

Abstract
The microvascular endothelium of the kidney glomerulus is injured in Shiga-like toxigenic bacterial infection, genetic or acquired loss of complement regulatory protein function, and allo-immune responses of solid-organ or bone marrow transplantation. Existing models of diseases with glomerular endothelial cell (EC) injury, collectively grouped as thrombotic microangiopathies, are problematic, impeding investigation of the mechanisms of microvascular defense and repair. To develop a model of glomerular endothelial injury in the mouse, we conjugated the M. oreades lectin to the cytotoxin, saporin, (LS) to selectively injure the glomerular endothelium. Injury of the microvasculature was evaluated by light, immunofluorescence, and electron microscopy, and by quantitative RT-PCR of cell-type specific transcripts. Renal function was evaluated by quantitation of serum creatinine. The toxin conjugate induced apoptosis of microvascular ECs in vitro, and subtle histologic features of thrombotic microangiopathy in vivo that were enhanced by co-injection of 50 μg/kg LPS. Among LS/LPS-treated animals, loss of glomerular EC staining correlated with decreased expression of EC-specific transcripts, and impaired kidney function. Selective injury of the glomerular microvasculature with LS toxin conjugate and LPS elicits histologic features of thrombotic microangiopathy and acute kidney failure.
Introduction
The vascular endothelium is the principal target of injury in a group of disorders collectively termed thrombotic microangiopathies (TMA). TMA is initiated by diverse processes including uncontrolled complement protein activity caused by inherited or acquired defects in complement regulatory proteins, cytotoxic drugs, or immune responses to allogeneic endothelium in the context of allogeneic bone marrow or solid organ transplantation. The most common cause of TMA is Shiga-toxigenic (Stx) E coli infection. TMA injury occurred in about 25% of infected cases to contribute to the high death rate observed in the recent German epidemic [1,2], and may be associated with chronic renal dysfunction among survivors [3,4]. Glomerular endothelial injury associated with the atypical hemolytic-uremic syndrome frequently leads to end stage kidney failure [5]. Similarly, transplant-associated TMA is a significant cause of morbidity, mortality, and kidney allograft loss [6,7].
In these disorders, acute microvascular thrombosis of the kidney glomerulus that compromises kidney function is a presenting feature in most cases. In vitro studies indicate Stx binds human microvascular endothelial cells (EC) to induce apoptosis [8–12], but a variety of subtle effects on endothelial gene transcription induced by sublethal toxin concentrations may also occur [13,14]. In addition, systemic activation of coagulation proteins both precedes and correlates with subsequent microvascular thrombosis in Shiga toxigenic infection, consistent with a parallel contribution of direct EC injury, and inflammation-driven prothrombotic effects to kidney pathology [15]. Study of the mechanisms of glomerular microvascular injury and repair following Stx administration to rodents, however, are confounded by predominant injury of the kidney tubular epithelial cells in vivo in the absence of glomerular EC injury [16,17].
In transplantation, both classical T cell-mediated and antibody-mediated (ABMR) allograft rejection target the microvasculature, recognized in clinical biopsy specimens by features such as subendothelial accumulation of lymphocytes in the allograft arterial intima, glomerulitis, and complement C4d labeling of the endothelium [18]. Endothelial injury in rejection can also be fulminant, typically associated with combined acute cell- and alloantibody-mediated attack, resulting in widespread loss of the endothelium, microvascular thrombosis, and parenchymal cell injury as a consequence of the disturbed microcirculation [19–23]. Isolated allo-antibody-mediated injury has proven difficult to model in rodents [24,25].
We describe a mouse model of acute microvascular endothelial injury selective for kidney glomerular endothelium with synchronized vascular damage and repair. Delivery of a toxin to the glomerular endothelium induces a wave of injury characterized by microvascular thrombosis and fulminant kidney failure. At sublethal doses, glomerular fibrin deposition, microvascular cell apoptosis and EC loss are evident.
Materials and Methods
Animals and ethics statement
Fourteen to 18 week old C57BL/6 female mice (Jackson Laboratory) were maintained according to Canadian Council for Animal Care (CCAC) guidelines under a protocol approved by the Health Sciences Animal Care and Use Committee of the University of Alberta.
Reagents
The following products were purchased: pure and biotinylated M. oreades lectin A (MOA) (EY labs; San Mateo, CA); LPS O55:B5, saporin, and the biotinylated L. esculentum lectin (Sigma-Aldrich; St. Louis, MO); rabbit antibody to cleaved caspase 3 (Cell Signaling Technology; Boston, MA); rat anti-mouse CD31 (BD Pharmingen; Mississuaga, ON); rabbit anti-mouse fibrinogen (GenWay; San Diego, CA); anti-mouse podocalyxin (R&D systems); anti-mouse podocin (Santa Cruz Biotechnology; Santa Cruz, CA); goat anti-rabbit, –rat, or -mouse IgG conjugated to FITC, or DyLight 549 (Jackson ImmunoResearch Laboratories, Inc.). RNA was isolated using RNAeasy Mini Kit (Qiagen; Toronto, ON). The cDNA was prepared using qScript cDNA SuperMix from Quanta Biosciences (Gaithersburg, MD).
Conjugation of MOA lectin with saporin
Five mg of pure MOA lectin and saporin were conjugated using sulfo-LC-SPDP as per manufacturer’s instructions (Thermo Scientific, Rockford, IL). The conjugate solution was processed by FPLC and a size-exclusion column (Superdex 75, GE Lifesciences). The resulting fractions of the lectin-saporin conjugate (LS) were analyzed for killing activity against mouse cardiac microvascular endothelial cells (MCEC; [26]); CELLutions Biosystems, Burlington, ON)) in vitro. The active fractions of pure LS were pooled, concentrated, and filter-sterilized using an 0.2 micron filter.
