

Abstract
It is currently not possible to predict which epitopes will be recognized by T cells in different individuals. This is a barrier to the thorough analysis and understanding of T-cell responses after vaccination or infection. Here, by combining mass cytometry with combinatorial peptide–MHC tetramer staining, we have developed a method allowing the rapid and simultaneous identification and characterization of T cells specific for many epitopes. We use this to screen up to 109 different peptide–MHC tetramers in a single human blood sample, while still retaining at least 23 labels to analyze other markers of T-cell phenotype and function. Among 77 candidate rotavirus epitopes, we identified six T-cell epitopes restricted to human leukocyte antigen (HLA)-A*0201 in the blood of healthy individuals. T cells specific for epitopes in the rotavirus VP3 protein displayed a distinct phenotype and were present at high frequencies in intestinal epithelium. This approach should be useful for the comprehensive analysis of T-cell responses to infectious diseases or vaccines.
T lymphocytes, key mediators of the adaptive immune response, are activated when their T-cell receptors (TCRs) interact with cognate antigenic peptides displayed by major histocompatibility complex molecules (MHC). T cells specific for any given antigen can be identified, enumerated and characterized either by stimulation with that antigen or by staining with peptide–MHC multimers1,2. Each approach has advantages and disadvantages. Because stimulation-based methods rely on T-cell proliferation or cytokine production, they detect only T cells that have those characteristics. In contrast, approaches based on peptide–MHC multimers can identify, enumerate and phenotypically assess specific T-cell populations even if they have no known function3 or are extremely rare3,4. The utility of the peptide–MHC multimer approach is especially apparent when combined with single-cell mass spectrometry5–7 (also called cytometry by time-of-flight, or CyTOF), which allows independent assessment of many more cellular parameters (currently over 40) than fluorescence-based flow cytometry7. However, unlike stimulation-based techniques, the identification of antigen-specific cells using peptide–MHC multimers requires knowledge of the precise identity of the peptide, or epitope, recognized by the TCR.
Most existing strategies for identifying T-cell epitopes are time consuming, require an abundance of cellular material and do not provide simultaneous phenotypic information about the T cells that are identified8. This limits the number of epitopes that can be identified and the number of MHC alleles that can be studied (MHC molecules bind and present the epitope to T cells, and show marked polymorphism throughout the human population). These limitations in turn restrict the number of antigens and human subjects that can be analyzed. Therefore, although algorithms for predicting peptide binding to MHC have improved9, predicting which peptide epitopes are actually recognized by T cells during an immune response is completely empirical from that point.
In particular, vaccine design would benefit from knowing which pathogen-derived peptides will be recognized by T cells in the individuals to be vaccinated. In the example we focus on here, oral rotavirus vaccines are much less effective in the developing world than in areas of higher socioeconomic status10 and studies aimed at identifying the reason for this discrepancy are currently hampered by the lack of tools available for analyzing the rotavirus-specific T-cell response. Only two rotavirus T-cell epitopes have been identified in humans11,12. In addition, little information exists on the relative dominance or distinguishing characteristics of rotavirus-specific CD8+ T cells in the peripheral blood or in the intestine13; the latter site is particularly important as it is the primary site of rotavirus infection.
By combining combinatorial14,15 and mass cytometry–based7 peptide–MHC multimer staining approaches, we developed a method that can be used to simultaneously screen for T-cell epitopes in any protein of known sequence and perform high-dimensional phenotypic analysis of human T cells specific for those epitopes. Using only 10 of the ∼40 currently available CyTOF heavy-isotope channels, together with three-dimensional antigen-specificity encoding (which involves assigning a unique combination of three metal tags to each antigen specificity), we probed, enumerated, and phenotypically characterized up to 109 candidate TCR specificities, and 20–30 additional surface and intracellular phenotypic markers, in a single human blood or intestinal lymphocyte sample. Using magnetic cellular enrichment in conjunction with this method, we detected rotavirus-specific cells present at frequencies as low as 1 in 105 CD8+ T cells. From a set of 77 candidate rotavirus epitopes, we identified T cells specific for six epitopes in the blood of 17 healthy donors and the jejunal tissue of 9 obese patients undergoing gastric-bypass surgery. T cells recognizing two of these epitopes were present at particularly high frequencies and displayed unique and potentially informative phenotypes.
Results
T-cell epitope discovery strategy
Our T-cell epitope discovery strategy (Fig. 1a) starts with a search for pathogen epitopes predicted to bind the HLA (the human version of MHC) molecule of interest; this search is performed by netMHC3.0 epitope prediction software16. For the project described here, we analyzed the sequences of the eight viral proteins (NSP1, 2, 5 and VP1, 2, 3, 6, 7) in the RV3 rotavirus vaccine strain (originally isolated from asymptomatic newborns17) for the presence of HLA-A*0201-binding motifs. From these, we selected the 132 candidate peptides (as well as 110 peptides predicted to bind HLA-B*0702 as negative controls) that are the most conserved among various rotavirus strains (depending on the peptide, we considered ∼30 strains)17. Next, we assessed the ability of each candidate peptide to bind to soluble HLA-A*0201 when forced to compete with a biotinylated peptide known to bind to HLA-A*0201 (Fig. 1b). None of the peptides with low predicted binding scores efficiently bound HLA-A*0201, showing that the prediction algorithm has a very low false-negative rate. Therefore, we chose peptides with binding scores higher than the highest-scoring negative control peptides (>30% displacement of the control peptide at 250-nM concentration); this left us with 77 candidate epitopes. Peptides with synthesis errors, poor solubility or weak affinity for MHC could have been missed by this approach, but this approach allows for rapid progress to the T-cell screening phase.
Figure 1.

T-cell epitope discovery strategy. (a) (1) Rotavirus (strain RV3) protein sequences are analyzed by netMHC3.0 epitope prediction software16; (2) peptides (a–d) that are predicted to bind HLA and that are conserved between rotavirus strains are selected; (3) these peptides are tested for HLA binding in a competition assay; and (4) the efficient HLA binders (peptides a, c and d) are used for combinatorial peptide–MHC tetramer staining–based screening. In (4) the three-color tags represent the three-metal codes for each peptide–MHC tetramer. (b) Peptide selection. Each point represents a peptide plotted according to its predicted HLA-binding score (x-axis) and its fraction displacement of a biotinylated competing peptide present at 250 nM (y-axis) from one experiment. The box indicates the approximate (30%) threshold used to select peptides used for subsequent peptide–MHC tetramer staining analysis.
Previously, we and others have described a combinatorial approach for labeling peptide–MHC tetramers that allows the analysis of many more TCR specificities than available flow cytometry color channels14,15. In this approach, instead of single labels for each tetramer, combinations of labels are used, greatly expanding the number of different peptide–MHC tetramers that can be employed simultaneously. For example, we routinely probed 15–32 TCR specificities at once using four or five fluorophores, and we showed that up to 64 TCR specificities could be probed using six fluorophores14. However, this approach suffers from a number of limitations, including spectral overlap between color channels (increasing noise), variable performance of fluorescently tagged reagents, nonspecific binding and the lack of remaining fluorescent channels with which to analyze the phenotypes of the antigen-specific cells. To circumvent these limitations, we combined the combinatorial peptide-MHC staining approach with mass cytometry, which offers many more channels and has much less cross-talk between channels. That is because mass cytometry uses isotopically purified metal conjugates as tags, which can be distinguished from one another with resolution better than a single atomic mass unit by time-of-flight mass spectrometry.
