

Abstract
The guanine nucleotide exchange factor C3G, in complex with the adaptor protein CrkII, mediates GTP loading of small GTPases Rap1 and R-Ras. Hence, C3G facilitates the activation of downstream signaling pathways, shown to be important in glomerulonephritis (GN). We evaluated glomerular expression of C3G in an experimental model of accelerated anti-GBM antibody induced GN. C3G expression (assessed by western blotting) was upregulated in glomeruli after induction of GN and was prominent in glomerular epithelial cells (assessed by immunostaining). In order to examine the consequences of upregulation of C3G expression in glomerular epithelial cells (GEC), we used adenovirus mediated gene transfer of C3G into cultured GEC and analyzed GTP-loading (activation) of Rap1 and R-Ras. Whereas activation of Rap1 was not affected by C3G, overexpression of C3G in GEC decreased the basal level of GTP-bound R-Ras and enhanced activation of R-Ras in response to endothelin. Furthermore C3G overexpression led to significant reduction in cultured GEC spreading and augmented cell migration accompanied by decreased E-cadherin and podocine expression. Taken together, these data represent the first report of pathologic renal C3G overexpression and suggest that it is involved in modulation of GEC morphology and behavior.
Keywords: C3G, glomerular epithelial cells, endothelin-1, small GTPases, cell spreading, migration, E-cadherin, glomerulonephritis
INTRODUCTION
In experimental models of proliferative (crescentic) glomerulonephritis (GN), induced by antibody against the glomerular membrane (GBM), and in human crescentic GN, injury to the glomerular visceral epithelial cells (GEC), also known as podocytes, appears to be of pivotal importance [1–3]. In order to support the glomerular filtration rate, the renal GEC develop microtubule-based thick processes with sophisticated branching morphology and have thin actin-based projections (podocyte foot processes), which spread and form cell-cell contacts in mature cells. Actin filaments are thought to play a central role in orchestrating the function of various molecules during the formation of podocyte foot processes. The molecular mechanism of foot process formation seems to include small GTP-binding proteins. One common mechanism utilized by small GTPases to regulate cellular function is to cycle between the inactive GDP-bound state and the active GTP-bound state. Guanine nucleotide exchange factors (GEFs) facilitate GDP dissociation and allow the more abundant GTP to rebind, while GTPase Activating Proteins (GAPs) accelerate GTP hydrolysis to complete the cycle. In a number of pathological processes, small GTPases are up-regulated due to overexpression, mutation, intensification of particular signaling pathways, or abnormal GEFs activity that are critical for the onset or progression of the disease [4]. This suggests that GEFs can be promising targets for the inhibition of small GTPase-dependent pathways that are amplified or diminished in diseases.
The GEF, C3G, predominantly catalyzes the guanine nucleotide exchange reaction for the Ras family of GTPases. Absence of C3G causes embryonic lethality in mice via the primary defect in vascular supporting cells of mesenchymal origin, and cells derived from such embryos demonstrate impaired cell adhesion, delayed spreading and enhanced cell migration [5]. However, the mechanisms by which C3G regulates these actin-dependent cellular functions are poorly understood. C3G-mediated Rap1 activation is required for cell adhesion and cell spreading, but suppresses cell migration [6]. Recently, a functional link was uncovered between C3G/Rap1 and the FGF2/Akt/Gsk3β/β-catenin pathway, where C3G plays a role as a self-limiting mechanism for proliferation signals [7]. Furthermore, York et al. proposed that the activation of Rap1 by C3G represents a common mechanism to induce sustained activation of the MAP kinase cascade in cells that express B-Raf [8]. Our previous studies in rat models of proliferative GN have demonstrated that both ERK and SAPK exhibit disease-associated activation, albeit with differing kinetics, and that ERK activation in glomeruli is accompanied by increased expression of its specific upstream activator, MEK1 [9]. Significant evidence has emerged that members of Ras family of small GTPases modulate ERK activation in a cell-context dependent manner [8;10–12]. Signaling through the small GTPases Rap1 and R-Ras contributes to the regulation of a wide variety of integrin-mediated processes including cell-cell and cell-matrix adhesion, actin polymerization, cell migration, and spreading [13;14]. Interestingly, enhanced Rap1 activity was found to be involved in p38 MAP kinase regulation, ERK suppression, increased cell adhesion, and was observed in samples from heart with right ventricular outflow tract obstruction and renal tumors [15;16]. By contrast, decreased R-Ras activity was linked to such biological effects as augmented cell motility and migration [14]. Taken together these data allow us to hypothesize that the Ras family of GTPases GEF, C3G, can be important for controlling the signaling cascades in proliferative glomerulonephritis. We have carried out a study of C3G expression in inflammatory kidney disease followed by analysis of C3G biological significance in cultured podocytes.
RESULTS
C3G is upregulated in glomeruli after induction of experimental GN
Western blot analysis of protein lysates obtained from isolated glomeruli revealed a significant increase in levels of GEF C3G 1, 3, and 7 days after induction the accelerated (crescentic) form of anti-GBM GN (Figure 1). Levels of the adaptor protein Grb2 were not different between glomeruli of control and experimental animals (Figure 1, lower panel).
Figure 1. Expression of C3G protein in anti-GBM antibody induced glomerulonephritis.

One control (−) and two experimental (+) animals were studied at 1, 3 and 7 days following injection of anti-GBM serum. Enhanced expression of C3G in experimental animals is identifiable by Western blotting as early as 1 day after induction of GN (upper panel). The nitrocellulose membrane was also probed with anti-Grb2 antibodies to confirm equal of loading (lower panel). Shown is a representative result. Experiment was repeated twice.
On day 14 following administration of anti-GBM antibody we observed significantly increased proteinuria (Figure 2), crescent formation (Figure 3A and 3B), glomerular sclerosis (Figure 3C), and periglomerular inflammatory cell infiltrates (Figure 3, compare A to D). C3G expression (assessed by immunostaining) was prominent in GEC (black arrows in Figure 3C), within cellular crescents (Figure 3B) and, occasionally, in tubular epithelium. Very little or no C3G staining was observed in glomerular cells of control animals. To confirm the localization of C3G staining we carried out double immunostaining for C3G and nestin as a mature podocytes marker, which is upregulated in response to injury [17] (Figure 4). Glomerulus from control animal was virtually free of brown C3G staining and was characterized by prominent blue signal from anti-nestin antibody (Figure 4A). After GN induction C3G staining was enhanced and co-localized with nestin forming grey to black color (Figure 4B).
Figure 2. Proteinuria 14 days after induction of glomerulonephritis.
