maurino T. J Biol Chem. 1996;271:12744–12748. doi: 10.1074/jbc.271.22.12744. [DOI] [PubMed] [Google Scholar]
- 21.Solís D, Romero A, Menédez M, Jiménez-Barbero J. In: The Sugar Code. Fundamentals of glycosciences. Gabius H-J, editor. Wiley-VCH; Weinheim, Gemany: 2009. pp. 233–245. [Google Scholar]
- 22.Diercks T, Ribeiro JP, Canada FJ, André S, Jiménez-Barbero J, Gabius HJ. Chem Eur J. 2009;15:5666–5668. doi: 10.1002/chem.200900168. [DOI] [PubMed] [Google Scholar]
- 23.André S, Cañada FJ, Shiao TC, Largartera L, et al. Eur J Org Chem. 2012;23:4354–4364. [Google Scholar]
- 24.O’Brien R, Ladbury JE, Chowdry BZ. In: Protein-Ligand Interactions: hydrodynamics and calorimetry. Harding SE, Chowdry BZ, editors. Oxford University Press; Oxford: 2001. pp. 263–86. [Google Scholar]; b) Brown A. Int J Mol Sci. 2009;10:3457–3477. doi: 10.3390/ijms10083457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bewley CA, Gustafson KR, Boyd MR, Covell DG, Bax A, Clore GM, Gronenborn AM. Nat Struct Biol. 1998;5:571–578. doi: 10.1038/828. [DOI] [PubMed] [Google Scholar]
- 26.Yang F, Bewley CA, Louis JM, Gustafson KR, Boyd MR, Gronenborn AM, Clore GM, Wlodawer A. J Mol Biol. 1999;288:403–412. doi: 10.1006/jmbi.1999.2693. [DOI] [PubMed] [Google Scholar]
- 27.Shenoy SR, Barrientos LG, Ratner DM, O’Keefe BR, Seeberger PH, Gronenborn AM, Boyd MR. Chem Biol. 2002;9:1109–1118. doi: 10.1016/s1074-5521(02)00237-5. [DOI] [PubMed] [Google Scholar]
- 28.Barrientos LG, Louis JM, Botos I, Mori T, Han Z, O’Keefe BR, Boyd MR, Wlodawer A, Gronenborn AM. Structure. 2002;10:673–686. doi: 10.1016/s0969-2126(02)00758-x. [DOI] [PubMed] [Google Scholar]
- 29.Bolmstedt AJ, O’Keefe BR, Shenoy SR, McMahon JB, Boyd MR. Mol Pharmacol. 2001;59:949–954. doi: 10.1124/mol.59.5.949. [DOI] [PubMed] [Google Scholar]
- 30.Barrientos LG, Gronenborn AM. Mini Rev Med Chem. 2005;5:21–31. doi: 10.2174/1389557053402783. [DOI] [PubMed] [Google Scholar]
- 31.Bewley CA, Kiyonaka S, Hamachi I. J Mol Biol. 2002;322:881–889. doi: 10.1016/s0022-2836(02)00842-2. [DOI] [PubMed] [Google Scholar]
- 32.Botos I, O’Keefe BR, Shenoy SR, Cartner LK, Ratner DM, Seeberger PH, Boyd MR, Wlodawer A. J Biol Chem. 2002;277:34336–34342. doi: 10.1074/jbc.M205909200. [DOI] [PubMed] [Google Scholar]
- 33.Sandström C, Berteau O, Gemma E, Oscarson S, Kenne L, Gronenborn AM. Biochemistry. 2004;43:13926–13931. doi: 10.1021/bi048676k. [DOI] [PubMed] [Google Scholar]
- 34.Sandström C, Hakkarainen B, Matei E, Glinchert A, Lahmann M, Oscarson S, Kenne L, Gronenborn AM. Biochemistry. 2008;47:3625–3635. doi: 10.1021/bi702200m. [DOI] [PubMed] [Google Scholar]
- 35.Barrientos LG, Matei E, Lasala F, Delgado R, Gronenborn AM. Protein Eng Des Sel. 2006;19:525–535. doi: 10.1093/protein/gzl040. [DOI] [PubMed] [Google Scholar]
- 36.Matei E, Furey W, Gronenborn AM. Structure. 2008;16:1183–1194. doi: 10.1016/j.str.2008.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matei E, Zheng A, Furey W, Rose J, Aiken C, Gronenborn AM. J Biol Chem. 2010;285:13057–13065. doi: 10.1074/jbc.M109.094938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dalvit C, Vulpetti A. ChemMedChem. 2011;6:104–114. doi: 10.1002/cmdc.201000412. [DOI] [PubMed] [Google Scholar]
- 39.Dalvit C, Vulpetti A. ChemMedChem. 2012;7:262–272. doi: 10.1002/cmdc.201100483. [DOI] [PubMed] [Google Scholar]
- 40.Vulpetti A, Dalvit C. Drug Discov Today. 2012;17:890–897. doi: 10.1016/j.drudis.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 41.Fromme R, Katiliene Z, Giomarelli B, Bogani F, Mc Mahon J, Mori T, Fromme P, Ghirlanda G. Biochemistry. 2007;46:9199–9207. doi: 10.1021/bi700666m. [DOI] [PubMed] [Google Scholar]
- 42.Perozzo R, Folkers G, Scapozza L. J Recept Sig Transd. 2004;24:1–52. doi: 10.1081/rrs-120037896. [DOI] [PubMed] [Google Scholar]
- 43.André S, Lahmann M, Gabius HJ, Oscarson S. Mol Pharm. 2010;7:2270–2279. doi: 10.1021/mp1002416. [DOI] [PubMed] [Google Scholar]
- 44.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 45.Johnson BA. Methods Mol Biol. 2004;278:313–352. doi: 10.1385/1-59259-809-9:313. [DOI] [PubMed] [Google Scholar]
- 46.Bodenhausen G, Wagner G, Rance M, Soerensen OW, Wuethrich K, Ernst RR. J Magn Reson. 1994;59:542–50. [Google Scholar]
- 47.Ernst RR, Bodenhausen G, Wokaun A. Principles of nuclear magnetic resonance in one and two dimensions. Claredon Press; Oxford: 1987. [Google Scholar]
- 48.Cheng Y, Prusoff WH. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.