

Abstract
Background
The hallmark of Trypanosoma cruzi infection is cardiomyopathy that leads to end-stage heart failure. We investigated whether G-CSF, known to induce heart tissue repair by bone marrow stem cell mobilization, ameliorates T. cruzi-induced myocarditis.
Methods and results
T. cruzi-infected C3H/He mice were treated with G-CSF and monitored for parasite burden, BMSC mobilization, cytokine profile and cardiac remodeling.
G-CSF increased the expression of CXCR4, CD34, and c-Kit, indicating mobilization and migration of BMSC, some of which differentiated to cardiomyocytes as evidenced by increased levels of GATA4+/MEF2C+ cells and desmin expression in chagasic hearts. G-CSF enhanced a mixed cytokine response (IL-10 + TGF-β > IFN-γ + TNF-α > IL-4) associated with increased heart inflammation and no beneficial effect on parasite control. Further, G-CSF controlled T. cruzi-induced extracellular-matrix alterations of collagens (Col I and Col llI), hydroxyproline, and glycosaminoglycan contents and promoted compensatory cardiac remodeling; however, these responses were not sufficient to control T. cruzi-induced cardiomyocyte atrophy. Benznidazole treatment prior to G-CSF resulted in the control of parasitism and parasite-induced inflammation, and subsequently, G-CSF was effective in executing the tissue repair, as evidenced by extracellular-matrix homeostasis and normalization of cardiomyocyte size in chagasic hearts. Conclusions: G-CSF treatment after T. cruzi infection enhanced migration and homing of BMSC, some of which differentiated to cardiomyocytes. Yet, G-CSF promoted a mixed (Treg > Th1 > Th2) immune response that contributed to persistent inflammation and limited improvement in cardiac homeostasis. Combinatorial therapy (BZ → G-CSF) was beneficial in arresting inflammatory processes and tissue damage in chagasic hearts.
Keywords: Chagas disease, Trypanosoma cruzi, G-CSF, Stem cell, Cardiac repair
1. Introduction
Chagas disease, caused by Trypanosoma cruzi, constitutes a major public health problem in Latin America due to its prevalence, morbidity, and mortality. The disease is characterized by cardiac and digestive abnormalities, developed in ~30–40% of T. cruzi-infected people several years after the acute phase has subsided [1]. In the acute phase, a clear causal relationship between the local presence of the parasite and tissue damage has been established, and treatment with benznidazole (BZ) leads to regression of clinical symptoms and parasitological cure in most human cases [2]. The exact mechanisms involved in the development of chronic myocarditis are, however, not fully understood, and, heart transplantation is the only available choice for the management of patients with chagasic end-stage heart failure [3].
Mice and rats experimentally infected with T. cruzi were demonstrated to reproduce several aspects of the human cardiomyopathy, and used as surrogate for human beings in studies of mechanisms of disease and treatment. We have shown that cardiac myocytes in proximity to inflamed tissue areas increase the expression of proliferating cell nuclear factor, probably as an attempt to compensate for damaged cells, whereas myocytes located farther from inflammatory foci experience hypertrophy, likely as an attempt to maintain homeostasis and heart function [4].
One of the mechanisms of tissue repair involves the mobilization of stem and progenitor cells from bone marrow in a multi-step process, initiated by the local release of various signaling molecules and proteolytic enzymes [5]. For example, G-CSF has been shown to signal cardiomyocyte proliferation during development [6], and mobilization and differentiation of postnatal bone marrow stem cells (BMSC) in mouse models of myocardial injury [7, 8]. G-CSF treatment decreased the infarct size by inducing the mobilization of CD34+CXCR4+ BMSC, with compensatory cardiomyocyte hypertrophy and decreased interstitial fibrosis [9, 10]. Stromal cell-derived factor-1 (SDF-1) has been identified as the major homing factor for stem cells (SC) that play a key role in regenerating damaged tissues [11]. Moreover, G-CSF was demonstrated to protect heart tissues from cardiomyocyte atrophy, fibrosis, and inflammatory cell infiltration in experimental models of drug-induced cardiomyopathy [12].
In this study, we aimed to evaluate the beneficial effects of G-CSF on mobilization and homing of progenitor cells into the heart of T. cruzi-infected mice, and to prevent development of chronic chagasic cardiomyopathy. Our findings suggest that exogenous administration of G-CSF only partially repaired the damage provoked by T. cruzi due to the high level of inflammation that persisted in this experimental model. Control of parasite-induced inflammatory responses by pre-treatment with BZ provided an opportunity to attain G-CSF-dependent repair of heart tissue damage in chagasic mice. We discuss the potential utility of G-CSF as an adjunct therapy in preventing progression of chronic chagasic cardiomyopathy.
2. Materials and methods
2.1. Mice, infection and treatment
Four-to-six-week-old male and female C3H/He mice were obtained from the Animal Breeding Facility of the Instituto Nacional de Parasitología “Dr. Mario Fatala Chabén”. T. cruzi trypomastigotes (Sylvio-X10/4 clone) were in vitro cultured in BESM cells. Mice were inoculated intraperitoneally with T. cruzi (106 trypomastigotes/mouse; n = 72 mice) orRPMI medium alone (n = 32 mice). Protocol 1 – Mice were treated with G-CSF (10 μg/kg/dose, five doses/week, ProSpec-Tany TechnoGene) or saline during four weeks, beginning day 21 (acute stage) or 1 year (chronic stage) post-infection (pi). Protocol 2 — Mice were orally given BZ (100 mg/kg/day, Roche, Radanil®) or saline for 30 days, beginning at 4 and 6 months (mo) pi. Later, administration of G-CSF was carried out at 10 mo pi, as in Protocol 1. Mice were harvested in 70% CO2 at the end of treatment All animals received humane care and that study protocols comply with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication no. 82–23, revised 1996) and was approved by the Institutional Committee for Care and Use of Laboratory Animals (CICUAL), University of Buenos Aires, School of Medicine, and Secretary of Science and Technology, Buenos Aires, Argentina, Resolution Number 015/2012. The manuscript complies with the Principles of Ethical Publishing in the International Journal of Cardiology.
2.2. Histology and immunohistochemistry
Heart tissue-sections were fixed in 10% formalin and embedded in paraffin. Tissue-sections (5 μm) were stained with hematoxylin/eosin (H&E), and inflammation was scored semiquantitatively according to distribution (focal, confluent or diffuse) and extent of inflammatory infiltrate in a double-blinded manner [13]. The presence and number of parasitized cells per section were recorded, and parasite density was estimated by the number of parasite nests over the total section area, measured with Image Tool software (http://ddsdx.uthscsa.edu/dig/itdeschtml), and standardized to 0.5 cm2.
The size of cardiac myocytes was estimated by the cell transversal diameter parameter [14]. Microphotographs of inflamed and non-inflamed myocardium were obtained at random from heart tissue sections stained with Periodic acid-Schiff, and diameter of randomly selected cells was determined across the nucleus using the Image Tool software.