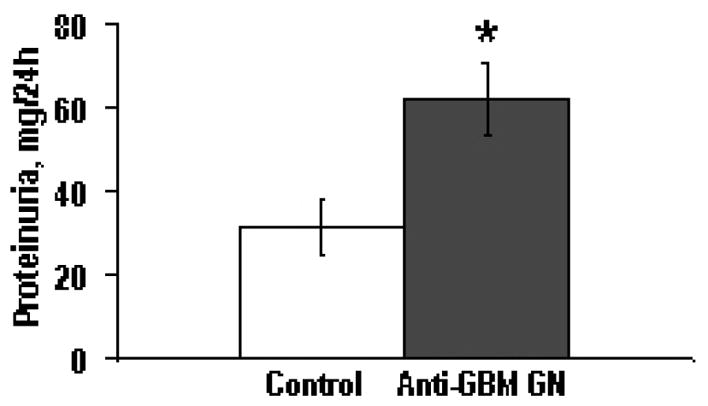
Urinary protein excretion was significantly (marked with star, p<0.05) increased as measured on day 14 following onset of anti-GBM glomerulonephritis (Anti-GBM GN) compared to control animals (Control).
Figure 3. Kidney morphology and C3G immunohistochemical detection in glomerulus after induction of glomerulonephritis.

Representative morphological (Masson’s Trichrom, A,D) and immunohistochemical staining for C3G (B,C,E) in kidney cortical sections obtained on day 14 following onset of anti-GBM glomerulonephritis (A,B,C,F) and in control animals (D,E). B and C represent two different animals with GN. In A, a crescentic glomerulus with periglomerular infiltrate is shown. In C, arrows indicate immunolocalization of C3G in glomerular epithelial cells. Also shown is a negative control (stained with an isotype-matched IgGs) (F), corresponding to the glomerulus shown in C. Cell nuclei in B, C, E and F are conterstained with hematoxylin, original magnifications 200×.
Figure 4. C3G and nestin immunohistochemical detection in glomerulus after induction of glomerulonephritis.
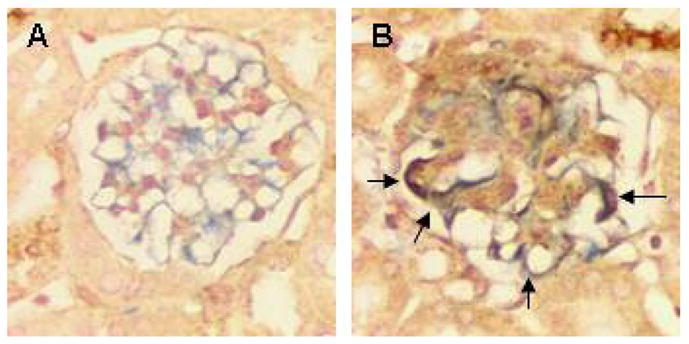
Double immunostainig for C3G (brown cyptoplasm) and for nestin (blue cytoplasm) showing changes after glomerulonephritis induction. (A) Normal glomerulus from control animal demonstrates very little if any C3G signal and many cells are nestin-positive. (B) Crescent glomerulus characterized by increased C3G staining. Majority of nestin-positive cells express C3G (gray to black color, marked with arrows). Cell nuclei are conterstained with Nuclear Fast Red, original magnifications 400×.
In order to investigate the possibility that C3G protein in glomeruli originates from infiltrating macrophages, levels of the macrophage marker ED-1 [18] were assessed in glomeruli isolated from control animals and animals with anti-GBM antibody-induced GN. Twelve hours following injection of anti-GBM antibody, C3G levels in glomeruli isolated from two experimental animals were indistinguishable from levels in glomeruli from a control animal (Figure 5A, upper panel). In contrast, ED-1 levels were greatly increased only in glomerular lysates of the two experimental animals (Figure 5A, lower panel). At later time points (24 and 48 hours and on days 3 and 7 following induction of GN) the differences in glomerular C3G expression between control and experimental animals were clearly manifested (Figures 1 and 5B). In contrast to the striking upregulation of C3G expression, Grb2 did not exhibit a disease-associated increase in expression levels (Figures 1 and 5B). Similarly, the level of expression of glomerular CrkII did not significantly change in animals with GN (Figure 5B). Immunohistochemistry data also did not reveal significant C3G staining in inflammatory infiltrates.
Figure 5. Glomerular levels of C3G protein and the macrophage marker ED-1 in anti-GBM glomerulonephritis.

(A) Twelve hours following administration of anti-GBM serum, C3G levels in glomeruli from two experimental rats (Figure 3A, lanes 2 and 3) were indistinguishable from the level in glomeruli of a control animal (lane 1) despite the prominent macrophage migration into glomeruli (Figure 3A, lower panel, lanes 2 and 3) as assessed by Western blot analysis of levels of the macrophage marker ED-1. One control (−) and two experimental animals (+) were studied. (B) Western blot analysis of C3G (upper panel) and Crk II (middle panel) expression was performed in glomeruli isolated 24 and 48 hours following administration of anti-GBM serum. One control (−) and two experimental animals (+) were studied for each time point. The nitrocellulose membrane was also probed with anti-Grb2 antibodies to confirm equal loading (lower panel). Shown is a representative result; the experiment was repeated two times.
Overexpression of C3G modulates activation of R-Ras in GEC
As detected by Rap1 affinity binding assay, Rap1 activation in response to ET-1 was not significantly affected in GEC infected with adenovirus encoding C3G (Figure 6A, upper panel). The amount of GTP-bound Rap1 from three independent experiments was further quantified using densitometry analysis (Figure 6B).
Figure 6. Adenoviral-mediated transfer of C3G into glomerular epithelial cells does not affect Rap1 activation in response to endothelin-1.

(A) Lysates from AdGFP- and AdC3G-infected rat glomerular epithelial cells, quiescent or stimulated with endothelin-1 (100 nM) for indicated periods of time (min), were subjected to a Rap1 affinity binding assay. GTP-bound active Rap1 was isolated by affinity precipitation with a GST-RBD RalGDS fusion protein followed by immunoblot analysis with an anti-Rap1 antibody (upper panels). Lysates from AdGFP- and AdC3G-infected rat glomerular cells were resolved by SDS-PAGE and immunoblotted with either anti-Actin antibodies for equal loading (lower panel) or anti-C3G antibodies (middle panel). AP: affinity precipitation. TCL: total cell lysates. (B) The amount of GTP-bound Rap1 was quantified using densitometry. Densitometry data were normalized to the AdGFP “0 min” point and presented as mean±S.E.M. In four separate experiments there was no statistically significant difference between AdGFP and AdC3G infected cells.
