

Abstract
Early observations of the patterns of neurofibrillary tangles and amyloid plaques in Alzheimer’s disease suggested a hierarchical vulnerability of neurons for tangles, and a widespread nonspecific pattern of plaques that nonetheless seemed to correlate with the terminal zone of tangle bearing neurons in some instances. The first neurofibrillary cortical lesions in Alzheimer’s disease occur in the entorhinal cortex, thereby disrupting the origin of the perforant pathway projection to the hippocampus, and amyloid deposits are often found in the molecular layer of the dentate gyrus, which is the terminal zone of the entorhinal cortex. We have modeled these anatomical changes in a transgenic mouse model that overexpresses both P301L tau (uniquely in the medial entorhinal cortex), and mutant APP/PS1 (in a widespread distribution), to examine the anatomical consequences of early tangles, plaques, or the combination. We find that tau uniformly occupies the terminal zone of the perforant pathway in tau expressing mice. By contrast, the addition of amyloid deposits in this area leads to disruption of the perforant pathway terminal zone and apparent aberrant distribution of tau containing axons. Moreover, human P301L tau containing axons appear to increase the extent of dystrophic axons around plaques. Thus the presence of amyloid deposits in the axonal terminal zone of pathological tau containing neurons profoundly impacts their normal connectivity.
Keywords: Van Hoesen, Alzheimer’s disease, perforant pathway
Introduction
Frequently the first, and nearly always the most pervasive, symptom in Alzheimer’s disease is memory impairment. Sophisticated neuroanatomical tract tracing studies in mammals, especially the nonhuman primate in pioneering studies by Van Hoesen and colleagues (Van Hoesen et al., 1972) demonstrated intimate connections of the limbic and association cortices with the hippocampal formation, creating a neural system devoted to memory that has extraordinarily specific anatomic underpinnings. As part of this special issue in The Journal of Comparative Neurology dedicated to the memory of Gary Van Hoesen, we present a description of mouse models of early Alzheimer disease showing that in the neural circuits described by Van Hoesen, amyloid plaques induce pathological changes in tau-containing axon terminals projecting from the entorhinal cortex to the dentate gyrus.
Van Hoesen and Pandya described in 1975 that the cortical input to the hippocampus is often not direct, but instead relayed via the layer II neurons of the entorhinal cortex in a major entorhinal-hippocampal projection called the perforant pathway, since it perforates the CA fields of the hippocampus and the hippocampal fissure on its way to a very discrete terminal zone in the molecular layer of the dentate gyrus (Van Hoesen and Pandya, 1975a). Conversely, efferent projections from hippocampal fields reciprocating those afferents arise from CA1/subicular fields, with a major projection to layer IV of the entorhinal cortex and a subsequent projection back to widespread limbic and association cortices (Rosene and Van Hoesen, 1977; Van Hoesen and Pandya, 1975b; Van Hoesen et al., 1979).
The observation that the entorhinal cortex contains the earliest cortical neurofibrillary tangles was made by Hyman, Damasio and Van Hoesen in 1984 (Hyman et al., 1984). Layer II of the entorhinal cortex (the neurons that give rise to the perforant pathway) and the large projection neurons of the CA1, subicular hippocampal fields and layer IV of entorhinal cortex (which accounted for the primary efferents of the hippocampal formation) were all selectively and severely affected by neurofibrillary tangles (Hyman et al., 1984; Hyman et al., 1986). Moreover, the perforant pathway terminal zone, an exquisitely specific region within the middle portion of the molecular layer of the dentate gyrus, was riddled with amyloid plaques and with tau containing dystrophic neurites (Hyman et al., 1988; Hyman et al., 1986; Van Hoesen et al., 1986), suggesting that this major projection that subserved cortical-hippocampal connections was anatomically disrupted early in Alzheimer’s disease. Since memory function depends extensively on the hippocampus, the conclusion was that these lesions caused, at least in large part, the early memory impairments of Alzheimer’s disease (Van Hoesen, 1985; Van Hoesen et al., 1986).
In addition to potentially providing a structure-function explanation for a clinical symptom in Alzheimer’s disease, these observations led to a series of questions about disease etiology and how it progresses. First was the question of hierarchical vulnerability of neuronal populations to tangles. Many other neurons in the brain develop neurofibrillary lesions in addition to the entorhinal cortex and CA1/subiculum, including many cell populations that appeared to be connected to these hippocampal structures (Arnold et al., 1991; Braak and Braak, 1991). Areas closely connected to the hippocampal formation appeared most vulnerable, with anatomically more distantly connected areas relatively spared. The reason for this selective vulnerability has been elusive. One likely possibility is that large projection neurons that are part of the same neural circuits and have similar functions have similar physiology, and so perhaps have similar pathophysiology. Another possibility is that the connections themselves are, at least in part, responsible for the pattern of hierarchical vulnerability as one moves farther away from limbic areas.
Second was the question of whether downstream targets are in fact “disconnected”, leading to isolation of network nodes, and the relative independent and synergistic roles of tangles and amyloid-beta deposits in this process (Hyman et al., 1984). In the experimental animal, loss of the perforant pathway is known to cause a robust sprouting response of remaining terminals in the molecular layer of the dentate gyrus (Chen et al., 1983; Lynch et al., 1972). The entorhinal pathology in human Alzheimer’s appears to be severe enough as to cause a quite analogous sprouting effect as noted with acetylcholinesterase histochemistry and glutamate receptor studies (Cabalka et al., 1992; Geddes et al., 1985; Hyman et al., 1987). Intriguingly, the neuritic dystrophies surrounding neuritic plaques are often acetylcholinesterase positive, giving rise to the possibility that they represent an aberrant sprouting phenomenon (Mesulam, 1998; 2000). Together with the observation that neuritic plaques frequently (although not always) occur in the terminal zone of neurons that have developed tangles (especially prominent in the perforant pathway terminal zone), the possibility exists that the neuritic component of senile plaques is a consequence of an interaction between amyloid deposits and sprouting of tau positive axons and dendrites.