For immunohistochemistry, tissue-sections were de-waxed, quenched with 20% acetic acid, blocked using a streptavidin/biotin blocking kit (Vector Laboratories) and incubated with goat serum for 30 min. Sections were treated with 10 mM citrate buffer (pH 6.0) for antigen retrieval, and incubated for 16 h at 4°C with 1:50 dilutions of rabbit anti-GATA-4 (Abcam) or anti-MEF2C (Sigma) antibodies. Slides were then incubated with biotinylated goat-anti-rabbit IgG (1:200) and streptavidin-alkaline phosphatase (AP) conjugate, and color developed using the nitrobluetetrazolium-5-bromo-4-chloro-3-indolyl-phosphate kit (Vector Laboratories).
2.3. Parasite burden
Total DNA was isolated from heart tissue sections, and used as template for PCR amplification of T. cruzi-specific 18SrDNA and murine GAPDH [15]. PCR products were run on 1.5% agarose gels, and densitometric quantification of amplicons performed using UN-SCAN-IT gel Version 6.1 (Orem, Utah, USA).
2.4. Real time RT-PCR
Total RNA was extracted from tissue sample (50 mg) using Trizol reagent (Invitrogen). Contaminating DNA was removed using DNase I (Applied Biosystems). First strand cDNA was synthesized using iScript cDNA Synthesis Kit (Bio-Rad), and used as a template for real-time PCR on an iCycler Thermal Cycler (Bio-Rad) using iQTM SYBR Green Supermix (Bio-Rad) and specific oligonucleotides listed in Supplement Table 1. The threshold cycle (Ct) values for each mRNA were normalized against β-actin mRNA, and the relative expression level of each target gene was calculated by the formula n-fold change = 2−ΔCt, where ΔCt represents Ct (target)infected − Ct (housekeeping)infected[16].
2.5. Western blotting
Tissue homogenates in RIPA buffer were resolved on denaturing 12% acrylamide gels (100-μg protein/well) in a mini-gel apparatus (Mini Protean 2, Bio-Rad), and proteins transferred to nitrocellulose membranes. Membranes were probed using antibodies against GATA-4 (1:300, Santa Cruz), MEF2C (1:1000, Abcam), G-CSF receptor (G-CSFR, 1:500, Santa Cruz), IL-4 (1:1000, BD Biosciences) and α-tubulin (1:1000, Santa Cruz), and signal was developed by chemiluminescence detection system (GE-Healthcare).
2.6. Extracellular matrix (ECM) components
Heart tissue homogenate was used for the detection of collagen, sulfated glycosaminoglycans (GAGs), hydroxyproline amino acids (HPR) and matrix metalloproteinases (MMPs), as described [17]. Briefly, for GAGs estimation, heart homogenates (30-μg protein) were digested with papain type III at 60°C for 1 h, transferred to 96-well plates and color developed with 1,9-dimethylmethylene blue (standard curve: 0.125–1.25 μg sodium chondroitin sulfate).
For the estimation of HPR, tissues were hydrolyzed with 6 N HCl at 95°C for 16 h. Precipitated hydrolyzed collagen samples were incubated with 1.75% chloramine-T in 0.1 M acetate–citrate pH 6.0 buffer (1:1, v/v) for 5 min followed by 0.2 ml of Ehrlich’s reagent for 20 min at 60°C (standard curve: 0.5–5 μg hydroxyproline) [18].
To examine the gelatinolytic activity of MMP2 (72 kDa) and MMP9 (92 kDa), tissue homogenates (100-μg protein) were electrophoresed through 8% polyacrylamide gels supplemented with type A porcine gelatin (2 mg/ml, Sigma). Gels were washed with 2.5% Triton X-100, and incubated with enzyme activation buffer (50 mM Tris–HCl pH 7.8, 150 mM NaCl and5 mMCaCl2) for24 h.Gels were stained with 0.05% Coomassie brilliant blue (G-250; Sigma), destained in 45% methanol/10% acetic acid, and areas of gelatinolytic activity detected as transparent bands on the blue background.
2.7. Cytokine levels
Serum levels of interleukin-10 (IL-10, BD Biosciences), IL-4 (BD Biosciences) and IFN-γ (eBioscience) were quantified using an enzyme-linked immunosorbent assay, according to the supplier’s instructions.
2.8. Statistical analysis
The results were expressed as the mean ± SD, derived from at least triplicate observations per sample (n > 6 mice/group). Data were analyzed using GraphPad InStat software. Comparisons between groups (equal variance) were made using two-tailed paired Student’s t test. Comparisons among multiple groups were made using one-way ANOVA followed by the Bonferroni test or Dunnet multiple comparisons test. Values of p < 0.05 were considered statistically significant.
3. Results
3.1. G-CSF fails to ameliorate T. cruzi-induced heart inflammation and cardiomyocyte atrophy
We first determined by histopathology if supplemental G-CSF modifies the magnitude of heart lesions induced by T. cruzi infection in C3H/He mice infected with Sylvio-X10/4 trypomastigotes. In this model system, myocardial inflammation is a constant finding, and fibrosis increases after heart parasitism decreases. Most mice survive long-term periods of infection, with a strong association between histopathological changes and electrocardiographic abnormalities [19]. We began G-CSF treatment during acute stage (21 dpi), when tissue parasitism and inflammation are known to be fully established, and in the chronic stage (1-yr pi) when heart inflammatory infiltrates are associated with tissue fibrosis [20].
In the present study, histopathological analysis of heart sections showed that the severity of inflammatory lesions in acutely infected mice was not significantly modified by G-CSF treatment (inflammation index: 2.38 ± 0.43-vs.-2.59 ± 0.44, I-vs.-I + GCSF, Fig. 1A). There were no significant treatment-related changes in the density of parasite nests in these mice (mean [range]: 0.43 [0–2]-vs.-0.26 [0–2] parasite nests/0.5 cm2, I-vs.-I + GCSF). Semi-quantitative PCR confirmed these observations; we found no effect of G-CSF on the Tc18SrDNA signal in the myocardium of acutely infected mice (Fig. 1B).
Fig. 1.