Cleaved caspase 3 assay
MCEC were washed in M199 media, then treated either with 20 μg/mL LS, 20 µg/mL unconjugated lectin, 10 ng/mL TNFα+ 3 μg/mL cycloheximide, PBS, or left untreated and incubated for 4 h at 37 °C and 5 % CO2. The cells were fixed and permeabilized, then stained with the anti-cleaved caspase 3 antibody at 1:3000 concentration for one hour. Goat anti-rabbit Dylight 549 secondary antibody was added at a 1:400 dilution. Finally, the caspase 3-positive cells were detected by flow cytometry.
MOA lectin administration in vivo
Five C57Bl/6 mice were injected via tail vein with 10 mg/kg biotinylated MOA lectin. The animals were euthanized by cervical dislocation after 3 h and the following organs were collected: heart, lung, kidney, liver, skeletal muscle, and brain. The organs were placed in OCT solution and snap-frozen in liquid nitrogen. The MOA lectin was visualized using Alexa Fluor 594-conjugated streptavidin (Jackson ImmunoResearch) and confocal microscopy. In subsequent experiments, where indicated, MOA lectin (500 μg/kg), LPS (50 μg/kg), or LS (200 or 500 μg/kg) ± LPS in 100 μL were introduced via retrograde carotid artery injection.
Histology and immunohistochemical staining
Kidney, heart, lung, liver, spleen, skeletal muscle, and brain were collected and either snap frozen in OCT or placed in IHC zinc fixative (BD Pharmingen). The tissues were stained with Hematoxylin & Eosin (H & E) and with periodic acid- Schiff (PAS). For immunofluorescence staining, 5 μm frozen sections were stained for fibrin, CD31, podocalyxin, podocin, CD42, CD45, or with L. esculentum lectin to specifically label glomerular and peritubular capillary endothelium, and imaged with a spinning disc confocal microscope (Quorum Wave FX-X1). For transmission electron microscopy, tissues ~1 mm2 from LPS/ LS or saline injected mice were fixed using Karnovsky fixative solution (Poly Scientific, New York, NY) and embedded in the low viscosity embedding Spurr's Kit (Electron Microscopy Sciences, Hatfield, PA) according to manufacturer's instructions. Ultra-thin sections were stained with uranyl acetate and lead citrate and viewed using a Philips 410 transmission electron microscope.
Real-time PCR
All PCR primers were designed using Primer Express software (ABI) and produced by Integrated DNA Technologies (San Diego, CA). See Table 1 for a list of the primers used in this study. One μg of total RNA was reversed transcribed into cDNA. SYBR green was used for the real-time PCR (7500 thermocycler, ABI). The data were analyzed using the δδCt relative quantitation method.
Table 1. Primers used for qRT-PCR.
| Gene | Forward | Reverse |
|---|---|---|
| CD31 | AGGACGATGCGATGGTGTATAA | AAGACCCGAGCCTGAGGAA |
| TIE2 | GGGCGAAAAAAGTTG TTTGG | CGAACTCGACCTTCACAGAAATAA |
| eNOS | TGTCTGCGGCGATGTCACTA | CATGCCGCCCTCTGTTG |
| Nephrin | GCGAGGCACTTCGTGAAAC | CACTTGCTCTCCCAGGAACTCT |
| KIM1 | CCGCAGAAAAACCCTACTAAGG | TGCTCAC AAGGAGCAGTAGCA |
Creatinine analysis
Serial serum samples were sent to the Metabolomics Innovation Center at the University of Alberta to apply a quantitative analysis of creatinine using a combination of direct injection mass spectrometry with reverse-phase LC-MS/MS (AbsoluteIDQ Kit; Biocrates Life Sciences AG, Austria). The serum samples were analyzed with the AbsoluteIDQ kit as directed by the manufacturer.
Semiquantitative analysis of the renal injury
The histology slides were evaluated by a renal pathologist, DCR, in a blinded fashion. Up to thirty consecutive glomeruli in equatorial section were scored per animal. The kidney sections were scored for glomerular injury, including fibrin and apoptotic cells within the capillary lumen, as a fraction of total glomeruli evaluated as described [27]. Tubular injury was scored using a semi-quantitative scale 0-4 (0- normal, 1- ≤ 25 %, 2- ≤ 50 %, 3- ≤ 75 %, and 4- ≥ 75 % fields affected) as described [28].
Statistical analysis
The data are expressed as mean ± SD, and analyzed using single or 2-way ANOVA with pairwise comparisons evaluated posthoc using Tukey’s test (GraphPad PRISM, La Jolla, CA). Kaplan-Meier curves were analyzed by the log-rank test. Nonparametric data were analyzed with the Mann-Whitney test. A p value < 0.05 was considered significant.
Results
To generate a potent compound to selectively injure microvascular endothelial cells, we conjugated the toxin, saporin, to the lectin A derived from M. oreades. Preliminary testing in vitro determined that the lectin-saporin (LS) conjugate, but not saporin or the lectin alone, bound and killed cultured mouse microvascular MCEC, determined by MTT assay of cell viability (data not shown). LS treatment of growth factor-starved MCEC induced apoptosis, indicated by activated caspase 3 staining, of 30.3 ± 5.0% of LS-treated cells versus 0.1 ± 0.1% of PBS-, 1.7 ± 0.4% of lectin-, or 17.5 ± 4.3% Tumor Necrosis Factor-α/cycloheximide-treated control EC (mean ± SEM; n=3 independent experiments; LS versus PBS p < 0.05) after 4 hours of treatment. This indicates that the lectin efficiently delivered the toxin to the microvascular EC to induce endothelial apoptosis in vitro in the absence of an additional pro-apoptotic stimulus.
