

1. Introduction
Arsenic is a trace element found in the earth’s crust at an average concentration of ∼5 μg/g (ppm). Although its relative abundance in the earth’s crust is about 54th, arsenic can become concentrated in some parts of the world because of natural mineralization. Arsenic is a component of 245 minerals, associated most frequently with other metals such as copper, gold, lead, and zinc in sulfidic ores.1−3 When disturbed by natural processes, such as weathering, biological activity, and volcanic eruption, arsenic may be released into the environment. Anthropogenic activities, such as combustion of fossil fuels, mining, ore smelting, and well drilling, also mobilize and introduce arsenic into the environment.
Chronic exposure to arsenic from groundwater has been recognized to cause the largest environmental health disaster in the world, putting more than 100 million people at risk of cancer and other arsenic-related diseases.4,5 Because of its prevalence in the environment, potential for human exposure, and the magnitude and severity of health problems it causes, the United States Agency for Toxic Substances and Disease Registry (ATSDR) has ranked arsenic as No. 1 on its Priority List of Hazardous Substances for many years. The recent priority list, posted in 2011 (http://www.atsdr.cdc.gov/SPL/index.html), shows arsenic as No. 1, ahead of lead, mercury, and polychlorinated biphenyls (PCBs).
Epidemiological studies of populations exposed to high levels of arsenic due to ingestion from water, including those from Taiwan,6−8 Argentina,9,10 Chile,11,12 West Bengal, India,13,14 Bangladesh,15−17 and Inner Mongolia, China,18,19 have repeatedly shown strong associations between the exposure to high concentrations of arsenic and the prevalence of several cancers,20−23 most severely bladder, lung, and skin cancers. Arsenic is classified as a human carcinogen by the International Agency for Research on Cancer (IARC) and the U.S. Environmental Protection Agency (EPA). Chronic exposure to elevated concentrations of arsenic has also been associated with the increased risk of a number of noncancerous effects.24−27 Although the adverse health effects arising from exposure to arsenic have been well-recognized, the mechanism(s) of action responsible for the diverse range of health effects are complicated and poorly understood.26−30
It is believed that inorganic arsenate (HAsO42-), which is a molecular analogue of phosphate (HPO42-), can compete for phosphate anion transporters and replace phosphate in some biochemical reactions.28 For example, generation of adenosine-5′-triphosphate (ATP) during oxidative phosphorylation can be inhibited by the replacement of phosphate with arsenate. Depletion of ATP by arsenate has been observed in cellular systems.28 However, the replacement of phosphate in DNA by arsenic is not firmly established.31−35
The toxicity of trivalent arsenicals likely occurs through the interaction of trivalent arsenic species with sulfhydryl groups in proteins. Arsenic binding to a specific protein could alter the conformation and function of the protein as well as its recruitment of and interaction with other functional proteins. Therefore, there has been much emphasis on studies of arsenic binding to proteins, for the purpose of understanding arsenic toxicity and developing arsenic-based therapeutics.
This review summarizes various aspects of arsenic binding to proteins. It discusses the chemical basis and biological implications and consequences of arsenic binding to proteins. It also describes analytical techniques and the characterization of arsenic binding, including the binding affinity, kinetics, and speciation.
2. Chemical Basis and Biological Implications of Arsenic Binding to Proteins
Trivalent arsenicals have high affinity for sulfhydryl groups and can bind to reduced cysteines in peptides and proteins.36−60 Figure 1 illustrates schematically the binding of inorganic arsenite (iAsIII), monomethylarsonous acid (MMAIII), and dimethylarsinous acid (DMAIII) to cysteine residues in proteins. This chemical basis has been well-known. For example, the toxicity of many chemical warfare agents in use during and after World War I, such as arsine halides, was based on their binding to protein dithiols.61−63 Lewisite (β-chlorovinyldichloroarsine) was known to react with some enzymes by forming a ring involving two thiol groups in the enzymes.64 To counteract the toxicity of these poison gases during World War I, the British developed dithioglycerol (British anti-lewisite, BAL), which could bind arsenic to form very stable complexes, preventing arsenic from binding to proteins.
Figure 1.
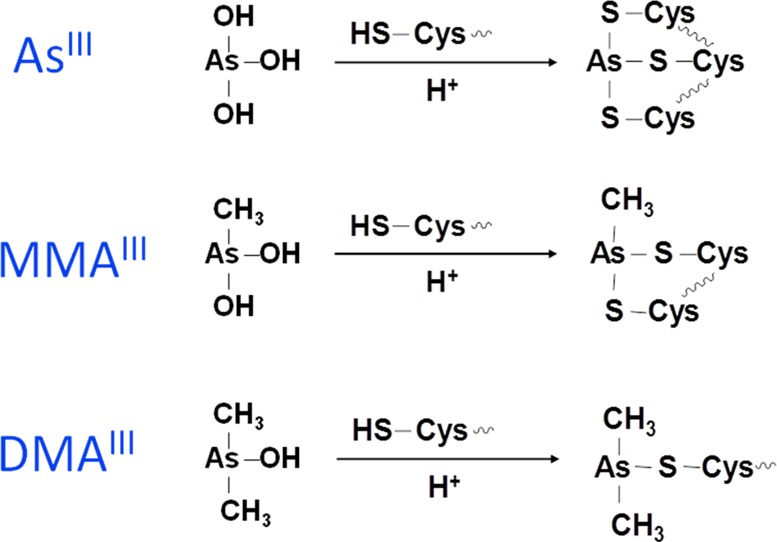
Binding of inorganic arsenite (iAsIII), monomethylarsonous acid (MMAIII), and dimethylarsinous acid (DMAIII) to cysteines in a protein.
Arsenic binding to a specific protein could alter the conformation of the protein, resulting in loss of its function, and affect its recruitment of and interaction with other proteins and DNA. Figure 2 shows a model of iAsIII binding to the Escherichia coli repressor protein ArsR. In each subunit, two cysteine residues are within an α-helix, which must unravel from one end in order to bind iAsIII. Melting of the helix produces the conformational change that dissociates the repressor from the DNA, inducing gene expression. This ArsR binds iAsIII at three cysteine residues, Cys32, Cys34, and Cys37. Cys32 and Cys34 are within the α1 helix in the DNA binding site. Their thiolates are oriented such that iAsIII cannot bind until the helix unwinds, bringing all three thiolates into position to form the three-coordinate iAsIII binding site. Unwinding the helix disrupts DNA binding, resulting in dissociation of the repressor from the operator site and hence gene expression.65
Figure 2.
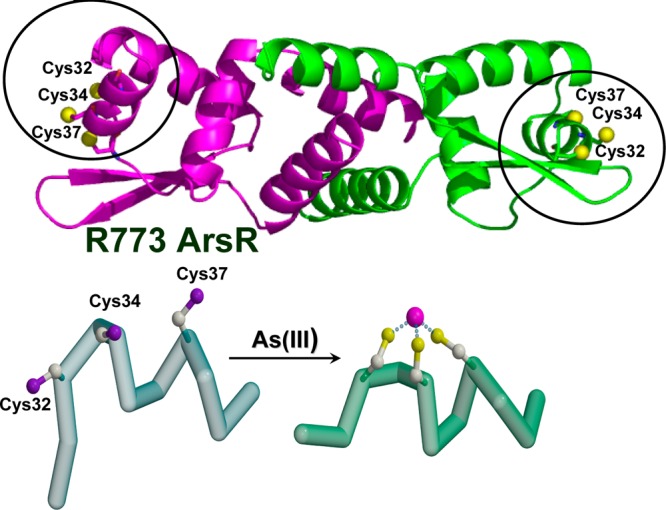
Binding of iAsIII to the ArsR repressor from E. coli plasmid R773 (P15905) results in conformational change of the repressor. iAsIII binds to Cys32, Cys34, and Cys37 of the ArsR repressor. Unwinding the helix disrupts DNA binding, resulting in dissociation of the repressor from the operator site. Dissociation of the repressor induces gene expression. (Adapted from ref (65).)
The importance of direct interaction of arsenic compounds with proteins has also been highlighted by the observation that the successful treatment of patients suffering from acute promyelocytic leukemia (APL) with As2O3 is likely due to the binding of arsenic to cysteine residues in zinc fingers of the aberrant promyelocytic leukemia-retinoic acid receptor (PML-RAR) fusion protein expressed by these patients.66,67
2.1. Arsenic Biomethylation
Biomethylation is the major metabolic pathway for inorganic arsenic (iAs) in nearly all organisms, including humans and most animal species.68 Arsenic biomethylation involves specific reductases27,30,69−73 and methyltransferases.50,68,74−78 The major methyltransferase responsible for arsenic biomethylation is arsenic (+3 oxidation state) methyltransferase (AS3MT, previously named Cty19). This enzyme catalyzes transfer of the methyl group of S-adenosylmethionine (SAM) to trivalent arsenic.50,68,74,75 The crystallographic structure of AS3MT from the thermophilic eukaryotic red alga Cyanidioschyzon merolae has recently been obtained.75,79 It reveals binding domains for arsenic and SAM (Figure 3). Although a crystal structure of human AS3MT is not available, Cys156 and Cys206, among the 14 cysteine residues, were demonstrated to be the active sites of human AS3MT.80−82
Figure 3.
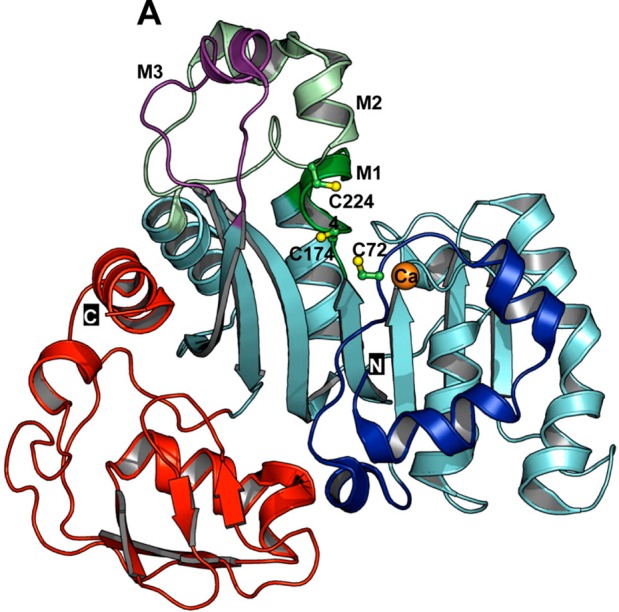
Ribbon representation of ligand-free AS3MT (CmArsM) from the thermophilic eukaryotic alga C. merolae. N and C indicate the N- and C-terminal domains and are colored blue and red, respectively. Cysteine residues are shown as balls and sticks and colored green (carbon) and yellow (sulfur). Three cysteine residues, C72, C174, and C224, are believed to be involved in arsenic binding. (Reprinted with permission from ref (75). Copyright 2012 American Chemical Society.)
2.2. Enzyme Inhibition
Arsenic may inactivate up to 200 enzymes.83,84 Most of the enzyme inhibition studies used iAsIII and some used organic arsenicals, particularly phenylarsine oxide (PAO). Enzymes that were shown to be inhibited by arsenic include glutathione reductase,49,85−87 glutathione S-transferase,88 glutathione peroxidase,88 thioredoxin reductase,89−91 thioredoxin peroxidase,92 DNA ligases,93 Arg-tRNA protein transferase,94,95 trypanothione reductase,86 IκB kinase β (IKKβ),96 pyruvate kinase galectin 1,97 protein tyrosine phosphatase,98−101 JNK phosphatase,102 Wip1 phosphatase,103 and E3 ligases c-CBL and SIAH1.104
2.2.1. Pyruvate Dehydrogenase (PDH)
The enzyme complex pyruvate dehydrogenase (PDH) is one of the most studied enzyme systems. A search for natural dithiol compounds to mimic BAL led to the discovery of its cofactor, lipoic acid.105 PDH complexes from prokaryotes and eukaryotes, which have molecular weights in excess of 106 Da, are formed by noncovalent interactions between multiple enzymes. Arsenite inhibits the PDH complex by binding to the lipoic acid moiety. MMAIII was shown to be a more potent inhibitor of the PDH complex than iAsIII.105,106 The PDH complex oxidizes pyruvate to acetyl-CoA, a precursor to intermediates of the citric acid cycle. The citric acid cycle degrades the intermediates, providing the mitochondria with reducing equivalents needed for electron transport and the subsequent production of ATP. Inhibition of the PDH complex can block the citric acid cycle and ultimately decrease the production of ATP, resulting in cell damage and death. On the basis of the fact that As2O3 was far more inhibitory to the intracellular PDH complex than to the purified PDH complex (actually similar phenomena were observed for other enzymes), the PDH complex (and perhaps other dithiol-containing enzymes) was proposed by Samikkannu et al.107 to be inhibited by arsenite via reactive oxygen species (ROS) production rather than direct thiol binding as in the case of PAO. However, a recent study105 conducted under conditions free of ROS generation suggested that all three trivalent arsenic species (iAsIII, MMAIII, and DMAIII) at biologically relevant concentrations inhibited the PDH complex and the 2-oxoglutarate dehydrogenase (KGDH) complex by direct binding. The reduced form of the lipoic acid moiety appeared to be essential for the binding. No pentavalent arsenicals (iAsV, MMAV, and DMAV) were found to inhibit the enzymes. All enzymes post-translationally modified by the addition of the lipoic acid cofactor are mitochondrial and are important for respiration.105 It is conceivable that trivalent arsenic binds to PDH or KGDH complexes and inhibits their activity, which depletes the mitochondrial NADH pool and leads to oxidative stress, producing ROS, which is otherwise suppressed or balanced. Thus, the binding of arsenic to those enzymes may be the initial point of action to inhibit the activity of the enzymes. However, subsequently generated ROS may have further effects on the enzymatic inhibition.
2.2.2. Thioredoxin
The direct binding between thioredoxin (Trx) and arsenic has been characterized using mass spectrometry.108 All of the four trivalent arsenic species under study (iAsIII, MMAIII, DMAIII, and PAO) bound to human Trx to form complexes at room temperature while only MMAIII, DMAIII, and PAO formed complexes with E. coli Trx, consistent with arsenic coordination chemistry and the availability of cysteine residues in these two proteins for binding. Trx proteins from both human and E. coli have the highly conserved −CysGlyProCys– region, but human Trx has three more cysteine residues (Cys62, Cys69, and Cys73). Human Trx showed slightly higher binding constants than E. coli Trx to the same arsenic species.108 Pentavalent arsenic species (iAsV, MMAV, and DMAV) could be reduced by excess Trx at 37 °C to their trivalent counterparts, which in turn could bind to Trx.