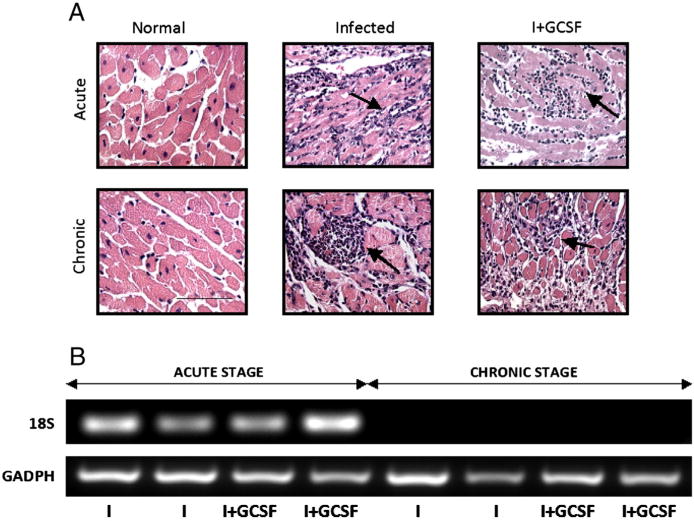
G-CSF effects on myocardial inflammatory lesions and parasite burden in T. cruzi-infected mice. C3H/He mice were infected with T. cruzi and treated with G-CSF at day 21 or 1 year post-infection. Mice were harvested after the completion of treatment. (A) Microphotographs of tissue-sections from acutely and chronically infected mice (±G-CSF) stained with H&E and examined for inflammation and intracellular parasites (black arrows, magnification: 200×; bar: 100 μm). (B) T. cruzi 18SrDNA sequence amplification by PCR (25 cycles) in heart tissue from infected mice. Murine GAPDH was amplified as a control. NI: uninfected control, I: infected, I + GCSF: infected/G-CSF-treated.
In chronically infected mice, G-CSF treatment resulted in a greater extent of myocardial infiltration of inflammatory cells (inflammation index: 1.75 ± 0.94-vs-2.50 ± 0.91, I-vs.-I + GCSF, Fig. 1A). Rare parasite nest were detected in chronically infected mice, irrespective of the G-CSF treatment (mean [range]: 0.066 [0–2]-vs.-0.040 [0–3], I-vs.-I + GCSF). PCR amplification failed to detect parasite DNA in heart tissue of chronically infected mice (Fig. 1B).
To evaluate the effect of G-CSF on cardiomyocyte size, we measured the MTD in morphologically preserved and inflamed myocardium of acutely and chronically infected mice. There were no statistically significant differences in the sizeof cardiomyocytesofacutely and chronically infected mice(±G-CSF), and controls when myocardial areas devoid of inflammatory cells were analyzed (Table 1). Acutely infected mice, irrespective of G-CSF treatment, exhibited a 20% cardiomyocyte size reductionin inflamed myocardial tissues(p < 0.05). Further, 15% and 25% reduction in cardiomyocyte size were noticed in inflamed myocardium in chronically infected- and infected/G-CSF-treated mice, respectively (p < 0.05). Altogether, these data show that G-CSF did not arrest inflammation, parasite burden, and atrophy of cardiomyocytes in T. cruzi infected mice.
Table 1.
Effect of G-CSF treatment on cardiac MTD in T. cruzi-infected mice.
| Infection stage | Morphology | Myocyte transversal diameter (μm)
|
|||
|---|---|---|---|---|---|
| NI | NI + G-CSF | I | I + GCSF | ||
| Acute | Unaffected area | 11.09 ± 0.86 | 11.09 ± 0.54 | 11.08 ± 0.77 | 11.09 ± 1.03 |
| Inflamed area | – | – | 8.87 ± 0.44*† | 8.27 ± 0.60*† | |
| Chronic | Unaffected area | 10.00 ± 0.85 | 10.33 ± 1.58 | 11.11 ± 1.38 | 10.65 ± 1.01 |
| Inflamed area | – | – | 8.56 ± 0.92*† | 8.08 ± 0.80*† | |
C3H/He uninfected and T. cruzi infected mice untreated or treated with G-CSF for four weeks, beginning day 21 (acute) or 1 year (chronic) post-infection were subjected to cardiomyocyte measurements by histo-morphometry. Data are presented as mean ± S.D. from analysis of >100 cells at 400× (>10 microscopic fields/slide, 3–4 tissue-sections/mouse, n ≥ 6 mice per group
p < 0.05, vs. normal
p < 0.05 vs. infected/unaffected area). NI: uninfected control, NI + GCSF: uninfected-treated, I: infected, I + GCSF: infected-treated.
3.2. Mobilization and homing of BMSC by G-CSF
To establish whether G-CSF promotes mobilization of BMSC to heart tissue damaged by T. cruzi infection, we evaluated expression levels of SC markers CXCR4, CD34, and c-Kit by real time RT-PCR. CD34 mRNA levels were decreased in acutely infected myocardium as compared to normal controls, whereas CXCR4 and c-Kit were either unaltered or marginally decreased in acutely and chronically infected mice (Fig. 2). Administration of G-CSF resulted in a significant increase in CXCR4, CD34, and c-Kit mRNA levels in acutely (83.50%, 83.77% and 71.55% increase, Fig. 2A) and chronically (66.55%, 32.26% and 48.32% increase, Fig. 2B) infected mice (p < 0.05). Measurement of SC homing factor SDF-1 showed similar expression levels in the myocardium of T. cruzi infected mice and controls; G-CSF treatment resulted in increased expression of SDF-1 in acutely infected myocardium (Fig. 2A). These results indicate that exogenous G-CSF stimulates mobilization, migration and homing of BMSC into myocardium in response to T. cruzi infection.
Fig. 2.
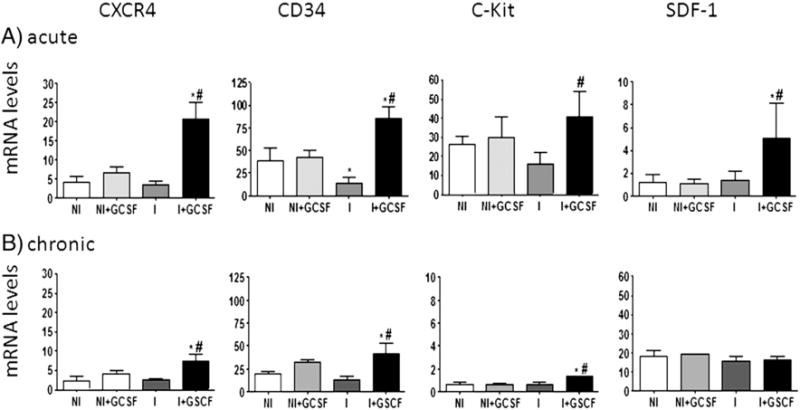
G-CSF induced mobilization and homing of progenitor cells. Mice were infected and treated with G-CSF as in Fig. 1. mRNA levels for CXCR4, CD34, c-Kit and SDF-1 in heart tissue of uninfected and infected mice harvested at acute (A) and chronic (B) stages of infection were determined by real time RT-PCR. Transcript levels were normalized against β-actin mRNA. Values are represent the mean ± SD, derived from ≥4-mice/group (*p < 0.05, vs. normal; #p b 0.05, vs. infected). NI: uninfected control, NI + GCSF: uninfected/G-CSF-treated, I: infected, I + GCSF: infected/G-CSF treated.