We observed a marked effect of C3G overexpression on ET-1 induced changes in R-Ras GTP loading (Figures 7A and 7B). Firstly, the basal level of R-Ras GTP-loading was decreased in cells overexpressing C3G (Figure 7A, upper panel, lane 6 versus lane 1 or 3). These differences were quantified by densitometry (Figure 7B). Statistically significant alterations between the two groups (AdGFP and AdC3G) were found at basal level (time 0 min, p<0.01) and following a 20 min exposure to ET-1 (p<0.05). Within the AdGFP group, R-Ras GTP loading following 5 min of ET-1 stimulation was significantly lower than that at 0 min (p<0.01). The same effect was observed in uninfected GEC control (Figure 7A, U). Within the AdC3G group, R-Ras GTP loading at 20 min was significantly higher than that at 0 min (p<0.001), or that at 5 min (p<0.02). In summary, GEC overexpressing C3G demonstrated a dramatic decrease in the basal level of GTP-bound R-Ras and a significant increase in the level of GTP-bound R-Ras 20 min following ET-1 stimulation.
Figure 7. Adenoviral-mediated transfer of C3G into glomerular epithelial cells modulates R-Ras GTP level.

(A) Lysates from uninfected (U), and from AdGFP- or AdC3G-infected epithelial cells, quiescent or stimulated with endothelin-1 (100 nM) for indicated periods of time (min), were subjected to R-Ras affinity binding assay. GTP-bound active R-Ras was isolated by affinity precipitation with a GST-RBD Raf fusion protein followed by immunoblot analysis with an anti-R-Ras antibody (upper panel). Lysates from AdGFP- or AdC3G-infected rat glomerular cells were resolved by SDS-PAGE and immunoblotted with either anti-R-Ras, anti-C3G or anti-Actin antibodies (for verification of equal loading). A representative analysis of five separate experiments is shown. AP: affinity precipitation. TCL: total cell lysates. (B) The amount of GTP-bound R-Ras was quantified using densitometry analysis. Densitometry data are normalized to AdGFP “0 min” point and presented as mean±S.E.M. Statistically significant differences between the two groups (AdGFP and AdC3G) were found at time 0 (marked with asterisk, *, p<0.01) and following a 20 min exposure to ET-1 (*, p<0.05). Within the AdGFP group, R-Ras GTP loading following 5 min of ET-1 stimulation was significantly lower than that at 0 min (#, p<0.01). R-Ras GTP-loading at 20 min was not different from that at 0 min or 5 min. Within the AdC3G group, R-Ras GTP loading at 20 min was significantly higher than that at 0 min (#, p<0.001) or that at 5 min (#, p<0.02). There was no significant difference in R-Ras GTP loading at 5 min compared to 0 min.
Overexpression of C3G affects spreading and migration in GEC
In our experiments C3G overexpression had no effect on cultured GEC attachment to fibronectin with or without ET-1 stimulation (data not shown). In accordance with R-Ras activation data, C3G upregulation significantly reduced cell size during spreading (Figure 8A). Additionally, GEC spreading under ET-1 treatment was significantly diminished in control cells, reflecting normal actin dynamic response to contractile stimulus, but remain unaffected in C3G overexpressing population (Figure 8A). To further explore changes in intracellular actin balance with or without C3G overexpression and ET-1 stimulation we labeled filamentous actin (F-actin) in cultured GEC with phalloidine, conjugated with fluorochrom (Figure 8B). After C3G overexpression and ET-1 stimulation amount of polymerized actin fibers was increased compare to unstimulated GFP-expressing control.
Figure 8. Adenoviral-mediated transfer of C3G into glomerular epithelial cells reduces cell spreading.

AdGFP- and AdC3G-infected rat glomerular epithelial cells were subjected to spreading assay (see Methods for details). (A) Histograms represent distribution of cell number (Y axis) of a given size (X axis) counted in the GFP- or C3G-expressing cell populations after spreading (1.5 h). Numbers near the histogram top represent the median values for each particular cell distribution. Arrows with stars indicate significant difference (p<0.05 based on the one way ANNOVA analysis) between marked cell populations. A representative example of three separate experiments is shown. (B) In AdGFP- and AdC3G-infected rat glomerular epithelial cells with or without 100 nM ET-1 stimulation F-actin was visualized with AlexaFluor568-phalloidine. Enhanced F-actin staining visible as separated cell-long lines was observed in C3G overexpressing cells and in cells after 24 h ET-1 stimulation.
After C3G upregulation cell migration was agumented compared to GFP control as measured by wound area closing (Figure 9A, B) and by number of migrating cells into the opened area (Figure 9C) in scratch assay. Next we analyzed proteins, involved into adherens and tight-junction formation and/or into migration signaling. In line with observed changes in cell morphology and behavior we detected significant decrease in E-cadherin and podocin expression after C3G upregulation (Figure 9D). ET-1 treatment for 24 h led to similar reduction of E-cadherin protein level in GFP expressing group compared to C3G counterpart (Figure 9D), correlating with spreading data (Figure 8A). We observed no change in vinculin and p130Cas expression. However, substrate binding domain activation of p130Cas (measured as Tyrosin 165 phosphorylation) was enhanced after C3G overexpression without any significant changes in response to 24 h ET-1 stimulation.
Figure 9. Adenoviral-mediated transfer of C3G into glomerular epithelial cells promotes cell migration and reduces E-cadherin expression.

AdGFP- and AdC3G-infected rat glomerular epithelial cells were subjected to scratch assays (see Methods for details). (A) Representative pictures of wound area in different cell populations at 0 h and at 24 h time points after scratch performed. (B) Quantification of per cent of unrecovered wound area 24 h after scratching the cell monolayer. (C) Number of migrated into the wound area cells presented. Star indicates significant difference (p<0.05) between experimental cell populations. Representative examples of four separate experiments are shown in A–C. (D) AdGFP- and AdC3G-infected rat glomerular epithelial cells were subjected to 24 h either starvation or 100 nM ET-1 stimulation. Lysates from AdGFP- or AdC3G-infected rat glomerular cells were resolved by SDS-PAGE and immunoblotted with either anti-E-cadherin, or anti-podocin, or anti-vinculin, or anti-p130CasY165, or anti-p130Cas, or anti-C3G, or anti-Actin antibodies (for verification of equal loading). A representative analysis of three separate experiments is shown.
DISCUSSION
We demonstrated for the first time that guanine nucleotide exchange factor C3G is upregulated in the model of anti-GBM glomerulonephritis. Little is known about regulation of C3G expression. There are few studies reporting down-regulation [19] as well as overexpression of C3G isoforms [20;21] during development of human cancers. However, mechanism of differentiated expression of C3G currently remains unclear.
Intense glomerular macrophage infiltration is known to occur during the early stages of the accelerated variant of anti-GBM antibody mediated crescentic GN employed in the present studies. Isolated glomeruli Western blotting results (Figures 1 and 5) indicate that glomerular C3G overexpression accompanies the course of anti-GBM antibody-induced GN and, although it could be triggered by macrophage infiltration in glomeruli, it did not result from infiltrating macrophages overexpressing C3G. Two weeks after disease induction when macrophage number inside the glomerulus mostly reverts to normal, based on immunolocalization of C3G (Figure 3C and 4B), we propose that GEC are the likely site where C3G overexpression takes place in the anti-GBM model of immune glomerular injury.
