

Abstract
The intracellular pathogen Legionella pneumophila is able to strike a balance between the death and survival of the host cell during infection. Despite the presence of high level of active caspase-3, the executioner caspase of apoptotic cell death, infected permissive macrophages are markedly resistant to exogenous apoptotic stimuli. Several bacterial molecules capable of promoting the cell survival pathways have been identified, but proteins involved in the activation of caspase-3 remain unknown. To study the mechanism of L. pneumophila-mediated caspase-3 activation, we tested all known Dot/Icm substrates for their ability to activate caspase-3. Five effectors capable of causing caspase-3 activation upon transient expression were identified. Among these, by using its ability to activate caspase-3 by inducing the release of cytochrome c from the mitochondria, we demonstrated that VipD is a phospholipase A2, which hydrolyzes phosphatidylethanolamine (PE) and phosphocholine (PC) on the mitochondrial membrane in a manner that appears to require host co-factor(s). The lipase activity leads to the production of free fatty acids and 2-lysophospholipids, which destabilize the mitochondrial membrane and may contribute to the release of cytochrome c and the subsequent caspase 3 activation. Furthermore, we found that whereas it is not detectably defectively in caspase 3 activation in permissive cells, a mutant lacking all of these five genes is less potent in inducing apoptosis in dendritic cells. Our results reveal that activation of host cell death pathways by L. pneumophila is a result of the effects of multiple bacterial proteins with diverse biochemical functions.
Keywords: Type IV secretion, effectors, mitochondrion, cell death, Cytochrome c
Introduction
Legionella pneumophila is a Gram negative, intracellularly replicating bacterial pathogen that is ubiquitous in the environment as a parasite of fresh water amoebae. Inhalation of this bacterium by immuno-compromised individuals can lead to an atypical acute pneumonia known as Legionnaires’ disease (Fields et al., 2002). Upon infection of alveolar macrophages, L. pneumophila utilizes the Dot (defect in organelle trafficking)/Icm (intracellular multiplication) secretion system to translocate at least 275 protein substrates into the host to modulate various host cell processes such as vesicle trafficking, innate immunity, and protein synthesis, to create an intracellular niche permissive for bacterial replication (Xu et al., 2013).
During apoptosis, activated caspases (cysteine-aspartic protease) causes DNA fragmentation, nuclear condensation, cell body shrinkage, and the formation of apoptotic bodies. This programmed cell death is a mechanism for efficient elimination of infected cells while eliciting little to no inflammatory response (Savill et al., 2002). Because activation of different forms of programmed cell death will result in different immunological consequences, many successful pathogens have evolved effective strategies to manipulate host apoptosis pathways (Lamkanfi et al., 2010). Permissive mammalian cells infected by L. pneumophila exhibit remarkable resistance to exogenous cell death stimuli, and such resistance requires a functional Dot/Icm system (Abu-Zant et al., 2007). Intriguingly, cells harboring actively replicating L. pneumophila also contain high levels of active caspase-3 (Gao et al., 1999, Abu-Zant et al., 2005), indicating the presence of a balance between the pro-death and pro-survival signaling. Clearly, in permissive mammalian cells, the activation of caspase-3 does not lead to restriction of L. pneumophila replication. However, the effect of such activation becomes apparent in the interactions between L. pneumophila and dendritic cells (DC). Infection of DCs by L. pneumophila causes extensive apoptotic cell death in a process that is dependent upon caspase 3 or Bax/Bak, two critical regulatory proteins in the mitochondrial cell death pathway (Nogueira et al., 2009).
A number of Dot/Icm substrates directly contribute to the apoptosis resistance in infected cells. Among these proteins, SidF functions by binding to two pro-death members of the Bcl-2 protein family and potentially by interfering with the host Akt signaling pathway via its phosphoinositide phosphatase activity (Banga et al., 2007, Hsu et al., 2012). Activation of cell survival pathways regulated by NFκB is essential for the anti-apoptotic resistance; and two effectors LegK1 and LnaB are implicated in such activation (Losick et al., 2006, Abu-Zant et al., 2007, Ge et al., 2009, Losick et al., 2010). SdhA contributes to the resistance by suppressing the damage caused by type I interferon by at least in part maintaining the integrity of the bacterial phagosomal membranes (Monroe et al., 2009, Creasey et al., 2012, Ge et al., 2012). In contrast, nothing is known about bacterial factors responsible for caspase-3 activation during L. pneumophila infection.
Patatins are a family of proteins that catalyze the hydrolysis of phospholipids at the sn-2 position, liberating a fatty acid and a 2-lysophospholipid molecule. Patatin-like-proteins (PLPs) are found in both eukaryotes and bacteria and share the conserved GxSxG and DGx (x: any amino acid) catalytic dyad motifs that are essential for their phospholipase activity (Banerji et al., 2004). PLPs play critical roles in diverse aspects of lipid metabolism and signal transduction, such as membrane trafficking, inflammatory response and apoptosis. Many pathogens code for PLPs to facilitate their infection and dissemination (Sitkiewicz et al., 2007). For example, ExoU, a phospholipase A2 from Pseudomonas aeruginosa plays an important role in diverse aspects of its interactions with hosts such as the inhibition of caspase-1 mediated inflammatory responses (Sutterwala et al., 2007). L. pneumophila is predicted to code for at least 11 PLPs, but the activity of most of these proteins has not yet been characterized (Banerji et al., 2008). The Dot/Icm substrate VipD is one such predicted PLP that was first identified in a screening for L. pneumophila Dot/Icm effectors by their ability to interfere with membrane trafficking in the budding yeast (Shohdy et al., 2005). This protein harbors a phospholipase A2 motif, which has been shown to be essential for the activity of ExoU from P. aeruginosa (VanRheenen et al., 2006). Unexpectedly, this motif is not essential for the interference of intracellular trafficking and yeast growth by VipD, suggesting the existence of an alternative function (Shohdy et al., 2005, VanRheenen et al., 2006).
To determine the bacterial factors responsible for caspase 3 activation during L. pneumophila infection, we performed a screening to identify Dot/Icm substrates capable of activating this enzyme upon transient expression in mammalian cells. These efforts led to the identification of five proteins with such activity. Here we present evidence that VipD is a phospholipase A2 that may contribute to the activation of caspase 3 during L. pneumophila infection.
Results
Identification of Dot/Icm substrates capable of activating caspase-3
The fact that permissive cells harboring actively replicating L. pneumophila contain active caspase-3 suggests that one or more Dot/Icm substrates directly activate this enzyme. To identify the Dot/Icm substrates capable of activating caspase-3, we transiently expressed GFP fusion of each of the 275 Dot/Icm substrates in 293T cells (Zhu et al., 2011). The presence of activated capase-3 in lysates of transfected cells was determined by the Caspase-Glo 3/7 Assay System (Promega). One protein Lpg0898 capable of activating caspase 3 was identified with this method. The identification of only one protein with the desired activity from the 275 candidates suggested that the sensitivity of bioluminescence assay was not sufficient for detecting the activity of other proteins. We thus employed an immunoblot-based method that detects the p17 and p19 fragments of processed caspase 3. Our efforts led to the identification of five Dot/Icm substrates, Lpg0716 (Zhu et al., 2011), Lpg0898(Ceg18) (Altman et al., 2008), Lpg1625 (Lem12) (Burstein et al., 2009), Lpg2176 (LegS2) (de Felipe et al., 2005), and Lpg2831 (VipD) (Shohdy et al., 2005, VanRheenen et al., 2006), which are able to induce caspase-3 activation in 293T cells upon transfection (Fig. 1). Although some of these proteins are toxic to mammalian cells, expression of GFP fusions of these proteins was readily detectable after transfection (Fig. 1, middle panel). Sequencing analyses revealed that Lpg2716(LegS2) is a sphingosine-1-phosphate lyase, which has been shown to target to the mitochondria during bacterial infection (Degtyar et al., 2009); Lpg2831(VipD) possesses a domain found in the catalytic motif of members of the patatin-like phospholipase A2 (Shohdy et al., 2005, VanRheenen et al., 2006). On the other hand, Lpg0716, Lpg0898(Ceg18), Lpg1625(Lem12) and Lpg1803 harbor no motif suggestive of any potential biochemical activity.
Fig. 1. Activation of caspase-3 by L. pneumophila Dot/Icm substrates.

Lysates of HEK293T cells transfected to express GFP or its fusions of indicated proteins for 24 hr were probed for active caspase-3 with a specific antibody (upper panel). The p-17 and p-19 bands are the processed mature form of caspase-3. The expression of the GFP fusions in these samples was probed with a GFP specific antibody (middle panel). Arrows indicate the band corresponding to the GFP fusion proteins. Tubulin was probed as a loading control (lower panel).