We designed a staining panel of 109 different peptide–MHC tetramers. In general, tetramers are formed by incubating a biotinylated peptide-MHC complex with streptavidin at a 4:1 ratio so that each streptavidin molecule is saturated by binding four peptide-MHC complexes. In our case, we used a modified form of recombinant streptavidin separately conjugated to ten different metals (ten different streptavidin conjugates). However, if one uses only ten metals individually one can distinguish between only ten peptide–MHC tetramers. In contrast, if each peptide–MHC tetramer is conjugated to three different metal-tagged streptavidins, and the differentially tagged peptide– MHC tetramers are mixed and used as a single staining reagent, one forms a three-metal staining code (Fig. 1a). With this system, there are 120 different unique combinations of three metals. Of the 109 peptide–MHC tetramers used here, 77 contained putative rotavirus epitopes and the other 32 contained positive- and negative-control peptides derived from previously identified HLA-A*0201–restricted T-cell epitopes (Table 1 and Supplementary Tables 1 and 2). These previously identified and characterized HLA-A*0201–restricted epitopes were derived from Epstein-Barr virus (EBV), cytomegalovirus (CMV), influenza virus (flu), mutated melanoma T-cell epitope18, (Mart1) and other tumor-associated antigens (EZH2, tyrosinase), GAD65 (a protein implicated in diabetes), HIV, hepatitis B and C viruses (HBV, HCV), measles virus, human papilloma virus (HPV), herpes simplex virus, tuberculosis, lymphocytic choriomeningitis virus, respiratory syncytial virus (RSV) and malaria (ref. 19, Table 1). All peptide–MHC tetramers were pooled and used simultaneously to stain each sample, making the use of these reagents relatively simple.
Table 1. Sequence and three-isotope codes of the 109 peptides tested in the rotavirus epitope screen.
| Rotavirus epitopes | Metal code | Rotavirus epitopes | Metal code | Previously characterized epitopes | Metal code | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
|
|||||||||
| Epitope | Sequence | 1 | 2 | 3 | Epitope | Sequence | 1 | 2 | 3 | Epitope | Sequence | 1 | 2 | 3 |
| NSP1_01 | KMYEHIEDV | 154 | 163 | 173 | VP2_21 | FLAGINSQA | 157 | 161 | 163 | CMV_01 | NLVPMVATV | 154 | 155 | 157 |
| NSP1_02 | FLDSEPHLL | 154 | 163 | 169 | VP2_22 | RLLSYNYET | 155 | 159 | 161 | CMV_02 | VLAELVKQI | 159 | 165 | 169 |
| NSP1_04 | FLDSEPHL | 154 | 169 | 175 | VP2_28 | FVTDSSVISL | 155 | 163 | 169 | CMV_03 | VLEETSVML | 159 | 169 | 175 |
| NSP1_06 | FLDSEPHLLRM | 154 | 157 | 163 | VP2_36 | FVSADTVEPI | 155 | 161 | 169 | EBV_01 | YVLDHLIVV | 159 | 163 | 175 |
| NSP1_07 | VLFNHEVNWDV | 154 | 161 | 165 | VP3_01 | YMLSDNTYV | 157 | 159 | 161 | EBV_02 | GLCTLVAML | 161 | 169 | 175 |
| NSP1_09 | KMYEHIEDVL | 154 | 163 | 175 | VP3_03 | YLHDYSYYT | 157 | 163 | 173 | EBV_03 | YLLEMLWRL | 154 | 155 | 161 |
| NSP2_01 | YLDHKLTPI | 155 | 157 | 165 | VP3_04 | YMLGSAPSYWI | 159 | 163 | 165 | EBV_04 | CLGGLLTMV | 161 | 165 | 173 |
| NSP2_02 | FTMWKLTYL | 154 | 173 | 175 | VP3_05 | FLISNLTTHNI | 157 | 169 | 175 | EBV_05 | YLQQNWWTL | 154 | 155 | 163 |
| NSP2_03 | YLDHKLTPIL | 154 | 157 | 169 | VP3_06 | YMLSDNTYVA | 155 | 159 | 163 | EZH2 | YMCSFLFNL | 154 | 165 | 175 |
| NSP2_04 | KMDEVSHVGV | 154 | 161 | 169 | VP3_07 | KLFPSIVNI | 155 | 161 | 173 | FluM1 | GILGFVFTL | 154 | 155 | 159 |
| NSP2_05 | KMDEVSHV | 154 | 159 | 173 | VP3_08 | FLPTDFEL | 157 | 159 | 163 | FluNP | KLGEFYNQMM | 154 | 155 | 169 |
| NSP2_10 | RIIWQNWYA | 155 | 157 | 159 | VP3_09 | YLLPGWKLTYV | 155 | 163 | 173 | GAD | VMNILLQYL | 159 | 163 | 173 |
| NSP5_01 | YMLSKSPEDI | 155 | 159 | 169 | VP3_11 | MLSDNTYVA | 157 | 163 | 175 | HBV_01 | FLLTRILTI | 163 | 165 | 169 |
| NSP5_05 | SLPSISSSI | 155 | 165 | 169 | VP3_13 | KLYNMFYRNYI | 155 | 159 | 165 | HBV_02 | WLSLLVPFV | 159 | 173 | 175 |
| VP1_01 | KIYSWSFHV | 159 | 161 | 163 | VP3_14 | YTYMLSDNTYV | 157 | 161 | 165 | HBV_03 | FLPSDFFPSV | 165 | 169 | 175 |
| VP1_02 | FILPYEYFI | 155 | 157 | 169 | VP3_15 | KLLHHPTTEV | 157 | 159 | 165 | HBV_04 | GLSPTVWLSV | 159 | 165 | 173 |
| VP1_03 | ILSEYLSFV | 155 | 161 | 175 | VP3_17 | FISDNMIHDV | 157 | 165 | 169 | HBV_05 | FLLSLGIHL | 163 | 169 | 173 |
| VP1_05 | LMDPAILTSL | 155 | 159 | 173 | VP3_18 | YLLPGWKL | 157 | 173 | 175 | HCV | ALYDVVTKL | 154 | 155 | 173 |
| VP1_06 | SLMDPAILTSL | 155 | 165 | 173 | VP3_19 | LLPGWKLTYV | 155 | 163 | 175 | HIV_01 | NVWATHACV | 161 | 173 | 175 |
| VP1_07 | KMWNITAL | 159 | 161 | 165 | VP6_01 | NIFPYSASFTL | 154 | 163 | 165 | HIV_02 | TLNAWVKVV | 169 | 173 | 175 |
| VP1_08 | KLWKKMWNI | 157 | 159 | 169 | VP6_02 | FLLNGQIINT | 154 | 169 | 173 | HIV_03 | FLGKIWPS | 161 | 165 | 175 |
| VP1_12 | YLVTWANSSI | 157 | 165 | 173 | VP6_03 | VLADANETL | 154 | 161 | 163 | HIV_04 | KLTPLCVTL | 163 | 165 | 175 |
| VP1_13 | NLMDSYVQI | 157 | 161 | 169 | VP6_04 | TLLANVTAV | 154 | 157 | 173 | HIV_05 | SLYNTVATL | 165 | 169 | 173 |
| VP1_14 | KLNSYAPVYL | 157 | 161 | 175 | VP6_05 | LLANVTAV | 154 | 161 | 175 | HIV_06 | LTFGWCFKL | 161 | 165 | 169 |
| VP1_20 | YLIAKELIIL | 159 | 161 | 169 | VP6_06 | GLLGTTLLNL | 154 | 159 | 163 | HIV_07 | GLADQLIHL | 161 | 169 | 173 |
| VP1_22 | FILPYEYFIA | 155 | 159 | 175 | VP6_07 | TLLNLDANYV | 154 | 159 | 175 | HIV_08 | ALVEMGHHA | 159 | 165 | 175 |
| VP1_26 | SLMDPAIL | 155 | 165 | 175 | VP7_01 | LLNYILKSV | 154 | 159 | 169 | HIV_09 | ILKEPVHGV | 163 | 173 | 175 |
| VP1_27 | NLNAVMFWL | 157 | 159 | 173 | VP7_02 | FVIYRFLFV | 154 | 161 | 173 | HPV | YMLDLQPETT | 154 | 157 | 161 |
| VP1_32 | FMPTLPDNV | 155 | 169 | 175 | VP7_03 | VIYRFLFVI | 154 | 159 | 161 | HSV1/2 | SLPITVYYA | 154 | 155 | 175 |
| VP1_39 | ILESYVYNL | 155 | 157 | 173 | VP7_04 | YTDVASFSV | 155 | 157 | 163 | LCMV | YLVSIFLHL | 154 | 165 | 169 |
| VP2_01 | MLTSNLTFTV | 157 | 165 | 175 | Unused metal codes (negative controls) | Malaria | YLNKIQNSL | 161 | 163 | 169 | ||||
| VP2_02 | ALVGALPFV | 157 | 159 | 175 | UNUSED | 161 | 163 | 175 | Mart1 | ELAGIGILTV | 154 | 155 | 165 | |
| VP2_03 | SLISGMWLLTV | 155 | 157 | 175 | UNUSED | 165 | 173 | 175 | MV | KLWCRHFCV | 154 | 157 | 159 | |
| VP2_04 | SLISGMWLL | 159 | 161 | 173 | UNUSED | 154 | 157 | 165 | RSV | KMLKEMGEV | 159 | 163 | 169 | |
| VP2_05 | HMLTSNLTFTV | 155 | 161 | 163 | UNUSED | 154 | 157 | 175 | TB_01 | GLPVEYLQV | 163 | 169 | 175 | |
| VP2_06 | QLMEALMQL | 155 | 161 | 165 | UNUSED | 154 | 165 | 173 | TB_02 | KLIANNTRV | 163 | 165 | 173 | |
| VP2_07 | FLNHQLVEPL | 157 | 169 | 173 | UNUSED | 155 | 169 | 173 | Tyr-ase | YMDGTMSQV | 155 | 157 | 161 | |