The thioredoxin system consists of two oxidoreductase enzymes, thioredoxin and thioredoxin reductase.109 The thioredoxin system complements the function of the glutathione system in maintaining reductive intracellular redox potential and protecting against toxicity. Thioredoxin is a small protein (∼12 kDa) containing a highly conserved sequence, −CysGlyProCys–. It was first identified in E. coli and subsequently found to be present in most eukaryotic and prokaryotic species.109 Thioredoxin is the major ubiquitous disulfide reductase responsible for maintaining proteins in their reduced state, in which process thioredoxin itself is oxidized and subsequently regenerated at the expense of NADPH via thioredoxin reductase. Thioredoxin has been implicated in a multitude of biological activities.110 As an electron carrier, thioredoxin activates the catalytic cycles of biosynthetic enzymes, such as ribonucleotide reductases, methionine sulfoxide reductases, and sulfate reductases. It protects cytosolic proteins from aggregation or inactivation via formation of intra- or intermolecular disulfides. Thioredoxin can regulate the activity of transcription factors such as NF-κB, AP-1, and p53. Thioredoxin plays a central role in thiol redox control of cell functions by modulation of the transcription events of target genes. It can interact with other proteins to form functional protein complexes. For example, thioredoxin is an essential processivity factor for bacteriophage T7 DNA polymerase. Its activity has been found outside of the cell, in the cytoplasm, in the nucleus, and in the mitochondria.111
As an important cellular dithiol redox enzyme, thioredoxin is required for the methylation of arsenic catalyzed by AS3MT.112,113 Trivalent arsenic compounds, especially the methylated metabolite MMAIII, were found to inhibit thioredoxin reductase in cultured rat hepatocytes.90 Arsenite was also shown to induce NF-kB activation and to affect AP-1 and NF-κB DNA binding activity by upregulating the expression of the Trx gene.114 Thioredoxin peroxidase II was identified as an arsenic-binding protein in mammalian cells.92 Thioredoxin stimulates cell growth and has antiapoptotic properties. Modulation of thioredoxin was implicated in the apoptosis of APL cancer cells treated with arsenic trioxide (As2O3).89
2.2.3. DNA Repair Enzymes
All three trivalent arsenicals (iAsIII, MMAIII, and DMAIII) can inhibit DNA repair.115−117 These trivalent arsenicals were found to inhibit the activities of several DNA repair proteins, including poly(ADP-ribose) polymerase-1 (PARP-1), formamidopyrimidine-DNA glycosylase (Fpg), and xeroderma pigmentosum group A protein (XPA), each containing a zinc finger DNA binding domain.118,119 The interactions between these repair proteins and arsenic compounds resulted in the release of zinc. As the first identified member of the PARP superfamily, PARP-1 is believed to mediate the major fraction of cellular poly(ADP-ribosyl)ation. Although its role is not fully understood, there is strong evidence that PARP-1 contributes to base excision repair (BER), perhaps also to nucleotide excision repair (NER)120,121 and to the nonhomologous end joining pathway of DNA double-strand break (DSB) repair.122 PARP-1 contains two zinc fingers, where each zinc is complexed through three cysteines and one histidine residue (C3H1). Zinc fingers are essential for the recognition of damaged DNA. Recently, the direct binding between arsenic and peptides derived from both zinc fingers of human PARP-1 (apo-PARPzf) was observed using matrix-assisted laser desorption ionization mass spectrometry (MALDI-MS).45,123 A 72-Da m/z shift corresponded to the covalent binding of iAsIII to the PARPzf peptides after the release of three hydrogen atoms from the three cysteine residues of the C3H1 zinc finger peptides. Displacement of Zn2+ from the PARPzf peptides by iAsIII was also demonstrated to be dependent on the iAsIII concentration.
XPA binds specifically to damaged DNA and plays a central role in the first step of NER. XPA contains a single C4 zinc finger domain and zinc is complexed by Cys105, Cys108, Cys126, and Cys129. Trivalent arsenicals (iAsIII, MMAIII, and DMAIII) were shown to release zinc from the zinc finger domain of the XPA protein (XPAzf). Methylated trivalent arsenicals were more efficient than inorganic arsenite in releasing zinc.115 It was further demonstrated that equimolar MMAIII could react with XPAzf and yield a mixture of mono- and diarsenical adducted XPAzf.124 The unprotected thiols in the MMAIII-XPAzf were further oxidized to form an intramolecular disulfide. There was no complex formation between iAsIII and XPAzf.
2.3. Resistance to Arsenic
All cells have regulatory mechanisms for controlling the intracellular concentrations of metals and metalloids. Metallothioneins, found in both eukaryotes and prokaryotes, are small proteins (2–7 kDa) high in thiolate sulfurs that bind metal ions for storage and/or for detoxification. Phytochelatins are present in plants and some fungi.125 They are cysteine-rich peptides that bind heavy metals and metalloids for detoxification, such as in the case of hyperaccumulation of arsenic.126−128 By contrast, metallochaperones constitute a different type of metal-binding protein, serving as an intracellular shuttles for metal ions. Metallochaperones sequester metals in the cytoplasm and deliver them to protein targets.129 The targets can be metalloenzymes, where metals are utilized as enzyme cofactors, or membrane transporters, which predominantly transport ATPases. These metallochaperones are involved in metal ion homeostasis and detoxification.129
2.3.1. Metallothionein
Metallothioneins (MT I and II) are highly inducible by metals (such as Zn2+, Cd2+, Co2+, Ni2+, Ag+, Hg2+, and Bi3+), alkylating agents, oxidizing agents, glulcocorticoids, and inflammatory signals.130−138 Arsenic was also reported to induce metallothioneins.139−144 The mechanism by which arsenic induces metallothioneins remained unclear until recently when metal-activated transcription factor 1 (MTF1), a member of the zinc finger family, was found to be involved in Mt1 induction.130 iAsIII and PAO covalently bound the C-terminal cysteine cluster (Cys632, Cys634, Cys636, Cys638, and Cys653) of MTF1, which in turn induced the binding of MTF1 to the metal response elements (MRE) of endogenous Mt1.
Metallothioneins have also been found to directly bind to many metals such as Zn2+, Cd2+, Co2+, Ni2+, Fe2+, Cu+, Ag+, Au+, Pt2+, Bi3+, Hg2+, and TcIVO, either in vivo or in vitro.145−148 The direct binding between metallothioneins and arsenic was studied by several research groups.149−159 In a large excess of arsenic over metal-free human metallothionein 2 (apo-hMT-2), up to six iAsIII could bind to hMT-2 (Figure 4), on the basis of results obtained from inductively coupled plasma atomic emission spectrometry (ICP-AES), UV absorption spectrometry, MALDI-TOF-MS, and electrospray ionization (ESI) TOF-MS.149,152,155 Evidence from MALDI-TOF-MS149 and ESI-TOF-MS151,152,155 also suggested that the binding of one iAsIII resulted in the loss of three protons from the protein. The binding stoichiometry was consistent with the fact that 20 cysteine residues are contained in mammalian metallothioneins and maximally six iAsIII can bind to the protein.
Figure 4.
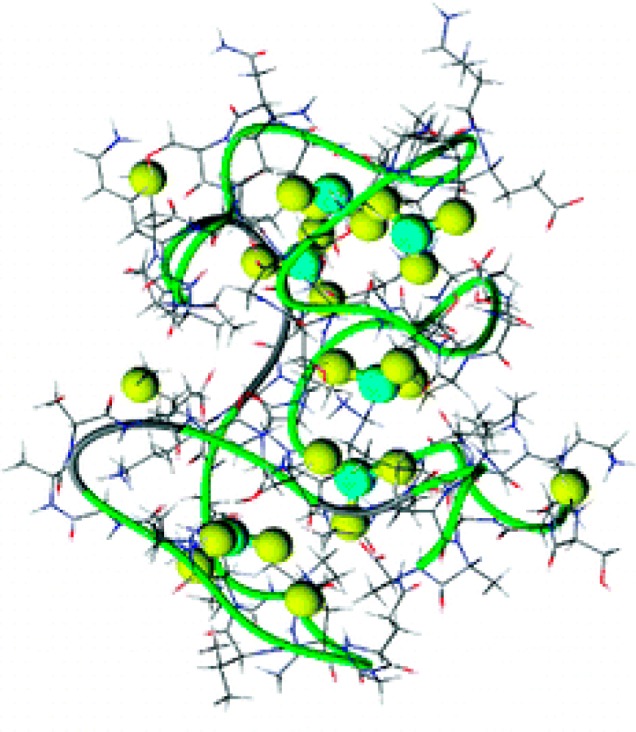
A model depicting six arsenic atoms bound to 18 cysteines in metallothionein. (Reprinted with permission from ref (155). Copyright 2008 American Chemical Society.)
A detailed binding study between six arsenic species (iAsIII, MMAIII, DMAIII, iAsV, MMAV, DMAV, and TMAOV) and rabbit metallothionein II (rMT II) was conducted by use of HPLC–ICP-MS and ESI-quadrupole-TOF-MS.151 The binding stoichiometry revealed by the complementary mass spectrometry data demonstrated that each apo-rMT II molecule could bind up to six iAsIII, 10 MMAIII, and 20 DMAIII, consistent with the coordinate chemistry of the individual trivalent arsenicals (Figure 5). Because the reduced product, trimethylarsine, from TMAOV, has no binding sites for thiol groups, no binding was observed from the reaction between MT and TMAOV. As expected, no binding occurred between apo-rMT II and pentavalent arsenic species. A downward mass shift of MT was observed in the presence of pentavalent arsenicals, however, implying the oxidation of −SH to disulfide in MT, associated with reduction of pentavalent arsenicals to their trivalent counterparts.
Figure 5.

Mass spectra showing the number of monomethylarsonous acid (MMAIII) molecules bound to metallothionein (MT). The solutions contained 7 μM metallothionein (rMT-IIa) and increasing concentrations of MMAIII. The concentration ratios of MMAIII to MT in the solutions were (a) 1:5, (b) 1:1, (c) 5:1, and (d) 50:1. The ions carrying 5+ and 4+ charges are shown. The numbers on the peaks represent the number of MMAIII bound to the MT molecule. For example, peak 6 represents MT-(CH3As)6. A maximum of 10 MMAIII molecules were bound to a single MT that contained 20 cysteines. (Adapted from ref (151).)
2.3.2. Ars Operon
Resistance to arsenite and antimonite in many prokaryotes is conferred by an ars operon, which often contains three genes, arsR, arsB, and arsC. Some operons have two extra genes, arsD and arsA, first identified in the E. coli plasmid R773 ars operon.160−162 Cells expressing the arsRDABC operon are more resistant to iAsV and iAsIII than those expressing the arsRBC operon. The first two genes, arsR and arsD, encode regulatory proteins. The arsA gene encodes the transport ATPase and the arsB gene encodes the membrane translocase subunit of an arsenic pump. The last gene of the operon, arsC, encodes an arsenate reductase that is required to catalyze the reduction of iAsV to iAsIII prior to extrusion. Figure 6 shows arsenic detoxification in E. coli by the plasmid R773 arsRDABC operon.
Figure 6.

Schematic representation of arsenic interaction with the arsRDABC operon and related enzymes in E. coli. ArsR is an AsIII-responsive transcriptional repressor that binds to the ars promoter, repressing transcription. Binding of iAsIII to ArsR results in dissociation of the repressor from the DNA and hence gene expression. iAsV is taken up by the phosphate transporter, while iAsIII is taken up by the aquaglyceroporin GlpF. iAsV is reduced to iAsIII by ArsC. Intracellular iAsIII is extruded from the cells by ArsB alone or by the ArsAB ATPase. (Modified from ref (163).)
ArsR is a homodimer of two 117-residue monomers with high affinity for the arsRDABC promoter. ArsR binds to the DNA and represses expression of the operon to a basal level in the absence of iAsIII. But in the presence of arsenic, the binding of iAsIII to ArsR changes its conformation and thus ArsR dissociates from the DNA, enabling transcription. ArsD also functions as a homodimer of two 120-residue subunits. ArsD and ArsR bind to the same operator site but the affinity of ArsD is 2 orders of magnitude lower than that of ArsR. Hence, ArsD has a weaker ars operon repressor activity and it binds to the ars operon when produced in high concentrations, such as after prolonged stimulation of transcription of the arsD gene following induction by the toxic iAsIII. It has been proposed that ArsD controls the upper level of expression of the operon, preventing the buildup of ArsB, which at high levels appears to be toxic to the cell. Although the affinity of ArsD for iAsIII is an order of magnitude lower than that of ArsR, ArsD binds to high levels of iAsIII and then dissociates from the DNA, allowing the operon to transcribe. Thus, ArsR and ArsD together form a regulatory circuit that controls the expression of arsRDABC operon at both high and low levels of iAsIII.162,163 Recently another role of arsD was identified.164 It serves as an arsenic metallochaperone that delivers iAsIII to the arsA ATPase. Interaction with ArsD increases the affinity of ArsA for iAsIII, resulting in increased efflux and resistance at environmental concentrations of arsenic.
In almost all the encoded proteins of the arsRDABC, arsenic–thiol interaction plays an essential role for their induction or function.161 ArsR, ArsD, and ArsA each contain critical cysteine residues that interact with AsIII. Coordination of AsIII with some critical cysteine thiolates in ArsR (Cys32, Cys34, and Cys37) and ArsD (Cys12, Cys13, Cys112, and Cys 113) results in a conformational change in the repressors that disrupts their interaction with the operator DNA. Three critical cysteine residues (Cys12, Cys13, and Cys18) were also reported to be required of ArsD to exert its metallochaperone activity.164 Cys113, Cys172, and Cys422 are involved in the metallostimulation of the ArsA ATPase. The strong arsenic–thiol interactions provide an excellent way to sense arsenic and to turn on the genes transcriptionally or the ArsA ATPase allosterically. In the ArsC protein, Cys12 is involved in forming a covalent thioester intermediate with arsenate. The thioester is further reduced by glutaredoxin and glutathione, producing a Cys12–S–AsIII intermediate, from which arsenite is released upon hydrolysis.165 ArsB contains a single cysteine residue that was shown not to be essential for its function. Low-affinity oxyanion binding instead of a very strong arsenic–thiol interaction seems to be appropriate for this membrane transporter protein to be effective, binding the substrate on one side of the membrane and releasing it on the other.162
2.4. Arsenic Binding to Hemoglobin and Accumulation of Arsenic in Rat Blood
There are noticeable differences in the half-life of arsenic in blood between animal species. Most arsenic can be rapidly eliminated from blood in humans, with a half-life of about 1 h. Studies on hemodialysis patients indicate that part of the arsenic is bound to transferrin.166 However, arsenic is retained in rat blood considerably longer than in blood of other species. The accumulation of arsenic in rat red blood cells was reported more than 50 years ago.167 The cat can also accumulate arsenic in blood, albeit to a lesser extent than does the rat.168 Binding of arsenic to hemoglobin in red blood cells was proposed as the site of accumulation in rat blood.169
Hemoglobin makes up to about 97% of the red blood cell’s dry content in mammals and, therefore, is a reasonable target protein of arsenic in red blood cells. Preferential accumulation of arsenic in rat blood was attributed to arsenic binding to hemoglobin.169 Rat hemoglobin showed higher affinity for trivalent arsenic species than human hemoglobin (Figure 7). Both hemoglobins are tetramers, each consisting of two α-chains and two β-chains. Rat hemoglobin has three cysteines in each α-chain (Cys13, Cys104, and Cys111) and two cysteines in each β-chain (Cys93 and Cys125). Human hemoglobin has only one cysteine in each α-chain (Cys104) and two cysteines in each β-chain (Cys93 and Cys112). Although previous in vivo studies170 showed that DMAV was detected in rat hemoglobin, further investigations revealed that DMAIII is the actual binding form of arsenic to rat hemoglobin and Cys13 is the preferential binding site for arsenic retention.60,171,172 It is interesting that cat hemoglobin also contains Cys13.60 Whether this coincidence is responsible for arsenic accumulation in the cat has not been investigated.
Figure 7.

Comparison of rat hemoblobin (rHb) and human hemoblobin (hHb) binding to three trivalent arsenicals (iAsIII, MMAIII, and DMAIII). Higher percentages of the trivalent arsenicals are bound to rHb than to hHb. (Reprinted with permission from ref (169). Copyright 2004 American Chemical Society.)