We demonstrate that C3G overexpression in cultured GEC results in modulation of ET-1 induced signaling via the small GTPase R-Ras. Our choice of ET-1 stimulation was based on knowledge that ET-1 play an important role in both the normal and the diseased kidney [22;23]. ET-1, acting in concert with other constrictor peptides, cytokines, and growth factors, contributes to proliferative renal diseases [22]. In immune complex glomerulonephritis there is increased glomerular ET-1 expression [24] and G-protein-coupled ET-A and ET-B receptor blockade reduces glomerular cell proliferation and proteinuria [25]. We observed diminished GTP-loading of R-Ras in cells overexpressing C3G, but increased level of GTP-bound R-Ras at later time-points after ET-1 stimulation (Figure 7). Recently, in wide range of cell lines, it was demonstrated that R-Ras regulates exocytosis [26]. Several reports describe that podocytes are capable of secretory exocytosis [27;28]. Endosomes, before recycling to membrane, contain R-Ras in GTP bound form. When endosomes are fused with the cell membrane, R-Ras is converted to GDP bound form by membrane-located GAPs. ET-1 could stimulate endosome recycling [29] to plasma membrane compartments containing GAPs and hence decrease R-Ras GTP binding. We speculate, that C3G overexpression in GEC promotes fusion of endosomes with the cell membrane, an event that may not require involvement of GEF activity of C3G, but could be controlled through C3G-mediated protein-protein interactions [30]. This could explain why C3G overexpression reduced the basal R-Ras GTP level in unstimulated GEC (Figure 5, lane 6 compared to lane 1). To the extent that ET-1 stimulates GEF activity of C3G in plasma membrane compartments, C3G would facilitate GTP binding of R-Ras in ET-1 stimulated GEC.
Both increased and decreased GTP-loading can play an important role in GTPase signaling [31]. Reduced R-Ras GTP level usually correlates with reduction in cell-cell and cell-matrix interactions presumably through the inactivation of integrins [32]. H-Ras and its downstream effector kinase Raf-1 suppress integrin activation in a manner that is independent of protein synthesis and mRNA transcription and correlates with activation of the ERK/MAP kinase pathway [33]. GTP-bound R-Ras antagonizes the Ras/Raf-initiated integrin suppression pathway in a manner that does not involve inhibition of the Ras/Raf-induced MAP kinase activation [34]. Although R-Ras signals through specific integrin alpha cytoplasmic domains to promote cell migration [35], the N-terminus of R-Ras plays a critical role for the efficient dissemination of R-Ras signals that govern cell shape and migration capacity. The N-terminus positively regulates Rac activation and cell spreading but negatively regulates R-Ras mediated cell migration [36]. Both reduced R-Ras GTP levels and its tyrosine 66 phosphorylation contribute to the ephrin inhibitory effects on COS cell migration and to ephrin-dependent growth cone collapse in primary neurons [37]. This suggests that the R-Ras inactivation or a non-GTPase function may be linked to the reduced GEC spreading and increased migration capacity. During GN progression podocytes reduce foot processes formation and detach from GBM [38]. Experiments addressing C3G-mediated spreading of cultured GEC are relevant for understanding intracellular actin dynamics which is important for podocyte foot process formation. Detached podocytes can migrate between parietal epithelial cells forming bridges between them and glomerular tuft. Bridge formation with denuded GBM was proposed to be a precursor to crescent development [39]. Together with observation of C3G-positive cells in glomerular crescents (Figures 3B and 4B) formed after induction of anti-GBM GN our findings allow us to speculate that possible C3G-R-Ras function is to enable cells to migrate in response to kidney injury.
In support of this hypothesis we found that C3G overexpression in cultured GEC resulted in the reduced spreading associated with enhanced filamentous actin formation and in the increased migration. Since endogenous C3G directly interacts with E-cadherin at initial cell-cell contacts [10], colocalizes with filametous actin, and is involved into cell adhesions formation and maturation, C3G overexpression indeed can modulate signaling involved in attachment to substrate and to neighboring cells. Adhesion of GEC within crescents is a regulated process as several adhesion molecules are expressed within forming crescents and their expression is modified during evolution of crescents [40]. Of the various adhesion molecules identified, PECAM-1 was shown to recruit Crk-C3G complexes [12] while crosslinking of ICAM-1 induces tyrosine phosphorylation of the cytoskeleton-associated protein p130Cas, which associates with Crk and C3G and is involved into filamentous actin assembling [41]. Formation of cadherin-based cell-cell junctions (adherens junctions) among GEC present within crescents also occurs [42;43]. Rap1 plays a key role in formation of cadherin-based cell-cell junctions [10]. It is, therefore, conceivable that the C3G overexpression in GEC observed in the present studies could modulate crescent formation, the likely mechanisms being: a) stabilization of Crk-C3G complexes by the increased C3G levels and modulation of phosphorylation status of scaffolding protein p130Cas, and b) C3G-mediated modulation of the level of GTP-bound R-Ras. The later effect would reduce cell spreading as we observed in cultured GEC overexpressing C3G (Figure 8A). However, we have not observed enhanced and prolonged Rap1 activation after C3G overexpression in response to ET-1 stimulation in GEC when compared to cells overexpressing reporter protein GFP. This could reflect cell specificity of Rap1 function in epithelial cells, whereby this small G protein and its GEF, C3G, are involved in maturation of cadherin-based cell-cell contacts [44]. Our observation of unchanged Rap1 activation in cultured GEC after C3G overexpression further suggests that Rap1 plays a minor role during formation of glomerular epithelial cell adherens-type junctions in crescentic glomeruli. Rather, the C3G-activated small GTPase R-Ras could be important in this process.
Interestingly, in our study C3G overexpression was associated with decreased E-cadherin and podocin protein levels in cultured GEC. Such reduction can partially explain pro-migratory effect of C3G upregulation in GEC. The mechanism of noticeable decline in adherent protein expression remains obscure. C3G acts as a part of self-limiting mechanism for proliferation signals due to a functional link between C3G/Rap1 and FGF2/Akt/Gsk3β/β-catenin pathways [7]. Thus, it is plausible that C3G can initiate negative feed back signaling aimed to reduce cell-cell contacts formation after overexpression in glomeruli. Weak C3G expression in normal glomerulus and dramatic increase of glomerular C3G protein level after GN induction were observed. Therefore, cadherin and podocin down-regulation may reflect GEC-specific signaling associated with not only pro-migratory effect of C3G, but also with known de-differentiation of GEC during kidney injury. Indeed, in several models of experimental renal disease, as well as in human biopsies, a decrease of manifestation of podocyte-specific markers (such as podocin, synaptopodin, nephrin) was detected [17;45]. The loss of these proteins, involved in slit diaphragm formation and function, results in podocyte de-differentiation contributing to glomerular crescents. Furthermore, observed ET-1-induced decrease in GEC E-cadherin expression finds similarity with known ET-1-induced disruption of intercellular communications and stimulation of cell migration in human cancers [46].