| VP2_10 | TLFHYYNVNV | 157 | 163 | 165 | UNUSED | 157 | 161 | 173 | ||||||
| VP2_11 | YMSLISGMWLL | 155 | 173 | 175 | UNUSED | 159 | 169 | 173 | ||||||
| VP2_12 | MLLNNQPVALV | 157 | 163 | 169 | UNUSED | 161 | 163 | 173 | ||||||
| VP2_16 | MLSQRTMSL | 159 | 161 | 175 | UNUSED | 161 | 169 | 175 | ||||||
| VP2_17 | RLLSYNYETL | 155 | 163 | 165 | UNUSED | 154 | 159 | 169 | ||||||
With 120 codes possible using ten metals, 11 codes act as internal negative controls (labeled ‘unused’). Metals associated with each isotope are listed in Supplementary Table 1. MV, measles virus; HPV, human papilloma virus; HSV1/2, herpes simplex virus; TB, tuberculosis; LCMV, lymphocytic choriomeningitis virus.
We stained a total or CD8+ T cell-enriched samples derived from healthy blood donors. After tetramer staining, a portion of the sample was subjected to tetramer-positive T-cell enrichment using magnetized columns. Both enriched and pre-enriched samples were analyzed by mass cytometry, allowing for an objective back-calculation of pre-enrichment antigen-specific T-cell frequencies in each donor sample. All cells were also stained using a cocktail of metal-tagged antibodies specific for 23–27 surface and intracellular markers that allow for the identification and phenotypic characterization of CD8+ T cells.
Identification of antigen-specific cells
After data acquisition, live CD8+ T-cell events were gated away from other cell types using FlowJo software (using heavy metal–labeled antibodies such as Nd-146–labeled anti-CD8, see Supplementary Fig. 1); these events were exported and custom scripts written in Matlab were used to identify the antigen-specific cells (Fig. 2a and Supplementary Matlab Scripts). Several parameters were optimized (Fig. 2a,b, Supplementary Fig. 2 and Supplementary Movie). After setting the threshold, slope and stringency parameters (Fig. 2b) for one sample, these values were held constant and used to analyze all of the samples in the study. For two of the experiments, each sample was split and analyzed separately, but in parallel, using an alternative and randomized metal-coding scheme. That is, each antigen specificity was stained with peptide–MHC tetramers bearing two different three-metal combinations. When we plotted the epitope frequency detected with one three-metal combination (order 1) against the epitope frequency detected with the other three-metal combination (order 2), we observed a very good correspondence across a wide range (>3 log) of frequencies (Fig. 2c,d). Because the signals became random below a frequency of 0.001%, for all subsequent analysis cells below this threshold were considered background. Note also that in these two examples, a good correspondence of T-cell frequencies were observed for flu-, EBV-, CMV-specific and a number of rotavirus-specific cells, controlling for possible artifacts associated with nonspecific streptavidin binding or T-cell cross-reactivity.
Figure 2.
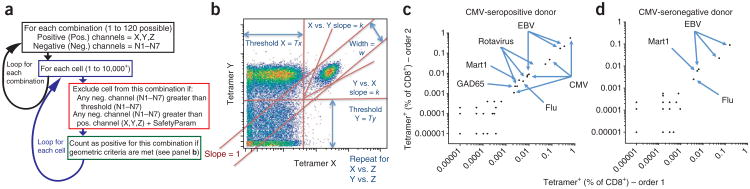
Auto-gating strategy and validation with scrambled three-metal coding. (a) The automated combinatorial peptide–MHC tetramer gating strategy (see also Supplementary Matlab Scripts). SafetyParam, see Online Methods. (b) The gating strategy and some of the parameters (threshold, slope, width) involved in automatically gating cells stained with a combination of three metals, X, Y and Z. As this is done for three channels, these criteria are evaluated for the X–Y, X–Z and Y–Z planes (Supplementary Fig. 2 and Supplementary Movie). (c,d) Two samples (c,d, respectively) were each split in half, and each half was stained with a panel of 96 peptide–MHC tetramers that was coded using one of two three-metal coding schemes (order 1 and order 2). The frequency of staining with each tetramer using order 1 (x axis) and order 2 (y axis) is plotted from two of the five donors tested in this way (Supplementary Table 2).
One potential limit of this approach is that the large numbers of peptide–MHC tetramers loaded with different peptides might compete heavily with any given tetramer. To determine whether this was a problem, we titrated an irrelevant HIV peptide–loaded MHC tetramer into staining mixes containing a constant low concentration of a CMV peptide–loaded MHC tetramer. Specific staining of CMV-specific T cells was maintained even in the presence of 1,000-fold excess irrelevant HIV peptide–MHC tetramer (Supplementary Fig. 3), indicating that competition from irrelevant tetramers would not be a major problem even if 1,000 antigen specificities were probed simultaneously by this approach. One thousand different peptide–MHC tetramers could, in theory, be screened if 20 metal labels were used in groups of three (1,140 possible combinations). Although such a staining scheme would result in loss of metal labels available for phenotypic and functional markers, we anticipate that the number of possible labels will soon grow to accommodate that loss.
Having optimized the combinatorial peptide–MHC tetramer staining process, we stained blood samples from 17 healthy donors and intestine intraepithelial lymphocyte (IEL) samples from nine obese patients undergoing gastric-bypass surgeries. All data were analyzed using the same Matlab script with the same set of optimized parameters. Common epitope ‘hits’ (defined as being detected in at least two donors at back-calculated frequencies of >0.001% of CD8+ T cells) are listed in Table 2 (for blood samples). Seventeen additional epitopes (ten derived from rotavirus, and seven derived from flu, RSV, HIV or GAD65) were each identified as ‘hits’ only in a single donor (data not shown). Among all epitopes tested, 8.82 ± 0.89 (mean ± s.e.m.) epitope hits were detected on average in each donor; 2.18 ± 0.38 of these epitopes were derived from rotavirus. It was not surprising that the most common hits detected were T cells recognizing Mart1 influenza, EBV and CMV epitopes. However, some rotavirus epitopes were identified as common hits, including the two previously characterized epitopes (VP6- and VP7-derived, refs. 11,12, respectively) and a number of novel epitopes derived from VP3, VP1, VP2 and NSP1. In 9 of 17 donors having significant numbers of VP3-specific T cells, the average frequency of these cells was relatively high (0.0198% of blood CD8+ T cells) for a memory T-cell population. We also detected VP3-specific cells in eight out of nine of the IEL samples tested, at an even higher average frequency (0.104 ± 0.032% mean ± s.e.m.). No other rotavirus epitopes were consistently detected in the IEL samples.