To begin to test these possibilities experimentally, we have recently developed the rTgTauEC line, a new transgenic model that allows the spread of tau pathology to be tracked. Taking advantage of a tta driver line that expresses primarily in the medial entorhinal cortex (Yasuda and Mayford, 2006), we created a line that expresses human P301L tau in the layer II entorhinal cortex neurons that are the most vulnerable for tangle formation in the human brain. As expected, these neurons developed tangles and the axonal projection of these neurons to the hippocampus, the perforant pathway, degenerates (de Calignon et al., 2012). Excitingly, as the animals age, human tau fibrillar inclusions - Gallyas positive neurofibrillary tangles - appear not only in the entorhinal cortex neurons that express human P301L tau, but also in the dentate gyrus granule cells, the neurons that receive the most robust entorhinal projection. Since mRNA encoding human tau could not be detected in granule cells isolated by laser capture microdissection, the most plausible conclusion is that human misfolded tau is released from the terminals of the entorhinal cortex, then taken up by the dendrites of the granule cells where they are translocated to the cell body, where it forms the nidus of a new tangle (de Calignon et al., 2012). This cell-to-cell propagation of misfolded tau suggests a mechanism for the spread of tangles across the cortical fields and ultimately could explain the way Alzheimer’s disease progresses over time to involve more and more neurons within specific vulnerable fields, and more and more cytoarchitectural fields. Similar results were observed in lines independently generated in two other laboratories (Harris et al., 2012; Liu et al., 2012).
We now use the neuropsin promoter to drive a fluorescent reporter containing a synaptophysin-GFP fusion protein and tdTomato to show the cell bodies, axons, and axonal terminals of the neurons that produce tta in the entorhinal cortex. Having established the anatomy of the tta positive subset of neurons in the medial entorhinal cortex and adjacent presubiculum and parasubiculum, we then examined the interactions between the tau-containing axons in the entorhinal cortex and dentate gyrus and amyloid deposits, by crossing the rTgTauEC line with a well-characterized line that widely overexpresses mutant APP and PS1delta9. This allowed us to experimentally address two issues: 1) does the presence of amyloid beta impact the patterns or rate of progression of tau propagation to the dentate gyrus? and (2) does the concurrent presence of abnormal tau containing neurites in the dentate gyrus molecular layer and the deposition of amyloid beta in plaques impact the distribution or characteristics of the tau containing axonal projections – i.e. is there evidence of disruption of the pattern of normal terminal zones and/or of axonal sprouting due to the presence of amyloid beta?
Materials and Methods
Animals
We used several transgenic mouse lines for this study: rTgTauEC, EC-tdTomato/Syp-GFP, APP/PS1, and crosses of (1) rTgTauEC with EC-tdTomato/Syp-GFP mice and (2) rTgTauEC mice with APP/PS1 mice were generated. The rTgTauEC line with reversible tau expression restricted to entorhinal cortex that was generated by crossing FVB-Tg(tetO-TauP301L) 4510 mice (Santacruz et al., 2005) with a transgenic line expressing tetracycline sensitive transcriptional activator controlled by the Klk8 neuropsin promoter (EC-tTa), developed by Yasuda and Mayford (Yasuda and Mayford, 2006). In rTgTauEC mice inheritance of both the responder and activator transgenes results in P301L mutant tau expression restricted to layer II of the EC and pre and para subiculum (de Calignon et al., 2012). Both male and female mice from the various lines were included in the present study.
EC-tdTomato/Syp-GFP mice were generated by crossing the EC-tTa mouse with the Tg(tetO-tdTomato-Syp/mut4EGFP)1.1Luo/J line obtained from Jackson Laboratory. This mouse expresses two fluorescent proteins under the control of the bi-directional tet-responsive promoter: a Myc-tagged tdTomato and a full-length mouse synaptophysin/mut4EGFP fusion protein (Syp-GFP) (Li et al., 2010; Miyamichi et al., 2010). This line is a fluorescent reporter with neurons expressing the transgene in the EC labeled with red cytoplasm and green synaptic terminals. Brains collected from three and six month-old mice were examined in the present study.
APP/PS1 mice expressing human APP with the Swedish mutation and human presenilin 1 with the dE9 mutation were obtained from Jackson laboratories (stock line B6.Cg-Tg(APPswe,PSEN1dE9)85Dbo/J). EC-tdTomato/Syp-GFP mice were crossed with FVB-Tg(tetO-TauP301L) to generate mice expressing both human P301L tau and tdTomato/Syp-GFP in EC neurons. APP/PS1 mice were crossed with EC-tTa mice and then crossed with the FVB-Tg(tetO-TauP301L) 4510 mice to generate animals expressing APP/PS1 in the entire forebrain and P301L tau restricted to the EC. Brains from 16 month-old APP/PS1 mice and APP/PS1 × rTgTauEC mice were used in the present study. Age-matched littermates expressing only the activator transgene, tdTomato/Syp-GFP, or APP/PS1 mice were used as human tau-negative controls.
PCR screening was used to genotype rTgTauEC mice using the primer pairs 5′-ACCTGGACATGCTGTGATAA-3′ and 5′-TGCTCCCATTCATCAGTTCC-3′ for the activator transgene, and 5′-TGAACCAGGATGGCTGAGCC-3′ and 5′-TTGTCATCGCTTCCAGTCCCCG-3′ for responder tau transgene. APP/PS1 mice were identified by PCR screening with the following primer pair: 5′-AGATGAGCCACGCAGTCC-3′ and 5′-TCACAGAAGATACCGAGACT-3′ for the PS1 transgene. EC-tdTomato/Syp-GFP mice were identified by PCR screening using the primer pairs 5′ CTT CAA GTC CGC CAT GCC CGA 3′ and 5′ TCC AGC AGG ACC ATG TGA TCG C 3′.
All animal experiments conformed to United States National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use Committees of Massachusetts General Hospital and McLaughlin Research Institute.