3.3. Cardiomyocyte differentiation and expression of hypertrophy markers
Next, we evaluated whether G-CSF modulates differentiation of BMSC into cardiomyocytes in T. cruzi infected myocardium. In order to determine cardiomyocyte differentiation, we measured the expression of GATA-4 and MEF2C transcription factors, which play pivotal roles in cardiomyocyte homeostasis, and of desmin, a sarcomeric protein regulated by these factors. The levels of GATA-4, MEF2C and desmin mRNA were substantially decreased in acutely infected hearts, and normalized or enhanced by G-CSF treatment (Fig. 3A). Nevertheless, Western blot revealed no changes in GATA-4 and MEF2C protein levels due to T. cruzi infection, and a significant decrease in GATA-4 and increase in MEF2C expression upon G-CSF treatment (Fig. 3B). A slight decrease in the number of GATA-4+ and MEF2C+ cardiomyocyte nuclei was observed in unaffected areas of the heart of acutely infected mice by immunohistochemistry, that was normalized or enhanced by G-CSF treatment (Supplement Fig. S1). In the chronic stage, no differences in GATA-4 mRNA and protein levels were detected between T. cruzi-infected mice and the controls (Fig. 3C & D); G-CSF treatment significantly decreased expression of GATA-4 in infected mice. Chronic infection induced a decline in MEF2C and desmin mRNA levels which were normalized or enhanced by G-CSF treatment (Fig. 3C).
Fig. 3.
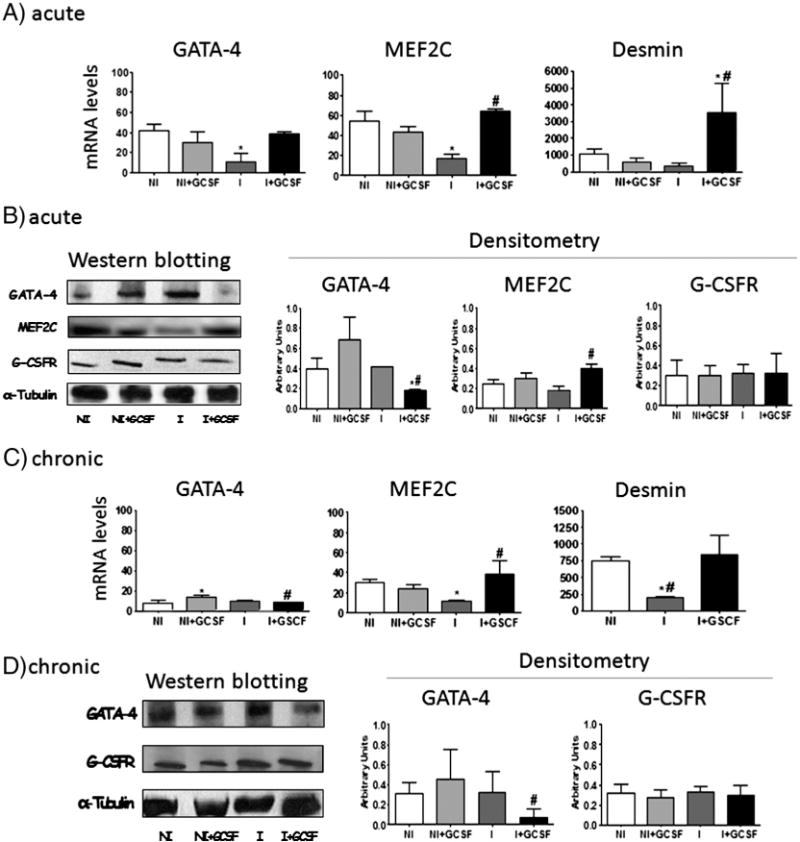
G-CSF effects on stem cell differentiation. T. cruzi-infected mice were treated with G-CSF during acute (A–B) and chronic (C–D) stages. (A & C) mRNA levels for GATA-4, MEF2C, and desmin were determined by real time RT-PCR, and normalized against β-actin. (B & D) Western blotting and densitometric analysis for GATA-4, MEF2C and G-CSFR in acutely (B) and chronically (D) infected myocardium. Expression levels were normalized against α-tubulin signal. Data are presented as mean ± SD (n = 4, *p < 0.05, vs. normal; #p < 0.05, vs. infected). NI: uninfected control, NI + GCSF: uninfected/G-CSF-treated, I: infected, I + GCSF: infected/G-CSF treated.
Then, we analyzed expression of G-CSFR in heart tissues, an indicator of early stages of cell differentiation and proliferation in response to G-CSF. Protein levels of G-CSFR were not significantly altered in the myocardium of acutely and chronically infected mice (±G-CSF) when compared to uninfected controls (Fig. 3D).
Together, these results suggest that G-CSF stimulates mRNA expression of cardiac differentiation markers GATA4, MEF2C and desmin as an attempt to repair the myocardial tissue damaged by T. cruzi. However, G-CSF-induced GATA-4 mRNA increases were not reflected at protein production level, suggesting that other factors prevented G-CSF’s ability to achieve differentiation of BMSCs into cardiomyocytes and restoration of heart tissue in chagasic mice.
3.4. G-CSF exert compensatory remodeling in myocardial ECM
In order to evaluate the effect of cardiac remodeling activity in T. cruzi infected mice, we determined components of ECM such as collagen, HPR, and GAGs, and ECM degradation enzymes (MMPs). We observed no significant change in collagen deposition as evidenced by histochemical staining with Masson’s Trichrome (Supplement Fig. S2) and mRNA levels of Col I and Col III (Fig. 4A) in the myocardium of acutely infected mice. Biochemical studies showed a 2-fold and 12-fold increase in HPR and GAGs, respectively, associated with a substantial increase in MMP2 and MMP9 activity (Fig. 4A). Treatment with G-CSF significantly enhanced the mRNA levels for Col I and Col III, and controlled the infection-induced increase in HPR and GAGs contents. We also observed a 12% and 42% decline in MMP2 and MMP9 activity in G-CSF-treated/acutely infected mice (Fig. 4A). In the chronic stage, infected mice exhibited a substantial increase in myocardial deposition of collagen, as evidenced by Masson’s Trichrome staining (score: 2.20 ± 0.97-vs-1.37 ± 0.47, I-vs.-I + GCSF Supplement Fig. S2); and 3-fold, 2.6-fold and 6-fold increases in Col I mRNA, and HPR and GAGs levels, respectively (Fig. 4B). G-CSF treatment increased Col III mRNA, while HPR content was normalized to control levels (Fig. 4B). These data suggest that G-CSF treatment promotes compensatory remodeling and ECM homeostasis, especially in chronic chagasic cardiomyopathy.
Fig. 4.