Some of the Legionella caspase 3 activators localize to the mitochondrion
Mitochondrion is the central regulator for the intrinsic apoptotic pathway. The sphingosine-1-phosphate lyaseLpg2176(LegS2) had been shown to localize to this organelle (Degtyar et al., 2009). We thus examined whether other identified caspase 3 activators also target to this organelle. Cells transfected to transiently express GFP fusions of each of these proteins were stained with an antibody specific Cyto c oxidase (COX4I1). We found that like GFP-Lpg2176, in cells expressing GFP-Lpg1625, the GFP signal exclusively localized to the mitochondria and the patterns of the signal were identical to those of COX4I1 (Fig. 2). Most, but not all of GFP-Lpg0898 localized to the mitochondria (Fig. 2). However, the expression level of GFP-0716 is low and the GFP signals appeared to localize to the cytosol. GFP-2831(VipD) mostly occupied the plasma membrane of transfected cells, and did not significantly colocalize with the COX4I1 staining signals (Fig. S1).
Fig. 2. Some of the caspase-3 activators localized to the mitochondrion.

Hela cells transfected to express GFP fusion of the indicated proteins were stained with antibody specific for the mitochondrial protein COX4I1. Samples were analyzed using an Olympus IX-81 fluorescence microscope for images acquisition. Images were pseudocolored with the IPLab software package. Bar: 20 μm.
We also found that protein Lpg0898 and Lpg2831(VipD) caused significant cell rounding on mammalian cells (Fig. S2). To rule out the possibility that the cytotoxicity of these proteins interferes with the proper cellular localization, we isolated a non-toxic mutant of Lpg0898, Lpg0898A151S; this mutant still partially localized to the mitochondria (Fig. S1). For Lpg2831, we determined whether the putative phospholipase A2 is responsible for the cell rounding phenotype by mutating serine 73 in the predicted catalytic motif to alanine. Although this mutation did not reveal the targeting of Lpg2831(VipD) to a specific organelle, it abolished its ability to cause the cell rounding phenotype in mammalian cells (Fig. S2). These results suggest that mitochondrion is the site of action for some of the caspase 3 activators. Our observations also suggest that the putative phospholipase A2 activity of VipD is important for its role in caspase 3 activation. Because of the potential biochemical activity and the availability of many useful reagents for VipD (VanRheenen et al., 2006), we chose to focus on this protein for further study.
VipD induces the release of Cyto c from mitochondria
The fact that VipDS73A is no longer toxic to mammalian cells prompted us to examine its capability in caspase 3 activation. Lysates of 293T cells transfected to express VipD or its mutant were probed for active caspase 3. Whereas expression of GFP-VipD consistently led to production of the p-17 and p-19 fragments of caspase 3, cells expressing GFP or GFP-VipDS73A did not produce detectable active caspase 3 (Fig. 3A). Thus, the putative PLPA2 activity of VipD is essential for its role in the activation of caspase 3.
Fig. 3. VipD-induced caspase 3 activation and Cyto c release require the phospholipase motif.

A. The phospholipase motif is essential for the activation of caspase 3. 100μg lysates of HEK293 cells transfected to express GFP (1st lane), GFP-VipD (2nd lane) or GFP-VipDS73A(3rd lane) for 24 hr were probed for active caspase 3. Note the high level expression of GFP-VipDS73A (3rd lane, middle panel). Tubulin was probed as loading controls (lower panel). B. 8 VipD-induced Cyto c release in vivo. 50μg soluble fraction and mitochondrial fraction of HEK293 cells transfected to express GFP-VipD or GFP-VipDS73A for 24 hr were probed for Cyto c. The expression of the GFP fusions was also analyzed. Tubulin present in the soluble fraction was detected as a loading control. C. VipD-induced Cyto c release from purified mitochondria. Indicated amounts of His6-VipD or His6-VipDS73A were incubated with 50μg mitochondria at 30°C for 2 hr. The presence of Cyto c in the soluble fractions was detected by immunoblotting. Note that little Cyto c release was induced by 40μg His6-VipDS73A (last lane).
In the intrinsic apoptotic pathway, the release of the cytochrome c from mitochondria is a prerequisite for caspase 3 activation; we then asked whether VipD is able to cause Cyto c release. Cells transfected with appropriate constructs were permeabilized with digitonin, which selectively disrupts plasma membrane while leaving mitochondrial membrane intact (Figueira et al., 2012). The resulting cytosolic fraction was separated from the mitochondria-rich membrane fraction and tested for the presence of Cyto c. Expression of wild type VipD but not the S73A mutant or GFP led to Cyto c release (Fig. 3B). We further examined the activity of VipD using a cell-free system. Recombinant VipD was added to purified mitochondria and the release of Cyto c was monitored after 2 hr of incubation. As little as 0.65 μg His6-VipD caused significant Cyto c release, and 2.5 μg protein was sufficient to disperse more than 90% of Cyto c from 100 μg mitochondria (Fig. 3C). Interestingly, addition of VipD also led to the release of SOD2, a mitochondria matrix resident protein, into the supernatant, suggesting the integrity of the mitochondrial inner membrane was also compromised in this process (Fig. 3C). Consistent with the in vivo results, as much as 40 μg VipDS73A failed to cause significant Cyto c release (Fig. 3C, last lane).
Cyto c release is often accompanied by the disruption of the mitochondrial outer membrane potential (Δψm) (Jiang et al., 2004). We thus examined whether VipD interferes with the integrity of the mitochondrial outer membrane potential. Hela cells transfected to produce VipD, VipDS73A, or GFP were stained with the mitochondrial transmembrane potential sensitive dye, tetramethylrhodamine methyl ester perchlorate (TMRM), and the fluorescence intensity of mitochondrion (Fm) and cytosol (Fc) reflected by the TMRM staining was quantified. The value of Δψm was obtained with the formula Δψm=[(Fm-(2/3)Fc)/(1/3) Fc]. Expression of VipD but not its S73A mutant compromised the integrity of the mitochondrial outer membrane (Fig. 4). Taken together, these results indicate that induction of caspase 3 activation by VipD is mediated by its putative PLA2 activity, which triggers the mitochondrial outer membrane permeabilization (MOMP) and the subsequent release of Cyto c.
Fig. 4. VipD compromises the mitochondrial outer membrane integrity.
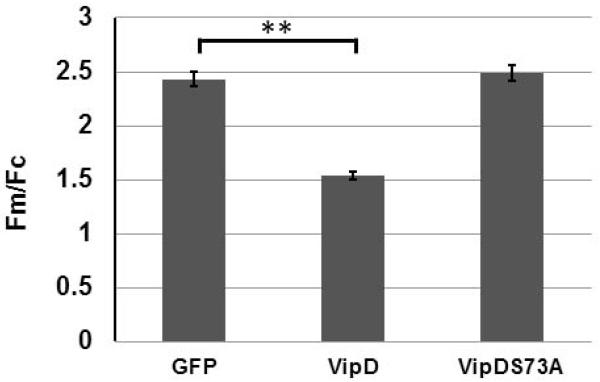
Culture medium of HEK293T cells transfected with indicated constructs for 18 hr was replaced with medium containing 20 nM TMRM for 20 min. Dye loaded samples were then mounted on a Teflon holder for image acquisition using a Nikon Eclipse Ti microscope (60x oil Plan Apo objective). TMRM images were thresheld to isolate the mitochondrial florescence (Fm), and the florescence intensity of areas of one square micrometer was measured using the software ImageJ (NIH). Cytoplasmic florescence (Fc) is determined in hand-drawn regions in close proximity to the mitochondria. Δψm is then determined as the ratio of Fm/Fc using the following formula: [(Fm-(2/3)Fc)/(1/3) Fc]. At least 300 mitochondria in each sample were analyzed and the statistical analysis was performed using SPSS 20.0 (IBM). **, p<0.001.
VipD-mediated mediated Cyto c release is independent of Bax and Bak
Since Bax and Bak, the two pro-death pore-forming proteins important for MOMP (Jiang et al., 2004) appear essential for the cell death induction during L. pneumophila infection (Nogueira et al., 2009), we determined whether these two proteins are necessary for the activity of VipD in caspase 3 activation. Transient expression of VipD in mouse embryonic fibroblasts (MEFs) deficient in Bax and Bak led to activation of caspase 3 (Fig. S3A upper left panel,). Of note is that the Mirus transfection reagent nonspecifically caused caspase 3 activation in this cell line (Fig. S3A, upper left panel, 1st lane). Although caspase 3 activation occurred in samples received no DNA or the catalytically inactive VipDS73A mutant, the amount of active caspase 3 in the sample expressing wild type VipD is consistently higher (Fig. S3 A-B). Importantly, in the Bax/Bak−/−MEFs, similar activation of caspase 3 was detected, but only in samples expressing wild type VipD (Fig. S3A, upper right panel). Similarly, purified His6-VipD but not the VipDS73A, was able to induce Cyto c release from mitochondria isolated from both wild type and bak/bax−/− MEFs (Fig. S3C). Thus, VipD appears to directly act on the mitochondrial membranes to compromise its integrity, leading to Cyto c release and the subsequent caspase 3 activation.