Antibody Characterization
Primary antibodies used and their characterization are shown in Table 1. Standard immunofluorescence controls were performed for each condition (no primary negative control). The antibodies used in the present study have been widely used and previously characterized. The anti-tau mouse monoclonal antibody Tau13 specifically recognizes residues 20 – 35 of the human form of tau. This antibody detects an 64 kDa band in immunoblots of human brain lysates, and has been used additionally in immunohistochemistry, immunoprecipitation and ELISA studies (Hoover et al., 2010). The mouse monoclonal IgM antibody Alz50 was a generous gift from Peter Davies at Albert Einstein College of Medicine, and was raised against homogenates collected from temporal cortex of human AD patients. This antibody recognizes human tau in a conformation-specific manner, at residues 2 – 10 and 312 – 342, and has been demonstrated to yield a band approximately 64-68kDa in size on immunoblots of human brain lysate collected from patients with Alzheimer’s disease (Carmel et al., 1996; Hyman et al., 1988; Jicha et al., 1997; Wolozin et al., 1986). Experiments involving immunohistochemistry and array tomography of brain tissue from mice expressing human tau, or from Alzheimer’s disease patients, have demonstrated that Alz50 labels pre-tangle tau, some mature NFTs, and neuropil threads (NT); minimal staining is observed in control brain tissue (Wolozin et al., 1986; Hyman et al., 1988; Kopeikina et al., 2013).
Table 1.
Antibody information
| Name | Immunogen/epitope | Manufacturer/ Catalog # |
Species | Mono/ polyclonal |
Characterization |
|---|---|---|---|---|---|
| Alz50 | Human AD homogenate; amino acid residues 2-10 & 312-342 (folded conformation) |
Peter Davies (NA) |
mouse | monoclonal | WB: 64-68kDa; IHC: pretangles, NFT, NT (Carmel et al., 1996; Hyman et al., 1988; Jicha et al., 1997; Wolozin et al., 1986) |
| GFP | Purified recombinant full length GFP |
Aves (GFP-1010) |
chicken | polyclonal | WB: 27kDa IHC: GFP, YFP, CFP, RFP, eGFP |
| GFP | Purified recombinant full length GFP |
AbD Serotec (4745-1051) |
sheep | polyclonal | WB: 27kDa IHC (Collins et al., 2010; Wu et al., 2011) |
| Tau13 | amino acid residues 20-35 |
Covance (MMS-520R-500) |
mouse | monoclonal | WB: 64kDa, IHC, IP and ELISA (Hoover et al., 2010) |
| AW7 | Synthetic aggregated Abeta |
Dominic Walsh (NA) |
rabbit | polyclonal | WB: 4 and 7kDa IHC and ELISA (McDonald et al., 2012) |
| SMI312 | Neurofilaments | Covance (SMI-312R) |
mouse | monoclonal | WB: heavy 200kDa, medium; 150kDa, light 68kDa. IHC, IP and ELISA (Bussiere et al., 2004; Chung et al., 2003) |
The rabbit polyclonal antibody against aggregated Aβ was generously provided by Dominic Walsh at Harvard Medical School. This anti-Aβ antibody (AW7) was raised against aggregated synthetic Aβ1-42 and recognizes this peptide by dot blot, immunoblot and ELISA. Immunohistochemistry experiments demonstrated the AW7 antibody recognizes both diffuse and neuritic plaques (McDonald et al., 2010) in human brain tissue.
The GFP antibodies used for experiments in the present study have been well-characterized in earlier works; further information can be found in the NIF Antibody Registry (Aves GFP Antibody ID: AB_10000240; Serotec GFP Antibody ID: AB_619712). These antibodies have been shown to recognize a 27 kDa band on immunoblot, and are suitable for immunohistochemistry (Collins et al., 2010; Wu et al., 2011). The Covance monoclonal antibody against axonal neurofilaments, noted in Table 1, has also been well-characterized (Antibody ID: AB_509993). This antibody labels heavy, medium and light chain neurofilaments on immunoblot (200, 150 and 68 kDa, respectively) and is also suitable for visualization of neurofilaments by immunohistochemistry (Bussiere et al., 2004; Chung et al., 2003).
Immunohistochemistry
Animals were sacrificed by CO2 inhalation. Brains from rTgTauEC mice, APP/PS1 mice, and the rTgTauECxAPP/PS1 were frozen in M1 mounting medium (Shandon, Thermo Scientific) for subsequent cryostat sectioning in the horizontal plane at (8–10 μm). Brains from EC-tdTomato/Syp-GFP mice and the EC-tdTomato/Syp-GFP mice crossed with rTgTauEC were fixed in 4% paraformaldehyde with 15% glycerol cryoprotectant for 48 hours and 50 μm horizontal floating sections were cut in the horizontal plane to allow visualization of the EC, perforant pathway, and hippocampal formation in the same sections. Sections were permeabilized by 20 min incubation in 0.1 % Triton solution, blocked in 5% normal goat serum (NGS) for 1 hour, and appropriate primary antibody was applied in 5% NGS overnight at 4°C (1:50 concentration for Alz50, 1:1000 for all other antibodies). Sections were subsequently washed in TBS and incubated in the appropriate secondary antibody in 5% NGS for 1 hr at room temperature. Sections were incubated in fluorescent Alexa Fluor 488, CY3-labeled and CY5-labeled secondary antibodies (Jackson ImmunoResearch Laboratories), then counterstained with DAPI. Images were collected on a Zeiss AxioImager epifluorescence microscope equipped with a Coolsnap digital camera and Axio-Vision software.
Array tomography
Tissue from 6 month-old mice expressing both tau and the tdTomato/Syp-GFP in the EC was fixed, dehydrated, and embedded in LRwhite resin (Koffie et al., 2009; Micheva et al., 2010; Micheva and Smith, 2007). An Ultracut microtome (Leica) was used to generate ribbons of 10-20 serial sections of 70 nm thickness. Ribbons were immunostained with antibodies for GFP (chicken polyclonal; Aves at 1:50 concentration) and Tau13 (mouse monoclonal, Covance at 1:50 concentration) and secondary anti-sheep 488 and anti mouse-Cy3 (Jackson ImmunoResearch Laboratories both at 1:50 concentration). Nuclei were counter-stained with DAPI (Sigma). Images were acquired with a Zeiss AxioImager Z2 microscope equipped with automated stage, digital camera, and array tomography automated imaging plugins in AxioVision software. The middle molecular layer of the dentate gyrus was identified at low magnification (10x) and images were taken of the same site on each section at 63x. Stacks were aligned in Image J and the GFP channel was processed using a threshold-based software as previously described (Koffie et al 2009) to exclude noise. Three-dimensional renderings of the image stacks were generated using the 3D Viewer plugin in ImageJ.