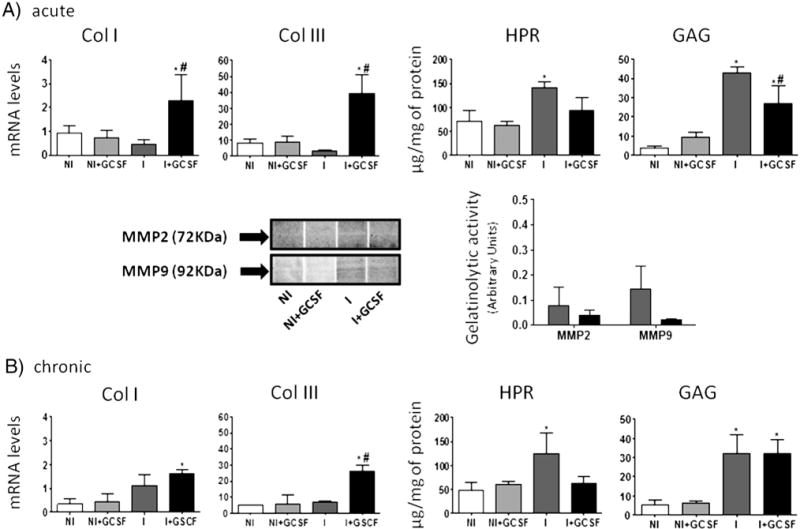
ECM components in T. cruzi-infected mouse heart tissues (±G-CSF). Mice were treated with G-CSF during acute (A) and chronic (B) stages of infection. Myocardial levels of mRNA for Col I and Col III (normalized against β-actin) were determined by real time RT-PCR. Myocardial levels of HPR amino acids and GAGs were determined by biochemical assays. The activity of matrix metalloproteinases was examined by gelatin zymography. Data are presented as mean ± S.D. (n ≥ 4, *p < 0.05, vs normal; #p < 0.05, vs. infected NI: uninfected control, NI + GCSF: uninfected/G-CSF-treated, I: infected, I + GCSF: infected/G-CSF treated.
3.5. G-CSF induces a mixed type of immune response in T. cruzi infected hearts
We analyzed the effect of G-CSF on the production of IFN-γ, TNF-α, IL-10, TGF-β and IL-4 and expression of transcription factors T-bet, FoxP3 and GATA-3 during T. cruzi infection. We observed a significant increase in serum levels of IFN-γ in acutely infected mice (p < 0.05), which were not modified by G-CSF (Fig. 5). Increased levels of IFN-γmRNA were also detected in heart tissues from chronically infected mice, which were restored to normal levels by G-CSF (p < 0.05, Fig. 5). The analysis of Th1 transcription factor T-bet showed a significant increase of T-bet mRNA levels in myocardial tissues during the acute and chronic stages of T. cruzi infection (Supplement Fig. S3); treatment with G-CSF was capable of restoring T-bet mRNA expression to control levels during the chronic but not the acute stage of the infection.
Fig. 5.
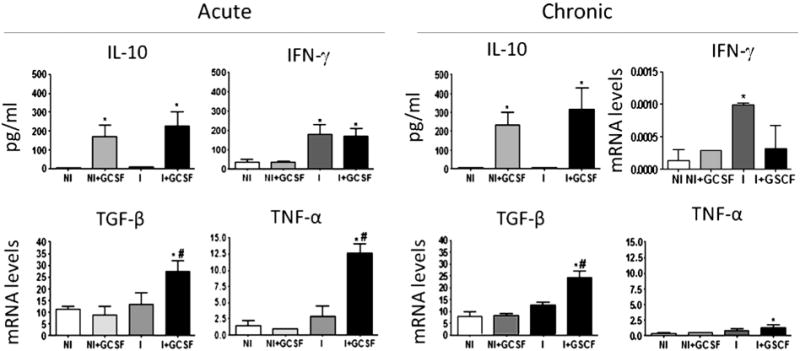
Effect of G-CSF on the immune response of T. cruzi-infected mice. Serum levels of IFN-γ and IL-10 in acutely and chronically infected mice were determined by ELISA. Myocardial levels of mRNA for IFN-γ, TGF-β and TNF-α were determined by real time RT-PCR and normalized against β-actin. Data are presented as mean ± SD (n ≥ 4, *p < 0.05, vs. normal; #p < 0.05, vs. infected). NI: uninfected control, NI + GCSF: uninfected/G-CSF-treated, I: infected, I + GCSF: infected/G-CSF treated.
No changes in serum levels of IL-10 were observed in infected mice; however, G-CSF treatment resulted in a significant increase in IL-10 levels in acutely and chronically infected mice, as well as in normal controls (Fig. 5). There were no significant differences in mRNA levels for TGF-β and TNF-α in the myocardium of acutely and chronically infected mice, and G-CSF treatment enhanced transcript levels for both cytokines (p < 0.05, Fig. 5). The analysis of the levels of mRNA for Treg transcription factor FoxP3 in heart tissues showed no significant differences among infected, infected-G-CSF treated and control groups, irrespective of the stage of infection (Supplement Fig. S3).
IL-4 was undetectable in the serum of mice tested by ELISA, regardless of infection and G-CSF treatment status. Nevertheless, increased levels of IL-4 and IL-4 mRNA were detected in cardiac tissues of infected mice, which remained unmodified after G-CSF treatment (Supplement Fig. S4). In contrast, expression of Th2 transcription factor GATA-3 mRNA was reduced in cardiac tissues of acutely infected- and infected-G-CSF-treated mice as compared to that noted in controls. These apparently opposed results suggest that other pathways might be involved in the modulation of this cytokine in T. cruzi infection. Overall, the data presented in Fig. 5, and Figs. S3 and S4 suggest that G-CSF modulated the T. cruzi-induced predominance of Th1 cytokine response toward a mixed Th1/Th2 cytokine response in both acute and chronic stages of infection and disease development.
3.6. Trypanocidal treatment helps G-CSF to maintain cardiac homeostasis
We hypothesized that G-CSF-dependent increase in immunoregulatory cytokine response prevents host’s ability to achieve parasite control, and, consequently, persistence of inflammatory responses hindered effective repair in the myocardium. To evaluate if such is the case, we treated T. cruzi-infected mice at 4- and 6-mo pi with BZ, prior to treatment with G-CSF at 10-mo pi. BZ-treatment of mice at 4-mo pi, when acute parasitemia but not the tissue parasite burden was controlled by natural immunity, resulted in undetectable Tc18SrDNA signal by PCR and real time PCR in the myocardium (data not shown). The control of acute tissue parasite burden in BZ-treated mice was associated with up to 80% reduction in inflammatory infiltrates, the reduction being more marked in BZ + G-CSF-treated mice (Fig. 6, p < 0.05). When treated with BZ at 6-mo pi, we noticed no significant changes in the extent of myocardial inflammation, suggesting factors other than the sole parasite presence contributing to chronic inflammation. Noticeably, BZ treatment at 6-mo pi followed by G-CSF led to a substantial decline in chronic persistence of inflammatory cell infiltration in mouse chagasic hearts (Fig. 6, p < 0.05). Estimation of cardiomyocyte size by histo-morphometry showed a 17.70%, 17.70% and 22.60% reduction in inflamed/fibrotic areas of the myocardium in infected, infected/G-CSF-treated and infected/BZ(6-mo pi)-treated mice, respectively (p < 0.05). When BZ-treated mice were given G-CSF, the size of cardiomyocytes size was restored to normal levels (Table 2). These results suggest that when inflammation is diminished by specific treatment, G-CSF helps to maintain cardiac homeostasis in Chagas heart disease.