Inhibition of VipD-mediated Cyto c release by two PLA2 inhibitors
The observation that the predicted PLA2 active site of VipD was important for the induction of Cyto c release strongly suggests that VipD is a phospholipase A2. We thus examined this activity by determining the effects of two specific phospholipase A2 inhibitors, Methyl arachidonyl fluorophosphonate (MAFP) (Deutsch et al., 1997) and (R)-Bromoenol lactone ((R)-BEL) (Balsinde et al., 1999), on VipD-mediated Cyto c release. As little as 0.2 μM MAFP almost completely abolished Cyto c release caused by VipD (Fig. 5); (R)-BEL also blocked VipD activity, but with a relatively lower potency; 10 μM of this compound was required to exert detectable inhibitory effects (Fig. 5). Because both MAFP and (R)-BEL are highly selective, active site directed, irreversible inhibitors of PLA2 (Deutsch et al., 1997, Balsinde et al., 1999), these results further suggest that VipD is a phospholipase A2.
Fig. 5. Inhibition of VipD-induced Cyto c release by two inhibitors for phospholipase A2.
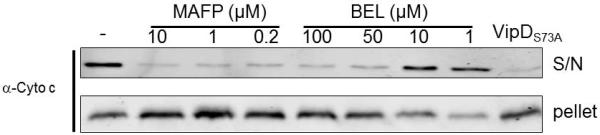
The inhibitors MAFP or (R)-BEL was added to suspensions of 50μg purified mitochondria at indicated concentrations simultaneously with 42 μg of His6-VipD. After incubation for 3 hr at 30°C, samples were subjected to centrifugation to obtain the soluble fraction and the insoluble membrane pellet and each fraction was analyzed for the presence of Cyto c. Samples receiving His6-VipD (first lane) or His6-VipDS73A (last lane) but no inhibitor were included as controls.
VipD does not indiscriminately disrupt biological membranes
Phospholipase A2s have relatively broad substrate spectrum and are capable of disrupting the integrity of membranes from different sources (Rehfeldt et al., 1993, Putz et al., 2007). To determine whether VipD-mediated Cyto c release is due to indiscriminate membrane disruption, we first examined whether VipD compromises the integrity of the plasma membrane. Hela cells were transfected with GFP-VipD for 24 hours and the integrity of the plasma membrane (PM) was assessed with ethidium bromide (EB), a dye impermeable to intact PM. Treatment with 0.2% TritonX-100 effectively facilitated the uptake of EB. However, although exhibiting the cell rounding phenotype, cells expressing GFP-VipD were not detectably permeable to this dye (Fig. S4A). We also tested the effects of exogenous VipD on PM integrity. Inclusion of His6-VipD in cell culture medium at 10 μM, which is 2-fold higher than the concentration necessary for effective Cyto c release, did not detectably disrupt the PM (Fig. S4B). Taken together, these results indicate that VipD specifically targets mitochondrial membranes but not the plasma membrane. VipD hydrolyzes two phospholipids on mitochondrial membranes The specific disruption of mitochondrial membranes by VipD suggests that its substrates are associated with this organelle. To identify these substrates, we extracted total lipids from the mitochondria treated with His6-VipD or His6-VipDS73A by the Folch method (Folch et al., 1957) and subjected these samples to mass spectrometry analysis. 2-lysophosphatidylcholine and 2-lysophosphatidylethanolamine, the end products of phospholipase A2, increased 18 and 16-fold, respectively in samples receiving VipD but not in those receiving VipDS73A, indicating that phosphatidylcholine (PC) and phosphatidylethanolamine (PE) are substrates of VipD (Fig. 6).
Fig. 6. Identification of the natural substrates of VipD.

Total lipids from mitochondria treated with His6-VipD or His6-VipDS73A were extracted using the Folch method. The composition of the lipids was profiled by mass spectrometry on an LTQ Orbitrap instrument. Shown are representative base peak chromatograms of mitochondrial lipid extracts obtained in positive (A) and negative (B) ion modes using reversed-phase chromatography. Lysophospholipid species with significant increase in His6-VipD treated samples detected by LC-MS/MS (C).
Despite extensive efforts, we were unable to detect the phospholipase activity of His6-VipD in biochemical assays using synthetic substrates, which was reported in a recent study on the structure of this protein (Ku et al., 2012) (Fig. S5A). To test the possibility that VipD needs co-factor(s) from the host, we treated isolated mitochondria with proteinase K prior to examining VipD-mediated Cyto c release. Exposure of mitochondria to 1 μg/ml of this proteinase abolished VipD-mediated Cyto c release (Fig. S5B). The loss of this activity is not caused by the potential residual proteinase activity as VipD recovered from this reaction was able to compromise membranes in fresh mitochondrial samples (Fig. S5B and data not shown). These results validate the potential need of host factors for the lipase activity of VipD.
Lysophospholipids and fatty acids can mimic the effect of VipD in inducing cytochrome c release
It is well-documented that phospholipids adopt a cone shape conformation when it is inserted in to the lipid bilayer (Barr et al., 2000). The 2-lysophospholipid and fatty acid molecules produced by phospholipase A2 from phospholipids adopt an inverted cone shape and cone shape, respectively, making them unsuitable in forming lipid bilayer (Caccin et al., 2009). Membrane damage and the subsequent Cyto c release caused by phospholipase A2 can result from direct lipid degradation (Grandbois et al., 1998) or from the formation of curvature or lesions on the mitochondrial membranes by the lipids and fatty acids (Rigoni et al., 2005). To determine whether the products of VipD contribute to such damage, we prepared a mixture of lysophosphoethanolamine and linolenic acid at a 1:1 ratio and examined the effects of the mixed compounds on purified mitochondria. This mixture is capable of recapitulating the effects of VipD on 3 mitochondria, and at least 9 μM of the mixed compounds were required to induce Cyto c release to the level induced by 40 μg of His6-VipD (Fig. 7). Thus, VipD-mediated production of lysophospholipids and fatty acids contributes to the disruption of mitochondrial membranes and the subsequent Cyto c release.
Fig. 7. A mixture of lyso-PE and linolenic acid induces Cyto c release from mitochondria.

5μl of linonelic acid and 7.5 mg of lysophosphoethanolamine were dispersed in 5-ml of L buffer. The mixture was brought to dryness under gentle nitrogen stream and was reconstituted with 5-ml L buffer; the indicated amounts of lipid mixture was then added to 50μg purified mitochondria, and incubated at 30°C for 2 hr. The samples were then separated by centrifugation, soluble or pellet fraction was resolved on SDS-PAGE and probed for Cyto c.
The caspase-3 activating effectors are required for maximal induction of cell death in dendritic cells
To determine whether these effectors proteins contribute to the caspase-3 activation during L. pneumophila infection, we constructed a series of mutants lacking one or more of these genes. To assess their roles in caspase-3 activation during L. pneumophilainfection, non-polar deletions of each of these genes and the combination of all of them are constructed. The deletion strain, along with WT L. pneumophila and a Dot/Icm mutant cultured to post-exponential phase were used to infect U937 at an MOI of 1 (Nogueira et al., 2009). Infected cells were lysed six hours after infection to assay for caspase 3 activity. Similarly prepared bacterial cells were used to infect mouse bone marrow-derived macrophages to evaluate intracellular replication. Deletion of single caspase-3 activator or all five of these genes (Lpg0898, Lpg1625, Lpg0716, Lpg2716, and Lpg2831) did not cause detectable defect in caspase-3 activation (Fig. S6). Consistent with this observation, the Δ5 mutant did not display any defect in intracellular bacterial growth (Fig. S7).
Because dendritic cells (DCs) are more sensitive to cell death induced by L. pneumophila, (Nogueira et al., 2009), we next examined whether the mutant missing these five genes is impaired in its ability to induce apoptosis in these cells. Although the Δ5 mutant did not productively replicate in DCs, comparing to the wild type strain, it was not efficiently killed by these cells (Fig. 8A). The mutant survived in rates very similar to those exhibited to the mutant deficient in the Dot/Icm transporter. Evaluation of the apoptotic status of infected DCs revealed that less than 5% of the cells harboring the Δ5 mutant stained positively by the TUNEL reagent, whereas larger than 25% of the cells infected by the wild type strains are apoptotic (Fig. 8B-C). Thus, deletion of the five genes only partially abolished the ability of L. pneumophila to induce cell death in DCs. Together with the fact that this mutant still grew robustly in macrophages, cells that are less sensitive to cell death induction by the bacteria, these data suggest the existence of yet unidentified caspase-3 activating effectors. Alternatively, activation of caspase-3 by L. pneumophila is a result of the collective effects of many effectors, which when expressed individually did not cause detectable activation.