Next, we determined the binding characteristics of MOA lectin in vivo. Earlier work identifies glomerular microvascular endothelial binding of this lectin [29]. Following iv injection into Bl/6 mice, we observed that biotinylated MOA lectin selectively labeled both heart and kidney glomerular microvascular EC (Figure 1). In contrast, when the biotinylated lectin was used as a staining reagent on frozen sections of normal kidney, both glomerular and peritubular capillary EC were uniformly labeled, suggesting that the circulating lectin was largely adsorbed from the blood during transit through the glomerulus. Since we wished to avoid injury to the heart microcirculation, we injected the biotinylated lectin intra-arterially, retrograde via the left carotid artery. We found glomerular but little heart EC labeling using this approach (Figure 1B, C, F). Within the glomerulus we found the lectin selectively bound the EC, but not the mesangial cells or podocytes (Figure 1C-H). We observed no staining of tissue sections from lung, liver, skeletal muscle or brain (data not shown).
Figure 1. MOA lectin binds the glomerular endothelium.

Intravenous injection of MOA lectin labels heart microvascular endothelium (A), but after intra-arterial injection, the kidney glomerular (C, F) but not heart endothelium (B) is labeled. Endothelial cells are labeled with anti-CD31 (green; B (right panel), D), or MOA lectin (red; A, B (left panel), C, F). Double-labeled merged images of CD31 and MOA lectin (E) demonstrate overlapping distribution in the glomerulus. No overlap is identified between MOA lectin (F) and the podocyte marker, podocin (G) in the merged image (H).
To emulate the pro-coagulant environment associated with many thrombotic microangiopathies, we opted to study mice co-treated with LPS at a threshold dose for tissue factor induction in the kidney [27]. In the first set of experiments, Bl/6 mice were treated with saporin, unconjugated MOA lectin, LS conjugate 500 μg/kg, LPS (50 μg/kg), LPS + LS, or saline, then tissues were harvested at 12 h and examined by a blinded observer (DCR) for evidence of thrombotic microvascular injury. No injury was observed among animals treated with saporin, MOA lectin, or LPS alone. Treatment with the LS conjugate alone, or as shown in Figure 2, treatment with LPS and the LS conjugate, elicited diffuse glomerular capillary thrombosis, but no injury of the heart microvasculature. Glomeruli from three LPS/ LS-injured mice were evaluated for quantitative analysis. We found 72 ± 14% of glomeruli from injured mice showed microvascular thrombosis compared to none from control mice (n=90 glomeruli; p<0.05). Glomerular capillaries stained for fibrin by immunohistochemistry, and fibrin clot was identified in glomerular capillary loops on transmission electron microscopy of LPS/ LS-treated mice (Figure 2C, D, E). The fenestrated EC was absent in many glomerular capillaries examined by electron microscopy (Figure 2D), and luminal cells adjacent to the basement membrane stain for activated caspase 3 by immunohistochemistry (Figure 2F). In contrast, intact podocyte foot process distribution was evident (Figure 2D). Occasional shistocytes could be identified on blood smears. No thrombosis of peritubular capillaries or injury to the tubular epithelial cells was evident at this early time-point. This data demonstrates that LS treatment induces rapid, selective injury of the glomerular endothelium resulting in thrombotic microangiopathy. However, at the 500 µg/kg LS dose, LPS/ LS and LS treatments were uniformly lethal, limiting the utility for experimentation (Figure 3).
Figure 2. Glomerular thrombotic microangiopathy develops after LPS/ LS treatment.

Kidneys were harvested 12 h after saline (A, C) or LPS/ LS (500 μg/kg) treatment (B, D, E, F). Hematoxylin and eosin staining reveals widespread amorphous eosinophilic glomerular capillary thrombi (B), whereas red blood cells are seen in patent capillaries in (A). Transmission electron microscopy shows focal loss of glomerular endothelium (D), and capillary thrombosis (D, E), but preserved podocyte foot processes similar to the control (D versus C). Occasional cells in glomerular capillary lumens are found to stain for activated caspase 3 (F; inset: irrelevant antibody control). RBC- red blood cell; GEC- glomerular endothelial cell; FP- podocyte foot process; F- fibrin clot; bm- basement membrane.
Figure 3. Dose-response effect of LPS/ LS or LS treatment on survival and glomerular injury.

Kaplan-Meier survival graph of mice treated with A) saline (n=8), LPS/ LS 500 μg/kg (n=38), or LPS/ LS 200 μg/kg LS (n=24), or B) LS alone 500 μg/kg (n=4), 200 μg/kg LS (n=14). p < 0.001, LS/LPS 500 versus LS/LPS 200 μg/kg; p < 0.001, LS 500 versus LS 200 μg/kg. c) The fraction of injured glomeruli at Day 4 (n=5 mice/ group; * p < 0.05).
In subsequent experiments, mice were treated with 200 μg/kg LS, or LPS/ LS, to characterize the functional consequence of sub-lethal toxin-induced glomerular endothelial injury. This lower dose of LS was tolerated (Figure 3A, B), and glomerular injury was evaluated (Figure 3C). Sub-lethal injury to the microvasculature is reflected at Day 4 by loss of microvascular ECs and regenerative changes most evident in the glomerular endothelial and tubular epithelial cell compartments of the kidney of LPS/ LS-treated mice. As shown in Figure 4, within the glomerulus we identified intralumenal apoptotic cells, frequently positioned adjacent to the capillary wall (Figure 4A). Mononuclear cells were seen in the glomerular capillaries (0.76 ± 0.24 cells/ glomerulus among LPS/LS treated mice versus 0.36 ± 0.17 cells/ glo among PBS controls; n=5 mice/ group; p = NS), and PMN were rarely seen. However, only rare CD45-positive leukocytes or CD42-positive platelets were identified in the glomerular microcirculation of LPS/LS-treated or control mice by immunofluorescence staining of frozen sections. Immunostaining for EC was discontinuous in glomerular capillaries of mice treated with the sub-lethal dose of LPS/ LS (Figure 5). Fibrin deposition was evident around the margin of the glomerular capillary loops in immunofluorescence microscopy (Figure 5). In contrast, LS 200 μg/kg given without LPS resulted in no mortality, and little change in glomerular immunostaining for EC or fibrin compared to controls (data not shown).