Here we report that, C3G upregulated in nephritic glomerulus and C3G overexpression modulates activity of R-Ras and contributes to migratory phenotype in podocytes. The increase in glomerular C3G expression observed in the present study represents a further example of transcriptional regulation of a molecule which can be involved in MAP kinase signaling via sustained activation of small GTPases. The data presented herein link the enhanced expression of C3G with dramatic change in GEC behavior, suggesting hypothetical mechanism explaining podocyte loss and de-differentiation during renal inflammation.
MATERIALS AND METHODS
Materials
The enhanced chemiluminescense system was obtained from Amersham Corp. (Arlington Heights, IL). Complete Freund’s adjuvant and all other reagents, unless specified differently, were from Sigma Chemical Co. (St. Louis, MO).
Animals
Male Sprague Dawley rats weighing 180 to 200 g (Charles River Laboratories, Wilmington, MA, and Harlan, Indianapolis, IN) were used for induction of accelerated proliferative GN, tissue collection, glomerular isolation, and cell culture. Male BALB/c mice were used for induction of accelerated proliferative GN and immunohistochemistry analysis. Animals were cared for according to NIH guidelines. All protocols were reviewed and approved by the Institutional Committee for care and use of animals at the Medical College of Wisconsin. Animals were maintained at the approved animal care facilities and had free access to food and water throughout the study.
Induction of accelerated proliferative GN
Accelerated anti-GBM proliferative glomerulonephritis was induced exactly as described previously [9]. Briefly, male Sprague-Dawley rats were immunized intraperitoneally with 1 mg rabbit IgG emulsified in complete Freund’s adjuvant. Five days after this immunization, experimental animals were injected in the tail vein with a subnephritogenic dose of rabbit immune serum raised against rat particulate GBM. This injection was repeated 24 h later. Control rats were pre-immunized with rabbit IgG in complete Freund’s adjuvant and subsequently given two intravenous injections of nonimmune rabbit serum. BALB/c mice (20–25 mg body weight) were subjected to the same protocol of immunization except they recieved sheep anti-rat GBM serum for induction of GN. Preimmunization was done with 1 mg of sheep non-immune serum. At the time of immunization, each mouse in the glomerulonephritis group received 1.5 mg of anti-GBM serum. Control mice were injected with the same amount of non-immune serum. Studies were performed at 12, 24 and 48 h and on days 3, 7 and 14 following the injection of anti-GBM serum or of non-immune serum. The day before study animals were placed in metabolic cages for urine collection to access urinary protein excretion. Upon completion of this collection, animals were killed and nephrectomized. Cortical tissue was used to generate a preparation of isolated glomeruli using standard sieving methods [47] before lysis in Triton X-100 containing lysis buffer.
Morphological and immunohistochemical analysis
Paraffin-embedded renal slices were cut at 4 μm on a standard microtome. Cross-sections included the full thickness of the cortex and medulla and were serially used for histopathological (Masson’s Trichrom) and immunohistochemical evaluation. Sections were deparaffinized in xylene and hydrated gradually through graded alcohols. For C3G and nestin detection, sections were incubated in 10 mmol/L citrate buffer pH 6.0 (InnoGenix, San Ramon, CA) at 98°C for 25 min and then cooled at room temperature for 10 min. Sections were then quenched in 3% H2O2 (DakoCytomation, Carpinteria, CA) for 20 min at room temperature, blocked using avidin/biotin blocking kit (Vector Laboratories, Inc., Burlingame, CA) and then blocked with Protein-free blocker (DakoCytomation, Carpinteria, CA) for 15 min at room temperature. Primary antibody for C3G (1:500, Santa Cruz Biotechnology, Santa Cruz, CA) was applied to the sections and incubated at room temperature for 45 min. Immunodetection for C3G antigen was performed by using the streptavidin-biotin peroxidase system (BiocareMedical, Concord, CA), diaminobenzidine as chromogen and counterstained with hematoxylin (both from DakoCytomation, Carpinteria, CA). For double immunostaining of C3G and nestin (1:100 antibody dilutuion, 1h incubation, Santa Cruz Biotechnology, Santa Cruz, CA) second antigen was visualized by using mouse-on-mouse polymer detection system conjugated with alkaline phosphotase (BiocareMedical, Concord, CA) and blue substrate with Nuclear Fast Red (Vector Laboratories, Inc., Burlingame, CA) counterstain. Control reactions were performed in the absence of primary antibody and in the presence of isotype-matched IgGs.
Cell culture
Rat glomerular epithelial cells were isolated and characterized based on formation of monolayers with cobblestone appearance with granular inclusions, presence of cilia on their surfaces, presence of receptors for complement (C3), and presence of angiotensin receptors as described [48–51].
Tissue and cell lysis, immunoprecipitation and Western blot analysis
Cell lysis, immunoprecipitation of samples normalized for protein content, SDS-PAGE analysis and Western blotting were performed as described previously [52]. Polyclonal anti-C3G, anti-Rap1, anti-R-Ras, anti-Grb2, anti-nestin and anti-CrkII antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-podocin antibody was a gift from Dr. C. Antignac (Paris, France). Antibody specific for machrophage ED-1 antigenic marker was from Serotec (Raleigh, NC). Secondary goat anti-rabbit or goat anti-mouse antibodies conjugated with HRP were from BioRad (Hercules, CA).
Expression cloning of mouse C3G and generation of adenovirus encoding C3G
Details of screening of a λEXlox library of 16-day mouse embryo (Novagene) in order to isolate mouse C3G cDNA and generation of adenovirus encoding C3G will be reported elsewhere. Infection of glomerular epithelial cells was done as follows: cells were incubated either with adenovirus encoding Green Fluorescent protein (AdGFP) or C3G at a multiplicity of infection of 100 plaque-forming units/cell for 1h at 37°C with periodic shaking, followed by addition of medium supplemented with 0.5 % serum. Fourty eight hours after infection, cells were stimulated and lysed for further analysis. Efficiency of gene transfer in glomerular epithelial cells was about 70–90% as determined by fluorescence microscopy visualization of AdGFP control.