Fig. 8. The five caspase 3 activating proteins are important for maximal cell death induction in dendritic cells.

A. Dendritic cells prepared from bone marrows of A/J mice used infected with indicated bacterial strains and the total bacterial counts were determined at indicated time points. Note the drastic drop in the number of survived wild type bacteria over the 48-hr infection duration. B. DCs similarly infected as panel A were subjected to immunostaining to label the cells, the bacteria and the apoptotic status of the infected cells with specific antibodies. Infected cells stained positively by the TUNEL reagent were enumerated under an Olympus IX-81 fluorescence microscope. At least 200 infected cells were scored from each sample done in triplicate. C. Representative images of the cells were acquired from these samples. Similar results were obtained in three experiments done with independently isolated DCs.
Discussion
It is well-established that L. pneumophila is able to maintain a balance between the induction and inhibition of host cell death in mammalian cells (Luo, 2011). Several lines of evidence suggest that induction of cell death occurs during L. pneumophila infection. First, active caspase 3 is present in infected cells (Gao et al., 1999, Abu-Zant et al., 2005). Second, macrophages lacking plasminogen activator inhibitor-2 (PAI-2), an anti-apoptotic protein whose expression is induced by L. pneumophila infection, are more apoptotic and less supportive for bacterial replication (Losick et al., 2006). Third, mouse dendritic cells undergo extensive apoptosis upon challenged by L. pneumophila, thus limiting intracellular bacterial replication. Further, this phenotype can be reversed by eliminating caspase 3 or Bax/Bak, two pro-death proteins, or by overexpressing the pro-survival protein Bcl-2 (Nogueira et al., 2009). In this study, we have identified several Dot/Icm substrates capable of inducing caspase activation when transiently expressed in mammalian cells. Although, the mechanism of action of most of these proteins is unknown, we showed that the induction of caspase 3 activation by VipD is a result of its putative phospholipase A2 activity, which causes damage in the mitochondrial membranes, leading to Cyto c release and subsequent caspase 3 activation.
VipD appears to have at least two functional domains: the N-terminal domain, which contains a consensus motif (G71xSxG) essential for the activity of some phospholipases, and a C-terminal domain important for inhibiting the yeast endocytic pathway (Shohdy et al., 2005, Ku et al., 2012). Both domains are required for the maximal toxicity of VipD to yeast (VanRheenen et al., 2006). Two lines of evidence suggest that VipD is a phospholipase A2, which contributes to the activation of caspase 3. First, substitution mutations disrupting the motif important for this family of enzymes abolished its ability to produce lysophospholipids and fatty acids from substrates in cell free samples. Second, two specific, established phospholipase inhibitors potently blocked its activity. Despite intensive efforts, we were unable to detect hydrolysis of phospholipids by VipD with synthetic substrates. For examples, although mass spectrometry analysis with the mitochondrial lipids revealed PE as one major substrate of VipD, incubation of synthetic PE with VipD did not yield detectable products (Fig. S5). ExoU, the phospholipase from Pseudomonas aeruginosa is not active in the absence of its co-factor ubiquitin or ubiquitinated proteins (Engel et al., 2009). Similarly, VipD may also need a eukaryotic co-factor for activation. Our observation that VipD failed to induce cytochrome c release from proteinase K-treated mitochondria strongly suggests that the co-factor(s) is of proteinaceous nature (Fig. S5B). The mild toxicity of VipD to yeast (VanRheenen et al., 2006) suggests that the co-factor(s) may be more specific or more abundant in mammalian cells; it will be interesting to identify such factor(s) and test whether its presence will increase the toxicity of VipD to yeast. Further, in intact cells VipD exhibits considerable specificity for the mitochondrion (Fig. S5), which may result from specific association of the co-factor(s) with this organelle.
The physiological role of cell death induction such as the activation of caspase-3 during L. pneumophila infection is not well understood. In human macrophages, blocking caspase 3 activity by specific or paninhibitor of caspases caused arrest in intracellular bacterial growth (Molmeret et al., 2004). Activated caspases may participate in cellular processes important for the intracellular life cycle of L. pneumophila. For example, the cleavage of Rab5 effector Rabaptin-5 by caspase-3 had been suggested to play a role in the evasion of the endocytic pathway by the bacterial phagosome (Molmeret et al., 2004). Caspases have been suggested to process bacterial effectors into forms capable of specific functions during Salmonella infection (Srikanth et al., 2010), it is possible that some of the Legionella Dot/Icm substrates undergo similar processing for specific activity. It is worth noting that the role of caspase 3 in L. pneumophila infection may differ among the hosts because primary macrophages deficient in this enzyme support normal bacterial replication (Zamboni et al., 2006). Alternatively, caspase inhibitors may block the activity of multiple such enzymes, leading to the strong defect of intracellular growth, whereas in caspase 3-deficient mouse macrophages, the activity can be compensated by other caspases.
The fact that an L. pneumophila mutant lacking all 5 caspase 3-activating effectors still fully activates this enzyme suggests the involvement of other Dot/Icm substrates in this process. This phenotype is consistent with proficient intracellular growth ability of this mutant (Fig. S7). However, the resistance to the killing by DCs exhibited by the Δ5 mutant suggests that these five proteins, do contribute to the induction of cell death during L. pneumophila infection. This notion is consistent with the fact that DCs infected by the Δ5 mutant is less apoptotic 8 hr after bacterial uptake. Because the induction of caspase 3 activation by VipD still occurs in the absence of Bax/Bak (Nogueira et al., 2009), two proteins that appear to critical for the cell death induced by L. pneumophila (Fig. S3), it is worth noting that the contribution of VipD to this process, if any, is achieved by a pathway that differs from the one caused by these two pore forming proteins.
That only Lpg0898 was identified from the 275 candidates (Zhu et al., 2011) in the Caspase-Glo 3/7 assay system indicated that this method is not as sensitive as the more labor-intensive immunoblot-based assay. Thus, it is likely more Dot/Icm substrates capable of activating caspase 3 remain to be identified. Alternatively, the observed caspase 3 activation by these effectors may be a byproduct of their yet unidentified biological functions; such functions can be different among the proteins and are not directly related to host cell death, which would explain the lack of defects in bacterial intracellular replication of our mutants. Nevertheless, future study aiming at obtaining a complete list of Dot/Icm substrate capable of inducing caspase 3 activation is required for determining the importance of cell death induction in L. pneumophila infection. Further study of the functions of these proteins may reveal the links between mitochondrial activity and some seemingly irrelevant cellular pathways directly targeted by these proteins.
Experimental Procedures
Bacterial strains, media and cell culture
All L. pneumophila strains (Table S1) are derivatives of the strain Lp02 (Berger et al., 1993); bacteria were grown on charcoal-yeast extract (CYE) solid medium or ACES-yeast extract (AYE). For infection experiments, L. pneumophila strains were grown to post-exponential phase (OD600=3.4-3.7) unless stated otherwise. E. coli strains were grown according to standard protocols (Luo et al., 2003).
Non-polar deletions of each or the combination of lpg0716, lpg0898, lpg1625, lpg2716, and lpg2831 were constructed as described elsewhere (Luo et al., 2004). In each case, primers were designed such that the open reading frame was replaced by a polypeptide consisting of 32 amino acids: the first and last 15 residues of the protein separated by two residues encoded by the restriction enzyme used for plasmid construction. The sequences of the primers used were 7 listed in Table S2.
293T cells and mouse embryonic fibroblasts (MEFs) were cultured in DMEM medium supplemented with 10% FBS; U937 cells were maintained in RPMI-1640 medium supplemented with 10% FBS prior to being induced by phorbol myristate acetate (PMA) (0.1 μg/ml) (Tilney et al., 2001). Bone marrow-derived macrophages were prepared according to an established protocol (Swanson et al., 1995). All cell lines were maintained at 37 °C within a humidified atmosphere and with 5% CO2.
Dendritic cells derived from A/J mouse bone marrow were cultured as previously described (Inaba et al., 2009). Maturing, nonadherent DCs were plated onto poly-lysine treated coverslips seeded in 24-well plates) at a concentration of 1×105 cells per well. 12 hr later, DCs were challenged with properly grown L. pneumophila strains at an MOI of 25. The plates were centrifuged at 150xg for 10 minutes. After two hours, extracellular bacterial growth was stopped by addition of 50 μg/ml gentamicin for 1 hour. DCs were then washed 3 times with PBS to remove residual antibiotic and cultured in fresh medium for the indicated length of time. At indicated time point, DCs were lysed in 0.02% saponin, and dilutions of lysate were plated on CYE agar to determine bacterial count.