Figure 4. Glomerular microvascular injury after sublethal LPS/ LS treatment.
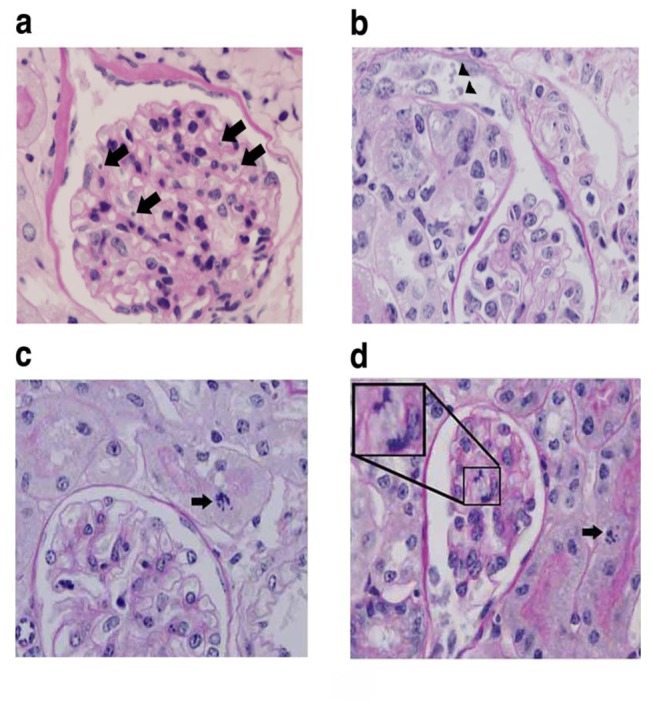
Mice were treated with LPS/ LS 200 μg/kg and tissues were harvested 4 days later and stained with PAS. Apoptotic cells (arrows) are seen in the glomerular capillaries (A) and tubules (B) after LS treatment. Regenerative mitotic changes are evident in the tubular (C) and glomerular capillary compartments (D).
Figure 5. Glomerular microvascular injury after sublethal LPS/ LS treatment.

L. esculentum lectin labeling of endothelium (upper panels, red) in glomerular (circled) and peritubular capillaries. Glomerular capillary fibrin immunostaining (lower panels, green) after LPS/ LS treatment. Glomerular and peritubular capillary endothelial staining at day 4 appears inhomogeneous, with glomerular fibrin accumulation, and normalizes at day 7. Representative of 5 mice/ group.
In agreement with these features of glomerular EC injury, the abundance of constitutively expressed EC-specific transcripts, CD31, TIE2, and NOS3, in the kidney cortex was reduced ~40% in sub-lethal LPS/ LS-treated versus control mice (Figure 6). Glomerular podocyte foot processes were focally effaced on EM images at Day 4 after injury, but the podocyte-specific transcript nephrin was unchanged during the acute phase of injury between Day 0-4 in LPS/LS-treated animals versus controls (Figure 6).
Figure 6. Endothelial gene abundance after glomerular EC injury.
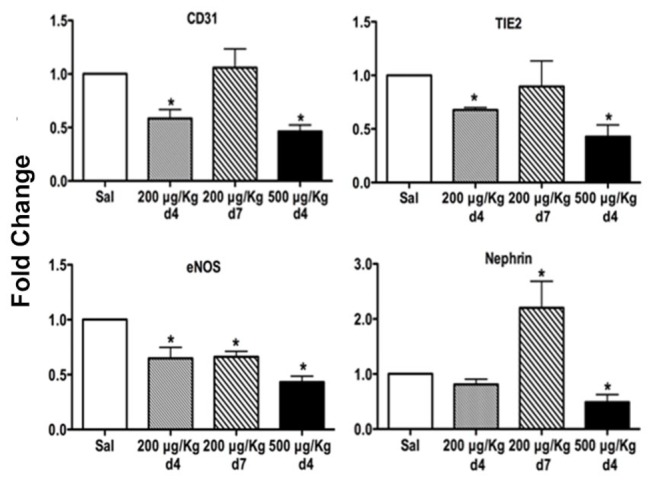
Mice were treated with saline or LPS/ LS 200 μg/kg to induce glomerular EC damage, then kidney cortex mRNA was isolated at day 4 or day 7 after injury. Constitutively expressed endothelial genes CD31, TIE2, and eNOS or the podocyte-specific gene nephrin were assessed by qRT-PCR (n = 5 mice/ group; * - p < 0.05 versus saline control).
Injury and regenerative change in the tubular cell compartment of the kidney was prominent at Day 4 as anticipated as a consequence of the disordered microcirculation in the sub-lethally injured animals (Figure 4B, C; Figure 7). Similarly, expression of KIM-1, a tubular epithelial cell stress gene, was elevated in both LS treatment groups at Day 4 after injury. At Day 7 after LPS/LS injury, pathologic features of tubular injury were resolving, and KIM-1 expression normalized (Figure 7). This data indicates that sub-lethal doses of LS induce microvascular endothelial loss that is followed by evidence of tubular epithelial injury and regeneration.
Figure 7. Tubular epithelial cell damage occurs after glomerular EC injury.

Tubular epithelial cell effacement and regeneration is evident at day 4 after LPS/ LS 200 μg/kg treatment (A). Features of tubular injury and repair are quantitated in (B) as described in Methods. Expression of the tubular epithelial cell stress gene KIM-1 is quantitated by qRT-PCR in (C). (n = 5 mice/ group; *- p<0.05 versus 200 μg/kg day 4 group; **- p<0.05 versus saline group).