2.5. Arsenic-Induced Apoptosis
Arsenic (in the form of As2O3) has been successfully used as a chemotherapeutic agent in treating certain types of cancers, especially relapsed or all-trans-retinoic acid (ATRA)-resistant acute promyelocytic leukemia (APL). As2O3-induced apoptosis was suggested to be the mechanism of the therapeutic effect of As2O3.173 Although apoptosis can be triggered by many different pathways, direct binding of iAsIII to cysteine residues in zinc fingers (ZFs) located in the promyelocytic leukemia protein (PML) was demonstrated to be one of the plausible modes of action leading to APL cell apoptosis.66,67 The conserved cysteines in both zinc fingers contained within PML-R and PML-B (Cys60, Cys77, Cys80 in ZF1 and Cys72, Cys88, Cys91 in ZF2) were indicated to be coordinated with iAsIII. Apo-PML-R (PML-R without zinc) could bind one or two arsenic atoms at a clinically relevant dose of As2O3, determined by MALDI-TOF-MS. Arsenic binding induces PML oligomerizaiton, which facilitates the SUMOylation (a reversible post-translational modification involving conjugation to small ubiquitin-related modifier proteins) and subsequent degradation of PML-RARα (a fusion protein of PML and retinoic acid receptor alpha) and/or PML proteins essential for the growth of APL cells (Figure 8).
Figure 8.
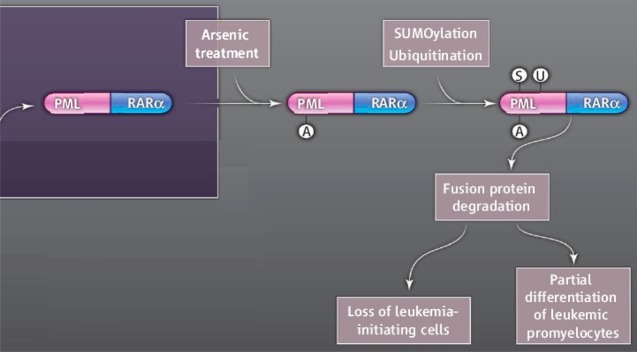
A proposed mechanism for the treatment of acute promyelocytic leukemia (APL) using inorgaic arsenic. Binding of arsenic to the promyelocytic leukemia (PML) retinoic acid receptor alpha (RARα) fusion protein triggers SUMOylation and ubiquitination and ultimately leads to the degradation of the oncoprotein and cell death. (Reprinted with permission from ref (66) and Kogan, S.C. 10.1126/science.1189198. Copyright 2010 American Association for the Advancement of Science.)
Mitochondria play a vital role in the cell, providing most of the cell’s energy and participating in Ca2+, redox, and pH homeostasis. A major mitochondrial dysfunction is likely to cause cell death.174,175 With few exceptions, mitochondria represent an essential component of many apoptotic pathways.176,177 Proteins present in the mitochondrial intermembrane space have been shown to induce apoptosis in response to a variety of apoptotic stimuli.174 Mitochondrial membrane permeabilization is critical for the release of proapoptotic factors from mitochondria. The mitochondrial permeability transition pore (MPTP), a multiprotein complex that is located at the contact sites of the inner and outer membranes, is responsible for mitochondrial permeabilization. It is comprised of the voltage-dependent anion channel (VDAC) and the adenine nucleotide translocator (ANT), the two most abundant MPTP components of the inner and outer mitochondrial membranes, respectively, in association with other proteins. ANT is a 30 kDa protein and has three cysteines (Cys57, Cys160, and Cys257). PAO and some trivalent arsenic–glutathione derivatives have been shown to cross-link the critical thiols (Cys160 and Cys257) in ANT and therefore lock ANT into an open configuration, leading to apoptosis.178−181 Since there are numerous redox-sensitive mitochondrial proteins that are arsenic binding targets,182−186 including PDH, VDAC, tubulin, glucocorticoid receptor, thioredoxin, thioredoxin reductase, thioredoxin peroxidase, and glutathione S-transferase, their contribution to arsenic-induced apoptosis cannot be precluded. Thiol modification/redox regulation in mitochondria was acknowledged to be relevant in inducing apoptosis.187,188 For instance, inhibition of thioredoxin reductase has been suggested to be responsible for APL cell apoptosis induced by As2O3.89 Both the N-terminal dithiols and the C-terminal selenothiol may participate in the reaction with the arsenic compound. Arsenic may also mimic glucocorticoids to induce apoptosis by binding to mitochondrial glucocorticoid receptor.189
2.6. Arsenic Binding to Other Proteins
Trivalent arsenicals also bind many other proteins, including tubulin,190,191 actin,192 glucocorticoid receptor,193,194 and estrogen receptor α.42,195 It is notable that iAsIII binds Kelch-like ECH-associated protein 1 (Keap1), an antioxidant sensing protein, at low Kd values.134 Actin-tethered Keap1 that binds to nuclear factor erythroid 2-related factor 2 (Nrf2) constitutes a protein triad that is indispensable for induction of the phase II response, a major cellular reaction to oxidative/electrophile stress. Nrf2 is involved in inducing phase II and antioxidative proteins and regulates both the basal and inducible expressions of many cytoprotective genes through a common antioxidant response element (ARE). Reaction of oxidative/electrophile inducers, such as iAsIII, with Keap1 releases Nrf2 for nuclear translocation and activation of ARE.196 The primary sensors for these inducers are the reactive cysteine thiols of Keap1.
Two cysteine residues are frequently found separated by two other amino acid residues (CXXC), a motif known to be present in a variety of metal-binding proteins and oxidoreductases.197 There is also a positive correlation between the occurrence of cysteine in proteins and the complexity of organisms,197 indicating that evolution takes advantage of increased use of cysteine residues. In humans, several hundred good binding sites for trivalent arsenicals were proposed to be present in each organ.42,198
3. Determination of Arsenic Species Bound to Proteins
Arsenic can exist in different oxidation states (commonly +V, +III, +I, 0, and −III), depending on the redox status and pH of the environment and biological activities. More than 50 arsenic species have been identified in the natural environment and biological systems.199 It has been recognized that environmental behavior, physical and chemical properties, toxicity, mobility, and biotransformation differ enormously between individual arsenic species.74 For example, Figure 9 shows cell viability as a function of the concentration of arsenic in the form of six arsenic species.200 The relative cytotoxicity of these six arsenic species varies by up to 5 orders of magnitude. Therefore, it is essential to identify and quantify individual arsenic species.
Figure 9.
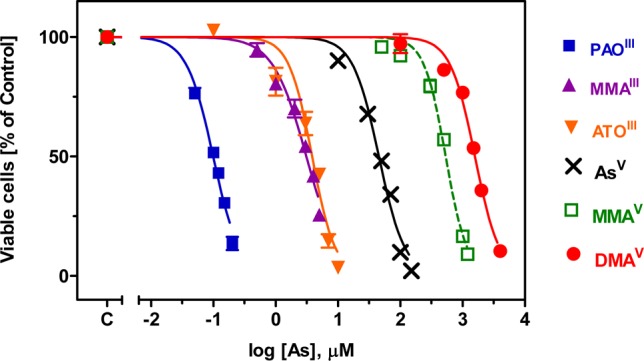
Toxicity of six arsenic compounds to HL-60 cells after a 48-h incubation. (Adapted from ref (200).)
3.1. Fractionation of Protein-Bound Arsenic
Dialysis, ultrafiltration, gel filtration chromatography, and protein precipitation have been employed to differentiate the protein-bound arsenic from the free arsenic species. Dialysis and ultrafiltration are both membrane-based techniques, aimed at separating high molecular weight molecules from small molecules. The molecular weight cutoff of the membranes is typically 10 or 25 kDa.201−206 After ultrafiltration or dialysis, the protein fraction is retained for analysis. Compared to ultrafiltration, which can be accelerated by centrifugation or pressurization, dialysis usually takes longer to complete and requires frequent change of the buffer outside of the dialysis sac. Moreover, dialysis usually results in sample dilution, and thus, subsequent sample concentration or redissolution after freeze-drying is sometimes needed to enable detection of trace amounts of arsenic.
Ultrafiltration is a convenient technique to evaluate the binding percentage when radiolabeled arsenic is used in an assay system, because the radioactivity retained in the ultrafiltration apparatus after washing can be taken as a measure of the extent of radiolabeled arsenic binding to proteins and expressed as a percentage of the total radionuclide in the assay system.207 It should be pointed out that washing residues and repeated ultrafiltration can remove free arsenic more efficiently, but this could result in a loss of arsenic-protein binding.166 Conceivably, centrifugation and filtration could be applied to studying arsenic binding to nonsoluble proteins, which would not be amenable to chromatographic methods of analysis.
Gel filtration chromatography enables separation of the unbound arsenic species from the protein–arsenic complexes. Absorbance at 280 nm is typically used for protein detection, while arsenic species are detected by either atomic spectrometry or radiochemical methods. Peaks appearing at the same elution times on both protein and arsenic detectors are assigned to the arsenic–protein complexes, although orthogonal separation techniques, each relying on a different separating mechanism, such as molecular size or charge, may be needed to avoid coelution and erroneous peak assignment.208,209 Gel filtration chromatography can be used to fractionate arsenic-binding proteins, giving additional information on the molecular weight range of the proteins. Both denaturing and nondenaturing polyacrylamide gel electrophoresis (PAGE) methods have been used as alternatives to determine the molecular weights of arsenic-binding proteins.201,210,211
Although generally regarded as a crude fractionation method, ammonium sulfate fractionation can also be used to maximize fractionation efficiency and partially purify arsenic-binding proteins prior to gel electrophoresis or gel filtration chromatography.211 The binding between radiolabeled arsenite and rabbit liver cytosolic proteins was examined, both in vitro and in vivo, by combining ammonium sulfate fractionation and gel electrophoresis. Protein precipitation is not only an effective way of separating proteins from contaminating small molecules but also serves as a preconcentration step for proteins from dilute solutions. Trichloroacetic acid (TCA) and acetone210,212−218 are commonly used to precipitate arsenic-bound proteins. Similar to nonspecific retention of arsenic on filter membranes during ultrafiltration,202,207,219,220 nonspecific adsorption of arsenic to sample tubes was also acknowledged in an in vitro binding study with radiolabeled arsenic219 and needs to be accounted for when determining the arsenic content in the protein pellet.211 Furthermore, the precipitating agents, such as 10% TFA, could affect the stability of arsenic–protein complexes.210,221
3.2. Release of Arsenic Species from Proteins
Treatment with thiol-reactive agents or chelators, such as N-ethylmaleimide, 5,5′-dithio-bis(2-nitrobenzoic acid), β-mercaptoethanol, dithiothreitol, and 2,3-dimercaptopropanol, failed to quantitatively release most protein-bound arsenicals.220 Because arsenic–protein complexes are sensitive to NaOH treatment,213 alkaline conditions have been explored to release arsenic from proteins. However, interconversion of iAsIII to iAsV was observed, during the treatment with a strong base, tetramethylammonium hydroxide (TMAH).222 Likewise, oxidation with concentrated H2O2 at room temperature was able to release all the protein-bound arsenic species as free pentavalent arsenicals.223−225
Acidic conditions have also been explored to release arsenic from proteins,213,226 although the use of TCA or HCl alone could not quantitatively release protein-bound arsenicals.213,226 By including CuCl in HCl (pH 1), Styblo et al.220 have developed a method of liberating protein-bound arsenicals. CuCl was originally used by Reinke et al.227 to reduce iAsV to iAsIII, which was subsequently extracted and analyzed polarographically for the determination of iAsIII and iAsV in fish and shellfish. This method was later modified202 by increasing the treatment temperature from 37 to 100 °C to shorten the treatment time. The reaction mechanism of how CuCl releases arsenic from proteins has not been investigated. It is possible that complexation of the cuprous ion with proteins is responsible for displacing arsenic from the cysteine residues in the proteins. Complexation of the cuprous ion with proteins is the basis for protein determination by the bicinchoninic acid (BCA) and Lowry methods.
A systematic study226 was conducted to examine release of arsenic species from liver proteins by four different acids (2 M HCl, 20% TCA, 2 M H2SO4, and 2 M HNO3), alone or in combination with heat treatment. The releasing capability of the four acids was in the descending order of HNO3 > H2SO4 > TCA > HCl. HNO3 (2 M) and 1 min heating at 110 °C proved to quantitatively (>99%) release all arsenic from the liver protein fraction. Similar to alkaline treatments, acidic treatments can change the oxidation status of arsenic species. For example, MMAIII and DMAIII were oxidized to their pentavalent counterparts upon TCA treatment, and iAsV was partially reduced to iAsIII upon TFA treatment.226 Although TFA at 4 °C did not influence the chemical form of DMAV and TMAO, a hot 2 M HNO3 treatment resulted in detectable loss of methyl groups from MMAV and DMAV. In the presence of proteins (rabbit liver cytosol), partial methylation of MMAV to DMAV was also observed after the 2 M HNO3 treatment.226 These observations suggest the importance of recognizing the integrity and artifacts of arsenic species during sample treatment.
Lu et al.228 have recently developed a method based on proteolytic digestion to release protein-bound arsenicals, aimed at preserving both the original methylation and oxidation status of the bound arsenic. Under mild alkaline conditions (pH 8.0), no oxidation of iAsIII and MMAIII was observed and up to 61% DMAIII (the least stable arsenic species) remained unoxidized.228 The recovery ranged from 93% to 106% with a digestion time as short as 30 min. Thus, the method not only quantitatively releases protein-bound arsenicals but also minimizes species conversion (at least in the AsIII state). The recent identification of a pentavalent arsenic–peptide complex (the glutathione conjugated form of dimethylmonothioarsinic acid) in cabbage emphasizes the importance of preserving the oxidation state of protein-bound arsenicals.229 The protease digestion method can be expected to achieve this goal because of its mildness toward arsenicals. However, it remains to be seen if this method can be extended from model proteins to complex protein samples from diverse biological sources, where protease inhibitors can be present. Removal of protease inhibitors will be necessary in those cases prior to protease digestion.
3.3. Determination of Arsenic Species Released from Proteins
After release of arsenicals from proteins, the arsenicals can be determined using a range of speciation techniques, as discussed in several reviews.230−233 The most commonly used techniques for speciation of arsenic in solution involve a combination of high-performance liquid chromatography (HPLC) separation with inductively coupled plasma mass spectrometry (ICP-MS) and/or electrospray ionization mass spectrometry (ESI-MS) detection.234−238
3.4. Direct Analysis of Arsenic Species by X-ray Spectroscopy
Direct speciation of arsenic protein fractions without releasing arsenic can be achieved by using X-ray absorption spectroscopy,239 especially EXAFS (extended X-ray absorption fine structure) and XANES (X-ray absorption near edge spectroscopy). These techniques can reveal the electronic environment around the arsenic atom and therefore provide information as to whether arsenic is trivalent or pentavalent and whether arsenic is bound to sulfur, oxygen, or carbon.240−247 Therefore, XAS is invaluable for probing the kinetically labile arsenic–protein complexes that are prone to dissociation or ligand exchange during sample handling.235 Complete identification of arsenic species is best achieved by complementing XAS information with structural information obtained by using other techniques.
4. Identification of Arsenic-Binding Proteins
4.1. Autoradiography
Radioactive isotopes of 73As and 74As (73As has a longer half-life and lower γ-emission) have been used, although organic arsenic compounds have labeling options with radioisotopic carbon.202,248 Whole-body autoradiography of mice after intravenous injection of 73As compounds provided information on the tissue distribution of arsenic.170,249 The studies on arsenic distribution and binding involving the use of radiolabeled arsenic compounds in animals and humans have been thoroughly reviewed.250 In general, keratin-rich tissues (such as the hair, nails, and skin) with high cysteine content exhibit the highest concentrations and longest retention. At the cellular level, arsenic-protein binding occurs mainly in the cytosol, most notably in liver and kidney. Greater retention of arsenic was observed with arsenite compared to arsenate, and differences in arsenic accumulation were noted in the liver, kidney, and skin. In the bloodstream, arsenic is distributed between erythrocytes and plasma and binds to proteins in both compartments.