For single cell analysis, DCs were infected by indicated strains of L. pneumophila for 8 hours. Infected samples were incubated with FITC-conjugated anti-CD11b antibody (Miltenyi Biotec, Auburn, CA) for 10 minutes in 4 °C to identify DCs. Infected samples were then fixed with 4% paraformaldehyde for 20 minutes at room temperature, washed 3 times with RPMI-1640 and permeabilized in RPMI-1640 containing 0.5% saponin for 15 min. L. pneumophila were stained as described elsewhere (Luo et al., 2004). Apoptotic DCs were identified using In Situ Cell Death Detection Kit (Roche) per manufacturer’s instruction.
Mutagenesis
The serine residue (S73) critical for the putative phospholipase of VipD is mutated to alanine using primers vipDS73AF and vipDS73AR (Table S2) with the Pfu UltraII (Agilent Technologies) DNA polymerase. Mutants of Lpg0898 no longer toxic to yeast were isolated by mutagenizing pGBKT7m-0898 with hydroxylamine using an established protocol (Luo et al., 2003). Treated DNA was introduced into yeast strain W303, colonies appeared on selective medium were screened for the presence of full-length Lpg0898 by immunoblot with a 5 Gal4 specific antibody (sc-577, Santa Cruz Biotechnology). All mutations were determined by double strand DNA sequencing.
Library construction and screen for caspase 3 activators
A library for expression of GFP fusions of experimentally confirmed Dot/Icm substrates in mammalian cells was constructed by inserting individual genes (Zhu et al., 2011) into vector pEGFP-C1 (Clontech). These genes were listed in Table S4 of reference (Zhu et al., 2011). 293T cells were transfected with individual plasmid harboring the GFP fusion using Lipofactamine-2000 (Invitrogen) per manufacturer’s instructions. Twenty-four hours after transfection, cells were lysed and the cellular caspase 3 activity was measured using the Caspase-Glo 3/7 Assay Systems (Promega, Madison, WI) according to protocols supplied by the manufacturer. Alternatively, transfected cells were lysed with the RIPA buffer (1% NP-40, 0.1% SDS, 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.5% sodium deoxycholate, 1 mM EDTA) supplemented with a protease inhibitor cocktail (Millipore, PA) 24 hr after transfection. The presence of activated caspase 3 (p17 and p19 fragments) was detected by immunoblot.
Antibodies, immunoblotting and immunostaining
For immunoblotting, the antibody for detecting cleaved caspase-3 was from Cell Signaling (#9664) and was used at 1:1000. The SOD2 antibody from Abcam was used at 1:2000. The Cyto c antibody from Santa Cruz Biotechnology (#sc-13560) was used at 1:3000. The VipD antibody (VanRheenen et al., 2006) was a kind gift from Dr. Ralph Isberg (Tufts Medical School). The antibody against GFP described earlier (Xu et al., 2010) was used at 1:20,000. Cyto c oxidase (COX4I1) was detected with an antibody from Cell Signaling at a 1:500 ratio for immunostaining. Mammalian cells seeded on glass coverslips were transfected to express the proteins of interest and were fixed with an established protocol (Conover et al., 2003). After permeabilized with 0.02% Triton X-100 for 5 min at room temperature, washed samples were stained with an antibody specific for COX4I1 followed by Texas-red-conjugated secondary antibody.
Measurement of mitochondrial transmembrane potential
Mitochondrial transmembrane potential (Δψm) is measured by determining the mitochondrial-to-cytoplasmic fluorescence ratio (Fm/Fc) using tetramethylrhodamine methyl ester perchlorate (TMRM) with established methods (Verburg et al., 2008). Briefly, Hela-60 cells were exposed to 20 nM of TMRM in DMEM+10% FBS medium for 20 minutes in a CO2 incubator of 37 °C to allow dye equilibration. The medium is then replaced with 5 nM of TMRM in DMEM+10% FBS medium for imaging. After loading with the mitochondrial dye, coverslips were mounted on a Teflon holder and the images were acquired using a Nikon Eclipse Ti microscope (60x oil Plan Apo objective). TMRM images were thresheld to isolate the mitochondrial florescence (Fm), and the florescence intensity of areas of one square micrometer was measured using the software ImageJ (NIH). Cytoplasmic florescence (Fc) is determined in hand-drawn regions in close proximity to the mitochondria. Δψm is then determined as the ratio of Fm/Fc using the following formula: [(Fm-(2/3)Fc)/(1/3) Fc]. At least 300 mitochondria in each sample were analyzed and the statistical analysis was performed using SPSS 20.0 (IBM).
In vivo cytochrome c release assay
293T cells transfected to express individual GFP fusion of testing proteins were harvested and washed twice with PBS. 1×107 Cells were then permeabilized for 5 min on ice with a mitochondrial buffer (20 mM HEPES, pH 7.4, 250 mM sucrose, 100 mM KCl, 5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, and 100 g/μl digitonin) at a concentration of about 3.5 x 107 cells/ml (Du et al., 2008). The cytosolic fraction was separated 1 from mitochondrial fraction by centrifugation at 600xg for 10 min at 4 °C. 50 μg of soluble cytosolic proteins (supernatant) and mitochondrial proteins (pellet) were then resolved on 15% SDS-PAGE gel. The presence of cytochrome c (Cyto c) in these samples was detected by immunoblot.
In vitro cytochrome c release assay
We isolated mitochondria from HEK293T cells using a Mitochondria Isolation Kit (Sigma) per manufacturer recommendations. 100 μg purified mitochondria were then incubated with VipD or its mutant proteins in a mitochondrial storage buffer (Sigma) at 30 °C for 2 hr. The samples were then cleared at 13,000xg for 10 min at 4 °C. The resulting supernatant and pellet were then resolved on 15% SDS-PAGE gels and Cyto c was detected by immunoblot. The matrix protein SOD2 was probed to monitor the integrity of mitochondrial inner membrane.
Lipid extraction
Lipids from 200 μg purified mitochondria that had been incubated with purified His6-VipD or His6-VipDS73A were extracted using the Folch method (Folch et al., 1957). Briefly, chloroform, methanol, and water with a volume ration of 8:4:3 were mixed in a separation funnel, allowed to stand to obtain a biphasic system. The upper phase was used as the pure-solvent-upper-phase. The mitochondria samples were placed into a glass homogenizer with 5 drops of ice-cold chloroform-methanol extraction solvent (2:1) and manually homogenized with a pestle. The volume of extraction solvent was slowly increased to 0.5-mL in order to achieve complete homogenization. Homogenates were filtered through a lipid-free paper and mixed with 0.2 volumes of H2O, and the mix is allowed to separate into 2 phases. After removing the upper phase, the lower phase was rinsed 3 times with the pure-solvent-upper-phase. The lower organic phase was collected, dried under a stream of nitrogen, and stored at -80°C.
Liquid chromatography mass spectrometry
Lipid extracts from samples treated by VipD or its S73A mutant were profiled using the LTQ Orbitrap instrument to determine their lipid contents. The analysis was performed using a Dionex 3000 Ultimate LC system (Dionex, Sunnyvale, CA) interfaced to an externally calibrated LTQ Orbitrap hybrid mass spectrometer with an electrospray ion source (Thermo Scientific, San Jose, CA). A 1-μL aliquot of lipid extract dissolved in 50 μL of methanol was injected onto a Kinetex™ C18 column (100 mm χ 2.1 mm, 2.6 μm particle size) equipped with an inline filter from Phenomenex (Phenomenex, Torrance, CA). Mobile phase A was 10 mM ammonium acetate in water:methanol (10%:90%, v:v), and mobile phase B was 10 mM ammonium acetate in isopropanol:methanol (50%:50%, v:v). The column was maintained at 25 °C and the gradient conditions were a linear ramp of 30% B to 100% B in 15 minutes using a flow rate of 250 μL/min.
The mass spectrometer was operated in an automated data dependent mode alternating between an FT-MS scan in the Orbitrap and a collision-induced dissociation (CID) scan in the linear ion trap. The scan in the Orbitrap was from m/z 300 to m/z 2000 at 15000 resolving power. The precursor ions were isolated in the linear trap using the data-dependent acquisition mode with a 2 m/z isolation width to select automatically and sequentially the three most intense ions from each survey scan. The isolated ions were collisionally activated in the linear trap at 35 % normalized collision energy. The cycle was continuously repeated throughout the entire separation with the dynamic exclusion set to 45 seconds for a repeat count of 2. Performing the initial mass scanning in the Orbitrap offered high mass accuracy of the selected precursor ions. The analysis was performed twice, once in positive ion mode and again in negative ion mode.