Fig. 6.
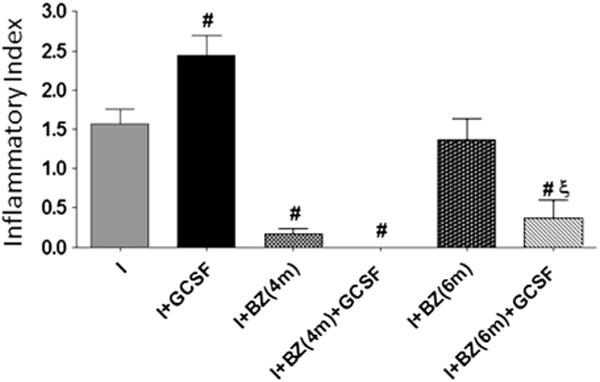
Effects of anti-T. cruzi therapy on G-CSF-mediated control of myocardial pathology. T. cruzi-infected mice were treated with BZ at 4 and 6 months post-infection, followed by administration of G-CSF at 10 months post-infection. Paraffin-embedded tissue-sections were subjected to H&E staining, and inflammatory index calculated as detailed in the Materials and methods (#p < 0.05, vs. infected, ξp < 0.05, vs. infected/BZ-treated (6-mo pi). I: infected, I + GCSF: infected/G-CSF treated. I + BZ(4 m)/I + BZ(6 M): infected/BZ-treated at 4 and 6 mo pi, respectively. I + BZ (4 m) + GCSF/I + BZ(6 m) + GCSF: infected/BZ-treated at 4 and 6 mo pi and then treated with G-CSF at 10 mo pi.
Table 2.
Pre-treatment with BZ resulted in G-CSF-dependent preservation of cardiomyocytes in T. cruzi-infected mice.
| Morphology | Myocyte transversal diameter (μm)
|
|||||||
|---|---|---|---|---|---|---|---|---|
| NI | NI + GCSF | I | I + GCSF | I + BZ(4 m) | I + BZ(4 m) + GCSF | I + BZ(6 m) | I + BZ(6 m) + GCSF | |
| Unaffected Area | 9.99 ± 0.85 | 10.33 ± 1.56 | 11.02 ± 0.77 | 11.37 ± 0.22 | 11.09 ± 0.58 | 11.66 ± 0.87 | 11.37 ± 0.22 | 10.02 ± 0.58 |
| Inflamed Area | – | 9.07 ± 0.62† | 9.36 ± 0.27*† | 9.40 ± 1.32 | – | 8.80 ± 0.81*† | 9.19 ± 0.8* | |
C3H/He uninfected mice and mice infected with T. cruzi were treated at 4 and 6 months post-infection with BZ for 30 days, and with G-CSF for 20 days at 10 months post-infection. Mice were harvested after completion of treatment, and heart tissue sections were subjected to cardiomyocyte measurement by histo-morphometry. Data are presented as mean ± S.D. from analysis of >10 microscopic fields/slide at 400× magnification (3–4 tissue-sections/mouse, n ≥ 6 per group
p < 0.05, vs. normal
p < 0.05 vs. infected/unaffected area). NI: uninfected control, NI + GCSF: uninfected/G-CSF-treated, I: infected, I + GCSF: infected/G-CSF treated. I + BZ(4 m)/I + BZ(6 m): infected/BZ-treated at 4 and 6 mo pi, respectively. I + BZ(4 m) + GCSF/I + BZ(6 m) + GCSF: infected/BZ-treated at 4 and 6 mo pi and then treated with G-CSF at 10 mo pi.
4. Discussion
In this study, we demonstrate that treatment with G-CSF enhanced the migration of BMSC to the myocardium of T. cruzi-infected mice, some of which have matured as cardiomyocytes, evidenced by increased expression of GATA-4 and MEF2C transcription factors and desmin. However, G-CSF treatment also resulted in increased heart inflammation that was ineffective in controlling tissue parasitism, likely due to the enhanced immunoregulatory IL-10/TGF-β production. Due to persistence of inflammatory infiltrates, G-CSF alone was not sufficient to prevent cardiac remodeling and the changes in ECM components observed in chagasic hearts. Different arms of the immune system are known to participate in the immune response against the invading pathogen and provide healing of the damage caused by injurious stimuli. As it relates to T. cruzi, IFN-γ, IL-12, and TNF-α participate in strengthening the activation of innate and adaptive effector immune responses, resulting in more efficient killing of the parasite and a strong inflammatory response in tissues where parasites replicate, including the myocardium [21]. Conversely, IL-10 and TGF-β counter-regulate the inflammatory processes to prevent immune-related tissue injuries, but might also indirectly favor parasite persistence within infected host cells. In our experimental model, G-CSF administration induced a significant increase in serum levels of IL-10 without affecting IFN-γ production. Similarly, G-CSF induced increased expression of TNF-α (Th1) and TGF-β (Treg) mRNA and infiltration of immune cells in the myocardium of T. cruzi-infected mice. These phenomena might be the result of G-CSF-induced mobilization of mononuclear and polymorphonuclear cells, besides progenitor cells, as described in the ischemic myocardium [22]. G-CSF is known to modulate cytokine production, increasing the secretion of IL-4 and IL-10 and decreasing IFN-γ (Th1) [23]. In the infarct healing process, inflammatory cells such as neutrophils and macrophages stimulate the reabsorption of the necrotic tissue and accelerate ECM replacement [24]. The correct balance of these events attenuates pathological growth of ventricular myocardium, preserving cardiac function and preventing ventricular dilatation [25]. We surmise that G-CSF enhanced the migration, maturation and translocation of immune cells to the heart in response to T. cruzi infection, some of which were proinflammatory (evidenced by increased TNF-α expression) while others were pro-healing (evidenced by increased IL-10 and TGF-β expression).
Experimental and clinical studies of dilated and ischemic cardiomyopathy indicate that native collagen is degraded by MMPs and replaced by fibrous intercellular deposits of poorly cross-linked collagen, changes associated with increased cardiac levels and activity of MMP-2 and MMP-9 [26, 27]. In our study, myocardial expression of ECM components responded to infection and G-CSF treatment. It is important to note that G-CSF treatment promoted a moderate architectural support for muscle cells, evidenced by a significant increase in Col III (indicates remodeling and repair), and marginally controlled the infection-induced increase in HPR and GAGs contents in the myocardium. These events were, however, not associated with compensatory heart hypertrophy as we observed no gain in myocyte size, thus, indicating that G-CSF made a poor attempt to achieve ECM homeostasis. Pre-treatment with BZ further enhanced the tissue repair efficacy of G-CSF, evidenced by normalization of cardiomyocyte size in chronic chagasic hearts. Noticeably, these benefits were afforded in site-specific manner, primarily near inflamed/fibrotic areasof the heart damagedby infection-induced events. Our observation is supported by others demonstrating that the effect of G-CSF on collagen proliferation can be site-specific and a well-controlled process [28].