Although the radiotracer can be monitored and its activity measured typically with a scintillation detector equipped with a thallium-activated sodium iodide crystal [NaI(Tl)],251 identification of arsenic-binding proteins requires more complementary techniques for separation and characterization. Since the heme moiety strongly absorbs light at 420 nm, simultaneous detection of both 74As and the heme moiety after size exclusion chromatography (SEC) revealed that arsenic was bound to hemoglobin in packed blood cells of rabbits intraperitoneally injected with 74As-labeled arsenate.204 This binding was also observed in the red blood cells of rats, as SEC showed that the 74As was predominantly associated with a protein that had a molecular mass of ∼60 kDa and a strong absorption band at 420 nm.209 To study potential arsenic binding partners in serum, cation and anion exchange separation schemes with fast protein liquid chromatrography were applied to serum that had been incubated in vitro with 74As-labeled arsenate. The protein fractions so isolated were further separated by isoelectric focusing and identified as asialotransferrin, sialotransferrin, and albumin.209
Other useful radiolabeled arsenic species include phenylarsine oxide derivatives. N-[3H]acetyl-4-aminophenylarsine oxide and 4-[125I]iodophenylarsine oxide were synthesized and employed as covalent affinity reagents to study arsenic–protein interaction.190
4.2. Fluorescent Arsenical Probes
Wang et al.252 identified two naturally occurring tetracysteine-containing sequences that bind to FlAsH [4,5-bis(1,3,2-dithioarsolan-2-yl)fluorescein] by selecting against the Shewanella oneidensis and E. coli proteomes. SlyD, a metallochaperone in the assembly of [Ni–Fe] clusters in the hydrogenase biosynthetic pathway, was identified as a high-affinity biarsenical binding protein in these two prokaryotic organisms. Fluorescent biarsenical affinity probes FlAsH and its many analogues, such as ReAsH (see structures in Table S1 of Supporting Information), were initially developed by Tsien and co-workers253,254 to selectively label a range of proteins in both eukaryotic and bacterial organisms fused to an engineered tetracysteine binding sequence, CysCysXXCysCys, where the central XX is preferentially ProGly. The biarsenical dyes undergo a dramatic enhancement of fluorescence upon binding to the proteins. Because endogenous proteins that would bind FlAsH are rare, fluorescent biarsenical probes permit the easy detection of the peptide-tagged proteins over the background. These fluorescent biarsenicals could also be used to detect endogenous cysteine-rich proteins.66,255,256
More recently, a naphthalimide-based fluorescent monoarsenical probe, NPE (see the structure in Table S1 of Supporting Information), has been developed by Huang et al.257 to detect and image vicinal dithiol proteins both in vitro and in living cells.
4.3. Affinity Chromatography
4.3.1. Protein-Specific Immunological Affinity Chromatography
Plasma from rabbits intraperitoneally injected with 74As-labeled arsenate was analyzed by transferrin-specific affinity chromatography by using an N-hydroxysuccinimide antirabbit transferrin HiTrap column.204 Up to 18% of total plasma 74As was found to bind to transferrin. Similarly, a transferrin-specific affinity column was used to further confirm the identity of transferrin in serum of patients on continuous ambulatory peritoneal dialysis.166 Transferrin is capable of binding to a wide range of synergistic anions at its iron binding site, and formation of an iron–arsenate–transferrin complex is presumably responsible for arsenate–transferrin binding.204
4.3.2. Arsenical Affinity Chromatography
Chromatography using trivalent arsenicals as affinity ligands was first devised by Hannestad et al.258 to retain and separate mono- and dithiols, making use of interactions between the thiols and trivalent arsenicals. The affinity adsorbent was prepared by directly binding p-aminophenylarsine oxide to CNBr-activated Sepharose 6B. The arsenic affinity chromatography was later modified by several research groups to study covalent arsenical–protein interaction or to purify proteins. In their investigation into the complex effects of the trivalent arsenical PAO on insulin-dependent hexose uptake in 3T3-L1 adipocytes, Hoffman and Lane190 prepared affinity resins by covalently coupling agarose, derivatized with a 10-atom spacer arm having a terminal hydroxysuccinimide active ester, to 4-aminophenyldithioarsine of β-mercaptoethanol (β-ME). The captured proteins were eluted off from this arsenic affinity column using β-mercaptoethanol in 1% Triton X-100 or SDS, further separated on SDS–PAGE, and detected by selective staining with 4-[125I]iodophenylarsine oxide. Several proteins from 3T3-L1 adipocytes were identified to be able to bind arsenic, including the insulin-responsive glucose transporter GLUT4 and tubulin. Zhou et al.259 covalently linked 4-aminophenylarsine oxide to carboxymethyl (CM)-Bio-Gel A activated through an N-hydroxysuccinimide ester. They used this column to purify human plasma lecithin:cholesterol acyltransferase (LCAT). LCAT is a dithiol enzyme that has two reduced cysteine residues (Cys31 and Cys184) within its catalytic site. Binding of these cysteines to the arsenical affinity column enabled the capture and subsequent elution (with a buffer containing 2,3-dimercaptopropanesulfonic acid), affording a resultant 13-fold increase in specific activity of the enzyme and an overall yield of 11%. Kalef et al.260 prepared affinity resins based on Sepharose 4B linked with aminohexanoyl-4-aminophenylarsine oxide to purify proteins containing vicinal dithiols from L1210 murine leukemia lymphoblasts. A one-step purification of the l-triiodothyronine recombinant rat c-erb Aβ1 T3 receptor synthesized in yeast was achieved with this affinity chromatography, resulting in a 62-fold increase in specific activity and a yield of 11%. CNBr-activated Sepharose 4B was derivatized with 6-aminohexanoic acid to constitute a spacer arm prior to coupling with freshly prepared 4-aminophenylarsine oxide (high solubility in DMSO) via carbodiimide. Elution was performed with β-mercaptoethanol or dithiothreitol (DTT) in buffers. The dithiol proteins were identified by specific labeling with N-iodoacetyl-3-[125I]-iodotyrosine ([125I]IAIT). A different method of preparing PAO-Sepharose 4B was reported by Berleth et al.261 to partially purify ubiquitin-protein ligase (E3). This method was subsequently adopted by Boerman and Napoli262 to partially purify a microsomal retinol dehydrogenase (RoDH).
Arsenic affinity chromatography was also used by Winski and Carter263 to characterize arsenic binding proteins in rat red blood cells. Proteins retained on the column were displaced by sequential washes with cysteine, glutathione, and dimercaptopropanesulfonate (DMPS) after removing nonspecific binding materials with buffer and ethylene glycol. Hemoglobin seemed to be the main arsenic-binding protein in the rat red blood cell, which was captured on the arsenic affinity column and eluted with 100 mM glutathione. Using the same arsenic affinity chromatographic strategy, Menzel et al.192 identified two arsenic-binding proteins to be tubulin and actin on the basis of their molecular weights. A highly arsenite-inducible protein, heme oxygenase 1 (HO1), failed to bind to the arsenic affinity column because it is devoid of cysteines. Chang et al.92 used commercial PAO-agarose resins to isolate AsIII-modulated proteins from Chinese hamster ovary (CHO) and SA7 (arsenic-resistant CHO) cells. Following washes with 2 M NaCl in Tris buffer to remove the nonspecific binding proteins, the specific binding proteins were sequentially eluted with 20 mM β-ME and 20 mM DTT. Galectin 1 (Gal-1; in the β-ME-eluted fraction from CHO cells), glutathione S-transferase P-form (GST-P), and thioredoxin peroxidase II (TPX-II) were identified by partial amino acid sequence analysis. TPX-II was preferentially expressed in SA7 cells (which were routinely cultured and maintained in AsIII-containing medium), but not in CHO or SA7N (a revertent of SA7 cells cultured in regular medium). In contrast, Gal-1 was specifically identified in CHO and SA7N cells, but not in SA7 cells. The specific binding of Gal-1 and TPX-II to AsIII was verified by both coimmunoprecipitation from mammalian cells treated with AsIII and coelution of recombinant Gal-1 and TPX-II with AsIII from arsenic-treated transfected E. coli.
Mizumura et al.264 prepared three different arsenic-bound Sepharose resins (AsV-immoblized Sepharose, type A; AsIII-diglutathione-immobilized Sepharose, type B; and AsIII-immobilized Sepharose, type C) to investigate the binding of hepatic cytosolic proteins to pentavalent, glutathione-conjugated, and trivalent arsenicals. They synthesized AsV-immoblized Sepharose by conjugating NHS-activated Sepharose 4 Fast Flow gel with p-arsanilic acid. This gel was further converted to AsIII-diglutathione-immobilized Sepharose gel after reduction with GSH, which could be stabilized in the presence of GSH. The AsIII-diglutathione-immobilized Sepharose gel was hydrolyzed to produce AsIII-immobilized Sepharose gel in the absence of GSH. Heptatic cytosolic proteins bound to each Sepharose were eluted with GSH, DTT, or LDS (lithium dodecyl sulfate) sample buffer. SDS–PAGE followed by EZBlue staining showed no proteins bound to pentavalent arsenic. MALDI-MS/MS was able to identify the binding of protein disulfide isomerase-related protein 5 (PDSIRP5) and peroxiredoxin 1/enhancer protein (PRX1/EP) to trivalent arsenic. Four proteins, peroxiredoxin 2, cytosolic pyrophosphatase, phosphoglycerate kinase 1, and KM-102-derived reductase-like factor (equivalent to thioredoxin reductase), were identified to specifically bind to glutathione-conjugated trivalent arsenic, possibly involving substitution of a cysteine residue of the proteins for glutathione at the As-SG site. He and Ma130 conjugated 4-amino-phenylarsine oxide to Affigel to make PAO affinity beads, which were used to probe binding of arsenic to Kelch-like ECH-associated protein 1 (Keap1), nuclear factor erythroid 2-related factor 2 (Nrf2), and the carboxyl half of metal-activated transcription factor 1 (MTF1). The PAO affinity materials have also been used to isolate some other enzymes, such as adenine nucleotide translocase (ANT) and protein tyrosine phosphatases (PTP), from cell lysates.188,265 In all these identified proteins, multiple cysteine residues are present for arsenic binding.
Reversible oxidation on proteins of vicinal thiols to form disulfides is believed to be an important means of redox-based regulation of cell signaling, metabolism, and gene expression. Gitler et al.266 introduced a general method to identify and enrich vicinal thiol proteins (VTPs) present in intact cells in the oxidized, disulfide state. They first blocked unoxidized thiols in situ by incubation of murine leukemia L1210 cells with cell permeable N-ethylmaleimide (NEM) and then reduced oxidized thiols with DTT. These proteins containing reduced thiols could be enriched by using PAO affinity chromatography. It was a challenge to the use of PAO affinity chromatography for the isolation of readily oxidizable VTPs because it required a delicate balance between maintaining the thiols in the reduced state necessary for PAO binding and the possible competition of DTT for the binding. Conceivably, the use of DTT could have led to an underestimation of the proteins able to bind PAO in the previous studies. By substituting tris(2-carboxyethyl)phosphine (TCEP), a nonthiol reducing agent, for DTT, Foley et al.267 improved the capture of VTPs from a Triton X-100-soluble rat brain extract by using PAO-affinity chromatography. After unoxidized thiols were alkylated with iodoacetamide (IAM), the oxidized thiols in proteins were reduced with TCEP prior to loading the protein sample onto a PAO affinity column. The eluted fractions were analyzed by SDS–PAGE, and protein bands were subjected to in-gel tryptic digestion and MALDI-MS/MS analysis for protein identification. The two most abundant proteins were identified as albumin and triose phosphate isomerase (TPI). The catalytic subunit of protein phosphatase 2A (PP2Ac) from rat brain was also revealed, by the improved method, to contain reversibly oxidized thiols.268 Similarly, protein phosphatase 2B (Calcineurin, CaN) and thioredoxin were isolated and identified from a cytosolic fraction of polymorphonuclear leukocytes (PMN) as involved in redox regulation, along with four other proteins (glutathione S-transferase P1-1, calgranulin, l-plastin, and cofilin).269
Our group270 has developed a targeted proteomic approach to identify arsenic-binding proteins in human cells involving arsenical affinity chromatography. The affinity resins were synthesized by reaction of Eupergit C beads with 4-aminophenylarsine oxide (NAPOIII). After being eluted with increasing levels of DTT in buffer with or without 1% SDS, the bound proteins were subjected to in-gel or in-solution digestion prior to LC–ESI-MS/MS analysis. This approach demonstrated that arsenic-binding proteins could be identified in the presence of a large excess of nonspecific proteins. Fifty proteins in the nuclear fraction and 24 proteins in the membrane/organelle fraction were identified in the A549 human lung carcinoma cell line.
4.4. Biotinylated Arsenical Pull-Down
Zhang et al.271 designed an in-solution pull-down approach to identify arsenic-binding proteins in human breast cancer MCF-7 cells. An arsenic–biotin conjugate was synthesized by coupling the pentafluorophenol ester of biotin with p-aminophenylarsine oxide. Improved synthesis of arsenic–biotin conjugates in a one-pot procedure was reported.272 Arsenic-binding proteins were pulled down with streptavidin resins and subsequently released, separated by SDS–PAGE, and identified by MALDI-MS/MS. Analyses of MCF-7 cells resulted in tentative identification of 50 proteins. Two proteins, β-tubulin and pyruvate kinase M2, were confirmed further by Western blotting and molecular modeling. The same approach was applied to demonstrate that arsenic bound to PML and PML-RARα fusion protein, but not to RARα alone.66 This direct binding was shown to be important in the anticancer activity of As2O3 in patients with acute promyelocytic leukemia (APL).