Repair of the injured vascular endothelium was evident on Day 4 and Day 7 tissue sections in the sublethal LS injured group, reflected by mitotic figures within the glomerular capillaries (Figure 4C, D), normalization of glomerular and peritubular capillary immunostaining for EC and fibrin (Figure 5), and endothelial-specific gene expression (Figure 6) at Day 7. Serum creatinine, as a measure of kidney function, was increased in the lethal LPS/LS dose group, consistent with acute renal failure associated with glomerular TMA injury. The creatinine showed an intermediate rise in the LS 200 μg/kg dose groups versus baseline, and was normalizing by Day 7 (Figure 8).
Figure 8. Kidney function is impaired in LS-treated mice.
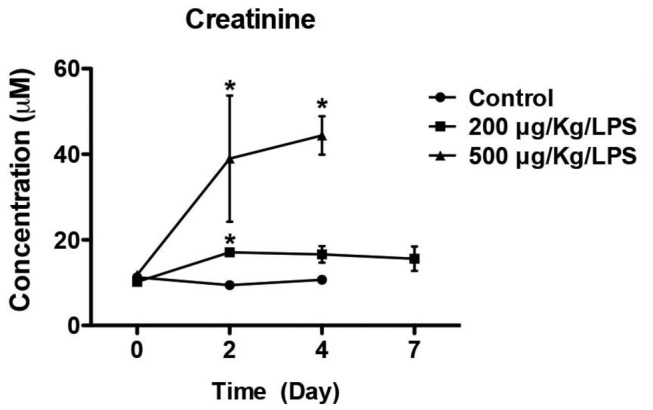
Serum creatinine was assayed by LC/MS/MS. (n = 3-6 mice/ group; p<0.01 LPS/ LS versus saline control in 2-way ANOVA. * - p<0.05 versus saline control in pairwise comparison).
Discussion
Endothelial injury of the kidney glomerular microvasculature is a key feature of diverse diseases resulting in thrombotic microangiopathy. The consequences of these disorders on organ function represent a considerable health burden to affected individuals. In particular, Stx-mediated glomerular EC injury in epidemic toxigenic E coli infections contributes to morbidity and mortality of both children and adults [1–4]. Moreover, among transplant recipients, cell-mediated allo-immune responses, and the more recently recognized antibody-mediated rejection, target the EC in kidney allografts [20]. Although low-grade injury is tolerated for a time, the cumulative burden of damage to the microvasculature ultimately limits kidney allograft survival [30–33]. Animal models of this disease are needed to develop an understanding of the mechanisms of endothelial defense, consequences of injury, and key repair mechanisms that might be exploited to minimize organ damage or prolong graft survival.
We describe a new model of temporally coordinated glomerular EC damage in the mouse, and characterize the immediate consequences of glomerular microvascular compromise on the kidney. With marked injury, widespread thrombosis of the glomerular capillaries is seen, followed by features of tubular injury and functional compromise of the kidney precipitating death in a few days. These features resemble the early phase of microvascular injury observed in clinical specimens with Stx- or alloantibody-mediated damage [20,21,23,34–37]. More limited endothelial injury, on the other hand, provokes moderate functional compromise associated with features of repair in the vascular and tubular cell compartments.
Other approaches to model thrombotic microangiopathy in the mouse have been developed. Mutations in complement regulatory proteins [38], or vascular endothelial growth factor [39] elicits asynchronous, chronic progressive microvascular thrombosis and ultimately death. Administration of Shiga-like toxins elicit acute kidney injury in the mouse [14,40], and directly damages kidney tubular epithelial, but not glomerular endothelial cells [16,17]. In a model of allo-immune EC injury, transplantation into CCR5-deficient mice sensitized to donor allo-antigen, elicits microvascular complement deposition and heart allograft rejection as a consequence of antibody binding to allogeneic EC and cardiac myocytes [24,25]. Selective injury of the microvasculature has been approached using lectins that label the endothelial glycocalyx after intravenous injection [41,42]. Concanavalin A, for example, binds endothelium from diverse microvascular beds in rodents, but selective injection into a renal artery followed by anti-concanavalin A antibody induces widespread microvascular injury associated with inflammatory changes in the glomerulus and peritubular capillaries [43].
These valuable models support investigations of the role of complement, alloantibody, and innate immune cell actions on endothelium, but often induce asynchronous complex injury that may not be selective for the kidney microvascular endothelium. Deficient expression of one or more components of the complement system in many inbred laboratory mouse strains may confound these approaches [44]. The current model has the strength of widespread, synchronized, and selective GEC injury resulting in reproducible TMA and compromised kidney function. The approach is less technically challenging than the concanavalin A model, since selective renal artery cannulation is not required. Like the Stx A subunit, the saporin moiety of the LS conjugate is known to inhibit ribosome-dependent translation, hence this injury model closely emulates the mechanism of the human hemolytic uremic disease, but lacks the confounding effect of direct Stx toxicity to tubular epithelial cells [16,17,45].
The MOA lectin has been characterized to specifically bind to the Gal-α(1,3)Gal epitope on glycoproteins displayed on the glycocalyx of mouse glomerular endothelium [29]. Injection of the MOA lectin alone at high doses was reported to elicit proteinuria, but not features of diffuse TMA, consistent with our observations. In contrast to this previous report, we observed the lectin binding to the heart microvascular EC in vitro and in vivo in Bl/6 mice after iv injection, but selective kidney targeting could be achieved by systemic intra-arterial injection. Injury of the heart microvasculature is under study to model the effects of microvascular injury and repair in that organ.
Injury of the glomerular endothelium was evident in both the high- and low-dose LS treatment groups. At early timepoints, we observed focal loss of the glomerular endothelium and EC injury reflected by loss of fenestration on transmission electron microscopy, inhomogeneous glomerular EC immunostaining, and focal fibrin deposition in glomerular capillaries after LPS/ LS treatment. This morphological data is supported by decreased transcript abundance of the characteristic constitutively-expressed endothelial genes CD31, TIE2, and NOS3. In contrast, injury to the podocyte, tubular cell, and peritubular capillary compartments is evident after the GEC injury. Taken together this data indicates primary GEC injury induced by LPS/ LS treatment, resulting in features of TMA.