Lipid preparation
Five μl of lysophosphoethanolamine (Avanti Lipids, Alabaster, AL) and 7.5 mg linolenic acid (Sigma, St. Louis, MI) were dissolved in 2-ml of chloroform: methanol 3:1 solvent, which are then dried to a thin film under a gentle nitrogen stream for at least 2 hours to remove residual organic solvent. The resulting thin film of lipids were then dispersed in 5-ml of L buffer (50 mM Tris·Cl 7.5, 150 mM NaCl) at 37 °C using a tip-sonicator with 20% vibration until optical clarity is achieved.
Phospholipase activity assay and thin-layered chromatography
Phosphatidylcholine (PC) and phosphatidylserine (PS) were purchased from NOF America Corp and phosphatidylethanolamine (PE) were obtained from Sigma. Lipids were dissolved in 100% chloroform to a final concentration of 100 mg/ml. To prepare PC or PE only liposomes, 40 μg of 2 each lipid was dried in a Speed-Vac and rehydrated with enzyme buffer (20 mM Tris·Cl pH 7.4, 150 mM NaCl, and 4 mM CaCl2) at RT for 30 min followed by two 30s incubation in bath sonicator (Branson 220). For PC:PS or PE:PS, liposomes were made in a ratio of 3:1 containing a total lipids of 40 μg.
For testing the phospholipase activity of His6-VipD, each reaction was prepared by mixing PC, PC:PS, PE, or PE:PS liposomes with 20 μg of enzyme and incubated for 3 hr at 30°C. The reaction was quenched with the addition of 4 volumes of chloroform/methanol (2:1 by volume). Total lipids were extracted from the lower organic phase and subjected to Speed-Vac drying. The dried pellet was resuspended in 10 μl of methanol/isopropanol/acetic acid (5/5/2) and spotted onto a TLC Silica gel 60 F254 (EMD) that was pretreated by soaking in methanol/water (3:2) containing 1% potassium oxalate and then 1 h drying in a 65°C oven. The TLC plates were developed in a solvent system consisting of 1-propanol/2 M acetic acid (65%:35%) and the final products were visualized using iodine vapor (Alfa Aesar). Phospholipase A2 from bovine pancreas (Sigma) was used as a positive control in the experiment.
Elimination of potential co-factors associated with mitochondria by proteinase K
We treated mitochondria (100 μg/sample) purified from HEK293T cells with proteinase K at a concentration of 1μg/ml or buffer at 25°C for 30 minutes. Proteinase K was then removed by washing three times with the storage buffer (Sigma) containing 2 mM PMSF. Mitochondria were then incubated with VipD or its mutant proteins in mitochondrial storage buffer (Sigma) at 30 °C for 2 hr. The release of cytochrome c from mitochondria was detected as described above.
Supplementary Material
Fig. S1 GFP-VipD localizes in the cytoplasm of mammalian cell. Hela cells transfected to express GFP, GFP-0898, GFP-0898A151S, GFP-VipD or GFP-VipDS73A for 18 hr were fixed and stained for the mitochondrial protein COX4I1. Samples were analyzed using an Olympus IX-81 fluorescence microscope for images acquisition. Images were pseudocolored with the IPLab software package. Bar: 20 μm.
Fig. S2 Cell rounding phenotype caused by Lpg0898 and VipD and their nontoxic mutants. 293T cells transfected to express GFP-Lpg0898 (A), GFP-Lpg0898 A151S (B), GFP-VipD (C) or GFP-VipDS73A (D) for 24 hr. Images were acquired with an Olympus IX-81 fluorescence microscope equipped with a CCD camera. Note the cell rounding phenotype caused by wild type Lpg0898 and VipD. Bar: 50 μm.
Fig. S3 VipD does not cause damage in plasma membrane. A. Hela cells transfected to express GFP or GFP-VipD for 24 hr were washed and stained with 4 μM EtBr. Retaining of the dye by the cells was analyzed by microscopic analysis using an Olympus IX-81 fluorescence microscope equipped with a CCD camera. Untransfected cells treated by Triton-100 were included as a control (lower panel). Note that the nuclei of cells treated by Triton-100 were stained by the dye. B. His6-VipD or BSA was added to culture medium at a final concentration of 8 μg/ml. After 3 hr of incubation, the integrity of the plasma membrane was assessed by the retaining of EtBr using an Olympus IX-81 fluorescence microscope equipped with a CCD camera. Bar: 60 μm.
Fig. S4 VipD-induced Cyto c release is independent of Bax and Bak. A. Induction of caspase 3 in MEFs by transient expression. Wild type and bax/bak−/− MEFs were transfected with plasmid DNA that allows the expression of GFP-VipD, GFP-VipDS73A and indicated controls for 3 24 h, and the lysates of these samples were analyzed for activated caspase 3 by immunoblot (upper panel). The levels of the GFP fusions were probed by an antibody specific for GFP (lower panel) and tubulin was detected as a loading control (middle panel). Note that in wild type MEFs, caspase 3 can be activated by the Mirus transfection reagent (Thermo Scientific, Waltham, MA), which is the only one that allows efficient transfection of these cells (lane 1). B. Quantitation of the caspase 3 in panel A. Quantitation was obtained by measuring the band intensity using the Odyssey imaging system. Similar results were obtained in two independent experiments. C. Induction of Cyto c release by VipD from mitochondria isolated from MEFs. 40μg His6-VipD or His6-VipDS73A was added to 50μg mitochondria purified from wild type mouse embryonic fibroblast (MEF) or from MEF of bak/bax−/− mouse. After incubation at 30°C for 3 hr, the release of Cyto c was evaluated by probing its presence in the soluble fraction by immunoblot.
Fig. S5 A. VipD did not detectably hydrolyze phospholipids in reactions with defined components. His6-VipD or commercial phospholipase A2 was added to liposome established with phosphoethanolamine and the reactions were allowed to proceed for 3 hr at 30°C. Total lipids extracted from the reactions were separated on thin-layer chromatograph (TLC) Silica gel 60 F254 (EMD) and lipids were detected using iodine vapor. B. Mitochondria were treated with proteinase K or buffer before incubated with VipD or its mutant. The release of cytochrome c from mitochondria was detected via immunoblot.
Fig. S6 Deletion of caspase 3 activators has little impact on the activation of this caspase by L. pneumophila. Differentiated U937 cells were infected by indicated L. pneumophila strains for 6 hr, lysates of infected samples resolved by SDS-PAGE were probed for active caspase 3. Strains: Δ2 (Lp02Δ0898, Δ1625); Δ3(Lp02Δ0898, Δ1625, Δ0716); Δ4(Lp02Δ0898, Δ1625, Δ0716, Δ2831); Δ5(Lp02Δ0898, Δ1625, Δ0716, Δ2831, Δ2176). The wild type strain Lp02 and Lp03, the mutant deficient for the Dot/Icm transporter was used as controls.
Fig. S7 A L. pneumophila deletion strain lacking more than 100 of the known Dot/Icm substrates still potently induced caspase 3 activation. Differentiated U937 cells were infected by indicated L. pneumophila strains for 6 hr, lysates of infected samples resolved by SDS-PAGE were probed for active caspase 3. Similar results were obtained in multiple independent experiments.
Acknowledgments
We thank Dr. Peter Hollenbeck (Purdue University, West Lafayette, IN, USA) for help in imaging analysis. Dr. Ralph Isberg (Tuft Medical School, Boston, MA, USA), Dr. Wei-Xing Zong (Stony Brook University, Stony Brook, New York) for bacterial strains, antibodies and the bax/bak−/− MEF cells, respectively. We also thank Dr. Chen Chen (University of California at Berkeley, Berkeley, CA) for instructions in the isolation of dendritic cells. This work was supported by NIH-NIAID grants R01AI069344, K02AI085403 and R21AI092043 (Z.-Q.L) and R01-GM094347 (Y.M.).