Results
Distribution of axons and terminal zones of rTgTauEC mice expressing tau and a fluorescent protein reporter transgene under neuropsin promotors
The rTgTauEC mouse line contains a transgene under the neuropsin promotor that allows for expression of human P301L tau in the layer II entorhinal cortex cells (de Calignon et al., 2012). In these mice, we have tracked the spread of human tau from the entorhinal cortex, through the axons of the perforant pathway, to the axon terminals in the dentate gyrus molecular layer (de Calignon et al., 2012). In order to further characterize the restricted expression of transgenes driven by the neuropsin promoter, and to examine the anatomy of the projections defined by these tta-positive neurons, we examined the brains of a responder mouse line that expresses synaptophysin-GFP fusion protein and tdTomato from the same transgene. We crossed these mice with the rTgTauEC mouse line, to obtain mice which express human P301L tau, synaptophysin-GFP and tdTomato in a restricted pattern determined by the neuropsin-tta driver. Brains from 3 month old mice were sectioned horizontally (50 μm) and immunostained with Tau13, an antibody directed against human tau and an anti-GFP antibody (Fig. 1). tdTomato fluorescence was preserved without immunolabeling. All three proteins are strongly expressed in the entorhinal cortex of these mice. Transgene expression in this region, particularly tdTomato, enabled clear visualization of the MEC somata and axons of the commissural fibers (Fig. 1) and perforant pathway (Fig. 2). Axons also contain high levels of human tau. Synaptophysin-GFP expression was enriched in axon terminals from local cortico-cortical connections in the EC and in the dentate gyrus. To examine the molecular layer of the dentate gyrus more closely (Fig. 2), array tomography, a high resolution imaging technique combining ultrathin sectioning, immunofluorescence, and three-dimensional reconstruction of image stacks (Micheva et al., 2010; Micheva and Smith, 2007), was used to visualize synaptophysin-GFP and human tau the axon terminals of 6 month old mice. Tau positive axons were observed that contained GFP positive terminal boutons and en passant boutons.
Figure 1.

Distribution of axons and terminal zones of 3 month old rTgTauEC mice expressing a fluorescent protein reporter transgene under the neuropsin promoter. A. The fluorescent reporter proteins are tdTomato (magenta) and synaptophysin-GFP (green). Horizontal sections (50 μm) of mouse brains revealed the expression pattern of these proteins. Strong protein expression is observed in the cell bodies of the entorhinal cortex (asterisk), the target of the neuropsin promoter transgene. Axons project from the entorhinal cortex through the commissural fibers (solid arrowhead) and the perforant pathway and terminate in the dentate gyrus (open arrowhead). tdTomato is located throughout the cell, strongly labeling neuronal soma, axons and terminals. Synaptophysin-GFP is largely present in neuronal soma in the entorhinal cortex and axon terminals in the dentate gyrus. Human P301L tau is strongly localized to axons and their terminals, and is observed to a lesser extent in the cell bodies in the entorhinal cortex. Scale bar = 500 μm.
Figure 2.

High-resolution anatomical specificity of projections. A. The schematic illustrates the locations of the high-resolution images in B-E. Cells expressing tdTomato/synGFP and tau in the MEC have tomato expression (magenta) throughout the cytoplasm and GFP in synapses. Neuronal somata in MEC layer II and local cortico-cortical synaptic connections in layers I and II (L1 and L2) are shown in B. Axons from these neurons project via the perforant pathway to terminate in the middle molecular layer (mm) of the dentate gyrus (C). D. Higher resolution images from the box in C show axons perforating the hippocampal fissure to enter the molecular layer of DG. Arrowheads indicate an axon traveling from the EC to the DG. Ultra-resolution images using three-dimensional reconstructions (1.5 μm in depth) of array tomography stacks from the middle molecular layer shows tau positive axons with both terminal synaptic boutons (arrows) and en passant boutons (arrowheads) (E). Scale bars represent 50 μm in B and D, 300 μm in C, 3 μm in E.
Distribution of human tau-positive axon terminals in the middle molecular layer of the dentate gyrus is disrupted by amyloid plaques in an rTgTauEC line crossed with APP/PS1 mice
The middle molecular layer (MML) of the dentate gyrus receives projections from neurons whose cell bodies are in layer II of MEC. In 16 month-old rTgTauEC animals, the distribution of these tau-expressing axons is uniform in the MML. In the presence of amyloid plaques in APP/PS1 × rTgTauEC mice, which have large amyloid deposits abundant throughout the hippocampus, the distribution of tau-containing axons and terminals in the MML appears patchy. Closer examination of these patchy areas of human tau revealed that this tau clustering occurred in the presence of amyloid plaques (Fig. 3). Furthermore, although the localization of human tau appeared restricted to the MML in the rTgTauEC mice, in areas immediately proximal to amyloid plaques (i.e., within 50 μm), tau-positive axons or axon terminals were visible in the outer molecular layer (OML) of the dentate gyrus, indicating alterations of the distribution of MEC neuron axons and their terminals in the presence of amyloid pathology.
Figure 3.

Distribution of axons containing human tau in middle molecular layer (MML) of dentate gyrus is altered in the presence of amyloid plaques. The MML of the dentate gyrus of (A) rTgTauEC and (B) APP/PS1 × rTgTauEC mice contains human tau-positive axons and terminals, labeled by Tau13 antibody (green), surrounding the densely-packed dentate granule cells (white). Scale bar = 100 μm. C, E. In the absence of amyloid plaques, human tau-positive (Tau13 antibody) axon terminals in the MML appear evenly distributed. D, F. However, in APP/PS1 × rTgTauEC mice, the distribution of tau appears patchy (open arrowheads) in regions positive for plaques. Furthermore, in rTgTauEC mice (C, E), the projection of human-tau positive (Tau13 labeled) neurons is limited to the MML; however, in APP/PS1 × rTgTauEC mice (D, F), tau-positive axons are visible in the OML (solid arrowheads), particularly in the vicinity of amyloid plaques (magenta). Scale bar = 50 μm. IML = inner molecular layer; MML = middle molecular layer; OML = outer molecular layer.