The addition of low-dose LPS to the LS toxin conjugate enhanced the reproducibility of glomerular injury. A low dose challenge with LPS was used to promote a prothrombotic environment [27,40,46], and we observed no effect of LPS alone on the kidney histology or function, consistent with previous reports [17,40]. Although LPS alone has been used as a model of kidney injury in lethal septicemia, the dose of LPS administered here is ~100 fold lower. Nevertheless, LPS in combination with cycloheximide, a ribosome inhibitory toxin similar in action to saporin, is known to induce EC apoptosis in vitro [47,48], hence we also expect that EC injury in LS-treated animals was enhanced by LPS. Together these agents reproduce the two important components, EC injury and hypercoagulability, of the pathophysiology thought to be involved in the development of human thrombotic microangiopathy [15].
In summary, we describe a mouse model of selective glomerular endothelial injury that elicits the pathological features of TMA similar to those seen in Stx-mediated acute kidney injury, and antibody-mediated allograft rejection. The synchronized endothelial injury is associated with kidney dysfunction and should facilitate investigation of mechanisms underlying defense and repair of the microvasculature, and recovery of organ function.
Acknowledgments
The authors acknowledge the technical assistance of Yue Huang.
Funding Statement
Funding for this work was obtained in part from the Canadian Institutes for Health Research operating grant (MOP 53319) to AGM. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Frank C, Werber D, Cramer JP, Askar M, Faber M et al. (2011) Epidemic profile of Shiga-toxin-producing Escherichia coli O104:H4 outbreak in Germany. N Engl J Med 365: 1771-1780. doi: 10.1056/NEJMoa1106483. PubMed: 21696328. [DOI] [PubMed] [Google Scholar]
- 2. Magnus T, Röther J, Simova O, Meier-Cillien M, Repenthin J et al. (2012) The neurological syndrome in adults during the 2011 northern German E. coli serotype O104:H4 outbreak. Brain 135: 1850-1859. doi: 10.1093/brain/aws090. PubMed: 22539260. [DOI] [PubMed] [Google Scholar]
- 3. Oakes RS, Kirkham JK, Nelson RD, Siegler RL (2008) Duration of oliguria and anuria as predictors of chronic renal-related sequelae in post-diarrheal hemolytic uremic syndrome. Pediatr Nephrol 23: 1303-1308. [DOI] [PubMed] [Google Scholar]
- 4. Siegler RL, Pavia AT, Christofferson RD, Milligan MK (1994) A 20-year population-based study of postdiarrheal hemolytic uremic syndrome in Utah. Pediatrics 94: 35-40. PubMed: 8008534. [PubMed] [Google Scholar]
- 5. Fremeaux-Bacchi V, Fakhouri F, Garnier A, Bienaimé F, Dragon-Durey MA et al. (2013) Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol 8: 554-562. doi: 10.2215/CJN.04760512. PubMed: 23307876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Colvin RB (2007) Antibody-mediated renal allograft rejection: diagnosis and pathogenesis. [Review ] [61 refs]. J Am Soc Nephrol 18: 1046-1056 doi: 10.1681/ASN.2007010073. PubMed: 17360947. [DOI] [PubMed] [Google Scholar]
- 7. Laskin BL, Goebel J, Davies SM, Jodele S (2011) Small vessels, big trouble in the kidneys and beyond: hematopoietic stem cell transplantation-associated thrombotic microangiopathy Blood 118: 1452-1462. doi: 10.1182/blood-2011-02-321315. PubMed: 21596850. [DOI] [PubMed] [Google Scholar]
- 8. Burns ER, Zucker-Franklin D (1982) Pathologic effects of plasma from patients with thrombotic thrombocytopenic purpura on platelets and cultured vascular endothelial cells. Blood 60: 1030-1037. PubMed: 7052160. [PubMed] [Google Scholar]
- 9. Pijpers AH, van Setten PA, van den Heuvel LP, Assmann KJ, Dijkman HB et al. (2001) Verocytotoxin-induced apoptosis of human microvascular endothelial cells. J Am Soc Nephrol 12: 767-778. PubMed: 11274238. [DOI] [PubMed] [Google Scholar]
- 10. Erwert RD, Winn RK, Harlan JM, Bannerman DD (2002) Shiga-like toxin inhibition of FLICE-like inhibitory protein expression sensitizes endothelial cells to bacterial lipopolysaccharide-induced apoptosis. J Biol Chem 277: 40567-40574. doi: 10.1074/jbc.M206351200. PubMed: 12189147. [DOI] [PubMed] [Google Scholar]
- 11. Brigotti M, Alfieri R, Sestili P, Bonelli M, Petronini PG et al. (2002) Damage to nuclear DNA induced by Shiga toxin 1 and ricin in human endothelial cells. FASEB J 16: 365-372. doi: 10.1096/fj.01-0521com. PubMed: 11874985. [DOI] [PubMed] [Google Scholar]
- 12. Fujii J, Wood K, Matsuda F, Carneiro-Filho BA, Schlegel KH et al. (2008) Shiga toxin 2 causes apoptosis in human brain microvascular endothelial cells via C/EBP homologous protein. Infect Immun 76: 3679-3689. doi: 10.1128/IAI.01581-07. PubMed: 18541659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Matussek A, Lauber J, Bergau A, Hansen W, Rohde M et al. (2003) Molecular and functional analysis of Shiga toxin-induced response patterns in human vascular endothelial cells. Blood 102: 1323-1332. doi: 10.1182/blood-2002-10-3301. PubMed: 12702508. [DOI] [PubMed] [Google Scholar]