Activated Rap1 and R-Ras affinity binding assay
Commercial kits (Upstate, Lake Placid, NY) were used to measure Rap1 and Ras GTP-loading. Aliquots (250–500 μg) of the supernatants from lysed cultured glomerular epithelial cells were mixed for 35 min at 4°C with glutathione-agarose containing 30 μg of Ral GDS-Rap binding domain (Ral-GDS-RBD), or the Ras binding domain of Raf (Raf-RBD) and were precipitated by centrifugation. Complexes were boiled in a Laemmli sample buffer and then separated on 4–15% SDS-polyacrylamide gels. The separated proteins were immunoblotted using corresponding antibody against R-Ras or Rap1 small GTPases.
Spreading assay
AdGFP- and AdC3G-infected glomerular epithelial cells were detached by trypsinization, and re-suspended in serum-free RPMI. The cell concentration was adjusted to180,000 cells/ml for re-plating in 35 mm tissue culture dishes. Serum-free RPMI with or without ET-1 in final concentration 100nM was poured into each culture dish prior to addition of the suspended cells. After incubation at 37°C for 1.5 h, cell spreading was stopped by removing the medium and incubating with 3.7% formaldehyde in PBS for 20 minutes. Cells were stained with AlexaFluor568-phalloidin. At least five images from different regions of the dish were captured by video camera interfaced with an inverted microscope. At least 300 individual cells were analyzed.
Scratch assay
AdGFP- and AdC3G-infected glomerular epithelial cells were starved for 24 h in serum-free RPMI to reduce cell proliferation. Scratch was made by yellow automatic pipette tip, and marked on the bottom of 60 mm tissue culture dish (time point 0 h). New serum free RPMI with or without 100 nM of ET-1 was added to each plate for additional incubation at 37°C for 24 h (time point 24 h). At least ten images from different regions of the scratch were captured by video camera interfaced with an inverted microscope at 0 h and at 24 h time points. Migrated into the wound area cells were counted and per cent of unrecovered area was calculated in each picture.
Statistical analysis
Unless otherwise indicated, results represent one of three identically performed experiments. Western blot data were subjected to densitometry evaluation and then expressed as relative units compared to unstimulated controls (mean ± SEM). Significance was tested by a two factor (time and treatment) ANOVA with corrections for within experiment repeated measures. For analysis of Rap1 activation, Tukey’s HSD was used for multiple comparisons adjustment. For analysis of R-Ras activation the Waller-Duncan multiple comparison adjustment was used. P values <0.05 were considered statistically significant. The digitized images from spreading and scratch experiments were analyzed in semi-automatic mode by MetaVue 5.0r1 software (Universal Imaging Corp.). Using SigmaStat 3.5 software (Systat Software, Inc.) one way ANOVA on ranks was used for further statistical analysis of spreading assay and unrecovered wound area changes in scratch assay. Student’s t-test was used for analysis of cell migration into the wound at 24 h.
Acknowledgments
This work was supported by National Institutes of Health Grants HL22563 and DK41684 to AS and American Heart Association 0525801Z Greater Midwest Affiliate Postdoctoral Fellowship to VR. We thank Dr. Raymond J. Hoffman (Division of Biostatistics, Department of Population Health, Medical College of Wisconsin, Milwaukee, WI) for the help with statistical analysis. We are gratefully acknowledging Lynn Gruman (Histology and Imaging Core, Medical College of Wisconsin, Milwaukee, WI) for teaching and consulting of our immunohistochemical part of presented research. We are grateful to Dr. Hasan Mukhtar and Dr. Naghma Khan (Department of Dermatology, University of Wisconsin Medical Science Center, Madison, WI) for generous providing us with positive control samples for our immunohistochemical detection of C3G.
Footnotes
DISCLOSURE
None of listed authors of this paper have any interests to disclose.
Reference List
- 1.Griffin SV, Petermann AT, Durvasula RV, Shankland SJ. Podocyte proliferation and differentiation in glomerular disease: role of cell-cycle regulatory proteins. Nephrol Dial Transplant. 2003;18(Suppl 6):vi, 8–13. doi: 10.1093/ndt/gfg1069. [DOI] [PubMed] [Google Scholar]
- 2.Bariety J, Bruneval P, Meyrier A, Mandet C, Hill G, Jacquot C. Podocyte involvement in human immune crescentic glomerulonephritis. Kidney Int. 2005;68:1109–1119. doi: 10.1111/j.1523-1755.2005.00503.x. [DOI] [PubMed] [Google Scholar]
- 3.Jefferson JA, Johnson RJ. Experimental mesangial proliferative glomerulonephritis (the anti-Thy-1.1 model) J Nephrol. 1999;12:297–307. [PubMed] [Google Scholar]
- 4.Zeghouf M, Guibert B, Zeeh JC, Cherfils J. Arf, Sec7 and Brefeldin A: a model towards the therapeutic inhibition of guanine nucleotide-exchange factors. Biochem Soc Trans. 2005;33:1265–1268. doi: 10.1042/BST0331265. [DOI] [PubMed] [Google Scholar]
- 5.Voss AK, Gruss P, Thomas T. The guanine nucleotide exchange factor C3G is necessary for the formation of focal adhesions and vascular maturation. Development. 2003;130:355–367. doi: 10.1242/dev.00217. [DOI] [PubMed] [Google Scholar]
- 6.Ohba Y, Ikuta K, Ogura A, Matsuda J, Mochizuki N, Nagashima K, Kurokawa K, Mayer BJ, Maki K, Miyazaki J, Matsuda M. Requirement for C3G-dependent Rap1 activation for cell adhesion and embryogenesis. EMBO J. 2001;20:3333–3341. doi: 10.1093/emboj/20.13.3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Voss AK, Krebs DL, Thomas T. C3G regulates the size of the cerebral cortex neural precursor population. EMBO J. 2006;25:3652–3663. doi: 10.1038/sj.emboj.7601234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.York RD, Yao H, Dillon T, Ellig CL, Eckert SP, McCleskey EW, Stork PJ. Rap1 mediates sustained MAP kinase activation induced by nerve growth factor. Nature. 1998;392:622–626. doi: 10.1038/33451. [DOI] [PubMed] [Google Scholar]