Exacerbated axonal dystrophies around amyloid plaques in the MML of the dentate gyrus in axons containing mutant human tau
Amyloid plaque-associated dystrophy of axons and dendrites is prominent in brains affected by Alzheimer’s disease, and is also observed in mouse models of the disease (Hsiao et al., 1996). In the present study, we examined axons found within plaques in 16 month old APP/PS1 or APP/PS1 × rTgTauEC mice, in order to determine whether the presence of human tau might affect axonal dystrophies. Initial assessment of these animals suggests that amyloid plaque burden is similar in both strains. We examined the areas of the MML occupied by amyloid plaques and found that punctate, dystrophic axons were present (Fig. 4). In APP/PS1 × rTgTauEC mice, the dystrophic puncta frequently colocalized with human tau (labeled by Alz50 antibody; green open arrowhead). Preliminary quantification suggests that the presence of human tau in axons in the MML may increase the number of dystrophic puncta per plaque by as much as 100% compared to mice without the human tau transgene, although further analysis is required. In addition, the mechanism by which human tau interacts with axonal morphology remains unclear; one possibility is that tau accumulation impairs normal intracellular transport of proteins and organelles, therefore leading to aggregates and eventually axonal dystrophy.
Figure 4.

Axonal dystrophies are associated with amyloid plaques in the MML of the dentate gyrus. Dystrophic axons were examined in dentate gyrus (DG) of brain slices (10 μm) from APP/PS1 × rTgTauEC (A - C), rTgTauEC (D - F), or APP/PS1 (G - I) mice, all 16 months old. In the MML of the DG, axons (labeled with SMI312 antibody: C, F, I) displayed punctate morphology (open arrowheads) in and around amyloid plaques (asterisks, labeled with anti-Aβ antibody: A, D, G). These dystrophic neurites were also positive for misfolded human tau (labeled by Alz50 antibody: B, E, H) and punctate tau was observed colocalizing with SMI312-positive neuritic dystrophies (open arrowheads). Alz50 tau-positive neurons were observed in the GC layer of the DG in both rTgTauEC and APP/PS1 × rTgTauEC mice (solid arrowhead). Scale bar = 50 μm. GC = granule cells; IML = inner molecular layer; MML = middle molecular layer.
Propagation of tau pathology does not appear different in the presence of amyloid pathology
Previous studies have demonstrated that in rTgTauEC mice, granule cells can accumulate human tau, most likely due to inter-cellular transfer of tau from entorhinal cortex neurons, which synapse onto these cells (de Calignon et al., 2012). However, it is not known whether the presence of amyloid plaques would affect the propagation of tau in this model. In order to investigate this question, we examined the granule cell layer of the dentate gyrus in 10 μm sections of brains from 16 month old rTgTauEC or APP/PS1 × rTgTauEC mice. Immunostaining of these neurons with an antibody directed against human tau (Tau13) revealed the presence of this tau in granule cells in both mouse lines (Fig. 5). Propagation of tau is variable in the rTgTauEC line at this age with 50-70% of animals demonstrating progression of pathology to the DG. Similarly, ~50% of rTgTauEC mice with amyloid pathology (crossed with APP/PS1 mice) have tau pathology in the DG at 16 months of age. The presence of amyloid plaques therefore did not substantially accelerate the spread of pathological tau through neural circuits in the rTgTauEC model.
Figure 5.
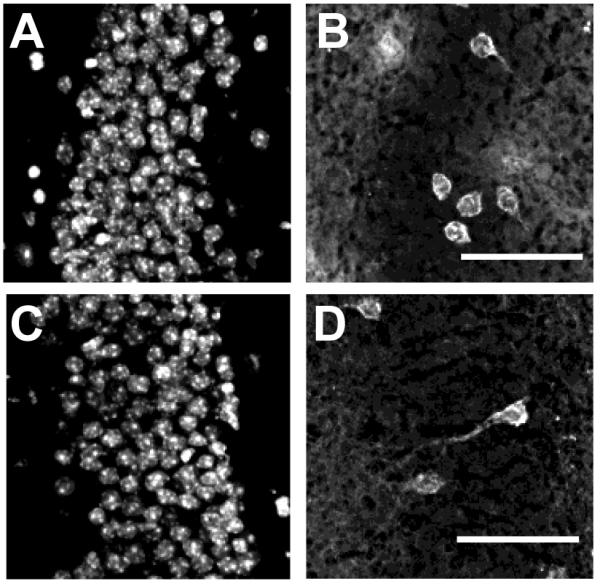
Accumulation of amyloid plaques in APP/PS1 × rTgTauEC mice does not accelerate the spread of tau pathology into granule cells of the dentate gyrus. Horizontal sections (10 μm) of either rTgTauEC (A, B) or APP/PS1 × rTgTauEC (C, D) mouse dentate gyrus were immunostained with an antibody against human tau (Tau13: B, D) and co-stained with DAPI to label cell nuclei (A, C). APP/PS1 × rTgTauEC mice displayed tau-positive (Tau13) neurons in the granule cell layer, similar to the neurons observed in the mice expressing rTgTauEC only. Scale bar = 50 μm.
Discussion
The development of the rTgTauEC line allows detailed evaluation of the anatomical consequences of expressing abnormal tau in the cortical neurons first affected in Alzheimer’s disease. The exquisitely precise anatomical projections of the entorhinal perforant pathway system provide an important experimental tool to evaluate the changes that occur in the earliest stages of Alzheimer’s disease, and to begin to dissect the factors that impact neural system degeneration, progression of disease, and potentially aberrant neurobiological phenomenon like abnormal sprouting in the setting of deafferentation and amyloid beta deposition.
Our current experiments focus on the interactions between amyloid beta and tau inclusions. While the distribution of tau inclusions and amyloid beta plaques does not show a particularly strong overlap in terms of cortical and subcortical structures in Alzheimer’s disease (Arnold et al., 1991), it is widely believed that amyloid in some way enhances tau based pathological changes since the lesions nearly always co-occur in the same brain in Alzheimer’s disease, including cases of autosomal dominant PS1 mutations and Down syndrome where the primary genetic lesion is mismetabolism of amyloid beta (Gomez-Isla et al., 1999; Hyman, 1996; Hyman et al., 1995). Additionally, tau containing dystrophic neurites occur in a halo around plaques that is rich in oligomeric amyloid beta in mouse models that develop plaques (Hsiao et al., 1996; Spires et al., 2005), and dystrophies develop in neurons exposed to amyloid beta in vitro (Wu et al., 2010). Moreover, the anatomical pattern of neuritic plaques often seems to line up with the projection zone of tangle bearing neurons (Hyman et al., 1986; Kromer Vogt et al., 1990).