Table 2. Epitopes detected in at least two blood donors.
| Source protein | Sequence | Hits (>0.001%) | Average (% of CD8+) | s.e.m. (%) | Average of hits (%) |
|---|---|---|---|---|---|
| CMV-IE1 | VLEETSVML | 5/17 | 0.2972 | 0.8933 | 1.0103 |
| CMV-pp65 | NLVPMVATV | 8/17 | 0.4592 | 1.1345 | 0.9755 |
| EBV-LMP2A | CLGGLLTMV | 12/17 | 0.1560 | 0.3527 | 0.2209 |
| EBV-BRLF1 | YVLDHLIVV | 9/11 | 0.0900 | 0.1654 | 0.1098 |
| EBV-BLMF1 | GLCTLVAML | 12/17 | 0.0687 | 0.0992 | 0.0973 |
| Rota-VP3 | YLLPGWKL | 9/17 | 0.0105 | 0.0163 | 0.0198 |
| Flu-M1 | GILGFVFTL | 14/17 | 0.0126 | 0.0246 | 0.0153 |
| EBV-LMP1-2 | YLQQNWWTL | 9/17 | 0.0058 | 0.0092 | 0.0109 |
| Flu-PB1 | NMLSTVLGV | 2/9 | 0.0024 | 0.0048 | 0.0107 |
| Rota-VP1 | SLMDPAILTSL | 2/8 | 0.0026 | 0.0055 | 0.0103 |
| EBV-LMP1-1 | YLLEMLWRL | 5/17 | 0.0025 | 0.0044 | 0.0080 |
| Rota-VP6 | TLLANVTAV | 8/17 | 0.0038 | 0.0084 | 0.0078 |
| CMV-IE1-2 | FMDILTTCVET | 2/9 | 0.0013 | 0.0030 | 0.0055 |
| Mart1 | ELAGIGILTV | 17/17 | 0.0047 | 0.0042 | 0.0047 |
| CMV-pp65-2 | QMWQARLTV | 5/9 | 0.0023 | 0.0026 | 0.0037 |
| Rota-VP2-1 | SLISGMWLL | 4/17 | 0.0008 | 0.0014 | 0.0028 |
| HIV-gag | TLNAWVKVV | 2/17 | 0.0007 | 0.0006 | 0.0021 |
| Rota-VP7-1 | LLNYILKSV | 2/17 | 0.0005 | 0.0006 | 0.0017 |
| CMV-pp65-3 | LMNGQQIFL | 2/9 | 0.0003 | 0.0005 | 0.0012 |
| Rota-NSP1 | FLDSEPHLL | 2/8 | 0.0005 | 0.0005 | 0.0011 |
| HBV-SAg | WLSLLVPFV | 2/8 | 0.0004 | 0.0005 | 0.0011 |
Each epitope was detected at a frequency of >0.001% of CD8+ T cells (hits) in at least two donors. Some epitopes were tested in <17 donors. The average frequency (and s.e.m.) observed in all donors tested, and the average frequency in donors with frequency >0.001% (average of hits), are indicated. Hits are boldface. Rota, rotavirus.
Analysis of antigen-specific cell phenotypes
Next, we compared the phenotypic profiles of these antigen-specific cells (using the other 23–27 antibodies included in the staining panel). To simplify our analysis, we averaged the intensity of each phenotypic marker for each T-cell-antigen specificity and compared it to the average of bulk tetramer-negative CD8+ T cells. We plotted these data as a heat plot for each donor (Fig. 3a and Supplementary Fig. 4). From these heat plots it was apparent that the phenotypes of cells specific for each well-characterized T-cell antigen were consistent with previous reports. For instance, Mart1-specific cells displayed a naive phenotype (CD45RA+CD27+CCR7+CD62L+), flu-specific cells showed central memory phenotypes (CD45RA−CD27+CCR7+CD62L+) and CMV-specific cells resembled late-stage effector cells (CD45RA+CD27−CC R7−CD62L−)20. In general EBV- and rotavirus-specific cells displayed more diverse phenotypes. To objectively compare the separate experiments, we calculated z-scores for each marker for each T-cell epitope for each donor, by normalizing within each 3- or 4-donor experiment. This normalization corrects for any differences in staining or machine performance between experiments, and the results act as internal validation. For instance, a heat plot of z-scores for all epitope-donor pairs for CMV-, flu- and Mart1-specific cells shows that the phenotypes of the antigen-specific T cells mostly cluster together (Supplementary Fig. 5). As mentioned above and described below, EBV- and rotavirus-specific cells displayed more heterogeneous phenotypic profiles.
Figure 3.

Analysis of the phenotype of antigen-specific T cells in the blood of 17 normal donors. (a) The average expression of each phenotypic marker (listed at bottom) on T cells specific for the indicated epitopes (listed at right) from one representative blood donor are plotted as a clustered heatmap. (b–d) The first two components obtained from principal component analysis (PCA) of z-score–normalized average expression levels for each marker are plotted for each hit (each point is a single epitope-donor combination, as listed in Table 2) for Mart1-, flu-, EBV-, CMV- and rotavirus-specific cells (b); and T cells specific for indicated EBV (c) and rotavirus peptides (d). (e) The principal component loading values are plotted for each parameter in the analysis for the first two components. These values are the PCA weightings that are used to obtain PC values for each population of cells, and provide insight about what these principal components represent. (f) Average z-score–normalized expression levels of integrin-β7 and CD103 (integrin-αE). Each point is a single epitope-donor combination (from 17 different blood donors). For rotavirus-specific cells, T cells targeting VP3- and non-VP3-derived epitopes were separated for this analysis. *P < 0.05, **P < 0.01, ***P < 0.001 by Dunn's multiple comparisons test.
We next used principal component analysis (PCA) to reduce the dimensionality and plot the relative similarity of each epitope-donor pair considering all donors and epitopes listed in Table 2. Mart1-specific T cells displaying naive-like phenotypes as defined above were on one end of the spectrum and CMV- specific (especially for dominant epitopes derived from pp65 and IE1; see Supplementary Fig. 6) late-stage effector-like T cells were on the other (Fig. 3b). Even when excluding Mart1-specific T cells from the PCA analysis, the overall relationship between the remaining specificities did not markedly change (Supplementary Fig. 7). PCA analysis also showed that some EBV- and rotavirus-specific T cells were central memory-like whereas others had varying degrees of effector-like phenotypes (gauged by their similarity to CMV-specific T cells, Fig. 3b). Upon deeper analysis of EBV- and rotavirus-specific T cells, we noted differences in phenotype that correlated with differences in peptide specificity. For example, EBV-specific cells specific for BRLF-1 and BLMF-1 peptides displayed varying degrees of effector-like phenotypes, whereas those specific for latency-associated antigens LMP-1 and LMP-2A peptides had phenotypes most similar to flu-specific cells (Fig. 3c). The difference in average PC1 values between BRLF-1/BLMF-1-specific (mean ± s.e.m. = 0.92 ± 0.25, n = 27 hits, with multiple hits per donor) and LMP- 1/LMP-2A-specific (mean ± s.e.m. = −0.99 ± 0.24, n = 23 hits) cells was statistically significant (two-sided t-test, P < 10−5). Among rotavirus-specific T cells, only those specific for VP3 peptides displayed effector-like phenotypes; some had PCA values even more extreme than CMV-specific cells (Fig. 3d). VP3- and non-VP3 peptide-specific T cells also significantly differed in their expression of trafficking receptor integrin-P7 (expressed on gut-trafficking intestinal resident lymphocytes21; P < 0.01, Fig. 3e,f) and integrin-αE (expressed on intestinal IEL cells; P < 0.05, Fig. 3e).