- 9.Bokemeyer D, Guglielmi KE, McGinty A, Sorokin A, Lianos EA, Dunn MJ. Activation of extracellular signal-regulated kinase in proliferative glomerulonephritis in rats. J Clin Invest. 1997;100:582–588. doi: 10.1172/JCI119568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kooistra MR, Dube N, Bos JL. Rap1: a key regulator in cell-cell junction formation. J Cell Sci. 2007;120:17–22. doi: 10.1242/jcs.03306. [DOI] [PubMed] [Google Scholar]
- 11.Hattori M, Minato N. Rap1 GTPase: functions, regulation, and malignancy. J Biochem. 2003;134:479–484. doi: 10.1093/jb/mvg180. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, Sheibani N. PECAM-1 isoform-specific activation of MAPK/ERKs and small GTPases: implications in inflammation and angiogenesis. J Cell Biochem. 2006;98:451–468. doi: 10.1002/jcb.20827. [DOI] [PubMed] [Google Scholar]
- 13.Bos JL, de Bruyn K, Enserink J, Kuiperij B, Rangarajan S, Rehmann H, Riedl J, de Rooij J, van Mansfeld F, Zwartkruis F. The role of Rap1 in integrin-mediated cell adhesion. Biochem Soc Trans. 2003;31:83–86. doi: 10.1042/bst0310083. [DOI] [PubMed] [Google Scholar]
- 14.Ada-Nguema AS, Xenias H, Sheetz MP, Keely PJ. The small GTPase R-Ras regulates organization of actin and drives membrane protrusions through the activity of PLCepsilon. J Cell Sci. 2006;119:1307–1319. doi: 10.1242/jcs.02835. [DOI] [PubMed] [Google Scholar]
- 15.Konstantinov IE, Coles JG, Boscarino C, Takahashi M, Goncalves J, Ritter J, Van Arsdell GS. Gene expression profiles in children undergoing cardiac surgery for right heart obstructive lesions. J Thorac Cardiovasc Surg. 2004;127:746–754. doi: 10.1016/j.jtcvs.2003.08.056. [DOI] [PubMed] [Google Scholar]
- 16.Habib SL, Phan MN, Patel SK, Li D, Monks TJ, Lau SS. Reduced constitutive 8-oxoguanine-DNA glycosylase expression and impaired induction following oxidative DNA damage in the tuberin deficient Eker rat. Carcinogenesis. 2003;24:573–582. doi: 10.1093/carcin/24.3.573. [DOI] [PubMed] [Google Scholar]
- 17.Thorner PS, Ho M, Eremina V, Sado Y, Quaggin S. Podocytes contribute to the formation of glomerular crescents. J Am Soc Nephrol. 2008;19:495–502. doi: 10.1681/ASN.2006101115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Floege J, Eng E, Young BA, Johnson RJ. Factors involved in the regulation of mesangial cell proliferation in vitro and in vivo. Kidney Int Suppl. 1993;39:S47–S54. [PubMed] [Google Scholar]
- 19.Okino K, Nagai H, Nakayama H, Doi D, Yoneyama K, Konishi H, Takeshita T. Inactivation of Crk SH3 domain-binding guanine nucleotide-releasing factor (C3G) in cervical squamous cell carcinoma. Int J Gynecol Cancer. 2006;16:763–771. doi: 10.1111/j.1525-1438.2006.00352.x. [DOI] [PubMed] [Google Scholar]
- 20.Gutierrez-Berzal J, Castellano E, Martin-Encabo S, Gutierrez-Cianca N, Hernandez JM, Santos E, Guerrero C. Characterization of p87C3G, a novel, truncated C3G isoform that is overexpressed in chronic myeloid leukemia and interacts with Bcr-Abl. Exp Cell Res. 2006;312:938–948. doi: 10.1016/j.yexcr.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 21.Hirata T, Nagai H, Koizumi K, Okino K, Harada A, Onda M, Nagahata T, Mikami I, Hirai K, Haraguchi S, Jin E, Kawanami O, Shimizu K, Emi M. Amplification, up-regulation and over-expression of C3G (CRK SH3 domain-binding guanine nucleotide-releasing factor) in non-small cell lung cancers. J Hum Genet. 2004;49:290–295. doi: 10.1007/s10038-004-0148-1. [DOI] [PubMed] [Google Scholar]
- 22.Kohan DE. Endothelins in the normal and diseased kidney. Am J Kidney Dis. 1997;29:2–26. doi: 10.1016/s0272-6386(97)90004-4. [DOI] [PubMed] [Google Scholar]
- 23.Simonson MS, Dunn MJ. Endothelin peptides and the kidney. Annu Rev Physiol. 1993;55:249–265. doi: 10.1146/annurev.ph.55.030193.001341. [DOI] [PubMed] [Google Scholar]
- 24.Wu H, Wang H, Wang Y, Liu T, Kuang Y. The role for endothelin in mesangial proliferative glomerulonephritis. J Tongji Med Univ. 1998;18:122–125. doi: 10.1007/BF02888483. [DOI] [PubMed] [Google Scholar]
- 25.Gomez-Garre D, Largo R, Liu XH, Gutierrez S, Lopez-Armada MJ, Palacios I, Egido J. An orally active ETA/ETB receptor antagonist ameliorates proteinuria and glomerular lesions in rats with proliferative nephritis. Kidney Int. 1996;50:962–972. doi: 10.1038/ki.1996.397. [DOI] [PubMed] [Google Scholar]
- 26.Takaya A, Kamio T, Masuda M, Mochizuki N, Sawa H, Sato M, Nagashima K, Mizutani A, Matsuno A, Kiyokawa E, Matsuda M. R-Ras regulates exocytosis by Rgl2/Rlf-mediated activation of RalA on endosomes. Mol Biol Cell. 2007;18:1850–1860. doi: 10.1091/mbc.E06-08-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rastaldi MP, Armelloni S, Berra S, Calvaresi N, Corbelli A, Giardino LA, Li M, Wang GQ, Fornasieri A, Villa A, Heikkila E, Soliymani R, Boucherot A, Cohen CD, Kretzler M, Nitsche A, Ripamonti M, Malgaroli A, Pesaresi M, Forloni GL, Schlondorff D, Holthofer H, D’Amico G. Glomerular podocytes contain neuron-like functional synaptic vesicles. FASEB J. 2006;20:976–978. doi: 10.1096/fj.05-4962fje. [DOI] [PubMed] [Google Scholar]
- 28.Roselli S, Moutkine I, Gribouval O, Benmerah A, Antignac C. Plasma membrane targeting of podocin through the classical exocytic pathway: effect of NPHS2 mutations. Traffic. 2004;5:37–44. doi: 10.1046/j.1600-0854.2003.00148.x. [DOI] [PubMed] [Google Scholar]
- 29.Bremnes T, Paasche JD, Mehlum A, Sandberg C, Bremnes B, Attramadal H. Regulation and intracellular trafficking pathways of the endothelin receptors. J Biol Chem. 2000;275:17596–17604. doi: 10.1074/jbc.M000142200. [DOI] [PubMed] [Google Scholar]
- 30.Guerrero C, Martin-Encabo S, Fernandez-Medarde A, Santos E. C3G-mediated suppression of oncogene-induced focus formation in fibroblasts involves inhibition of ERK activation, cyclin A expression and alterations of anchorage-independent growth. Oncogene. 2004;23:4885–4893. doi: 10.1038/sj.onc.1207622. [DOI] [PubMed] [Google Scholar]