We find that overexpressed mutant P301L tau is transported down axons and occupies the normal terminal zone of the perforant pathway, reflecting the same anatomical specificity observed with a fluorescent reporter protein and matching well with previous data demonstrating the projection zone of the medial perforant pathway using classical tract tracing techniques (Van Hoesen and Pandya, 1975a; Van Hoesen and Pandya, 1975b; Witter, 2007). However, the presence of amyloid beta deposits disrupts this pattern. Intriguingly, in addition to generating a large number of neuritic dystrophies that are human tau immunoreactive (as expected), we note that the pattern of the projection becomes disrupted - instead of a smooth and evenly distributed set of projections to the middle molecular layer of the dentate, the human tau containing axons become patchy, focused on amyloid deposits both within the middle molecular layer of the dentate and in adjacent areas. These results suggest that the anatomical projection to the dentate is further disrupted by the presence of amyloid deposits, highlighting the complex interplay between these two types of lesions, and suggesting that amyloid beta - induced changes in axons may lead both to sprouting and, somewhat paradoxically, worsened deafferentation as the axons are directed away from their natural targets. The mistargeting of axonal projections due to amyloid beta is reminiscent of our recent observations in another transgenic model system in which sparse amyloid beta was generated in the olfactory system, leading to unequivocal mistargeting of axonal projections (Cao et al., 2012) as well as altered plasticity responses in a developmental visual deprivation paradigm (William et al., 2012). Taken together, the data strongly support a role for amyloid beta in collaboration with abnormal tau containing neurites to lead to disrupted axonal connectivity even in the adult brain. Whether this is due to fibrillar amyloid beta deposits, to the “halo” of oligomeric amyloid beta that surrounds plaques (Koffie et al., 2009), or to soluble species is still uncertain.
We also examined whether the presence of amyloid-beta would enhance the propagation phenomenon of tau protein being taken up and accumulating as aggregates in downstream neurons. While the number of animals examined to date is modest, and we have examined only a single time point, no clear difference in the propagation patterns of tau to the dentate gyrus granule cells was observed. Consistent with this observation, in human Alzheimer’s disease tissue, there is not a clear concordance in the number of amyloid beta deposits and either the number or distribution of tangles (see for example, (Gomez-Isla et al., 1997; Hyman and Tanzi, 1995). Thus the primary interaction between tau related lesions and amyloid beta related lesions appears to be in disrupting the fine details of exquisitely refined projections that subserve neural systems, thereby supporting the broad hypothesis that neural system disruption is at the heart of the effects of tangles and plaques in mediating clinical manifestations of Alzheimer’s disease (Koffie et al., 2009; Kuchibhotla et al., 2008; Van Hoesen and Hyman, 1990).
Supplementary Material
Acknowledgments
We thank Mark Mayford for providing neuropsin-tTA mice and Dominic Walsh and Peter Davies for providing antibodies.
Funding: This work was supported by R00AG033670, R01AG026249-07 and grants from the American Health Assistance Foundation and Alzheimer’s Research UK.
Footnotes
The authors declare no competing financial interests.
Conflict of Interest None.
ROLE OF AUTHORS All authors had full access to the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: A.M.P., M.P., T.L.S.J., B.T.H. Acquisition of data: A.M.P., M.P., S.K.W., K.R.K., L.S., T.L.S.J. Analysis and interpretation of data: A.M.P., M.P., S.K.W.,T.L.S.J., B.T.H. Drafting of article: A.M.P., M.P., T.L.S.J., B.T.H. Critical revision of article for important intellectual content: R.P., G.A.C., T.G.I., M.W.A., T.L.S.J., B.T.H. Statistical analysis: A.M.P. Obtained funding: T.L.S.J., B.T.H. Administrative, technical, and material support: K.R.K., L.S., R.P., G.A.C. Study supervision: T.L.S.J., B.T.H.
Literature Cited
- Arnold SE, Hyman BT, Flory J, Damasio AR, Van Hoesen GW. The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer’s disease. Cereb Cortex. 1991;1(1):103–116. doi: 10.1093/cercor/1.1.103. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Bussiere T, Bard F, Barbour R, Grajeda H, Guido T, Khan K, Schenk D, Games D, Seubert P, Buttini M. Morphological characterization of Thioflavin-S-positive amyloid plaques in transgenic Alzheimer mice and effect of passive Abeta immunotherapy on their clearance. The American journal of pathology. 2004;165(3):987–995. doi: 10.1016/s0002-9440(10)63360-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabalka LM, Hyman BT, Goodlett CR, Ritchie TC, Van Hoesen GW. Alteration in the pattern of nerve terminal protein immunoreactivity in the perforant pathway in Alzheimer’s disease and in rats after entorhinal lesions. Neurobiol Aging. 1992;13(2):283–291. doi: 10.1016/0197-4580(92)90041-u. [DOI] [PubMed] [Google Scholar]
- Cao L, Schrank BR, Rodriguez S, Benz EG, Moulia TW, Rickenbacher GT, Gomez AC, Levites Y, Edwards SR, Golde TE, Hyman BT, Barnea G, Albers MW. Abeta alters the connectivity of olfactory neurons in the absence of amyloid plaques in vivo. Nat Commun. 2012;3:1009. doi: 10.1038/ncomms2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmel G, Mager EM, Binder LI, Kuret J. The structural basis of monoclonal antibody Alz50’s selectivity for Alzheimer’s disease pathology. The Journal of biological chemistry. 1996;271(51):32789–32795. doi: 10.1074/jbc.271.51.32789. [DOI] [PubMed] [Google Scholar]