Reference
- Abu-Zant A, Jones S, Asare R, Suttles J, Price C, Graham J, Kwaik YA. Anti-apoptotic signalling by the Dot/Icm secretion system of L. pneumophila. Cell Microbiol. 2007;9:246–264. doi: 10.1111/j.1462-5822.2006.00785.x. [DOI] [PubMed] [Google Scholar]
- Abu-Zant A, Santic M, Molmeret M, Jones S, Helbig J, Abu Kwaik Y. Incomplete activation of macrophage apoptosis during intracellular replication of Legionella pneumophila. Infect Immun. 2005;73:5339–5349. doi: 10.1128/IAI.73.9.5339-5349.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman E, Segal G. The response regulator CpxR directly regulates expression of several Legionella pneumophila icm/dot components as well as new translocated substrates. J Bacteriol. 2008;190:1985–1996. doi: 10.1128/JB.01493-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balsinde J, Balboa MA, Insel PA, Dennis EA. Regulation and inhibition of phospholipase A2. Annu Rev Pharmacol Toxicol. 1999;39:175–189. doi: 10.1146/annurev.pharmtox.39.1.175. [DOI] [PubMed] [Google Scholar]
- Banerji S, Aurass P, Flieger A. The manifold phospholipases A of Legionella pneumophila - identification, export, regulation, and their link to bacterial virulence. Int J Med Microbiol. 2008;298:169–181. doi: 10.1016/j.ijmm.2007.11.004. [DOI] [PubMed] [Google Scholar]
- Banerji S, Flieger A. Patatin-like proteins: a new family of lipolytic enzymes present in bacteria? Microbiology. 2004;150:522–525. doi: 10.1099/mic.0.26957-0. [DOI] [PubMed] [Google Scholar]
- Banga S, Gao P, Shen X, Fiscus V, Zong WX, Chen L, Luo ZQ. Legionella pneumophila inhibits macrophage apoptosis by targeting pro-death members of the Bcl2 protein family. Proc Natl Acad Sci U S A. 2007;104:5121–5126. doi: 10.1073/pnas.0611030104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr FA, Shorter J. Membrane traffic: do cones mark sites of fission? Curr Biol. 2000;10:R141–144. doi: 10.1016/s0960-9822(00)00326-2. [DOI] [PubMed] [Google Scholar]
- Berger KH, Isberg RR. Two distinct defects in intracellular growth complemented by a single genetic locus in Legionella pneumophila. Mol Microbiol. 1993;7:7–19. doi: 10.1111/j.1365-2958.1993.tb01092.x. [DOI] [PubMed] [Google Scholar]
- Burstein D, Zusman T, Degtyar E, Viner R, Segal G, Pupko T. Genome-scale identification of Legionella pneumophila effectors using a machine learning approach. PLoS Pathog. 2009;5:e1000508. doi: 10.1371/journal.ppat.1000508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccin P, Rossetto O, Montecucco C. Neurotoxicity of inverted-cone shaped lipids. Neurotoxicology. 2009;30:174–181. doi: 10.1016/j.neuro.2008.11.013. [DOI] [PubMed] [Google Scholar]
- Conover GM, Derre I, Vogel JP, Isberg RR. The Legionella pneumophila LidA protein: a translocated substrate of the Dot/Icm system associated with maintenance of bacterial integrity. Mol Microbiol. 2003;48:305–321. doi: 10.1046/j.1365-2958.2003.03400.x. [DOI] [PubMed] [Google Scholar]
- Creasey EA, Isberg RR. The protein SdhA maintains the integrity of the Legionella-containing vacuole. Proc Natl Acad Sci U S A. 2012;109:3481–3486. doi: 10.1073/pnas.1121286109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Felipe KS, Pampou S, Jovanovic OS, Pericone CD, Ye SF, Kalachikov S, Shuman HA. Evidence for acquisition of Legionella type IV secretion substrates via interdomain horizontal gene transfer. J Bacteriol. 2005;187:7716–7726. doi: 10.1128/JB.187.22.7716-7726.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degtyar E, Zusman T, Ehrlich M, Segal G. A Legionella effector acquired from protozoa is involved in sphingolipids metabolism and is targeted to the host cell mitochondria. Cell Microbiol. 2009;11:1219–1235. doi: 10.1111/j.1462-5822.2009.01328.x. [DOI] [PubMed] [Google Scholar]
- Deutsch DG, Omeir R, Arreaza G, Salehani D, Prestwich GD, Huang Z, Howlett A. Methyl arachidonyl fluorophosphonate: a potent irreversible inhibitor of anandamide amidase. Biochem Pharmacol. 1997;53:255–260. doi: 10.1016/s0006-2952(96)00830-1. [DOI] [PubMed] [Google Scholar]
- Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med. 2008;14:1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel J, Balachandran P. Role of Pseudomonas aeruginosa type III effectors in disease. Curr Opin Microbiol. 2009;12:61–66. doi: 10.1016/j.mib.2008.12.007. [DOI] [PubMed] [Google Scholar]
- Fields BS, Benson RF, Besser RE. Legionella and Legionnaires’ disease: 25 years of investigation. Clin Microbiol Rev. 2002;15:506–526. doi: 10.1128/CMR.15.3.506-526.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueira TR, Melo DR, Vercesi AE, Castilho RF. Safranine as a fluorescent probe for the evaluation of mitochondrial membrane potential in isolated organelles and permeabilized cells. Methods Mol Biol. 2012;810:103–117. doi: 10.1007/978-1-61779-382-0_7. [DOI] [PubMed] [Google Scholar]
- Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- Gao LY, Abu Kwaik Y. Activation of caspase 3 during Legionella pneumophila- induced apoptosis. Infect Immun. 1999;67:4886–4894. doi: 10.1128/iai.67.9.4886-4894.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge J, Gong YN, Xu Y, Shao F. Preventing bacterial DNA release and absent in 2 melanoma 2 inflammasome activation by a Legionella effector functioning in membrane trafficking. Proc Natl Acad Sci U S A. 2012;109:6193–6198. doi: 10.1073/pnas.1117490109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge J, Xu H, Li T, Zhou Y, Zhang Z, Li S, et al. A Legionella type IV effector activates the NF-kappaB pathway by phosphorylating the IkappaB family of inhibitors. Proc Natl Acad Sci U S A. 2009;106:13725–13730. doi: 10.1073/pnas.0907200106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandbois M, Clausen-Schaumann H, Gaub H. Atomic force microscope imaging of phospholipid bilayer degradation by phospholipase A2. Biophys J. 1998;74:2398–2404. doi: 10.1016/S0006-3495(98)77948-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu F, Zhu W, Brennan L, Tao L, Luo ZQ, Mao Y. Structural basis for substrate recognition by a unique Legionella phosphoinositide phosphatase. Proc Natl Acad Sci U S A. 2012;109:13567–13572. doi: 10.1073/pnas.1207903109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba K, Swiggard WJ, Steinman RM, Romani N, Schuler G, Brinster C. Isolation of dendritic cells. Curr Protoc Immunol. 2009:7. doi: 10.1002/0471142735.im0307s86. Chapter 3, Unit 3. [DOI] [PubMed] [Google Scholar]
- Jiang X, Wang X. Cytochrome C-mediated apoptosis. Annu Rev Biochem. 2004;73:87–106. doi: 10.1146/annurev.biochem.73.011303.073706. [DOI] [PubMed] [Google Scholar]
- Ku B, Lee KH, Park WS, Yang CS, Ge J, Lee SG, et al. VipD of Legionella pneumophila Targets Activated Rab5 and Rab22 to Interfere with Endosomal Trafficking in Macrophages. PLoS Pathog. 2012;8:e1003082. doi: 10.1371/journal.ppat.1003082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamkanfi M, Dixit VM. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe. 2010;8:44–54. doi: 10.1016/j.chom.2010.06.007. [DOI] [PubMed] [Google Scholar]
- Losick VP, Haenssler E, Moy MY, Isberg RR. LnaB: a Legionella pneumophila activator of NF-kappaB. Cell Microbiol. 2010;12:1083–1097. doi: 10.1111/j.1462-5822.2010.01452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losick VP, Isberg RR. NF-kappaB translocation prevents host cell death after low-dose challenge by Legionella pneumophila. J Exp Med. 2006;203:2177–2189. doi: 10.1084/jem.20060766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo ZQ. Striking a balance: modulation of host cell death pathways by legionella pneumophila. Front Microbiol. 2011;2:36. doi: 10.3389/fmicb.2011.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo ZQ, Isberg RR. Multiple substrates of the Legionella pneumophila Dot/Icm system identified by interbacterial protein transfer. Proc Natl Acad Sci U S A. 2004;101:841–846. doi: 10.1073/pnas.0304916101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo ZQ, Smyth AJ, Gao P, Qin Y, Farrand SK. Mutational analysis of TraR. Correlating function with molecular structure of a quorum-sensing transcriptional activator. J Biol Chem. 2003;278:13173–13182. doi: 10.1074/jbc.M210035200. [DOI] [PubMed] [Google Scholar]
- Molmeret M, Zink SD, Han L, Abu-Zant A, Asari R, Bitar DM, Abu Kwaik Y. Activation of caspase-3 by the Dot/Icm virulence system is essential for arrested biogenesis of the Legionella-containing phagosome. Cell Microbiol. 2004;6:33–48. doi: 10.1046/j.1462-5822.2003.00335.x. [DOI] [PubMed] [Google Scholar]