As mentioned above, among rotavirus-specific T cells, only those specific for VP3 peptides were consistently detected in the jejunal IEL samples analyzed here (mean ± s.e.m. = 0.104 ± 0.032%, n = 8 donors with detectable VP3-specific cells). For four out of nine of these, we ran a sufficient number of blood samples in parallel (blood and IEL samples were from different donors) to allow reasonable comparison of phenotypic marker expression using z-scores. VP3-specific IEL T cells displayed phenotypes overlapping those of bulk tetramer-negative IEL CD8+ T cells (Fig. 4a). In contrast, VP3-specific blood T cells were relatively unique among blood T cells in that they expressed markers of an effector-phenotype (CD45RA−CD62L−) and high amounts of integrin-αE (Fig. 4a). PCA or hierarchical clustering considering all markers analyzed confirmed that VP3-specific IEL T cells were indistinguishable from bulk IEL CD8+ T cells and that compared to blood T cells of other antigen-specificities, VP3-specific blood T cells more closely resembled IEL T cells (Fig. 4b,c). The phenotypic distinction between rotavirus-specific T cells targeting this VP3-derived epitope versus other epitopes may reflect differences in trafficking and/or function in the intestinal epithelium.
Figure 4.

Phenotypic comparison of IEL and blood-derived rotavirus-specific CD8+ T cells. (a) Expression levels of integrin-β7, CD103 (integrin-αE), CD45RA and CD62L on representative samples from one blood donor (representative of nine donors with detectable VP3-specific cells) and one IEL donor stained in parallel (representative of eight donors with detectable VP3-specific cells). Top panels show total CD8+ IEL T cells as a black density plot overlaid with a red colored contour plot gated on VP3-specific IEL T cells identified by tetramer analysis. The bottom panels show total CD8+ blood T cells as a black density plot overlaid with a blue colored contour plot gated on VP3-specific blood T cells identified by tetramer analysis. (b) PCA values and (c) hierarchical clustering analysis (as in Fig. 3a–d) for total (tetramer-negative) and VP3- or VP6-specific rotavirus-specific IEL and blood T cells. For hierarchical clustering analysis, relative expression of each marker is plotted as z-score calculated for each experiment (Online Methods).
Discussion
The limits of combinatorial peptide–MHC tetramer staining14,15 are quickly reached when fluorescence-based flow cytometry is used because of the wide overlap between fluorescent channels as well as nonspecific binding that depends on the fluorophore species being used. Therefore, to meaningfully increase the number of T-cell epitopes we could analyze in a single sample, we turned to mass cytometry. Although the signal/noise ratio for a given tetramer is usually lower in mass cytometry than in flow cytometry7, the combinatorial strategy used here mitigates this particular type of background noise by exploiting the large number and limited cross-talk between mass cytometry channels. To be more specific, for a given cellular event acquired using mass cytometry, background noise arises from nonspecific tetramer binding, detector noise, or metal ions from cellular debris or other sources entering coincidentally with the cell. Because the background signal derived from these latter two sources is stochastic, for a given cell the chances of observing a high background signal on two or three unrelated isotopic channels is much lower than for one channel alone. That is, if “P” equals the probability of high background for one channel, the probability of coincident high background on two or three channels equals P2 or P3, respectively. For example, if at a given threshold, 1% of cells are positive due to background noise in one channel, only 0.01% will be coincidentally positive for two channels ([0.01 × 0.01] × 100%) and only 0.001% will be positive for three channels. Here, we labeled each species of peptide–MHC tetramer with a combination of three different isotopic metal tags. By creating a Matlab automatic gating script that identifies cells that are coincidentally positive (with similar intensities) for those three and only those three channels, we identified rare antigen-specific T cells in the midst of background noise.
To further validate this approach and to rule out the possibility of artifactual labeling, we stained the same samples with two different mixtures of tetramers, each with a different combinatorial three-metal code. The fact that we saw a very good correspondence between the two mixtures shows that the identity of the peptide is the critical parameter and allows for an objective measure of assay sensitivity. This correspondence between the two staining panels was lost for tetramers staining at a frequency <0.001% of CD8+ cells, indicating that this was the effective limit of detection. This level of sensitivity for rare cells was achieved in part by using large numbers of input cells and magnet-based tetramer enrichment as described3,4. For samples where the frequencies of antigen-specific T cells are expected to be higher (for example, samples from donors infected with a pathogen or cells isolated from tissues with T-cell infiltrates), we expect that this approach would work with considerably fewer cells. The results of analysis of the two staining panels also suggest that cross-reactivity is not a major problem, at least with the number of epitopes analyzed here. It is possible that our analysis missed rare occasions of T cells binding to multiple peptide–MHC tetramers. However, these cells would be excluded from subsequent analysis by the nature of the auto-gating script. In future experiments where some instances of T-cell cross-reactivity might be expected, this script can be modified to accommodate and quantify these cells. Additional validation of the accuracy of this method in identifying antigen-specific cells is found in the phenotypic profiles of the cells. For instance, Mart1-, flu- and CMV-specific cells displayed naive, central memory and effector phenotypes as expected based on previously published work7,22,23.
Our screen for HLA-A*0201–restricted rotavirus epitopes was successful in that we independently identified T cells specific for both of the previously identified HLA-A2–restricted rotavirus epitopes11,12 as well as T cells specific for several previously uncharacterized epitopes. As the gut is the primary site of rotavirus infection, it was reassuring that, relative to T cells specific for other epitopes, the rotavirus-specific T cells we identified express higher levels of mucosal trafficking receptors CD103 (integrin-αE) and/or integrin-β7 (ref. 21). Compared to T cells specific for other rotavirus epitopes, VP3-specific T cells displayed hallmarks of more differentiated cells, expressed little or no integrin-β7, and expressed abundant integrin-αE. It is not clear why this might be the case and it is unclear what protein(s) integrin-αE pairs with in the absence of detectable integrin-β7. As has been reported for integrin-α4 (ref. 24), some other integrin-β family member may compete for binding to integrin-αE to prevent functional expression of the typical αEβ7 heterodimer that promotes trafficking to the gut. Nevertheless, VP3-specific T cells were detected in the IEL at fivefold higher frequency than in the blood, and displayed a phenotypic profile indistinguishable from bulk CD8+ IEL; this phenotype includes high expression of both integrin-β7 and integrin-αE. Non-VP3–specific blood T cells displayed a more central memory–like phenotype, expressed high levels of integrin-β7 and were mostly absent from the IEL. Although the reason for this distinction is not clear, the presence of VP3-specific T cells displaying effector-like phenotypes implies that these cells may have encountered antigen more recently compared to central memory–like cells such as flu- or VP6-specific cells, perhaps within intestinal tissue. In contrast, non-VP3–specific T cells with a central memory phenotype may not have encountered antigen as recently and may be poised to respond to antigen in lymph nodes during responses to systemic infections. Although we can not formally rule out the possibility that VP3-specific T cells cross-react with an unrelated epitope, the VP3 peptide sequence matches (identically) a wide range of rotavirus sequences but does not perfectly match any other sequence in the NCBI nonredundant protein sequence database. It is conceivable that because VP3 is a part of the viral transcription complex, it may be differentially expressed by the virus or differentially processed by the infected cell relative to other viral proteins. For example, VP6-specific antibodies have been shown to function intracellularly during transcytosis to block virion transcription, and may somehow interfere with antigen processing and presentation25. Lastly, it is possible that these epitopes originate in different strains of rotavirus that are more or less common in the healthy adult population. Indeed, the dominant VP3 epitope we identified is conserved in all three major types of rotavirus that have been isolated from humans (Wa-, DS- and Au-like strains). In contrast, the VP6 epitope is altered in the DS- and Au-like strains of rotavirus26; thus more recent subclinical infections of DS- or Au-like strains in healthy adults could lead to the observed differential phenotypes and locations of VP3- and VP6-specific T cells. Future work on active rotavirus infections in children and adults may shed light on the reason for the dichotomy of T-cell phenotypes that we observe in this small cohort of healthy American adults.
In summary, by combining single-cell mass cytometry analysis with combinatorial peptide–MHC tetramer staining, we developed a method that allows for simultaneously screening a large number of T-cell epitopes (up to 109 demonstrated here and potentially many more) in a single sample, together with extraction of a great deal of phenotypic and functional information. We used this method to validate two and discover four HLA-A*0201–restricted rotavirus T-cell epitopes, but this method could in theory be applied to discover T-cell epitopes in any pathogen, tumor or self protein whose sequence is known. The method we describe here should also be useful for evaluating the effects of an infectious disease or vaccine or any situation in which a comprehensive analysis of a T-cell response may be useful.