- Chen LL, Van Hoesen GW, Barnes CL, West JR. Enhanced acetylcholinesterase staining in the hippocampal perforant pathway zone after combined lesions of the septum and entorhinal cortex. Brain research. 1983;272(2):354–359. doi: 10.1016/0006-8993(83)90583-8. [DOI] [PubMed] [Google Scholar]
- Chung RS, Vickers JC, Chuah MI, West AK. Metallothionein-IIA promotes initial neurite elongation and postinjury reactive neurite growth and facilitates healing after focal cortical brain injury. J Neurosci. 2003;23(8):3336–3342. doi: 10.1523/JNEUROSCI.23-08-03336.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins RT, Linker C, Lewis J. MAZe: a tool for mosaic analysis of gene function in zebrafish. Nature methods. 2010;7(3):219–223. doi: 10.1038/nmeth.1423. [DOI] [PubMed] [Google Scholar]
- de Calignon A, Polydoro M, Suarez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA, Spires-Jones TL, Hyman BT. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron. 2012;73(4):685–697. doi: 10.1016/j.neuron.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geddes JW, Monaghan DT, Cotman CW, Lott IT, Kim RC, Chui HC. Plasticity of hippocampal circuitry in Alzheimer’s disease. Science. 1985;230(4730):1179–1181. doi: 10.1126/science.4071042. [DOI] [PubMed] [Google Scholar]
- Gomez-Isla T, Growdon WB, McNamara MJ, Nochlin D, Bird TD, Arango JC, Lopera F, Kosik KS, Lantos PL, Cairns NJ, Hyman BT. The impact of different presenilin 1 andpresenilin 2 mutations on amyloid deposition, neurofibrillary changes and neuronal loss in the familial Alzheimer’s disease brain: evidence for other phenotype-modifying factors. Brain. 1999;122(Pt 9):1709–1719. doi: 10.1093/brain/122.9.1709. [DOI] [PubMed] [Google Scholar]
- Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol. 1997;41(1):17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- Harris JA, Koyama A, Maeda S, Ho K, Devidze N, Dubal DB, Yu GQ, Masliah E, Mucke L. Human P301L-Mutant Tau Expression in Mouse Entorhinal-Hippocampal Network Causes Tau Aggregation and Presynaptic Pathology but No Cognitive Deficits. PLoS One. 2012;7(9):e45881. doi: 10.1371/journal.pone.0045881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan LL, Ashe KH, Liao D. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68(6):1067–1081. doi: 10.1016/j.neuron.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274(5284):99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Hyman BT. Alzheimer’s disease or Alzheimer’s diseases? Clues from molecular epidemiology. Ann Neurol. 1996;40(2):135–136. doi: 10.1002/ana.410400202. [DOI] [PubMed] [Google Scholar]
- Hyman BT, Kromer LJ, Van Hoesen GW. Reinnervation of the hippocampal perforant pathway zone in Alzheimer’s disease. Ann Neurol. 1987;21(3):259–267. doi: 10.1002/ana.410210307. [DOI] [PubMed] [Google Scholar]
- Hyman BT, Kromer LJ, Van Hoesen GW. A direct demonstration of the perforant pathway terminal zone in Alzheimer’s disease using the monoclonal antibody Alz-50. Brain research. 1988;450(1-2):392–397. doi: 10.1016/0006-8993(88)91582-x. [DOI] [PubMed] [Google Scholar]
- Hyman BT, Tanzi R. Molecular epidemiology of Alzheimer’s disease. N Engl J Med. 1995;333(19):1283–1284. doi: 10.1056/NEJM199511093331910. [DOI] [PubMed] [Google Scholar]
- Hyman BT, Van Hoesen GW, Damasio AR, Barnes CL. Alzheimer’s disease: cell-specific pathology isolates the hippocampal formation. Science. 1984;225(4667):1168–1170. doi: 10.1126/science.6474172. [DOI] [PubMed] [Google Scholar]
- Hyman BT, Van Hoesen GW, Kromer LJ, Damasio AR. Perforant pathway changes and the memory impairment of Alzheimer’s disease. Ann Neurol. 1986;20(4):472–481. doi: 10.1002/ana.410200406. [DOI] [PubMed] [Google Scholar]
- Hyman BT, West HL, Rebeck GW, Lai F, Mann DM. Neuropathological changes in Down’s syndrome hippocampal formation. Effect of age and apolipoprotein E genotype. Arch Neurol. 1995;52(4):373–378. doi: 10.1001/archneur.1995.00540280059019. [DOI] [PubMed] [Google Scholar]
- Jicha GA, Bowser R, Kazam IG, Davies P. Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. Journal of neuroscience research. 1997;48(2):128–132. doi: 10.1002/(sici)1097-4547(19970415)48:2<128::aid-jnr5>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, Micheva KD, Smith SJ, Kim ML, Lee VM, Hyman BT, Spires-Jones TL. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(10):4012–4017. doi: 10.1073/pnas.0811698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopeikina KJ, Polydoro M, Tai HC, Yaeger E, Carlson GA, Pitstick R, Hyman BT, Spires-Jones TL. Synaptic alterations in the rTg4510 mouse model of tauopathy. J Comp Neurol. 2013;521(6):1334–53. doi: 10.1002/cne.23234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kromer Vogt LJ, Hyman BT, Van Hoesen GW, Damasio AR. Pathological alterations in the amygdala in Alzheimer’s disease. Neuroscience. 1990;37(2):377–385. doi: 10.1016/0306-4522(90)90408-v. [DOI] [PubMed] [Google Scholar]
- Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron. 2008;59(2):214–225. doi: 10.1016/j.neuron.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Tasic B, Micheva KD, Ivanov VM, Spletter ML, Smith SJ, Luo L. Visualizing the distribution of synapses from individual neurons in the mouse brain. PLoS One. 2010;5(7):e11503. doi: 10.1371/journal.pone.0011503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, Duff K. Trans-synaptic spread of tau pathology in vivo. PLoS One. 2012;7(2):e31302. doi: 10.1371/journal.pone.0031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch G, Matthews DA, Mosko S, Parks T, Cotman C. Induced acetylcholinesterase-rich layer in rat dentate gyrus following entorhinal lesions. Brain research. 1972;42(2):311–318. doi: 10.1016/0006-8993(72)90533-1. [DOI] [PubMed] [Google Scholar]