- 31.Riedl JA, Brandt DT, Batlle E, Price LS, Clevers H, Bos JL. Down-regulation of Rap1 activity is involved in ephrinB1-induced cell contraction. Biochem J. 2005;389:465–469. doi: 10.1042/BJ20050048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Elowe S, Holland SJ, Kulkarni S, Pawson T. Downregulation of the Ras-mitogen-activated protein kinase pathway by the EphB2 receptor tyrosine kinase is required for ephrin-induced neurite retraction. Mol Cell Biol. 2001;21:7429–7441. doi: 10.1128/MCB.21.21.7429-7441.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hughes PE, Renshaw MW, Pfaff M, Forsyth J, Keivens VM, Schwartz MA, Ginsberg MH. Suppression of integrin activation: a novel function of a Ras/Raf-initiated MAP kinase pathway. Cell. 1997;88:521–530. doi: 10.1016/s0092-8674(00)81892-9. [DOI] [PubMed] [Google Scholar]
- 34.Sethi T, Ginsberg MH, Downward J, Hughes PE. The small GTP-binding protein R-Ras can influence integrin activation by antagonizing a Ras/Raf-initiated integrin suppression pathway. Mol Biol Cell. 1999;10:1799–1809. doi: 10.1091/mbc.10.6.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keely PJ, Rusyn EV, Cox AD, Parise LV. R-Ras signals through specific integrin alpha cytoplasmic domains to promote migration and invasion of breast epithelial cells. J Cell Biol. 1999;145:1077–1088. doi: 10.1083/jcb.145.5.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holly SP, Larson MK, Parise LV. The unique N-terminus of R-ras is required for Rac activation and precise regulation of cell migration. Mol Biol Cell. 2005;16:2458–2469. doi: 10.1091/mbc.E03-12-0917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dail M, Richter M, Godement P, Pasquale EB. Eph receptors inactivate R-Ras through different mechanisms to achieve cell repulsion. J Cell Sci. 2006;119:1244–1254. doi: 10.1242/jcs.02842. [DOI] [PubMed] [Google Scholar]
- 38.Petermann A, Floege J. Podocyte damage resulting in podocyturia: a potential diagnostic marker to assess glomerular disease activity. Nephron Clin Pract. 2007;106:c61–c66. doi: 10.1159/000101799. [DOI] [PubMed] [Google Scholar]
- 39.Kriz W, LeHir M. Pathways to nephron loss starting from glomerular diseases-insights from animal models. Kidney Int. 2005;67:404–419. doi: 10.1111/j.1523-1755.2005.67097.x. [DOI] [PubMed] [Google Scholar]
- 40.Patey N, Lesavre P, Halbwachs-Mecarelli L, Noel LH. Adhesion molecules in human crescentic glomerulonephritis. J Pathol. 1996;179:414–420. doi: 10.1002/(SICI)1096-9896(199608)179:4<414::AID-PATH601>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 41.Etienne S, Adamson P, Greenwood J, Strosberg AD, Cazaubon S, Couraud PO. ICAM-1 signaling pathways associated with Rho activation in microvascular brain endothelial cells. J Immunol. 1998;161:5755–5761. [PubMed] [Google Scholar]
- 42.Ng YY, Fan JM, Mu W, Nikolic-Paterson DJ, Yang WC, Huang TP, Atkins RC, Lan HY. Glomerular epithelial-myofibroblast transdifferentiation in the evolution of glomerular crescent formation. Nephrol Dial Transplant. 1999;14:2860–2872. doi: 10.1093/ndt/14.12.2860. [DOI] [PubMed] [Google Scholar]
- 43.Usui J, Kanemoto K, Tomari S, Shu Y, Yoh K, Mase K, Hirayama A, Hirayama K, Yamagata K, Nagase S, Kobayashi M, Nitta K, Horita S, Koyama A, Nagata M. Glomerular crescents predominantly express cadherin-catenin complex in pauci-immune-type crescentic glomerulonephritis. Histopathology. 2003;43:173–179. doi: 10.1046/j.1365-2559.2003.01660.x. [DOI] [PubMed] [Google Scholar]
- 44.Hogan C, Serpente N, Cogram P, Hosking CR, Bialucha CU, Feller SM, Braga VM, Birchmeier W, Fujita Y. Rap1 regulates the formation of E-cadherin-based cell-cell contacts. Mol Cell Biol. 2004;24:6690–6700. doi: 10.1128/MCB.24.15.6690-6700.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Asano T, Niimura F, Pastan I, Fogo AB, Ichikawa I, Matsusaka T. Permanent genetic tagging of podocytes: fate of injured podocytes in a mouse model of glomerular sclerosis. J Am Soc Nephrol. 2005;16:2257–2262. doi: 10.1681/ASN.2004121134. [DOI] [PubMed] [Google Scholar]
- 46.Rosano L, Spinella F, Di CV, Decandia S, Nicotra MR, Natali PG, Bagnato A. Endothelin-1 is required during epithelial to mesenchymal transition in ovarian cancer progression. Exp Biol Med (Maywood) 2006;231:1128–1131. [PubMed] [Google Scholar]
- 47.Misra RP. Isolation of glomeruli from mammalian kidneys by graded sieving. Am J Clin Pathol. 1972;58:135–139. doi: 10.1093/ajcp/58.2.135. [DOI] [PubMed] [Google Scholar]
- 48.Liebau MC, Lang D, Bohm J, Endlich N, Bek MJ, Witherden I, Mathieson PW, Saleem MA, Pavenstadt H, Fischer KG. Functional expression of the renin-angiotensin system in human podocytes. Am J Physiol Renal Physiol. 2006;290:F710–F719. doi: 10.1152/ajprenal.00475.2004. [DOI] [PubMed] [Google Scholar]
- 49.Sharma R, Lovell HB, Wiegmann TB, Savin VJ. Vasoactive substances induce cytoskeletal changes in cultured rat glomerular epithelial cells. J Am Soc Nephrol. 1992;3:1131–1138. doi: 10.1681/ASN.V351131. [DOI] [PubMed] [Google Scholar]
- 50.Sharma R, Sharma M, Vamos S, Savin VJ, Wiegmann TB. Both subtype 1 and 2 receptors of angiotensin II participate in regulation of intracellular calcium in glomerular epithelial cells. J Lab Clin Med. 2001;138:40–49. doi: 10.1067/mlc.2001.115493. [DOI] [PubMed] [Google Scholar]
- 51.Kreisberg JI, Hoover RL, Karnovsky MJ. Isolation and characterization of rat glomerular epithelial cells in vitro. Kidney Int. 1978;14:21–30. doi: 10.1038/ki.1978.86. [DOI] [PubMed] [Google Scholar]
- 52.Foschi M, Chari S, Dunn MJ, Sorokin A. Biphasic activation of p21ras by endothelin-1 sequentially activates the ERK cascade and phosphatidylinositol 3-kinase. EMBO J. 1997;16:6439–6451. doi: 10.1093/emboj/16.21.6439. [DOI] [PMC free article] [PubMed] [Google Scholar]