Online Methods
Peripheral blood cells
Leukocyte reduction system cones (LRS) containing peripheral blood mononuclear cells (from platelet aphaeresis donors) were obtained from the Stanford Blood Center according to Institutional Review Board (IRB) (“Minimal Risk Research Related Activities at Stanford Blood Center”, Protocol #13942) protocol. For the purposes of peptide–MHC tetramer staining experiments, HLA–A*0201 positive anonymous donor samples from residents of the Palo Alto, California, area were typed and identified by the Stanford Blood Center. No information about age, gender or prior exposure to rotavirus is currently available for these donors. These samples were also serotyped for cytomegalovirus (CMV) infection status. For CD8+ or total T-cell enrichment by negative selection, the LRS samples were diluted to 20 ml phosphate buffered saline (PBS) + 2% FCS before adding 750 μL of CD8+ or whole T-cell RosetteSep cocktail (Stemcell Technologies). The cells were further diluted with PBS + 2% FCS and Ficoll-separated (Ficoll-Paque PLUS, GE Healthcare) from red blood cells and negatively selected cells before cryopreservation in 90% FCS + 10% DMSO. In some cases, blood cells were used fresh without freezing (experiments A, D and E, see Supplementary Table 2).
Intraepithelial lymphocyte isolation
Deidentified proximal jejunum tissue resections were obtained from anonymous patients undergoing bariatric surgery in accordance with Stanford University IRB (“Study of tissue lymphocyte responses to rotavirus using intestinal resections”, Protocol #1726) protocols. No information about, age, gender or prior exposure to rotavirus is available for these donors. Intraepithelial lymphocytes (IEL) were isolated using an adapted version of previously published protocols27. Specimens were washed four times with cold Ca2+ and Mg2+ free Hank's Balanced Salt Solution (HBSS) containing 100 U/ml penicillin and 100 μg/ml streptomycin to remove erythrocytes and debris and to reduce contamination. The muscle layer was removed and the resections were dissected into 0.3–0.5-cm2 fragments. To isolate IEL, tissue fragments were washed with 0.15% dithiothreitol (DTT, Invitrogen) in HBSS at 37 °C with constant rotation, followed by two washes each for 30 min at 37 °C in 2 mM EDTA in HBSS and HBSS, until the resulting supernatant was clear. Additional washes with HBSS were done if necessary. Supernatants from EDTA and HBSS treatments were pooled, centrifuged, resuspended in 44% Percoll (GE Healthcare), and subjected to 70/44/20% Percoll density centrifugation. IEL were harvested at the 70/44% interface, washed twice with complete RPMI and filtered using a 40 μM cell strainer. All IEL samples were then cryopreserved in 90% FCS + 10% DMSO. After thawing, the cells were incubated for 2–4 h in RPMI media + 10% FCS (37 °C) before use of the Miltenyi dead-cell removal kit (Miltenyi Biotec) to remove dead cells and debris from the sample.
Epitope prediction
Epitope prediction was done using NetMHC3.0 epitope prediction software16 that was downloaded and interfaced with python scripts to evaluate the relative conservation score of each candidate epitope. First, predicted MHC binding scores (score >0.4 predicting weak binding, scores >0.6 predicting strong binding) were evaluated for all possible 8-, 9-, 10- or 11-mer peptides derived from eight different RV3 strain rotavirus proteins (NSP1, 2, 5 and VP1, 2, 3, 6, 7)17. Next, a conservation score was calculated based on the “difflib.SequenceMatcher” function in python using accession numbers listed for each protein17 (scores ranged between ∼0.4 for low conservation and 1 for high conservation). A composite score with 1 × weighting on conservation and 2 × weighting on predicted binding was calculated for each peptide. Peptides with the highest composite scores for each protein were synthesized and used for subsequent analysis (10 from NSP1, 11 from NSP2, 6 from NSP5, 31 from VP1, 21 from VP2 and 21 from VP3). For VP6 and VP7, conservation analysis was not used; instead using the RV3 rotavirus strain sequence, all predicted strong binders (binding score >0.6) were synthesized (13 for VP6 and 19 for VP7). In total, 132 peptides were synthesized and tested for MHC binding.
Peptide competition binding assay
Relative peptide binding of peptides was assessed using a competition binding assay. In these assays 0.2 μM biotinylated peptide with sequence SLYNTVATLGGGSGGGK-biotin (based on a known HIV-derived HLA-A*0201-binding peptide) and 10 μg/ml UV-exchangeable nonbiotinylated peptide-MHC (which was cleared with streptavidin-agarose (Sigma/Aldrich) before purification) was mixed with varying amounts of the test peptide (250 nM, 2.5 μM and 25 μM), UV-irradiated to allow peptide exchange, incubated overnight and then loaded on an enzyme-linked immunosorbent assay (ELISA)-plate that was coated with 2 μg/ml anti-HLA-A,B,C (Biolegend) and blocked with 10% FCS. After MHC was bound, the plates were washed and probed with Europium-labeled streptavidin using the Perkin-Elmer DELFIA ELISA system to detect presence of the biotinylated HIV peptide. Inhibition of biotinylated peptide binding was used to assess the relative binding of the test peptide at each concentration. Peptides with >30% inhibition at 250 nM were used for subsequent tetramer-staining experiments.
Streptavidin expression and heavy metal labeling
Streptavidin with six free cysteines separated by short flexible linkers on each subunit was produced in order to magnify the mass cytometry signal. Six free cysteine residues separated by glycine linkers were introduced to the C terminus of streptavidin (with amino acid sequence: SGGCCGGGCCGGGCCK) for recombinant expression, refolding and purification as described28. Using this construct we observed more robust peptide–MHC tetramer staining (higher signal/noise, data not shown) compared to streptavidin with a single free cysteine7; however, we did not attempt to measure the absolute number of metal-loaded polymers conjugated to each streptavidin protein. After purification by size-exclusion chromatography, the streptavidin protein was stored in 10 mM TCEP in 20 mM HEPES (pH 7.2) buffered saline and 50% glycerol before coupling. For heavy metal coupling, 50 μg of streptavidin protein was exchanged into fresh 10 mM TCEP solution in DVS Sciences R buffer using a 30-kDa cutoff concentrator (Millipore) and incubated at 37 °C for 30 min. After this incubation, the streptavidin was exchanged into C and W buffer as recommended for the DVS antibody labeling procedure, using the same 30-kDa cutoff concentrator. Two aliquots of DVS Sciences DN3 polymers (free of heavy metals) were loaded with metals in ‘L’-buffer as recommended (some metals not available from DVS including Gd-155, Gd-157, Dy-161, Dy-163 and Yb-173 were obtained from Trace Sciences International, Inc. and dissolved at 100 mM in ‘L’-buffer and used in a similar manner as metal solutions obtained from DVS) and washed in additional L buffer after loading using a single 3-kDa cutoff (Pall) concentrator. After washes, the reduced and exchanged streptavidin in ∼100 μL of W buffer was added to the 3-kDa cutoff concentrator containing the metal-loaded polymer and centrifuged to <10 μL volume to drive the reaction with high protein and polymer concentrations. Unfortunately, we do observe some variability in the quality of metal-labeled streptavidin produced by this procedure, and we are still investigating this. Thus, each batch was tested with a positive control to ensure sufficient tetramer staining signal for the experiment.
Peptide–MHC tetramer production
To produce the various biotinylated peptide-MHC molecules, HLA-A*0201 was refolded with a UV-cleavable peptide, biotinylated and purified as described29,30. After purification, the protein stock was stored in PBS + 50% glycerol at −20 °C. For each peptide specificity, peptide exchange reactions were setup in 100 μL volumes each containing 10 μM peptide and 100 μg/ml HLA-A2 or protein in PBS. Peptide sequences used in this study are listed in Table 1 and Supplementary Table 2. After a 20-min exposure to 365 nm UV irradiation using a Stratagene UV Stratalinker 2400, the protein was stored at 4 °C overnight to complete the exchange.