- McDonald JM, Savva GM, Brayne C, Welzel AT, Forster G, Shankar GM, Selkoe DJ, Ince PG, Walsh DM. The presence of sodium dodecyl sulphate-stable Abeta dimers is strongly associated with Alzheimer-type dementia. Brain. 2010;133(Pt 5):1328–41. doi: 10.1093/brain/awq065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JM, Cairns NJ, Taylor-Reinwald L, Holtzman D, Walsh DM. The levels of water-soluble and triton-soluble Abeta are increased in Alzheimer’s disease brain. Brain research. 2012;1450:138–147. doi: 10.1016/j.brainres.2012.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesulam MM. Some cholinergic themes related to Alzheimer’s disease: synaptology of the nucleus basalis, location of m2 receptors, interactions with amyloid metabolism, and perturbations of cortical plasticity. J Physiol Paris. 1998;92(3-4):293–298. doi: 10.1016/s0928-4257(98)80036-3. [DOI] [PubMed] [Google Scholar]
- Mesulam MM. A plasticity-based theory of the pathogenesis of Alzheimer’s disease. Ann N Y Acad Sci. 2000;924:42–52. doi: 10.1111/j.1749-6632.2000.tb05559.x. [DOI] [PubMed] [Google Scholar]
- Micheva KD, Busse B, Weiler NC, O’Rourke N, Smith SJ. Single-Synapse Analysis of a Diverse Synapse Population: Proteomic Imaging Methods and Markers. Neuron. 2010;68(4):639–653. doi: 10.1016/j.neuron.2010.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheva KD, Smith SJ. Array tomography: a new tool for imaging the molecular architecture and ultrastructure of neural circuits. Neuron. 2007;55(1):25–36. doi: 10.1016/j.neuron.2007.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamichi K, Amat F, Moussavi F, Wang C, Wickersham I, Wall NR, Taniguchi H, Tasic B, Huang ZJ, He Z, Callaway EM, Horowitz MA, Luo L. Cortical representations of olfactory input by trans-synaptic tracing. Nature. 2010;472(7342):191–196. doi: 10.1038/nature09714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosene DL, Van Hoesen GW. Hippocampal efferents reach widespread areas of cerebral cortex and amygdala in the rhesus monkey. Science. 1977;198(4314):315–317. doi: 10.1126/science.410102. [DOI] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309(5733):476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires TL, Meyer-Luehmann M, Stern EA, McLean PJ, Skoch J, Nguyen PT, Bacskai BJ, Hyman BT. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci. 2005;25(31):7278–7287. doi: 10.1523/JNEUROSCI.1879-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hoesen G, Pandya DN. Some connections of the entorhinal (area 28) and perirhinal (area 35) cortices of the rhesus monkey. I. Temporal lobe afferents. Brain Res. 1975a;95(1):1–24. doi: 10.1016/0006-8993(75)90204-8. [DOI] [PubMed] [Google Scholar]
- Van Hoesen GW. Neural systems of the non-human primate forebrain implicated in memory. Ann N Y Acad Sci. 1985;444:97–112. doi: 10.1111/j.1749-6632.1985.tb37582.x. [DOI] [PubMed] [Google Scholar]
- Van Hoesen GW, Hyman BT. Hippocampal formation: anatomy and the patterns of pathology in Alzheimer’s disease. Prog Brain Res. 1990;83:445–457. doi: 10.1016/s0079-6123(08)61268-6. [DOI] [PubMed] [Google Scholar]
- Van Hoesen GW, Hyman BT, Damasio AR. Cell-specific pathology in neural systems of the temporal lobe in Alzheimer’s disease. Prog Brain Res. 1986;70:321–335. doi: 10.1016/s0079-6123(08)64313-7. [DOI] [PubMed] [Google Scholar]
- Van Hoesen GW, Pandya DN. Some connections of the entorhinal (area 28) and perirhinal (area 35) cortices of the rhesus monkey. III. Efferent connections. Brain research. 1975b;95(1):39–59. doi: 10.1016/0006-8993(75)90206-1. [DOI] [PubMed] [Google Scholar]
- Van Hoesen GW, Pandya DN, Butters N. Cortical afferents to the entorhinal cortex of the Rhesus monkey. Science. 1972;175(4029):1471–1473. doi: 10.1126/science.175.4029.1471. [DOI] [PubMed] [Google Scholar]
- Van Hoesen GW, Rosene DL, Mesulam MM. Subicular input from temporal cortex in the rhesus monkey. Science. 1979;205(4406):608–610. doi: 10.1126/science.109926. [DOI] [PubMed] [Google Scholar]
- William CM, Andermann ML, Goldey GJ, Roumis DK, Reid RC, Shatz CJ, Albers MW, Frosch MP, Hyman BT. Synaptic plasticity defect following visual deprivation in Alzheimer’s disease model transgenic mice. J Neurosci. 2012;32(23):8004–8011. doi: 10.1523/JNEUROSCI.5369-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witter MP. The perforant path: projections from the entorhinal cortex to the dentate gyrus. Prog Brain Res. 2007;163:43–61. doi: 10.1016/S0079-6123(07)63003-9. [DOI] [PubMed] [Google Scholar]
- Wolozin BL, Pruchnicki A, Dickson DW, Davies P. A neuronal antigen in the brains of Alzheimer patients. Science. 1986;232(4750):648–650. doi: 10.1126/science.3083509. [DOI] [PubMed] [Google Scholar]
- Wu HY, Hudry E, Hashimoto T, Kuchibhotla K, Rozkalne A, Fan Z, Spires-Jones T, Xie H, Arbel-Ornath M, Grosskreutz CL, Bacskai BJ, Hyman BT. Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J Neurosci. 2010;30(7):2636–2649. doi: 10.1523/JNEUROSCI.4456-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Sonner PM, Titus DJ, Wiesner EP, Alvarez FJ, Ziskind-Conhaim L. Properties of a distinct subpopulation of GABAergic commissural interneurons that are part of the locomotor circuitry in the neonatal spinal cord. J Neurosci. 2011;31(13):4821–4833. doi: 10.1523/JNEUROSCI.4764-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda M, Mayford MR. CaMKII activation in the entorhinal cortex disrupts previously encoded spatial memory. Neuron. 2006;50(2):309–318. doi: 10.1016/j.neuron.2006.03.035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.