Donoghue et al.273 devised a membrane-impermeable trivalent arsenical to identify closely spaced thiols in mammalian cell-surface proteins undergoing redox reactions. Because GSH is constitutively secreted by mammalian cells but is not taken up by these cells, an arsenic probe for the cell-surface proteins could be developed by conjugating GSH to arsenic. Aminophenylarsine oxide was therefore coupled to the thiol of GSH to produce 4-(N-(S-glutathionylacetyl)amino)-p-phenylarsine oxide (p-GSAO), which was shown to bind tightly to dithiols but not to monothiols. To identify cell-surface proteins that contain closely spaced thiols, they attached a biotin moiety through a spacer arm to the primary amino group of the γ-glutamyl residue of GSAO to afford GSAO-B. The biotin moiety was expected to prevent cleavage of the γ-glutamyl residue by membrane-bound γ-glutamyl transpeptidases.274 Cells were labeled with GSAO-B in the absence or presence of 2,3-dimercaptopropanol (DMP), lysed, and incubated with streptavidin-agarose beads to collect biotin-labeled proteins. The bound proteins were eluted with 50 mM DTT in buffer, resolved on SDS–PAGE, transferred to PVDF membranes, and blotted with specific antibodies against candidate proteins. Alternatively, after labeling with GSAO-B, proteins were resolved on SDS–PAGE, transferred to PVDF membranes, and blotted with streptavidin-peroxidase to detect the GSAO-B label. There were 10 distinct proteins on the surface of bovine aortic endothelial (BAE) cells and 12 on the surface of human fibrosarcoma (HT1080) cells that incorporated GSAO-B. Protein disulfide isomerase (PDI) was labeled with GSAO-B on the cell surfaces of both cell types. The pattern of labeling of the cell lysate with GSAO-B was very different from the pattern of labeling of the cell surface proteins, reflecting the membrane-impermeable nature of GSAO-B. The interaction of GSAO-B with PDI and thioredoxin was also demonstrated in vitro with purified proteins. The adenine nucleotide translocase (ANT) was captured by GSAO-B, from isolated rat liver mitochondria, when the arsenical moiety of GSAO was either at the ortho (o-GSAO-B) or para (p-GSAO-B) position on the phenyl ring.275 The GSAO-B-bound proteins were then pulled down by streptavidin- or avidin-coated beads and eluted with DTT. By a similar strategy, GSAO-B has been demonstrated to be retained in the cytosol predominantly by covalent reaction with a dithiol in the 90 kDa heat shock protein (Hsp90), followed by eukaryotic translation elongation factor 2 and filamin A.276 Apart from GASO-B, three novel biotinylated PAO compounds with spacers of varying length were also synthesized as bifunctional reagents to examine spatially close thiols in Torpedo nicotinic receptors.277
Aside from identifying arsenic binding proteins, arsenical probes and PAO affinity chromatography have been employed to confirm the interaction of arsenic and the proteins in question.66,134,196,278,279 They can further be used to ascertain the critical thiols for binding in proteins after site-directed mutagenesis.134,161,280
4.5. Metalloproteomic Approaches
General metalloproteomic approaches combining efficient separation of metalloproteins by liquid chromatography or electrophoresis with highly sensitive elemental detection can be applied to identification of arsenic-binding proteins in biological samples.201,281 Several excellent review papers have been dedicated to the analytical chemistry approaches in metalloproteomic or metallomic research.232,282−288 Proteomics approaches using mass spectrometry (with MALDI and ESI) remain the state-of-the-art for high-throughput protein identification.289,290
5. Characterization of Protein Binding to Arsenic Species
Although numerous cysteine-containing proteins that can bind arsenic have been identified, not many arsenic-binding proteins have been thoroughly characterized in terms of their binding sites, binding stoichiometry, and binding affinity. Part of the reason for this paucity is that purified proteins are not readily available in sufficient amounts for structural determination and binding characterization. One early study that employed a fractional occupancy assay with a 73As tracer indicated that the average dissociation constant (Kd) for all proteins capable of binding arsenite in rabbit liver was 18.4 μM.211
5.1. Binding Sites
Cysteines and histidines are known to be most frequently involved in binding metals such as iron, zinc, and copper.291,292 The binding between arsenic and histidines, however, has never been unequivocally established. In an NMR spectroscopic study of complexation of arsenicals to rabbit hemoglobin,293 a direct interaction of AsIII with histidine was discounted because there was no NMR spectral modification observed to a buffered solution of histidine when AsIII was added. AsIII seemed to bind to Cys93 but not to other neighboring amino acids of Hb. Similarly, histidine tags in recombinant Trx and ArsD were shown not to bind iAsIII.108,164 Rosen and co-workers162,294−296 frequently substituted serine residues for cysteine residues in their site-directed mutagenesis studies to define the role of the cysteine residues in arsenic binding, on the premise that the interaction of serine with arsenite is weak. So far, the only binding ligand of arsenic identified in the ArsRDABC operon proteins is cysteine thiolates. In a recent examination of amino acid constituents that can potentially bind arsenic, Kitchin and Wallace42 showed that amino acids other than cysteine in their synthetic peptides did not bind arsenite at all. Adequate cysteine content and vicinal cysteine residues were shown to be required for high-affinity arsenite binding with the peptides.42,43 Arsenite was shown to preferentially bind zinc finger peptides containing three or four cysteine residues and interact selectively with zinc finger proteins containing a C3H1 or C4 but not with a C2H2 motif in releasing zinc and reducing DNA-binding capacity of the proteins.45 In sum, cysteine (and selenocysteine as in selenoproteins, considering its lower pKa and therefore increased nucleophilicity297) is the only naturally occurring amino acid that has been demonstrated to be responsible for trivalent arsenic–protein binding. Other amino acid residues in close proximity to cysteine may serve as proton acceptors and enhance thiol reactivity toward arsenic.298
Li and Pickart94 demonstrated that vicinal thiols were not involved in the binding of phenylarsine oxide to Arg-tRNA protein transferase. They proposed that structural features other than cysteines and/or serines were responsible for the binding. Arg-tRNA protein transferase is an enzyme containing 15 cysteine residues, and multiple cysteine residues are essential for its activity. Despite using site-directed mutagenesis to mutate specific cysteine residues, it was challenging to rule out the role in the arsenic binding of any remaining cysteine residues that could be brought into spatial proximity, even in the presence of urea because “unfolded proteins are conformationally mobile and structurally accommodating”.299 The final possibility of “redundancy in the abilities of cysteine residues of the transferase to bind arsine oxide” could not be excluded but was dismissed as unlikely by the authors.94 However AsIII has lately been demonstrated to be able to distort a polypeptide structure so that AsIII can capture thiols in the reorganized structure in order to satisfy its desire for thiolate coordination.299−301 Therefore, it is possible that the inhibition of Arg-tRNA protein transferase occurs due to conformational distortion of the enzyme induced by arsenic cross-linking of thiol groups.
Besides sulfhydryl groups, hydroxyl groups in small molecules were demonstrated to have some potential to form a ring structure with phenylarsine oxide in the gas phase.258 In a crystal structure of the arsenite–dithiothreitol complex, AsIII bound with two sulfur thiolates and one hydroxyl oxygen.302 Therefore, in proteins, hydroxyl-containing serine appears to be another possible arsenic binding ligand. In support of this possibility, several studies303−305 have implicated the role of the active site serine bound by arsenic in the inhibition of enzymes, including those containing no cysteine. In these studies the enzymes were all serine hydrolases and the arsenicals interacting with the active site serine were all pentavalent. The only exception is cholinesterases, of which arsenite but not arsenate is a potent inhibitor.306,307 A tripartite oxyanion hole in proximity to the active site is usually required to stabilize the transition state complex between pentavalent arsenic and the catalytic serine.305
5.1.1. X-ray Crystallography and Nuclear Magnetic Resonance (NMR) Spectroscopy
X-ray crystallography and NMR spectroscopy are the two primary techniques employed to determine three-dimensional structures of the metal binding sites of proteins. The first example of covalent modification of serine by pentavalent arsenic was observed with X-ray crystallography.304 In the study, the crystal structures of the acetyl esterase HerE and its complexes with an inhibitor, dimethylarsinic acid, were determined and a covalent bond via the Oγ of Ser160 seemed to be formed between the enzyme and arsenic. For this experiment, HerE was crystallized in the presence of 100 mM cacodylate (dimethylarsinic acid) without DTT. This was different from the DTT-mediated reaction of cysteine with the arsenic center of the cacodylate present in the crystallization buffer (usually containing100 mM cacodylate and 5 mM DTT) observed with several proteins. In the latter case, the presence of DTT results in the reduction of cacodylate by DTT and subsequent reaction with exposed cysteine residues.308−312 X-ray crystallography was also employed by Rosen and co-workers to study the mechanism of arsenic binding in arsenate reductase ArsC.165 The crystal structures of ArsC and its complexes with arsenate (substrate) and arsenite (product) were determined. Arsenate forms a thioester with the catalytic residue Cys12. Arsenite also binds the Cys12 thiol to form an unusual bicoordinate thiarsahydroxyl intermediate ArsC-Cys12-As+-OH, which is essential to prevent the suicide inhibition of the enzyme by its reaction product (arsenite). Rosen and co-workers have also recently obtained the X-ray crystallographic and NMR information313,314 about the ArsD arsenic metallochaperone in attempts to examine the structural changes in the protein in response to metalloid binding and upon interaction with ArsA. Molecular modeling was additionally performed to probe the metal binding sites of ArsD to incorporate induced changes upon arsenic binding since the apoprotein (without bound arsenic) was used in their structural determinations. Conserved cysteine residues Cys12, Cys13, and Cys18 form a tricoordinate binding site for AsIII, which corroborated their previous determination of AsIII binding sites on this protein by using site-directed mutagenesis combined with arsenic analysis.315 The coordination of AsIII with three sulfur atoms in ArsD was further demonstrated by X-ray absorption spectroscopy.164
Compared with X-ray crystallography, NMR generally offers a lower-resolution structure, but it does not require crystallization. After transmetalation to CdII, which served as an NMR spectroscopic probe, the ZnII enzyme alkaline phosphatase was studied on its binding with arsenate.316 Arsenate, an enzyme inhibitor, bridged Ser102 and zinc-binding sites. This binding geometry was confirmed by a subsequent X-ray crystallographic study of alkaline phosphatase.317
5.1.2. Molecular Modeling and Site-Directed Mutagenesis
Molecular modeling, a term for computational protein structure prediction, can contribute to the protein determination process and deal with large proteins that are impossible with either X-ray crystallography or NMR.318,319 Many review articles and monographs318,320,321 have been dedicated to the topic of molecular modeling, and it has been used to determine the arsenic binding site in the example of ArsD mentioned above.313
The site-directed mutagenesis approach is generally applicable to identify the roles of specific residues in ligand binding, specificity, and catalysis, which in turn is guided by knowledge of the 3-D structures of proteins. The importance of site-directed mutagenesis and subsequent quantitative kinetic analysis of mutant enzymes has been highlighted in structural biology in the case of adenylate kinase, where a discrepancy between the X-ray model and the NMR model existed.322 Site-directed mutagenesis has been intensively used by Rosen and co-workers to identify arsenic binding sites of Ars operon proteins. For instance, a combination of site-directed mutagenesis and gel permeation chromatography analysis revealed that ArsA had a high-affinity metalloid binding site composed of Cys113 and Cys422.323 The 2.3 Å crystal structure of ArsA crystallized in the presence of SbIII and MgADP showed that ArsA had two other bound metalloid atoms, one bound to Cys172 and His453 and the other bound to His148 and Ser420. Further site-directed mutagenesis analysis revealed that His418 and Ser420 had little effect on metalloid binding while the C172A single mutant and C172A/H453A double mutant exhibited significantly decreased affinity for SbIII, suggesting that C172 is the third residue that participates in high-affinity binding.296 A site-directed mutagenic approach was also employed in search for a third arsenic ligand in ArsR, Cys37, which forms a tricoordinate complex with AsIII together with highly conserved Cys32 and Cys34.161 The As-S coordination environment in ArsR was confirmed by X-ray absorption spectroscopy of ArsR stoichiometrically treated with arsenite. Cys95, Cys96, and Cys102, the only three cysteine residues in an ArsR family protein in Acidithiobacillus ferrooxidans, were also identified to form a tricoordinate AsIII binding site.295
5.1.3. Electrospray Ionization Mass Spectrometry (ESI-MS)
The development of soft ionization methods (MALDI and ESI) for mass spectrometry has facilitated the study of metal ion binding to macromolecules, including proteins. With ESI-MS, noncovalent protein–metal interactions in solution can be maintained during desolvation and transfer to the gas phase. Therefore, solution-phase complex formation can be detected and monitored by mass spectrometry in simple “mix-and-measure” experiments, which makes ESI-MS a more natural choice than MALDI-MS.324 Binding stoichiometries can be determined directly from the measured mass. In favorable cases binding affinities and binding kinetics may be obtained from the measured intensity ratios of the free and bound protein ions.324 Structural information such as binding sites may also be obtained using tandem MS (MS/MS) or top-down strategies. In combination with chemical modifications, the capability of binding site determination by ESI-MS can be extended.325 As further evidence that trivalent arsenicals have a strong preference to bind to cysteine residues in proteins, ESI-MS studies revealed that trivalent arsenicals failed to bind purified hemoglobin and thioredoxin after reactive cysteine sulfhydryl groups were blocked with N-ethylmaleimide (NEM) or monobromobimane.108,169
ESI-MS has enabled identification of a preferential binding cysteine in rat hemoglobin (rHb).60 In vitro binding of trivalent arsenicals to hemoglobin generated the corresponding arsenic–hemoglobin complexes with all possible stoichiometries depending on the relative concentrations of arsenicals and hemoglobin.169 In vivo, however, a DMAIII–rHb complex was exclusively observed on the α-chain with a stoichiometry of 1:1 in rats exposed to iAsV, MMAV, or DMAV. A mass shift of 104 Da was indicateive of the binding of one DMAIII [(CH3)2AsOH, FW 122] to the α-unit with the loss of one H2O molecule. No complexes of iAsIII or MMAIII with rHb were detected from any of the treated rats. It has been known that in the rat iAsIII and MMAIII can be excreted into the bile in the form of As(GS)3 and MeAs(GS)2, respectively, while DMAIII is released into the bloodstream and efficiently taken up by red blood cells.326,327 The identity of DMAIII bound to rHb was confirmed by HPLC analysis of arsenic species released from the protein: only DMAIII and its oxidation product DMAV were observed.60 Collision-induced dissociation (CID) tandem mass spectrometry172 revealed two characteristic fragment ions associated with a DMAIII-tagged cysteine residue that were equally observable from DMAIII complexes with cysteine and glutathione. From measuring the accurate masses of the DMAIII-tagged internal ions (di-, tri-, and tetrapeptides) and comparing with the theoretical masses for peptides encompassing the three cysteine residues (Cys13, Cys104, and Cys111) in the rHb α-unit, Cys13 was determined to be the only binding site of DMAIII. The observation of exclusively DMAIII-tagged fragment ions at Cys13 was not due to differential production by CID fragmentation, because the internal ions from all three cysteine residues could be detected from rHbα–(DMAIII)3 that was formed by incubating rHb with a 100-fold molar excess of DMAIII. The unbiased fragmentation was further confirmed by CID MS/MS analysis of DMAIII complexes with three synthetic nonapeptides embedding each of the cysteine residues in the α-chain.172 The relative reactivity of cysteine residues in rHb α-chain was determined to be in the order of Cys13 ≫ Cys111 > Cys104. The preferential binding of Cys13 to DMAIII is probably because Cys13 in rat Hb is located in an open hydrophobic pocket that provides easy access and a favorable binding microenvironment for moderately hydrophobic DMAIII.60 In the β-chain, Cys125 is more reactive than Cys93, and glutathione preferentially binds Cys125.