An important mechanism by which G-CSF exerts beneficial effects in myocardial infarction is suggested to be through mobilization of hematopoietic cells that differentiate into myofibroblasts, improving the healing process [9]. In addition, circulating CXCR4+ cells, which include CD34+ cells, lymphocytes and monocytes, express a variety of repair-related cytokines, thereby mediating angiogenesis and fibrolysis [29, 30]. Cardiac cells expressing c-Kit exhibit the characteristics of stem cells, being clonogenic, self-renewing, multi-potent, and having substantial regenerative potential [31]. Zhang et al. [32] reported that G-CSF mobilized c-Kit+ SC from the marrow into the peripheral blood and promoted the “homing” to the heart infarct. Our observations of increased expression of CD34, CXCR4, and c-kit in G-CSF-treated/infected mice support the role of G-CSF in mobilization of hematopoietic stem cells to the heart tissue damaged by T. cruzi infection. At least some of these progenitor cells were destined to be cardiac myocytes or myofibroblasts, evidenced by increased mRNA and protein levels of MEF2C transcription factors as well as GATA-4+ and MEF2C+ cell numbers in the myocardium of G-CSF-treated/infected mice. GATA-4 is required for the onset of cardiac myocyte gene expression and implicated in early heart development [33]. Both GATA-4 and MEF2C were shown to be involved in differentiation and hypertrophy of adult cardiomyocytes [34, 35]. Desmin, one of the earliest protein markers of muscle tissue in embryogenesis or differentiation, was also enhanced in the myocardium of G-CSF-treated/infected mice. We surmise that G-CSF stimulated hematopoietic cell migration and differentiation into myofibroblasts as an attempt to repair the myocardium damaged by T. cruzi. These beneficial effects were more pronounced when T. cruzi-mediated inflammatory responses were controlled by BZ administration, indicating that G-CSF mightily be useful as an adjunct therapy with anti-parasite drugs in controlling the chronic progression of Chagas heart disease.
The main limitation of our study was the lack of data on cardiac function. We circumvented this issue by using a well-defined and reliable animal model for human Chagas heart involvement, for which extensive histopathological and electrocardiographical data is available [19, 20]. Also, we focused our analysis on the fully developed acute and chronic stages of T. cruzi-induced myocarditis, and studying tissue remodeling in a slowly evolving illness may require evaluation of gene expression and histopathology at multiple levels of the heart disease.
In summary, we have shown that complex inflammatory and cardioregenerative processes are present in the heart of T. cruzi-infected mice, and an imbalance in these processes leads to chronic progression of chagasic cardiomyopathy. G-CSF treatment enhanced the expression of molecules involved in heart reparation, in an attempt to heal the damage provoked by the parasite. Treatment with BZ was effective in controlling the parasite persistence and associated inflammation, but was not sufficient to prevent loss of ECM homeostasis in the chronic stage of disease development. Our results suggest that a combination of G-CSF, capable of modulating cardiac regeneration, and anti-parasite drugs capable of parasite clearing and abolishing parasite-associated inflammation in the myocardium, would prove beneficial in preventing cardiac pathology and loss of LV function in Chagas disease.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.ijcard.2013.03.049.
Supplementary Material
Acknowledgments
We are thankful to Dr. Monisha Dhiman (real time RT-PCR, ECM analysis) and Dr. Shivali Gupta (immunohistochemical staining) for help in standardization of various techniques.
Funding
This work was supported by grants from the Argentine Ministry of Science, Technology and Product Innovation (BID-PICT 2007 No 01732 and 2010 No 2148) to MP and the National Institutes of Health/National Institute of Allergy and Infectious Diseases (2R01AI054578-07) to NJG. MNG was awarded a CONICET fellowship.
Footnotes
Authors take responsibility for all aspects of the reliability and freedom from bias of the data presented and their discussed interpretation.
References
- 1.World Health Organization. Report of the Scientific Working Group on Chagas Disease. Buenos Aires: Argentina UNDP/World Bank/WHO2006; Apr, 2005. pp. 17–20. [Google Scholar]
- 2.Viotti R, Vigliano C, Lococo B, et al. Long-term cardiac outcomes of treating chronic Chagas disease with benznidazole versus no treatment: a nonrandomized trial. Ann Intern Med. 2006;144:724–34. doi: 10.7326/0003-4819-144-10-200605160-00006. [DOI] [PubMed] [Google Scholar]
- 3.From the Centers for Disease Control and Prevention Chagas disease after organ transplantation—United States, 2001. JAMA. 2002;287:1795–6. [PubMed] [Google Scholar]
- 4.Arnaiz MR, Fichera LE, Postan M. Cardiac myocyte hypertrophy and proliferating cell nuclear antigen expression in Wistar rats infected with Trypanosoma cruzi. J Parasitol. 2002;88:919–25. doi: 10.1645/0022-3395(2002)088[0919:CMHAPC]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 5.Lapidot T, Dar A, Kollet O. How do stem cells find their way home? Blood. 2005;106:1901–10. doi: 10.1182/blood-2005-04-1417. [DOI] [PubMed] [Google Scholar]
- 6.Shimoji K, Yuasa S, Onizuka T, et al. G-CSF promotes the proliferation of developing cardiomyocytesin vivo and in derivation from ESCs and iPSCs. Cell Stem Cell. 2010;6:227–37. doi: 10.1016/j.stem.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 7.Orlic D, Kajstura J, Chimenti S, et al. Mobilized bone marrow cells repair the infarcted heart, improving function and survival. Proc Natl Acad Sci U S A. 2001;98:10344–9. doi: 10.1073/pnas.181177898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Srinivas G, Anversa P, Frishman WH. Cytokines and myocardial regeneration: a novel treatment option for acute myocardial infarction. Cardiol Rev. 2009;17:1–9. doi: 10.1097/CRD.0b013e31817bd7ab. [DOI] [PubMed] [Google Scholar]
- 9.Misao Y, Takemura G, Arai M, et al. Importance of recruitment of bone marrow-derived CXCR4+ cells in post-infarct cardiac repair mediated by G-CSF. Cardiovasc Res. 2006;71:455–65. doi: 10.1016/j.cardiores.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 10.Li Y, Takemura G, Okada H, et al. Treatment with granulocyte colony-stimulating factor ameliorates chronic heart failure. Lab Invest. 2006;86:32–44. doi: 10.1038/labinvest.3700367. [DOI] [PubMed] [Google Scholar]