- 14. Petruzziello-Pellegrini TN, Yuen DA, Page AV, Patel S, Soltyk AM et al. (2012) The CXCR4/CXCR7/SDF-1 pathway contributes to the pathogenesis of Shiga toxin-associated hemolytic uremic syndrome in humans and mice. J Clin Invest 122: 759-776. doi: 10.1172/JCI57313. PubMed: 22232208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chandler WL, Jelacic S, Boster DR, Ciol MA, Williams GD et al. (2002) Prothrombotic coagulation abnormalities preceding the hemolytic-uremic syndrome. N Engl J Med 346: 23-32. doi: 10.1056/NEJMoa011033. PubMed: 11777999. [DOI] [PubMed] [Google Scholar]
- 16. Psotka MA, Obata F, Kolling GL, Gross LK, Saleem MA et al. (2009) Shiga toxin 2 targets the murine renal collecting duct epithelium. Infect Immun 77: 959-969. doi: 10.1128/IAI.00679-08. PubMed: 19124603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Paixão-Cavalcante D, Botto M, Cook HT, Pickering MC (2009) Shiga toxin-2 results in renal tubular injury but not thrombotic microangiopathy in heterozygous factor H-deficient mice. Clin Exp Immunol 155: 339-347. doi: 10.1111/j.1365-2249.2008.03826.x. PubMed: 19040606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Solez K, Colvin RB, Racusen LC, Haas M, Sis B et al. (2008) Banff 07 classification of renal allograft pathology: updates and future directions. Am J Transplant 8: 753-760. doi: 10.1111/j.1600-6143.2008.02159.x. PubMed: 18294345. [DOI] [PubMed] [Google Scholar]
- 19. Kissmeyer-Nielsen F, Olsen S, Petersen VP, Fjeldborg O (1966) Hyperacute rejection of kidney allografts, associated with pre-existing humoral antibodies against donor cellls. Lancet 288: 662-665. doi: 10.1016/S0140-6736(66)92829-7. [DOI] [PubMed] [Google Scholar]
- 20. Trpkov K, Campbell P, Pazderka F, Cockfield S, Solez K et al. (1996) Pathologic features of acute renal allograft rejection associated with donor-specific antibody. Transplant 61: 1586-1592. doi: 10.1097/00007890-199606150-00007. [DOI] [PubMed] [Google Scholar]
- 21. Mauiyyedi S, Crespo M, Collins AB, Schneeberger EE, Pascual MA et al. (2002) Acute humoral rejection in kidney transplantation: II. Morphology, immunopathology, and pathologic classification. J Am Soc Nephrol 13: 779-787. PubMed: 11856785. [DOI] [PubMed] [Google Scholar]
- 22. Magro CM, Calomeni EP, Nadasdy T, Shusterman BD, Pope-Harman AL et al. (2005) Ultrastructure as a diagnostic adjunct in the evaluation of lung allograft biopsies. Ultrastruct Pathol 29: 95-106. doi: 10.1080/019131290924108. PubMed: 16028666. [DOI] [PubMed] [Google Scholar]
- 23. Lipták P, Kemény E, Morvay Z, Szederkényi E, Szenohradszky P et al. (2005) Peritubular capillary damage in acute humoral rejection: an ultrastructural study on human renal allografts. Am J Transplant 5: 2870-2876. doi: 10.1111/j.1600-6143.2005.01102.x. PubMed: 16302999. [DOI] [PubMed] [Google Scholar]
- 24. Bickerstaff A, Pelletier R, Wang JJ, Nadasdy G, DiPaola N et al. (2008) An experimental model of acute humoral rejection of renal allografts associated with concomitant cellular rejection. Am J Pathol 173: 347-357. doi: 10.2353/ajpath.2008.070391. PubMed: 18583312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nozaki T, Rosenblum JM, Schenk AD, Ishii D, Fairchild RL (2009) CCR5 is required for regulation of alloreactive T-cell responses to single class II MHC-mismatched murine cardiac grafts. Am J Transplant 9: 2251-2261. doi: 10.1111/j.1600-6143.2009.02786.x. PubMed: 19656127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Barbieri SS, Ruggiero L, Tremoli E, Weksler BB (2008) Suppressing PTEN activity by tobacco smoke plus interleukin-1beta modulates dissociation of VE-cadherin/beta-catenin complexes in endothelium. Arterioscler Thromb Vasc Biol 28: 732-738. doi: 10.1161/ATVBAHA.107.159434. PubMed: 18202321. [DOI] [PubMed] [Google Scholar]
- 27. Yamamoto K, Loskutoff DJ (1997) The kidneys of mice with autoimmune disease acquire a hypofibrinolytic/procoagulant state that correlates with the development of glomerulonephritis and tissue microthrombosis. Am J Pathol 151: 725-734. PubMed: 9284821. [PMC free article] [PubMed] [Google Scholar]
- 28. Matsumoto M, Tanaka T, Yamamoto T, Noiri E, Miyata T et al. (2004) Hypoperfusion of peritubular capillaries induces chronic hypoxia before progression of tubulointerstitial injury in a progressive model of rat glomerulonephritis. J Am Soc Nephrol 15: 1574-1581. doi: 10.1097/01.ASN.0000128047.13396.48. PubMed: 15153568. [DOI] [PubMed] [Google Scholar]
- 29. Warner RL, Winter HC, Speyer CL, Varani J, Oldstein IJ et al. (2004) Marasmius oreades lectin induces renal thrombotic microangiopathic lesions. Exp Mol Pathol 77: 77-84. doi: 10.1016/j.yexmp.2004.04.003. PubMed: 15351229. [DOI] [PubMed] [Google Scholar]
- 30. Gloor JM, Sethi S, Stegall MD, Park WD, Moore SB et al. (2007) Transplant glomerulopathy: subclinical incidence and association with alloantibody. Am J Transplant 7: 2124-2132. doi: 10.1111/j.1600-6143.2007.01895.x. PubMed: 17608832. [DOI] [PubMed] [Google Scholar]