5.2. Binding Stoichiometry
In order to obtain information on metal-binding sites from X-ray spectroscopy, it is necessary to have access to well-defined structures of stoichiometric preparations of metal–protein complexes. For X-ray crystallography, sufficient amounts of the ligand have to be present to ensure the formation of a fully occupied stoichiometric complex obtained either by cocrystallization or by soaking the ligand into a fully grown crystal.328 Mass spectrometry offers a rapid and precise determination of metal–protein binding stoichiometry, providing integer numbers of bound metal ions per protein molecule, even when not all protein molecules are fully metal-loaded. Titration-based methods, isothermal titration calorimetry included, may yield noninteger numbers of metal ions (the average metal content) per protein molecule under similar circumstances.329 The binding stoichiometries of AsIII were shown by using either ESI-MS or MALDI-MS to be consistent with the number of available cysteines in proteins such as metallothionein (shown in Figures 4 and 5) and hemoglobin examined at saturating arsenic concentrations.149,152−155,169
5.3. Binding Affinity
The binding affinity (association constant, Ka; dissociation constant, Kd) of arsenic–protein/peptide binding can be determined by measuring changes in either the properties of the protein/peptide or in the partition of arsenic. Commonly adopted procedures to determine Kd include absorbance shift of protein/peptide or arsenical,254,330,331 intrinsic tryptophan fluorescence quenching,164,299,332 the reduction of thiol content due to arsenic binding,273 and the changes in the protein electrophoretic mobility measured by capillary electrophoresis.97 Alternatively, partition of arsenic in free and protein/peptide-bound forms after reaching binding equilibrium can be quantified to determine the binding affinity after separating the two forms by using size exclusion liquid chromatography,108,169 dialysis,248,333−335 or filtration.43,198 Use of radioactive or fluorescently labeled arsenic compounds offers a fast and convenient determination of arsenic-protein binding affinity.254,336 As a technique that directly measures the heat released or absorbed during a biomolecular binding event, isothermal titration calorimetry (ITC) can simultaneously determine several binding parameters (binding stoichiometry, binding constant, binding enthalpy, and entropy) in one experiment and has been used to study arsenic–protein binding.337 Information readily obtained from ITC, such as formation of multiple titration species fitting a sequential-binding model, was proven to be useful for data modeling, distinguishing this technique from other titration methods. For example, a stability constant (Ka = 1/Kd) of 2 × 106 was determined with ITC for the 1:3 arsenite complex with GSH,338 for which unusually high values of 2 × 1033 and 1 × 1032 were reported by Rey et al.339 on the basis of their near-UV absorption and potentiometric data, respectively. The binding affinities between arsenic and several thiol-containing biomolecules were also determined with ESI-MS by Schmidt and co-workers,212,340−342 but their values were inconsistent. One issue with quantifying both the unbound and bound species using ESI-MS is that these species may have different ESI-MS responses and their intensities may not be calibrated against a single species. Thus, ESI-MS should be used to determine relative binding affinities rather than absolute values.341,343,344Kd values obtained by MS should be used with caution in the absence of validation from other solution-phase techniques.343
The arsenic–protein binding affinity can also be determined indirectly in inhibition studies by measuring the inhibitory effect of arsenic on ligand binding or enzyme activity. When measured this way, the dissociation constant of binding of the inhibitor is termed the inhibition constant, Ki (equivalent to Kd). The inhibition of enzyme activity by arsenic compounds was shown to be either competitive or noncompetitive (mixed),345−356 demonstrating the ability of arsenic to bind to free enzymes.357 A majority of the binding constants for arsenic–protein binding were obtained indirectly. IC50, the half-maximal inhibitory concentration, which can often be related to Ki by the Cheng–Prusoff equation [Ki = IC50/(1 + [S]/Km)],358 is not a direct indicator of affinity and may vary with experimental conditions such as substrate concentrations. Therefore, IC50 values reported in the literature are not included in this review. When arsenic acts as the substrate for an enzyme, Km, the Michalis constant, can be determined using Michaelis–Menten kinetics. Km is the concentration of substrate at which enzyme activity is half-maximal. Its values include not only the affinity of the substrate for the enzyme but also the rate at which the enzyme-bound substrate is converted to the product in the catalytic reaction. Thus, Km can be interpreted as a crude measurement of the affinity of the substrate for the enzyme.359 A large Km indicates weak affinity. Km values reported in the literature for arsenic-containing compounds are included in Table S3 of the Supporting Information. A schematic comparison with statistical analysis is shown in Figure 10 on the Kd (or Ki) and Km values for trivalent and pentavalent arsenicals separately. Kd values of the binding between trivalent arsenicals and small peptides/thiols are included as well (see Supporting Information, Table S4). Strict comparison of the listed binding constants cannot always be made since different arsenic compounds, methods, and assay conditions have been used. For example, in the absence of tetrahedral oxyanions, such as sulfate, oxidative inactivation of ArsC (arsenate reductase) encoded by Staphylococcus aureus, a highly oxygen sensitive redox enzyme, was observed during the course of the enzymatic assay, leading to significantly lower Vmax and Km values.355,360
Figure 10.

A comparison of binding affinity of trivalent and pentavalent arsenicals to proteins.
Several interesting inferences can be made from the enzymatic studies showing Km or Ki values: (A) Reflective of product inhibition in enzymatic reactions, arsenite is a potent inhibitor of arsenate reductase, for which arsenate is the substrate. Similarly, MMAIII is an inhibitor of iAsIII methylation by arsenic methyltransferase from rabbit liver, suggesting a possibility that MMAIII is the enzymatic reaction product of arsenic methyltransferase from iAsIII.361,362 (B) Arsenic reductases (arsenate reductase and GSTO1-1) generally have higher Km values than arsenic methyltransferases (including AS3MT), indicating that the reduction step is rate-limiting for the overall process of arsenic metabolism. (C) Pentavalent arsenicals are substrates (showing Km) or inhibitors (showing Ki) of many enzymes, as shown in Table S3 (Supporting Information), probably as a result of their mimicking a tetrahedral phosphorus atom in substrate or product analogues. These enzymes include phosphorylases,363−368 phosphatases,335,348,369−375 phosphoenolpyruvate mutase,376 glycerol-3-phosphate dehydrogenase,377 adenylate kinase,378 enolase,379 ethanolamine-phosphate cytidylytransferase,380 triosephosphate isomerase,381−383 ornithine carbamoyltransferase,384,385 acetyl esterase,304 chymotrypsin,305,386 subtilisin,305,386 and angiotensin-converting enzyme.387 Ubiquitin, which has esterase, carbonic anhydrase, and phosphatase activities, was also inhibited by arsenate.388 The majority of the enzymes mentioned above fall into the category of serine hydrolases. Serine hydrolases form covalent intermediates with their substrate through the hydroxyl group of the active site serine, and the catalytic action can be nearly irreversibly inhibited by organophosphorus compounds, accounting for the notoriously toxic effects of organophosphorus compounds.389 The anomaly manifested by cholinesterases (acetylcholinesterase and butylrylcholinesterase) for which arsenite but not arsenate is an inhibitor, may be related to their unique structures compared to other serine hydrolases. They contain a deep and narrow active site groove with two separate ligand binding sites: an acylation site or A-site at the base of the groove and a peripheral anionic site or P-site at the groove mouth, with a strong electrostatic dipole aligned with the groove leading to its active site.390,391 Ligand binding to the P-site can result in steric blockade of the A-site and inhibit substrate hydrolysis.392 The anionic nature of the P-site393 allows entry of cationic but not anionic (e.g., arsenate) ligands to the active site groove. Inhibition of acetylcholinesterase by arsenite was shown to be pH-dependent,394 possibly reflecting the change with pH of the dissociated state of arsenious acid (neutral). Arsenobetaine (AsB) and arsenocholine (AsC) were shown to inhibit butyrylcholinesterase.395 (D) Arsenate is a potent inhibitor of alkaline or acid phosphatase (Ki < 1 mM) while arsenite inhibits the same enzyme with Ki ≥ 17 mM, indicating that reasonably low Ki (or Kd) values can differentiate specific from nonspecific binding between arsenic and proteins. However, it is difficult to establish a universal benchmark Ki (or Kd) below which a specific binding is defined because of different methods and assay conditions used across the studies. (E) With regard to possible arsenic–DNA interactions, the dissociation constant (Kd) of arsenite–DNA binding was determined spectroscopically to be around 8–90 μM396,397 and can be even lower in the case of arsenic binding to DNA aptamers.398 However, no specific binding of either arsenate or arsenite to DNA was observed by using vacuum filtration binding assays,198 and the nature of arsenic–DNA binding is noncovalent.399
5.4. Binding Kinetics
A few studies have reported rate constants (association rate constant, kon; dissociation rate constant, koff) for the binding between arsenicals and proteins/peptides (Table S5 in Supporting Information). Various kinetic methods can be employed for the determination of rate constants. Radioactive arsenic offers an extremely sensitive yet discontinuous assay,44 requiring off-line separation of bound from free arsenic, which needs large sample size, excessive analysis time, and time profile reconstruction. Assays based on UV–vis absorption or fluorescence spectroscopy are most convenient since the reaction can be monitored continuously and thus remain the major methods for studying the reaction kinetics between arsenic and proteins/peptides. The application of these assays requires optical changes accompanying the reaction. These changes can result from ligand to metal charge-transfer,330,400,401 use of chromophoric substrates,87,254,301,331,336,394 or intrinsic tryptophan quenching.299 Obviating the need for chromophoric probes, time-resovled ESI-MS has been shown to be a powerful tool for kinetic studies in online experiments.402,403 Capable of simultaneous monitoring of multiple intermediate species, ESI-MS affords accurate rate parameters of biochemical reactions and provides insights into reaction mechanism.403 Stillman and co-workers have acquired kon values and depicted mechanisms for the binding between arsenite and metallothioneins using time-resolved ESI-MS.152−155 Figure 11 shows an example of reaction kinetics between iAsIII and metallothionein. Current restriction(s) with this technique lie(s) in the incompatibility of the electrospray process with nonvolatile components, such as high concentrations of salts, that may be required in some biochemical reactions. Under such circumstances online microdialysis for rapid desalting of the reaction mixture immediately prior to ESI-MS analysis is necessary and can be achieved with an automated microfluidic system, without significantly compromising the time resolution.404
Figure 11.

Kinetic data showing the relative abundance of the various arsenic–metallothionein species detected using electrospray mass spectrometry. The solution was analyzed following reaction of 9 μM apo-rfMT with 108 μM arsenite at 23.5 °C. The experimental data have a relative standard error of 7%. (Reprinted with permission from ref (153).)
The kinetics of the binding between arsenic and protein thiols have also been investigated at the single molecule level. By constructing transmembrane protein pores formed by mutated α-hemolysin in a planar bilayer, Shin et al.405,406 studied the interaction of 4-sulfophenylarsonous acid with a strategically positioned cysteine in the protein. The kon, koff, and Kd values were derived from recorded fluctuations in the ionic current flowing through a single transmembrane protein pore after the arsenical was added to the trans side of the bilayer.
Generally speaking, arsenic binds both proteins and peptides with koff values of similar orders of magnitude (10–3–10–6 s–1). Arsenic binds to proteins with much lower kon values than to small peptides, possibly reflecting that an additional energy barrier has to be overcome in restructuring proteins for arsenic binding. This energetic expense to disrupt preorganized conformations has been evidenced in the case of arsenite binding to human metallothionein α-domain with a slower than expected kon for the first arsenite binding event when koff was assumed to be constant and insignificant.155
6. Summary and Perspectives
The biological effects of arsenic are diverse and the mechanisms of arsenic toxicity and carcinogenicity are complicated. Arsenic–sulfur interactions are the chemical basis of most of these effects. Trivalent arsenicals bind to thiols that are contained in numerous intracellular and cell-surface proteins, and this arsenic–protein binding often triggers cellular responses. For example, arsenic binding to PML or some mitochondrial proteins is a key step leading to cell differentiation or apoptosis induced by As2O3. A low baseline cellular glutathione content and a high expression level of the primary membrane transporter aquaglyceroporin 9 that mediates uptake of As2O3 into cells predetermine the selectivity of APL toward As2O3.407 Generation of cellular ROS is often implicated in the inhibition of various enzymes by arsenic; however, it cannot supplant the role of direct protein binding by trivalent arsenicals. ROS generation itself may result from thiol chelation or complexation in dithiol-containing enzymes, such as pyruvate dehydrogenase (PDH), by trivalent arsenic metabolites.
Direct protein binding by pentavalent arsenicals is mainly manifested in phosphate utilizing enzymes that normally alkylate, acylate, or phosphorylate the phosphate,408 with conceivable biological implications. Serine hydrolases are often the proteins that bind pentavalent arsenicals at the active site serine residue: sulfhydryl groups do not seem to play an important role in their enzymatic activities. Whether pentavalent arsenicals bind to these proteins in a covalent way or as a coordination complex of certain thermodynamic and kinetic properties needs to be examined. Future arsenic–protein binding studies should include a variety of arsenicals on selected proteins in order to fully understand the nature and diversity of arsenic binding to proteins.
The issue of nonspecific association needs to be addressed in protein binding to arsenicals. Stable and specific interaction between arsenic and biomolecules may be necessary to sustain the biological impact of arsenic. For example, arsenite binds peptides containing three (C3) or four (C4) cysteine residues more stably than those containing two cysteine (C2) residues,45,46 which may explain the finding that cellular arsenic exposure resulted in loss of zinc from C3 or C4 (PARP-1 and XPA), but not C2 (SP-1, APTX, and PARP-1 zinc finger mutants) zinc finger proteins.45 Similarly, nonspecific association between arsenite and DNA198 demonstrates that arsenic does not directly react with DNA and may account for the fact that inorganic arsenic is inactive or extremely weak in its ability to induce gene mutations.409,410
With the development of integrated metallomic approaches, more arsenic-binding proteins are expected to be identified under physiologically and pathologically relevant conditions. However, binding specificity should be enshrined as the key concept and scrutinized when identifying novel arsenic-binding proteins with putative arsenic-binding domains demonstrated.
Acknowledgments
The authors thank the Canadian Institutes of Health Research, the National Sciences and Engineering Research Council of Canada, the Canada Research Chairs Program, Alberta Cancer Foundation, Alberta Health, and Alberta Innovates for funding this work. The authors also thank Dr. Barry P. Rosen (Florida International University) for helpful discussion and sharing unpublished data.
Biographies

Shengwen Shen obtained his Ph.D. from the Department of Public Health Sciences at the University of Alberta, Edmonton, Canada, in 2006. He performed his postdoctoral research in the Department of Laboratory Medicine and Pathology at the University of Alberta from 2006 to 2008. He then worked in the Department of Agricultural, Food and Nutritional Science (2008-2010), and is currently a research associate in the Department of Laboratory Medicine and Pathology at the University of Alberta (2010–present). His current research focuses on the cocarcinogenicity of arsenic, with an emphasis on the roles of arsenic in protein binding and inhibition of DNA repair.

Xing-Fang Li is Professor of Analytical and Environmental Toxicology in the Faculty of Medicine and Dentistry, University of Alberta, Edmonton, Canada. She received her B.Sc. (1983) from Hangzhou University (Hangzhou, China), M.Sc. (1986) from the Research Centre for Eco-Environmental Sciences, Chinese Academy of Sciences (Beijing, China), M.Sc. (1990) from Brock University (St. Catharines, Canada), and Ph.D. (1995) from the University of British Columbia (Vancouver, Canada). She joined the Faculty of Medicine and Dentistry at the University of Alberta in 2001 and was promoted through the ranks to Professor in 2010. She received the W.A.E. McBryde Medal from the Canadian Society for Chemistry in 2010 and Killam Annual Professorship from the University of Alberta in 2012. Her research interests include studies of protein interaction with small molecules, selection of DNA aptamers, detection of microbial pathogens, characterization of new drinking water disinfection byproducts, and studies of toxicity of water contaminants and human health effects.

William R. Cullen received a Ph.D. from Cambridge University (Cambridge, U.K.) in 1959. He joined the Faculty of the Chemistry Department, University of British Columbia (Vancouver, Canada) in 1958. He was promoted to Professor in 1969. In 1998 he became Professor Emeritus at UBC and Adjunct Professor at the Royal Military College of Canada (Kingston, Canada) in 2000. He is a Fellow of the Royal Society of Canada. His research has focused on the chemistry of arsenic with increasing emphasis on biological and environmental problems. His book Is Arsenic an Aphrodisiac: The Sociochemistry of an Element was published by the Royal Society of Chemistry in 2008.

Michael Weinfeld received his B.A. degree in Chemistry from Oxford University (Oxford, U.K.) in 1978 and his Ph.D. from the University of London in 1982. After postdoctoral research at the Chalk River Nuclear Laboratories (Ontario, Canada) and the Cross Cancer Institute (Edmonton, Canada), he joined the Faculty of Medicine and Dentistry at the University of Alberta and Cross Cancer Institute in 1989. His current research interests include DNA damage and repair, with a special focus on the repair of DNA strand breaks and arsenic carcinogenesis.