For combinatorial tetramer cocktails, 96-well plates were used to premix metal-labeled streptavidins in the 120 possible combinations listed in Table 1 and Supplementary Table 2. These mixtures were then added to each well of exchanged peptide-MHC (described above) stepwise in four additions with 5-min incubations at room temperature between each addition to achieve a final ratio of 1:4 (total streptavidin:peptide-MHC) in each well. After the last incubations, 10 μM free biotin in 10 μL of cytometry buffer was added to each tetramer to quench unbound streptavidin before mixing14. We then combined all tetramers, and this mixture was concentrated using a 10 kDa cut-off concentrator. This cocktail was then used to stain cells with each peptide–MHC tetramer at a concentration of ∼100 nM.
Antibody labeling
Purified antibodies (lacking carrier proteins) were purchased from BD Biosciences, Biolegend, eBioscience, Abcam, Invitrogen or R&D systems, and clone names are listed in Supplementary Table 1. The antibodies were labeled 100 μg at a time according to instructions provided by DVS Sciences with heavy metal-loaded maleimide-coupled DN3 MAXPAR chelating polymers using the recommended labeling procedure. For all metal conjugations done in this study, including conjugations to streptavidin, free DN3 MAXPAR polymers were purchased from DVS Sciences and loaded with metal before use.
Staining and data acquisition
Cryopreserved peripheral blood cells were thawed and washed with complete RPMI before overnight recovery at 37 °C. In cases when freshly isolate cells were used, a recovery incubation with complete RPMI at 37 °C was also performed. Our input cell numbers ranged between 16 and 84 million in instances where CD8+ enriched cells were used and between 42 and 120 million (except for one sample where we had 302 million cells) in instances where total T cells were used. However, in several cases these cells were split into two pools to test two different tetramer-coding schemes on the same sample. Similar numbers of cryopreserved IEL (mostly CD8+) cells were thawed and rested for 2–4 h before dead cells were removed using the Miltenyi-dead-cell removal kit (Miltenyi Biotec). Cells were then transferred to tubes, washed and resuspended in cytometry buffer (PBS + 0.05% sodium azide + 2 mM EDTA + 2% FCS) for staining as previously described7. In brief, the cells were stained with the tetramer cocktail in the presence of 50 nM dasatinib (Sprycel), which may reduce T-cell receptor internalization and improve tetramer staining31, before antibody staining. Antibody dilution factors used for staining cocktails were tested and adjusted for each batch of antibody conjugated as described above. After setting aside an aliquot of cells for analysis “pre-enrichment”, tetramer enrichment was performed using Miltenyi anti-Myc MACS particles. Dead-cell discrimination and DNA-staining was also performed as described7. For the dead cell discrimination, cell impermeable, metal-labeled Maleimido-mono-amine-DOTA (Macrocyclics Inc., #B-272) was used as previously reported6,7; this stain is based on the same principles as the cell-impermeable amine-reactive LIVE/DEAD stain sold by Invitrogen Inc. and cisplatin-based methods for identifying dead cells32. Unlike live cells, which are not stained by metal-conjugated DOTA-maleimide, fixed and permeabilized cells, like dead cells, are robustly labeled33.
Data analysis
After built-in cell identifying software creates an FCS file, mass cytometry data was analyzed using FlowJo software (Treestar, Inc.) as described7. We gated on live CD8+ T cells using a heavy-metal labeled cell-viability marker and a heavy-metal labeled anti-CD8 antibody. These events were exported to a tab-delimited text file using FlowJo v9 and further analyzed using scripts to identify tetramer-stained cells and tabulate their phenotypic profiles (as described below, see supplemental text) written in Matlab. After, transformation of data into Logicle biexponential scaling as described34 (to allow comparison with FlowJo, which also using the Logicle display method), an automated gating script was used. As illustrated in Figure 2a, several parameters were optimized to allow accurate identification of tetramer stained cells. For each combination of three metals and for each cell, several criteria were used to identify epitope-specific T cells: (i) Each of the relevant three metals must have higher staining level than all seven other metals by a ‘safety’ factor parameter, which was optimized by trial and error with one training data set. (ii) The geometrical parameters illustrated in Figure 2b must be met to ensure correlated levels of each metal were measured for the cells. (iii) None of the other seven metals could have a higher than threshold level of staining, with thresholds set based on cells manually gated as negative for all ten tetramers. Once each parameter was set for the training set, the same parameters were used for analysis for all other samples. For subsequent analysis of expression patterns and frequencies of hits as listed in Table 2, frequencies and average expression levels of all stained markers were compiled in Excel spreadsheets. In some cases, however, due to high background values or bleed-over from high-intensity antibody channels (through oxidation of the heavy metal leading to detection of <3% metal-oxygen adducts at 16 atomic mass units greater than the expected molecular weight for the metal), the script was modified to allow high values for one of the ten tetramer channels. Because the antigen-specific cells were redundantly coded by the triple coding scheme, this was tolerable but not ideal for maintaining optimal stringency. Thus, staining panels for subsequent experiments were adjusted. In experiments where two cocktails of alternatively coded tetramers were used (Fig. 2b,c), the lesser frequency obtained by the combinatorial analysis was used to further reduce false positive hits. Finally, only epitopes with hits in two or more donors were considered for Table 2. To verify that this gating procedure was working properly and to visualize the relevance of the parameters chosen, 3D plots were exported as pdb files and plotted in PyMol as described7 (Supplementary Movie).
To back-calculate frequencies of antigen-specific T cells before tetramer enrichment, two samples mass cytometry samples were acquired for each donor, one taken before the tetramer enrichment procedure and one after. An enrichment factor was obtained by dividing the frequency cells specific for one epitope in the enriched sample by the frequency of cells specific for the same epitope in the pre-enriched fraction. This method requires that at least one population of antigen-specific cells be detectable in the pre-enrichment fraction (for example, specific for EBV, CMV). One donor blood sample failed these criteria and was omitted from the analysis. This enrichment factor was then used to back-calculate frequencies for all antigen-specific cells detected in the enriched sample.
To compare phenotypes of tetramer stained cells, to control for variation in antibody and instrument sensitivity between experiments (3–4 blood donors plus one or two IEL donors tested per experiment), we calculated z-scores for each phenotypic marker for each epitope-donor combination by subtracting the average and dividing by the s.d. obtained by aggregating all blood-derived hits (the average of the tetramer negatives for each donor as one point for normalization as well) within each experiment. Thus, for each donor-epitope combination, a vector of z-scores representing each phenotypic marker was created. In cases when markers were missing due to differences in antibody staining panels, zero z-scores were assigned. This did not appear to interfere to any great extent with the analysis. These vectors were used for subsequent PCA to reduce the dimensionality of the average phenotypic profile for each epitope-donor combination (first 2 dimensions plotted account for ∼40% of variation within the phenotypic profiles of all hits obtained from all blood samples).
Statistical analysis
Although the identities of all donor samples were anonymous, all were considered normal so the analysis done here was not blinded in any way. All t-tests used were two-sided. In cases where mean expression values were compared between populations of antigen-specific cells, Dunn's multiple comparisons test was used to evaluate P values using Prism software (Graphpad).
Supplementary Material
Acknowledgments
We thank members of the Davis, Holden Maecker and Garry Nolan labs for sharing advice and experience concerning mass cytometry and antibody clone usage, especially M. Leipold for help with the mass cytometry instrument, and X. He, F. Wen, W. O'Gorman, A. Han, S. Bendall, O. Goldberger and Y.-H. Chien for helpful discussions. This work was supported by the Bill and Melinda Gates Foundation Grand Challenges Exploration phase I and II grants, National Institutes of Health grants U19-AI057229 and U19-AI090019, and The Howard Hughes Medical Institute. E.W.N. was supported by The American Cancer Society's Steven Stanley and Edward Albert Bielfelt Post-Doctoral Fellowship and by funding through the Singapore Immunology Network.