- 31. Gaston RS, Cecka JM, Kasiske BL, Fieberg AM, Leduc R et al. (2010) Evidence for antibody-mediated injury as a major determinant of late kidney allograft failure. Transplantation 90: 68-74. doi: 10.1097/00007890-201007272-00129. PubMed: 20463643. [DOI] [PubMed] [Google Scholar]
- 32. Sellarés J, de Freitas DG, Mengel M, Sis B, Hidalgo LG et al. (2011) Inflammation lesions in kidney transplant biopsies: association with survival is due to the underlying diseases. Am J Transplant 11: 489-499. doi: 10.1111/j.1600-6143.2010.03415.x. PubMed: 21342447. [DOI] [PubMed] [Google Scholar]
- 33. Smith JD, Banner NR, Hamour IM, Ozawa M, Goh A et al. (2011) De novo donor HLA-specific antibodies after heart transplantation are an independent predictor of poor patient survival. Am J Transplant 11: 312-319. doi: 10.1111/j.1600-6143.2010.03383.x. PubMed: 21219570. [DOI] [PubMed] [Google Scholar]
- 34. John HD, Thoenes W (1982) The glomerular lesions in endotheliotropic hemolytic nephroangiopathy (hemolytic uremic syndrome, malignant nephrosclerosis, post partum renal insufficiency). Pathol Res Pract 173: 236-259. doi: 10.1016/S0344-0338(82)80087-3. PubMed: 6889731. [DOI] [PubMed] [Google Scholar]
- 35. Mitra D, Kim J, MacLow C, Karsan A, Laurence J (1998) Role of caspases 1 and 3 and Bcl-2-related molecules in endothelial cell apoptosis associated with thrombotic microangiopathies. Am J Hematol 59: 279-287. doi: 10.1002/(SICI)1096-8652(199812)59:4. PubMed: 9840908. [DOI] [PubMed] [Google Scholar]
- 36. Lajoie G (1997) Antibody-mediated rejection of human renal allografts: an electron microscopic study of peritubular capillaries. Ultrastruct Pathol 21: 235-242. doi: 10.3109/01913129709021919. PubMed: 9183824. [DOI] [PubMed] [Google Scholar]
- 37. Ishii Y, Sawada T, Kubota K, Fuchinoue S, Teraoka S et al. (2005) Injury and progressive loss of peritubular capillaries in the development of chronic allograft nephropathy. Kidney Int 67: 321-332. doi: 10.1111/j.1523-1755.2005.00085.x. PubMed: 15610258. [DOI] [PubMed] [Google Scholar]
- 38. Pickering MC, de Jorge EG, Martinez-Barricarte R, Recalde S, Garcia-Layana A et al. (2007) Spontaneous hemolytic uremic syndrome triggered by complement factor H lacking surface recognition domains. J Exp Med 204: 1249-1256. doi: 10.1084/jem.20070301. PubMed: 17517971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M et al. (2008) VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med 358: 1129-1136. doi: 10.1056/NEJMoa0707330. PubMed: 18337603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Keepers TR, Psotka MA, Gross LK, Obrig TG (2006) A murine model of HUS: Shiga toxin with lipopolysaccharide mimics the renal damage and physiologic response of human disease. J Am Soc Nephrol 17: 3404-3414. doi: 10.1681/ASN.2006050419. PubMed: 17082244. [DOI] [PubMed] [Google Scholar]
- 41. Belloni PN, Nicolson GL (1988) Differential expression of cell surface glycoproteins on various organ-derived microvascular endothelia and endothelial cell cultures. J Cell Physiol 136: 398-410. doi: 10.1002/jcp.1041360303. PubMed: 3170638. [DOI] [PubMed] [Google Scholar]
- 42. Thurston G, Baluk P, Hirata A, McDonald DM (1996) Permeability-related changes revealed at endothelial cell borders in inflamed venules by lectin binding. Am J Physiol 271: H2547-H2562. PubMed: 8997316. [DOI] [PubMed] [Google Scholar]
- 43. Hohenstein B, Braun A, Amann KU, Johnson RJ, Hugo CP (2008) A murine model of site-specific renal microvascular endothelial injury and thrombotic microangiopathy. Nephrol Dial Transplant 23: 1144-1156. PubMed: 18045820. [DOI] [PubMed] [Google Scholar]
- 44. Choo JK, Seebach JD, Nickeleit V, Shimizu A, Lei H et al. (1997) Species differences in the expression of major histocompatibility complex class II antigens on coronary artery endothelium: implications for cell-mediated xenoreactivity. Transplantation 64: 1315-1322. doi: 10.1097/00007890-199711150-00014. PubMed: 9371674. [DOI] [PubMed] [Google Scholar]
- 45. Barbieri L, Valbonesi P, Bonora E, Gorini P, Bolognesi A et al. (1997) Polynucleotide:adenosine glycosidase activity of ribosome-inactivating proteins: effect on DNA, RNA and poly(A). Nucleic Acids Res 25: 518-522. doi: 10.1093/nar/25.3.518. PubMed: 9016590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pawlinski R, Wang JG, Owens AP 3rd, Williams J, Antoniak S et al. (2010) Hematopoietic and nonhematopoietic cell tissue factor activates the coagulation cascade in endotoxemic mice. Blood 116: 806-814. doi: 10.1182/blood-2009-12-259267. PubMed: 20410508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Daniel S, Arvelo MB, Patel VI, Longo CR, Shrikhande G et al. (2004) A20 protects endothelial cells from TNF-, Fas-, and NK-mediated cell death by inhibiting caspase 8 activation. Blood 104: 2376-2384. doi: 10.1182/blood-2003-02-0635. PubMed: 15251990. [DOI] [PubMed] [Google Scholar]
- 48. Li JH, D'Alessio A, Pober JS (2009) Lipopolysaccharide can trigger a cathepsin B-dependent programmed death response in human endothelial cells. Am J Pathol 175: 1124-1135. doi: 10.2353/ajpath.2009.090113. PubMed: 19661440. [DOI] [PMC free article] [PubMed] [Google Scholar]