- Monroe KM, McWhirter SM, Vance RE. Identification of host cytosolic sensors and bacterial factors regulating the type I interferon response to Legionella pneumophila. PLoS Pathog. 2009;5:e1000665. doi: 10.1371/journal.ppat.1000665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogueira CV, Lindsten T, Jamieson AM, Case CL, Shin S, Thompson CB, Roy CR. Rapid pathogen-induced apoptosis: a mechanism used by dendritic cells to limit intracellular replication of Legionella pneumophila. PLoS Pathog 5. 2009:e1000478. doi: 10.1371/journal.ppat.1000478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putz T, Ramoner R, Gander H, Rahm A, Bartsch G, Bernardo K, et al. Bee venom secretory phospholipase A2 and phosphatidylinositol-homologues cooperatively disrupt membrane integrity, abrogate signal transduction and inhibit proliferation of renal cancer cells. Cancer Immunol Immunother. 2007;56:627–640. doi: 10.1007/s00262-006-0220-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehfeldt W, Resch K, Goppelt-Struebe M. Cytosolic phospholipase A2 from human monocytic cells: characterization of substrate specificity and Ca(2+)-dependent membrane association. Biochem J. 1993;293(Pt 1):255–261. doi: 10.1042/bj2930255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigoni M, Caccin P, Gschmeissner S, Koster G, Postle AD, Rossetto O, et al. Equivalent effects of snake PLA2 neurotoxins and lysophospholipid-fatty acid mixtures. Science. 2005;310:1678–1680. doi: 10.1126/science.1120640. [DOI] [PubMed] [Google Scholar]
- Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. 2002;2:965–975. doi: 10.1038/nri957. [DOI] [PubMed] [Google Scholar]
- Shohdy N, Efe JA, Emr SD, Shuman HA. Pathogen effector protein screening in yeast identifies Legionella factors that interfere with membrane trafficking. Proc Natl Acad Sci U S A. 2005;102:4866–4871. doi: 10.1073/pnas.0501315102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitkiewicz I, Stockbauer KE, Musser JM. Secreted bacterial phospholipase A2 enzymes: better living through phospholipolysis. Trends Microbiol. 2007;15:63–69. doi: 10.1016/j.tim.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Srikanth CV, Wall DM, Maldonado-Contreras A, Shi HN, Zhou D, Demma Z, et al. Salmonella pathogenesis and processing of secreted effectors by caspase-3. Science. 2010;330:390–393. doi: 10.1126/science.1194598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J Exp Med. 2007;204:3235–3245. doi: 10.1084/jem.20071239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson MS, Isberg RR. Association of Legionella pneumophila with the macrophage endoplasmic reticulum. Infect Immun. 1995;63:3609–3620. doi: 10.1128/iai.63.9.3609-3620.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilney LG, Harb OS, Connelly PS, Robinson CG, Roy CR. How the 9 parasitic bacterium Legionella pneumophila modifies its phagosome and transforms it into rough ER: implications for conversion of plasma membrane to the ER membrane. J Cell Sci. 2001;114:4637–4650. doi: 10.1242/jcs.114.24.4637. [DOI] [PubMed] [Google Scholar]
- VanRheenen SM, Luo ZQ, O’Connor T, Isberg RR. Members of a Legionella pneumophila family of proteins with ExoU (phospholipase A) active sites are translocated to target cells. Infect Immun. 2006;74:3597–3606. doi: 10.1128/IAI.02060-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verburg J, Hollenbeck PJ. Mitochondrial membrane potential in axons increases with local nerve growth factor or semaphorin signaling. J Neurosci. 2008;28:8306–8315. doi: 10.1523/JNEUROSCI.2614-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Luo ZQ. Cell biology of infection by Legionella pneumophila. Microbes Infect. 2013;15:157–167. doi: 10.1016/j.micinf.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Shen X, Bryan A, Banga S, Swanson MS, Luo ZQ. Inhibition of host vacuolar H+-ATPase activity by a Legionella pneumophila effector. PLoS Pathog. 2010;6:e1000822. doi: 10.1371/journal.ppat.1000822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamboni DS, Kobayashi KS, Kohlsdorf T, Ogura Y, Long EM, Vance RE, et al. The Birc1e cytosolic pattern-recognition receptor contributes to the detection and control of Legionella pneumophila infection. Nat Immunol. 2006;7:318–325. doi: 10.1038/ni1305. [DOI] [PubMed] [Google Scholar]
- Zhu W, Banga S, Tan Y, Zheng C, Stephenson R, Gately J, Luo ZQ. Comprehensive identification of protein substrates of the Dot/Icm type IV transporter of Legionella pneumophila. PLoS One. 2011;6:e17638. doi: 10.1371/journal.pone.0017638. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 GFP-VipD localizes in the cytoplasm of mammalian cell. Hela cells transfected to express GFP, GFP-0898, GFP-0898A151S, GFP-VipD or GFP-VipDS73A for 18 hr were fixed and stained for the mitochondrial protein COX4I1. Samples were analyzed using an Olympus IX-81 fluorescence microscope for images acquisition. Images were pseudocolored with the IPLab software package. Bar: 20 μm.
Fig. S2 Cell rounding phenotype caused by Lpg0898 and VipD and their nontoxic mutants. 293T cells transfected to express GFP-Lpg0898 (A), GFP-Lpg0898 A151S (B), GFP-VipD (C) or GFP-VipDS73A (D) for 24 hr. Images were acquired with an Olympus IX-81 fluorescence microscope equipped with a CCD camera. Note the cell rounding phenotype caused by wild type Lpg0898 and VipD. Bar: 50 μm.
Fig. S3 VipD does not cause damage in plasma membrane. A. Hela cells transfected to express GFP or GFP-VipD for 24 hr were washed and stained with 4 μM EtBr. Retaining of the dye by the cells was analyzed by microscopic analysis using an Olympus IX-81 fluorescence microscope equipped with a CCD camera. Untransfected cells treated by Triton-100 were included as a control (lower panel). Note that the nuclei of cells treated by Triton-100 were stained by the dye. B. His6-VipD or BSA was added to culture medium at a final concentration of 8 μg/ml. After 3 hr of incubation, the integrity of the plasma membrane was assessed by the retaining of EtBr using an Olympus IX-81 fluorescence microscope equipped with a CCD camera. Bar: 60 μm.
Fig. S4 VipD-induced Cyto c release is independent of Bax and Bak. A. Induction of caspase 3 in MEFs by transient expression. Wild type and bax/bak−/− MEFs were transfected with plasmid DNA that allows the expression of GFP-VipD, GFP-VipDS73A and indicated controls for 3 24 h, and the lysates of these samples were analyzed for activated caspase 3 by immunoblot (upper panel). The levels of the GFP fusions were probed by an antibody specific for GFP (lower panel) and tubulin was detected as a loading control (middle panel). Note that in wild type MEFs, caspase 3 can be activated by the Mirus transfection reagent (Thermo Scientific, Waltham, MA), which is the only one that allows efficient transfection of these cells (lane 1). B. Quantitation of the caspase 3 in panel A. Quantitation was obtained by measuring the band intensity using the Odyssey imaging system. Similar results were obtained in two independent experiments. C. Induction of Cyto c release by VipD from mitochondria isolated from MEFs. 40μg His6-VipD or His6-VipDS73A was added to 50μg mitochondria purified from wild type mouse embryonic fibroblast (MEF) or from MEF of bak/bax−/− mouse. After incubation at 30°C for 3 hr, the release of Cyto c was evaluated by probing its presence in the soluble fraction by immunoblot.
Fig. S5 A. VipD did not detectably hydrolyze phospholipids in reactions with defined components. His6-VipD or commercial phospholipase A2 was added to liposome established with phosphoethanolamine and the reactions were allowed to proceed for 3 hr at 30°C. Total lipids extracted from the reactions were separated on thin-layer chromatograph (TLC) Silica gel 60 F254 (EMD) and lipids were detected using iodine vapor. B. Mitochondria were treated with proteinase K or buffer before incubated with VipD or its mutant. The release of cytochrome c from mitochondria was detected via immunoblot.
Fig. S6 Deletion of caspase 3 activators has little impact on the activation of this caspase by L. pneumophila. Differentiated U937 cells were infected by indicated L. pneumophila strains for 6 hr, lysates of infected samples resolved by SDS-PAGE were probed for active caspase 3. Strains: Δ2 (Lp02Δ0898, Δ1625); Δ3(Lp02Δ0898, Δ1625, Δ0716); Δ4(Lp02Δ0898, Δ1625, Δ0716, Δ2831); Δ5(Lp02Δ0898, Δ1625, Δ0716, Δ2831, Δ2176). The wild type strain Lp02 and Lp03, the mutant deficient for the Dot/Icm transporter was used as controls.
Fig. S7 A L. pneumophila deletion strain lacking more than 100 of the known Dot/Icm substrates still potently induced caspase 3 activation. Differentiated U937 cells were infected by indicated L. pneumophila strains for 6 hr, lysates of infected samples resolved by SDS-PAGE were probed for active caspase 3. Similar results were obtained in multiple independent experiments.