Footnotes
Note: Supplementary information is available in the online version of the paper.
Author Contributions: E.W.N. conceived and designed the experiments, wrote the manuscript, made and validated CyTOF reagents, wrote analysis scripts, helped with tetramer staining experiments, helped adapt IEL cell preparations for mass cytometry, adapted the epitope prediction algorithm and performed all data analysis. N.S. helped conceive and design the experiments, made and validated CyTOF reagents, made all multiplex tetramer-staining cocktails and performed all tetramer-staining experiments. N.N. optimized the IEL processing procedure for use of IEL samples with mass cytometry and processed all IEL samples for the experiments. B.A.K. adapted the epitope prediction algorithm and performed all epitope predictions. H.B.G. helped conceive and design the experiments. M.M.D. helped conceive and design the experiments, and wrote the manuscript. All authors made corrections and provided critical feedback on the manuscript.
Competing Financial Interests: The authors declare no competing financial interests.
References
- 1.Altman JD, et al. Phenotypic analysis of antigen-specific T lymphocytes. Science. 1996;274:94–96. [PubMed] [Google Scholar]
- 2.Davis MM, Altman JD, Newell EW. Interrogating the repertoire: broadening the scope of peptide-MHC multimer analysis. Nat Rev Immunol. 2011;11:551–558. doi: 10.1038/nri3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Day CL, et al. Ex vivo analysis of human memory CD4 T cells specific for hepatitis C virus using MHC class II tetramers. J Clin Invest. 2003;112:831–842. doi: 10.1172/JCI18509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moon JJ, et al. Naive CD4(+) T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity. 2007;27:203–213. doi: 10.1016/j.immuni.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ornatsky O, Baranov VI, Bandura DR, Tanner SD, Dick J. Multiple cellular antigen detection by ICP-MS. J Immunol Methods. 2006;308:68–76. doi: 10.1016/j.jim.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 6.Bendall SC, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332:687–696. doi: 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Newell EW, Sigal N, Bendall SC, Nolan GP, Davis MM. Cytometry by time-of-fight shows combinatorial cytokine expression and virus-specific cell niches within a continuum of CD8(+) T cell phenotypes. Immunity. 2012:142–152. doi: 10.1016/j.immuni.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang CX, et al. Sources of diversity in T cell epitope discovery. Front Biosci. 2011;16:3014–3035. doi: 10.2741/3895. [DOI] [PubMed] [Google Scholar]
- 9.Lundegaard C, Lund O, Nielsen M. Prediction of epitopes using neural network based methods. J Immunol Methods. 2011;374:26–34. doi: 10.1016/j.jim.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Babji S, Kang G. Rotavirus vaccination in developing countries. Curr Opin Virol. 2012 doi: 10.1016/j.coviro.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 11.Wei J, et al. A naturally processed epitope on rotavirus VP7 glycoprotein recognized by HLA-A2.1-restricted cytotoxic CD8+ T cells. Viral Immunol. 2009;22:189–194. doi: 10.1089/vim.2008.0091. [DOI] [PubMed] [Google Scholar]
- 12.Wei J, et al. Identification of an HLA-A*0201-restricted cytotoxic T-lymphocyte epitope in rotavirus VP6 protein. J Gen Virol. 2006;87:3393–3396. doi: 10.1099/vir.0.82031-0. [DOI] [PubMed] [Google Scholar]
- 13.Ward R. Mechanisms of protection against rotavirus infection and disease. Pediatr Infect Dis J. 2009;28:S57–S59. doi: 10.1097/INF.0b013e3181967c16. [DOI] [PubMed] [Google Scholar]
- 14.Newell EW, Klein LO, Yu W, Davis MM. Simultaneous detection of many T-cell specificities using combinatorial tetramer staining. Nat Methods. 2009;6:497–499. doi: 10.1038/nmeth.1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hadrup SR, et al. Parallel detection of antigen-specific T-cell responses by multidimensional encoding of MHC multimers. Nat Methods. 2009;6:520–526. doi: 10.1038/nmeth.1345. [DOI] [PubMed] [Google Scholar]
- 16.Lundegaard C, et al. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Res. 2008;36:W509–W512. doi: 10.1093/nar/gkn202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rippinger CM, Patton JT, McDonald SM. Complete genome sequence analysis of candidate human rotavirus vaccine strains RV3 and 116E. Virology. 2010;405:201–213. doi: 10.1016/j.virol.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cole DK, et al. Germ line-governed recognition of a cancer epitope by an immunodominant human T-cell receptor. J Biol Chem. 2009;284:27281–27289. doi: 10.1074/jbc.M109.022509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vita R, et al. The immune epitope database 2.0. Nucleic Acids Res. 2010;38:D854–D862. doi: 10.1093/nar/gkp1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. 2004;22:745–763. doi: 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- 21.Gorfu G, Rivera-Nieves J, Ley K. Role of beta7 integrins in intestinal lymphocyte homing and retention. Curr Mol Med. 2009;9:836–850. doi: 10.2174/156652409789105525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ravkov EV, Myrick CM, Altman JD. Immediate early effector functions of virus-specific CD8+CCR7+ memory cells in humans defined by HLA and CC chemokine ligand 19 tetramers. J Immunol. 2003;170:2461–2468. doi: 10.4049/jimmunol.170.5.2461. [DOI] [PubMed] [Google Scholar]
- 23.Pittet MJ, et al. High frequencies of naive Melan-A/MART-1-specific CD8(+) T cells in a large proportion of human histocompatibility leukocyte antigen (HLA)-A2 individuals. J Exp Med. 1999;190:705–716. doi: 10.1084/jem.190.5.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DeNucci CC, Pagan AJ, Mitchell JS, Shimizu Y. Control of alpha4beta7 integrin expression and CD4 T cell homing by the beta1 integrin subunit. J Immunol. 2010;184:2458–2467. doi: 10.4049/jimmunol.0902407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng N, et al. Inhibition of rotavirus replication by a non-neutralizing, rotavirus VP6-specific IgA mAb. J Clin Invest. 2002;109:1203–1213. doi: 10.1172/JCI14397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matthijnssens J, et al. Recommendations for the classification of group A rotaviruses using all 11 genomic RNA segments. Arch Virol. 2008;153:1621–1629. doi: 10.1007/s00705-008-0155-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fiocchi C, Youngman KR. Isolation of human intestinal mucosal mononuclear cells. Curr Prot Immunol. 2001;Chapter 7(19):7.30. doi: 10.1002/0471142735.im0730s19. [DOI] [PubMed] [Google Scholar]
- 28.Ramachandiran V, et al. A robust method for production of MHC tetramers with small molecule fluorophores. J Immunol Methods. 2007;319:13–20. doi: 10.1016/j.jim.2006.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Toebes M, et al. Design and use of conditional MHC class I ligands. Nat Med. 2006;12:246–251. doi: 10.1038/nm1360. [DOI] [PubMed] [Google Scholar]
- 30.Bakker AH, et al. Conditional MHC class I ligands and peptide exchange technology for the human MHC gene products HLA-A1, -A3, -A11, and -B7. Proc Natl Acad Sci USA. 2008;105:3825–3830. doi: 10.1073/pnas.0709717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lissina A, et al. Protein kinase inhibitors substantially improve the physical detection of T-cells with peptide-MHC tetramers. J Immunol Methods. 2009;340:11–24. doi: 10.1016/j.jim.2008.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fienberg HG, Simonds EF, Fantl WJ, Nolan GP, Bodenmiller B. A platinum-based covalent viability reagent for single-cell mass cytometry. Cytometry A. 2012;81:467–475. doi: 10.1002/cyto.a.22067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bodenmiller B, et al. Multiplexed mass cytometry profiling of cellular states perturbed by small-molecule regulators. Nat Biotechnol. 2012;30:858–867. doi: 10.1038/nbt.2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parks DR, Roederer M, Moore WA. A new “Logicle” display method avoids deceptive effects of logarithmic scaling for low signals and compensated data. Cytometry A. 2006;69:541–551. doi: 10.1002/cyto.a.20258. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.