- 11.Lapidot T, Petit I. Current understanding of stem cell mobilization: the roles of chemokines, proteolytic enzymes, adhesion molecules, cytokines, and stromal cells. Exp Hematol. 2002;30:973–81. doi: 10.1016/s0301-472x(02)00883-4. [DOI] [PubMed] [Google Scholar]
- 12.LiL, Takemura G, Li Y, et al. Granulocyte colony-stimulating factor improves left ventricular function of doxorubicin-induced cardiomyopathy. Lab Invest. 2007;87:440–55. doi: 10.1038/labinvest.3700530. [DOI] [PubMed] [Google Scholar]
- 13.Martin DL, Postan M, Lucas P, Gress R, Tarleton RL. TGF-beta regulates pathology but not tissue CD8+ T cell dysfunction during experimental Trypanosoma cruzi infection. Eur J Immunol. 2007;37:2764–71. doi: 10.1002/eji.200737033. [DOI] [PubMed] [Google Scholar]
- 14.Loud AV, Anversa P, Giacomelli F, Wiener J. Absolute morphometric study of myocardial hypertrophy in experimental hypertension. I. Determination of myocyte size. Lab Invest. 1978;38:586–96. [PubMed] [Google Scholar]
- 15.Garg N, Popov VL, Papaconstantinou J. Profiling gene transcription reveals a deficiency of mitochondrial oxidative phosphorylation in Trypanosoma cruzi-infected murine hearts: implications in chagasic myocarditis development. Biochim Biophys Acta. 2003;1638:106–20. doi: 10.1016/s0925-4439(03)00060-7. [DOI] [PubMed] [Google Scholar]
- 16.Bhatia V, Sinha M, Luxon B, Garg N. Utility of Trypanosoma cruzi sequence database for the identification of potential vaccine candidates: in silico and in vitro screening. Infect Immun. 2004;72:6245–54. doi: 10.1128/IAI.72.11.6245-6254.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dhiman M, Garg NJ. NADPH oxidase inhibition ameliorates Trypanosoma cruzi-induced myocarditis during Chagas disease. J Pathol. 2011;225:583–96. doi: 10.1002/path.2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoemann CD. Molecular and biochemical assays of cartilage components. Methods Mol Med. 2004;101:127–56. doi: 10.1385/1-59259-821-8:127. [DOI] [PubMed] [Google Scholar]
- 19.Postan M, Bailey JJ, Dvorak JA, McDaniel JP, Pottala EW. Studies of Trypanosoma cruzi clones in inbred mice. III. Histopathological and electrocardiographical responses to chronic infection. Am J Trop Med Hyg. 1987 Nov;37(3):541–9. doi: 10.4269/ajtmh.1987.37.541. [DOI] [PubMed] [Google Scholar]
- 20.Postan M, Cheever AW, Dvorak JA, McDaniel JP. A histopathological analysis of the course of myocarditis in C3H/He mice infected with Trypanosoma cruzi clone Sylvio-X10/4. Trans R Soc Trop Med Hyg. 1986;80:50–5. doi: 10.1016/0035-9203(86)90193-8. [DOI] [PubMed] [Google Scholar]
- 21.Golgher D, Gazzinelli RT. Innate and acquired immunity in the pathogenesis of Chagas disease. Autoimmunity. 2004;37:399–409. doi: 10.1080/08916930410001713115. [DOI] [PubMed] [Google Scholar]
- 22.Minatoguchi S, Takemura G, Chen XH, et al. Acceleration of the healing process and myocardial regeneration may be important as a mechanism of improvement of cardiac function and remodeling by postinfarction granulocyte colony-stimulating factor treatment. Circulation. 2004;109:2572–25780. doi: 10.1161/01.CIR.0000129770.93985.3E. [DOI] [PubMed] [Google Scholar]
- 23.Pan L, Delmonte J, Jr, Jalonen CK, Ferrara JL. Pretreatment of donor mice with granulocyte colony-stimulating factor polarizes donor T lymphocytes toward type-2 cytokine production and reduces severity of experimental graft-versus-host disease. Blood. 1995;86:4422–9. [PubMed] [Google Scholar]
- 24.Lambert JM, Lopez EF, Lindsey ML. Macrophage roles following myocardial infarction. Int J Cardiol. 2008;130:147–58. doi: 10.1016/j.ijcard.2008.04.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsuruda T, Costello-Boerrigter LC, Burnett JC., Jr Matrix metalloproteinases: pathways of induction by bioactive molecules. Heart Fail Rev. 2004;9:53–61. doi: 10.1023/B:HREV.0000011394.34355.bb. [DOI] [PubMed] [Google Scholar]
- 26.Jane-Lise S, Corda S, Chassagne C, Rappaport L. The extracellular matrix and the cytoskeleton in heart hypertrophy and failure. Heart Fail Rev. 2000;5:239–50. doi: 10.1023/A:1009857403356. [DOI] [PubMed] [Google Scholar]
- 27.Altieri P, Brunelli C, Garibaldi S, et al. Metalloproteinases 2 and 9 are increased in plasma of patients with heart failure. Eur J Clin Invest. 2003;33:648–56. doi: 10.1046/j.1365-2362.2003.01187.x. [DOI] [PubMed] [Google Scholar]
- 28.Baldo MP, Rodrigues SL, Mill JG. Granulocyte colony-stimulating factor for ischemic heart failure: should we use it? Heart Fail Rev. 2010;15:613–23. doi: 10.1007/s10741-010-9188-0. [DOI] [PubMed] [Google Scholar]
- 29.Badorff C, Brandes RP, Popp R, et al. Transdifferentiation of blood-derived human adult endothelial progenitor cells into functionally active cardiomyocytes. Circulation. 2003;107:1024–32. doi: 10.1161/01.cir.0000051460.85800.bb. [DOI] [PubMed] [Google Scholar]
- 30.Kawamoto A, Tkebuchava T, Yamaguchi J, et al. Intramyocardial transplantation of autologous endothelial progenitor cells for therapeutic neovascularization of myocardial ischemia. Circulation. 2003;107:461–8. doi: 10.1161/01.cir.0000046450.89986.50. [DOI] [PubMed] [Google Scholar]
- 31.Beltrami AP, Barlucchi L, Torella D, et al. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003;114:763–76. doi: 10.1016/s0092-8674(03)00687-1. [DOI] [PubMed] [Google Scholar]
- 32.Zhang JS, He QY, Huang T, Zhang BX. Effects of panax notoginseng saponinson homing of C-kit + bone mesenchymal stem cells to the infarction heart in rats. J Tradit Chin Med. 2011;31:203–8. doi: 10.1016/s0254-6272(11)60043-5. [DOI] [PubMed] [Google Scholar]
- 33.Molkentin JD, Olson EN. GATA4: a novel transcriptional regulator of cardiac hypertrophy? Circulation. 1997;96:3833–5. [PubMed] [Google Scholar]
- 34.Zhao R, Watt AJ, Battle MA, Li J, Bondow BJ, Duncan SA. Loss of both GATA4 and GATA6 blocks cardiac myocyte differentiation and results in acardia in mice. Dev Biol. 2008;317:614–9. doi: 10.1016/j.ydbio.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oka T, Maillet M, Watt AJ, et al. Cardiac-specific deletion of Gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability. Circ Res. 2006;98:837–45. doi: 10.1161/01.RES.0000215985.18538.c4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.