X. Chris Le is Distinguished University Professor and Director of the Analytical and Environmental Toxicology Division in the Faculty of Medicine and Dentistry, University of Alberta (Edmonton, Canada). He holds a Canada Research Chair (Tier 1) in Bioanalytical Technology and Environmental Health. He is an elected Fellow of the Royal Society of Canada (Academy of Science). He received a B.Sc. in Chemistry from Wuhan University (Wuhan, China) in 1983, an M.Sc. in Environmental Chemistry from the Chinese Academy of Sciences (Beijing, China) in 1986, a second M.Sc. in Analytical Chemistry from Brock University (St. Catharines, Canada) in 1988, and a Ph.D. in Analytical Environmental Chemistry from the University of British Columbia (Vancouver, Canada) in 1993. With the support of a Killam Postdoctoral Fellowship, he pursued postdoctoral research in Bioanalytical Chemistry at the University of Alberta (Canada) in 1994. His research on arsenic speciation in biological and environmental systems focuses on arsenic binding to proteins, human exposure to arsenic species, and mechanisms of arsenic metabolism and health effects. Another area of his research deals with bioanalytical chemistry, focusing on the development of DNA and protein binding assays for measuring proteins, DNA damage, and molecular interactions. Dr. Le is an Associate Editor for Environmental Health Perspectives.
Supporting Information Available
Tables S1–S5 that list the structures of arsenic species, structures of common thiols, Kd values, and Kon and Koff values. This material is available free of charge via the Internet at http://pubs.acs.org/.
The authors declare no competing financial interest.
Supplementary Material
References
- Cullen W. R.Is Arsenic an Aphrodisiac? The Sociochemistry of an Element; RSC Publishing: Cambridge, U.K., 2008; 412 pp. [Google Scholar]
- Oremland R. S.; Stolz J. F. Science 2003, 300, 939. [DOI] [PubMed] [Google Scholar]
- Nordstrom D. K. Science 2002, 296, 2143. [DOI] [PubMed] [Google Scholar]
- Jain C. K.; Ali I. Water Res. 2000, 34, 4304. [Google Scholar]
- Bhattacharya P.; Welch A. H.; Stollenwerk K. G.; McLaughlin M. J.; Bundschuh J.; Panaullah G. Sci. Total Environ. 2007, 379, 109. [DOI] [PubMed] [Google Scholar]
- Chen C.; Chuang Y.; You S.; Lin T.; Wu H. Br. J. Cancer 1986, 53, 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.; Chen C.; Wu M.; Kuo T. Br. J. Cancer 1992, 66, 888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou H. Y.; Hsueh Y. M.; Liaw K. F.; Horng S. F.; Chiang M. H.; Pu Y. S.; Shinn-Nan Lin J.; Huang C. H.; Chen C. J. Cancer Res. 1995, 55, 1296. [PubMed] [Google Scholar]
- Hopenhayn-Rich C.; Biggs M. L.; Fuchs A.; Bergoglio R.; Tello E. E.; Nicolli H.; Smith A. H. Epidemiology 1996, 117. [DOI] [PubMed] [Google Scholar]
- Steinmaus C.; Yuan Y.; Kalman D.; Rey O. A.; Skibola C. F.; Dauphine D.; Basu A.; Porter K. E.; Hubbard A.; Bates M. N. Toxicol. Appl. Pharmacol. 2010, 247, 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A. H.; Marshall G.; Yuan Y.; Liaw J.; Ferreccio C.; Steinmaus C. Am. J. Epidemiol. 2011, 173, 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A. H.; Goycolea M.; Haque R.; Biggs M. L. Am. J. Epidemiol. 1998, 147, 660. [DOI] [PubMed] [Google Scholar]
- Chowdhury T. R.; Mandal B. K.; Samanta G.; Basu G. K.; Chowdhury P.; Chanda C.; Karan N.; Lodh D.; Dhar R.; Das D. In Arsenic Exposure and Health Effects; Abernathy C. O., Calderon R. L., Chappell W. R., Eds.; Chapman & Hall: London, 1997; p 93. [Google Scholar]
- Chatterjee A.; Das D.; Mandal B. K.; Chowdhury T. R.; Samanta G.; Chakraborti D. Analyst 1995, 120, 643. [DOI] [PubMed] [Google Scholar]
- Argos M.; Kalra T.; Rathouz P. J.; Chen Y.; Pierce B.; Parvez F.; Islam T.; Ahmed A.; Rakibuz-Zaman M.; Hasan R. Lancet 2010, 376, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohel N.; Persson L. Å.; Rahman M.; Streatfield P. K.; Yunus M.; Ekström E. C.; Vahter M. Epidemiology 2009, 20, 824. [DOI] [PubMed] [Google Scholar]
- Lindberg A. L.; Sohel N.; Rahman M.; Persson L. Å.; Vahter M. Environ. Health Perspect. 2010, 118, 533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z. D.; Zhang Y. M.; Ma L.; Zhang G. Y.; He X.; Wilson R.; Byrd R. W.; Griffiths J. G.; Lai S.; He L.; Grumski K.; Lamm S. H. In Arsenic Exposure and Health Effects; Abernathy C. O., Calderon R. L., Chappell W. R., Eds.; Chapman & Hall: London, 1997; p 55. [Google Scholar]
- Mo J.; Y. Xia Y.; Wade T. J.; Schmitt M.; Le X. C.; Dang R.; Mumford J. L. Environ. Health Perspect. 2006, 114, 835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor K. P.; Lubin J. H. Toxicol. Appl. Pharmacol. 2007, 222, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celik I.; Gallicchio L.; Boyd K.; Lam T. K.; Matanoski G.; Tao X.; Shiels M.; Hammond E.; Chen L.; Robinson K. A. Environ. Res. 2008, 108, 48. [DOI] [PubMed] [Google Scholar]
- Heck J. E.; Andrew A. S.; Onega T.; Rigas J. R.; Jackson B. P.; Karagas M. R.; Duell E. J. Environ. Health Perspect. 2009, 117, 1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A. H.; Ercumen A.; Yuan Y.; Steinmaus C. M. J. Expo. Sci. Environ. Epidemiol. 2009, 19, 343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. J.; Wang S. L.; Chiou J. M.; Tseng C. H.; Chiou H. Y.; Hsueh Y. M.; Chen S. Y.; Wu M. M.; Lai M. S. Toxicol. Appl. Pharmacol. 2007, 222, 298. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Graziano J. H.; Parvez F.; Liu M.; Slavkovich V.; Kalra T.; Argos M.; Islam T.; Ahmed A.; Rakibuz-Zaman M. BMJ 2011, 342, d2431. 10.1136/bmj.d2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Research Council. Arsenic in Drinking Water; National Academy Press: Washington, DC, 1999. [Google Scholar]
- National Research Council. Arsenic in Drinking Water: Update; National Academy Press: Washington, DC, 2001. [Google Scholar]
- Hughes M. F. Toxicol. Lett. 2002, 133, 1. [DOI] [PubMed] [Google Scholar]
- Hughes M. F.; Beck B. D.; Chen Y.; Lewis A. S.; Thomas D. J. Toxicol. Sci. 2011, 123, 305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aposhian H. V.; Aposhian M. M. Chem. Res. Toxicol. 2006, 19, 1. [DOI] [PubMed] [Google Scholar]
- Reaves M. L.; Sinha S.; Rabinowitz J. D.; Kruglyak L.; Redfield R. J. Science 2012, 337, 470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erb T. J.; Kiefer P.; Hattendorf B.; Günther D.; Vorholt J. A. Science 2012, 337, 467. [DOI] [PubMed] [Google Scholar]
- Fekry M. I.; Tipton P. A.; Gates K. S. ACS Chem. Biol. 2011, 6, 127. [DOI] [PubMed] [Google Scholar]
- Denning E. J.; MacKerell A. D. Jr. J. Am. Chem. Soc. 2011, 133, 5770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Gu J.; Leszczynski J. Chem.Commun. 2012, 48, 3626. [DOI] [PubMed] [Google Scholar]
- Summers A. O. Curr. Opin. Microbiol. 2009, 12, 138. [DOI] [PubMed] [Google Scholar]
- Thompson D. J. Chem. Biol. Interact. 1993, 88, 89. [DOI] [PubMed] [Google Scholar]
- Wiberg N.; Holleman A.. Holleman-Wiberg’s Inorganic Chemistry; Academic Press: New York, 2001. [Google Scholar]
- Kitchin K. T.; Wallace K.; Andrewes P. In Arsenic Exposure and Health Effects V; Chapell W. R., Abernathy C. O., Calderon R. L., Thomas D. J., Eds.; Elsevier B.V.: Amsterdam, 2003; p 345 [Google Scholar]
- Cullen W.; McBride B.; Reglinski J. J. Inorg. Biochem. 1984, 21, 179. [Google Scholar]
- Thayer J. S. Appl. Organomet. Chem. 2002, 16, 677. [Google Scholar]
- Kitchin K. T.; Wallace K. Toxicol. Appl. Pharmacol. 2005, 206, 66. [DOI] [PubMed] [Google Scholar]
- Kitchin K. T.; Wallace K. J. Biochem. Mol. Toxicol. 2006, 20, 35. [DOI] [PubMed] [Google Scholar]
- Kitchin K. T.; Wallace K. J. Biochem. Mol. Toxicol. 2006, 20, 48. [DOI] [PubMed] [Google Scholar]
- Zhou X.; Sun X.; Cooper K. L.; Wang F.; Liu K. J.; Hudson L. G. J. Biol. Chem. 2011, 286, 22855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L.; Chen S.; Jia L.; Shu S.; Zhu P.; Liu Y. Metallomics 2012, 4, 988. [DOI] [PubMed] [Google Scholar]
- Lopez S.; Miyashita Y.; Simons S. Jr. J. Biol. Chem. 1990, 265, 16039. [PubMed] [Google Scholar]
- Bellacchio E.; Paggi M. G. J. Cell. Physiol. 2008, 214, 681. [DOI] [PubMed] [Google Scholar]
- Styblo M.; Serves S. V.; Cullen W. R.; Thomas D. J. Chem. Res. Toxicol. 1997, 10, 27. [DOI] [PubMed] [Google Scholar]
- Marapakala K.; Qin J.; Rosen B. P. Biochemistry 2012, 51, 944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fluharty A.; Sanadi D. Proc. Natl. Acad. Sci. U. S. A. 1960, 46, 608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fluharty A. L.; Sanadi D. J. Biol. Chem. 1961, 236, 2772. [PubMed] [Google Scholar]
- Fletcher M. J.; Sanadi D. Arch. Biochem. Biophys. 1962, 96, 139. [DOI] [PubMed] [Google Scholar]
- Craven P. A.; DeRubertis F. R. Biochim. Biophys. Acta Enzymol. 1978, 524, 231. [DOI] [PubMed] [Google Scholar]
- Bagui T. K.; Ghosh M.; Datta A. K. Biochem. J. 1996, 316, 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi S.; Hughes J. B. J. Biol. Chem. 1981, 256, 11112. [PubMed] [Google Scholar]
- Spiegel A. M.; Brown E. M.; Aurbach G. D. J. Cyclic Nucleotide Res. 1976, 2, 393. [PubMed] [Google Scholar]
- González-Segura L.; Mújica-Jiménez C.; Muñoz-Clares R. A. Chem. Biol. Interact. 2009, 178, 64. [DOI] [PubMed] [Google Scholar]
- Chang Y. Y.; Kuo T. C.; Hsu C. H.; Hou D. R.; Kao Y. H.; Huang R. N. Arch. Toxicol. 2012, 86, 911. [DOI] [PubMed] [Google Scholar]
- Lu M.; Wang H.; Li X.; Arnold L. L.; Cohen S. M.; Le X. C. Chem. Res. Toxicol. 2007, 20, 27. [DOI] [PubMed] [Google Scholar]
- Stocken L. A.; Thompson R. Physiol. Rev. 1949, 29, 168. [DOI] [PubMed] [Google Scholar]
- Stocken L.; Thompson R. Biochem. J. 1946, 40, 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocken L.; Thompson R. Biochem. J. 1946, 40, 535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson K. J.; Hale G.; Perham R. N. Biochemistry 1978, 17, 2189. [DOI] [PubMed] [Google Scholar]
- Ordóñez E.; Thiyagarajan S.; Cook J. D.; Stemmler T. L.; Gil J. A.; Mateos L. M.; Rosen B. P. J. Biol. Chem. 2008, 283, 25706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X. W.; Yan X. J.; Zhou Z. R.; Yang F. F.; Wu Z. Y.; Sun H. B.; Liang W. X.; Song A. X.; Lallemand-Breitenbach V.; Jeanne M. Science 2010, 328, 240. [DOI] [PubMed] [Google Scholar]
- Jeanne M.; Lallemand-Breitenbach V.; Ferhi O.; Koken M.; Le Bras M.; Duffort S.; Peres L.; Berthier C.; Soilihi H.; Raught B. Cancer Cell 2010, 18, 88. [DOI] [PubMed] [Google Scholar]
- Styblo M.; Drobná Z.; Jaspers I.; Lin S.; Thomas D. J. Environ. Health Perspect. 2002, 110, 7667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aposhian H. V. Enzymatic. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 397. [DOI] [PubMed] [Google Scholar]
- Board P. G.; Coggan M.; Chelvanayagam G.; Easteal S.; Jermiin L. S.; Schulte G. K.; Danley D. E.; Hoth L. R.; Griffor M. C.; Kamath A. V. J. Biol. Chem. 2000, 275, 24798. [DOI] [PubMed] [Google Scholar]
- Zakharyan R. A.; Tsaprailis G.; Chowdhury U. K.; Hernandez A.; Aposhian H. V. Chem. Res. Toxicol. 2005, 18, 1287. [DOI] [PubMed] [Google Scholar]
- Messens J.; Martins J. C.; Van Belle K.; Brosens E.; Desmyter A.; De Gieter M.; Wieruszeski J. M.; Willem R.; Wyns L.; Zegers I. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 8506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messens J.; Silver S. J. Mol. Biol. 2006, 362, 1. [DOI] [PubMed] [Google Scholar]
- Cullen W. R.; Reimer K. J. Chem. Rev. 1989, 89, 713. [Google Scholar]
- Ajees A. A.; Marapakala K.; Packianathana C.; Sankaran B.; Rosen B. P. Biochemistry 2012, 51, 5476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Waters S. B.; Drobna Z.; Devesa V.; Styblo M.; Thomas D. J. Toxicol. Appl. Pharmacol. 2005, 204, 164. [DOI] [PubMed] [Google Scholar]
- Fomenko D. E.; Xing W.; Adair B. M.; Thomas D. J.; Gladyshev V. N. Science 2007, 315, 387. [DOI] [PubMed] [Google Scholar]
- Hamdi M.; Yoshinaga M.; Packianathan C.; Qin J.; Hallauer J.; McDermott J. R.; Yang H.; Tsai K.; Liu Z. Toxicol. Appl. Pharmacol. 2012, 162, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J.; Lehr C. R.; Yuan C.; Le X. C.; McDermott T. R.; Rosen B. P. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 5213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X.; Geng Z.; Li X.; Zhao Q.; Hu X.; Zhang X.; Wang Z. Biochimie 2010, 93, 369. [DOI] [PubMed] [Google Scholar]
- Song X.; Geng Z.; Zhu J.; Li C.; Hu X.; Bian N.; Zhang X.; Wang Z. Chem. Biol. Interact. 2009, 179, 321. [DOI] [PubMed] [Google Scholar]
- Song X.; Geng Z.; Li X.; Hu X.; Bian N.; Zhang X.; Wang Z. Biochimie 2010, 92, 1397. [DOI] [PubMed] [Google Scholar]
- Ratnaike R. N. Postgrad. Med. J. 2003, 79, 391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb J. L. In Enzyme and Metabolic Inhibitors Academic Press: New York, 1966; Vol. III, p 595. [Google Scholar]
- Muller S.; Walter R. D.; Fairlamb A. H. Mol. Biochem. Parasitol. 1995, 71, 211. [DOI] [PubMed] [Google Scholar]
- Cunningham M. L.; Zvelebil M. J. J. M.; Fairlamb A. H. Eur. J. Biochem. 1994, 221, 285. [DOI] [PubMed] [Google Scholar]
- Knowles F. C. Arch. Biochem. Biophys. 1985, 242, 1. [DOI] [PubMed] [Google Scholar]
- Chouchane S.; Snow E. T. Chem. Res. Toxicol. 2001, 14, 517. [DOI] [PubMed] [Google Scholar]
- Lu J.; Chew E. H.; Holmgren A. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 12288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S.; Del Razo L. M.; Styblo M.; Wang C.; Cullen W. R.; Thomas D. J. Chem. Res. Toxicol. 2001, 14, 305. [DOI] [PubMed] [Google Scholar]
- Lin S.; Cullen W. R.; Thomas D. J. Chem. Res. Toxicol. 1999, 12, 924. [DOI] [PubMed] [Google Scholar]
- Chang K. N.; Te Chang Lee M. F. T.; Chen Y. C.; Lee L. W.; Lee S. Y.; Lin P. J.; Huang R. N. Biochem. J. 2003, 371, 495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow E. T.; Hu Y.; Yan C. C.; Chouchane S.. Modulation of DNA Repair and Glutathione Levels in Human Keratinocytes by Micromolar Arsenite. In Arsenic Exposure and Health Effects; Chappell W. R., Abernathy C. O., Calderon R. L., Eds.; Elsevier: Oxford, UK. 1999; p 243. [Google Scholar]
- Li J.; Pickart C. M. Biochemistry 1995, 34, 15829. [DOI] [PubMed] [Google Scholar]
- Li J.; Pickart C. M. Biochemistry 1995, 34, 139. [DOI] [PubMed] [Google Scholar]
- Kapahi P.; Takahashi T.; Natoli G.; Adams S. R.; Chen Y.; Tsien R. Y.; Karin M. J. Biol. Chem. 2000, 275, 36062. [DOI] [PubMed] [Google Scholar]
- Lin C. H.; Huang C. F.; Chen W. Y.; Chang Y. Y.; Ding W. H.; Lin M. S.; Wu S. H.; Huang R. N. Chem. Res. Toxicol. 2006, 19, 469. [DOI] [PubMed] [Google Scholar]
- Zhang X. Y.; Bishop A. C. Biochemistry 2008, 47, 4491. [DOI] [PubMed] [Google Scholar]
- Walton Z. E.; Bishop A. C. Bioorg. Med. Chem. 2010, 18, 4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis O.; Bishop A. C. Bioconjugate Chem. 2012, 23, 272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman K.; Chen Z.; Wang W. W.; Wang Y. W.; Sakamoto A.; Zhang Y. F.; Suzuki N.; Naranmandura H. Toxicol. Appl. Pharmacol. 2012, 263, 273. [DOI] [PubMed] [Google Scholar]
- Cavigelli M.; Li W. W.; Lin A.; Su B.; Yoshioka K.; Karin M. EMBO J. 1996, 15, 6269. [PMC free article] [PubMed] [Google Scholar]
- Yoda A.; Toyoshima K.; Watanabe Y.; Onishi N.; Hazaka Y.; Tsukuda Y.; Tsukada J.; Kondo T.; Tanaka Y.; Minami Y. J. Biol. Chem. 2008, 283, 18969. [DOI] [PubMed] [Google Scholar]
- Mao J. H.; Sun X. Y.; Liu J. X.; Zhang Q. Y.; Liu P.; Huang Q. H.; Li K. K.; Chen Q.; Chen Z.; Chen S. J. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 21683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergquist E. R.; Fischer R. J.; Sugden K. D.; Martin B. D. J. Organomet. Chem. 2009, 694, 973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrick J. S.; Jagadish B.; Mash E. A.; Aposhian H. V. M. Chem. Res. Toxicol. 2001, 14, 651. [DOI] [PubMed] [Google Scholar]
- Samikkannu T.; Chen C. H.; Yih L. H.; Wang A. S. S.; Lin S. Y.; Chen T. C.; Jan K. Y. Chem. Res. Toxicol. 2003, 16, 409. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Zhang H.; Li X. F.; Le X. C. Rapid Commun. Mass Spectrom. 2007, 21, 3658. [DOI] [PubMed] [Google Scholar]
- Powis G.; Montfort W. R. Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 421. [DOI] [PubMed] [Google Scholar]
- Arnér E. S. J.; Holmgren A. Eur. J. Biochem. 2000, 267, 6102. [DOI] [PubMed] [Google Scholar]
- Gromer S.; Urig S.; Becker K. Med. Res. Rev. 2004, 24, 40. [DOI] [PubMed] [Google Scholar]
- Waters S. B.; Devesa V.; Del Razo L. M.; Styblo M.; Thomas D. J. Chem. Res. Toxicol. 2004, 17, 404. [DOI] [PubMed] [Google Scholar]
- Thomas D. J. Toxicol. Sci. 2009, 107, 309. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Jin X.; Snow E. T. Toxicol. Lett. 2002, 133, 33. [DOI] [PubMed] [Google Scholar]
- Schwerdtle T.; Walter I.; Hartwig A. DNA Repair 2003, 2, 1449. [DOI] [PubMed] [Google Scholar]
- Shen S.; Lee J.; Weinfeld M.; Le X. C. Mol. Carcinog. 2008, 47, 508. [DOI] [PubMed] [Google Scholar]
- Shen S.; Wang C.; Weinfeld M.; Le X. C. Chin. Sci. Bull. 2013, 58, 214. [Google Scholar]
- Walter I.; Schwerdtle T.; Thuy C.; Parsons J. L.; Dianov G. L.; Hartwig A. DNA Repair 2007, 6, 61. [DOI] [PubMed] [Google Scholar]
- Hartwig A.; Blessing H.; Schwerdtle T.; Walter I. Toxicology 2003, 193, 161. [DOI] [PubMed] [Google Scholar]
- Flohr C.; Bürkle A.; Radicella J. P.; Epe B. Nucleic Acids Res. 2003, 31, 5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luijsterburg M. S.; Lindh M.; Acs K.; Vrouwe M. G.; Pines A.; van Attikum H.; Mullenders L. H.; Dantuma N. P. J. Cell Biol. 2012, 197, 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audebert M.; Salles B.; Calsou P. J. Biol. Chem. 2004, 279, 55117. [DOI] [PubMed] [Google Scholar]
- Ding W.; Liu W.; Cooper K. L.; Qin X. J.; de Souza Bergo P. L.; Hudson L. G.; Liu K. J. J. Biol. Chem. 2009, 284, 6809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pia̧tek K.; Schwerdtle T.; Hartwig A.; Bal W. Chem. Res. Toxicol. 2008, 21, 600. [DOI] [PubMed] [Google Scholar]
- Cobbett C.; Goldsbrough P. Plant Biol. 2002, 53, 159. [DOI] [PubMed] [Google Scholar]
- Raab A.; Feldmann J.; Meharg A. A. Plant Physiol. 2004, 134, 1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.; Cai Y. Anal. Chem. 2003, 75, 7030. [DOI] [PubMed] [Google Scholar]
- Ma L. Q.; Komar K. M.; Tu C.; Zhang W.; Cai Y.; Kennelley E. D. Nature 2001, 409, 579. [DOI] [PubMed] [Google Scholar]
- Lin Y. F.; Walmsley A. R.; Rosen B. P. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 15617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X.; Ma Q. J. Biol. Chem. 2009, 284, 12609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews G. K. Biochem. Pharmacol. 2000, 59, 95. [DOI] [PubMed] [Google Scholar]
- Klaassen C. D.; Liu J.; Choudhuri S. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 267. [DOI] [PubMed] [Google Scholar]
- Margoshes M.; Vallee B. L. J. Am. Chem. Soc. 1957, 79, 4813. [Google Scholar]
- He X.; Ma Q. J. Pharmacol. Exp. Ther. 2010, 332, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stillman M. J.; Shaw C. F.; Suzuki K. T. (Eds.) Metallothionein: Synthesis, Structure, and Properties of Metallothioneins, Phytochelatins, and Metal–Thiolate Complexes; Wiley-VCH: New York. 1992. [Google Scholar]
- Suzuki K. T.; Imura N.; Kimura M. (Eds.) Metallothionein III: Biological Roles and Medical Implications; Birkhauser: Boston, 1993. [Google Scholar]
- Henkel G.; Krebs B. Chem. Rev. 2004, 104, 801. [DOI] [PubMed] [Google Scholar]
- Sutherland D. E.; Willans M. J.; Stillman M. J. Biochemistry 2010, 49, 3593. [DOI] [PubMed] [Google Scholar]
- Albores A.; Koropatnick J.; Cherian M. G.; Zelazowski A. J. Chem. Biol. Interact. 1992, 85, 127. [DOI] [PubMed] [Google Scholar]
- Kreppel H.; Bauman J. W.; Liu J. I. E.; McKim J. M.; Klaassen C. D. Toxicol. Sci. 1993, 20, 184. [DOI] [PubMed] [Google Scholar]
- Falnoga I.; Zelenik Pevec A.; Šlejkovec Z.; Žnidarič M. T.; Zajc I.; Mlakar S. J.; Marc J. Biol. Trace Elem. Res. 2012, 149, 331. [DOI] [PubMed] [Google Scholar]
- Macias-Barragán J.; Huerta-Olvera S.; Ramos-Márquez M. E.; Armendáriz-Borunda J.; Siller-López F. Toxicol. Lett. 2006, 164, S137. [DOI] [PubMed] [Google Scholar]
- Zhou X. D.; Sens D. A.; Sens M. A.; Namburi V. B.; Singh R. K.; Garrett S. H.; Somji S. Toxicol. Sci. 2006, 91, 467. [DOI] [PubMed] [Google Scholar]
- Ng J.; Seawright A.; Moore M. Toxicology 2001, 164, 59. [Google Scholar]
- Kaegi J. H. R.; Schaeffer A. Biochemistry 1988, 27, 8509. [DOI] [PubMed] [Google Scholar]
- Singh S.; Mulchandani A.; Chen W. Appl. Environ. Microbiol. 2008, 74, 2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T.; Ohata S.; Togawa T.; Himeno S.; Tanabe S. J. Toxicol. Sci. 2007, 32, 321. [DOI] [PubMed] [Google Scholar]
- Langdon C.; Winters C.; Stürzenbaum S.; Morgan A.; Charnock J.; Meharg A.; Piearce T. G.; Lee P.; Semple K. T. Environ. Sci. Technol. 2005, 39, 2042. [DOI] [PubMed] [Google Scholar]
- Toyama M.; Yamashita M.; Hirayama N.; Murooka Y. J. Biochem. 2002, 132, 217. [DOI] [PubMed] [Google Scholar]
- Chen C. L.; Whanger P. D. J. Inorg. Biochem. 1994, 54, 267. [DOI] [PubMed] [Google Scholar]
- Jiang G.; Gong Z.; Li X. F.; Cullen W. R.; Le X. C. Chem. Res. Toxicol. 2003, 16, 873. [DOI] [PubMed] [Google Scholar]
- Ngu T. T.; Stillman M. J. J. Am. Chem. Soc. 2006, 128, 12473. [DOI] [PubMed] [Google Scholar]
- Ngu T. T.; Lee J. A.; Rushton M. K.; Stillman M. J. Biochemistry 2009, 48, 8806. [DOI] [PubMed] [Google Scholar]
- Ngu T. T.; Lee J. A.; Pinter T. B. J.; Stillman M. J. J. Inorg. Biochem. 2010, 104, 232. [DOI] [PubMed] [Google Scholar]
- Ngu T. T.; Easton A.; Stillman M. J. J. Am. Chem. Soc. 2008, 130, 17016. [DOI] [PubMed] [Google Scholar]
- Merrifield M.; Ngu T.; Stillman M. Biochem. Biophys. Res. Commun. 2004, 324, 127. [DOI] [PubMed] [Google Scholar]
- Ngu T. T.; Dryden M. D.; Stillman M. J. Biochem. Biophys. Res. Commun. 2010, 401, 69. [DOI] [PubMed] [Google Scholar]
- Ngu T. T.; Stillman M. J. IUBMB Life 2009, 61, 438. [DOI] [PubMed] [Google Scholar]
- Guo Y.; Ling Y.; Thomson B. A.; Siu K. W. M. J. Am. Soc. Mass Spectrom. 2005, 16, 1787. [DOI] [PubMed] [Google Scholar]
- Rosen B. P. FEBS Lett. 2002, 529, 86. [DOI] [PubMed] [Google Scholar]
- Shi W.; Dong J.; Scott R. A.; Ksenzenko M. Y.; Rosen B. P. J. Biol. Chem. 1996, 271, 9291. [DOI] [PubMed] [Google Scholar]
- Xu C.; Zhou T.; Kuroda M.; Rosen B. P. J. Biochem. 1998, 123, 16. [DOI] [PubMed] [Google Scholar]
- Fu H.; Jiang X.; Rosen B. P. In Biological Chemistry of Arsenic, Antimony and Bismuth; Sun H., Ed.; John Wiley & Sons: Hoboken, NJ, 2009; p 181. [Google Scholar]
- Yang J.; Rawat S.; Stemmler T. L.; Rosen B. P. Biochemistry 2010, 49, 3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin P.; DeMel S.; Shi J.; Gladysheva T.; Gatti D. L.; Rosen B. P.; Edwards B. F. P. Structure 2001, 9, 1071. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Cornelis R.; De Kimpe J.; Mees L.; Lameire N. Clin. Chem. (Washington, D. C.) 1998, 44, 141. [PubMed] [Google Scholar]
- Hunter F.; Kip A.; Irvine J. Jr J. Pharmacol. Exp. Ther. 1942, 76, 207. [Google Scholar]
- Lanz H. Jr; Wallace P.; Hamilton J. Univ. Calif. (Berkeley) Pubs. Pharmacol. 1949, 2, 263. [PubMed] [Google Scholar]
- Lu M.; Wang H.; Li X. F.; Lu X.; Cullen W. R.; Arnold L. L.; Cohen S. M.; Le X. C. Chem. Res. Toxicol. 2004, 17, 1733. [DOI] [PubMed] [Google Scholar]
- Vahter M.; Marafante E.; Dencker L. Arch. Environ. Contam. Toxicol. 1984, 13, 259. [DOI] [PubMed] [Google Scholar]
- Lu M.; Li X. F.; Le X. C.; Weinfeld M.; Wang H. Rapid Commun. Mass Spectrom. 2010, 24, 1523. [DOI] [PubMed] [Google Scholar]
- Lu M.; Wang H.; Wang Z.; Li X. F.; Le X. C. J. Proteome Res. 2008, 7, 3080. [DOI] [PubMed] [Google Scholar]
- Waxman S.; Anderson K. C. Oncologist 2001, 6, 3. [DOI] [PubMed] [Google Scholar]
- Wang X. Genes Dev. 2001, 15, 2922. [PubMed] [Google Scholar]
- Wang C.; Youle R. J. Annu. Rev. Genet. 2009, 43, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S. Int. J. Oncol. 2003, 22, 15. [PubMed] [Google Scholar]
- Desagher S.; Martinou J. C. Trends Cell Biol. 2000, 10, 369. [DOI] [PubMed] [Google Scholar]
- Don A. S.; Kisker O.; Dilda P.; Donoghue N.; Zhao X.; Decollogne S.; Creighton B.; Flynn E.; Folkman J.; Hogg P. J. Cancer Cell 2003, 3, 497. [DOI] [PubMed] [Google Scholar]
- Belzacq A. S.; El Hamel C.; Vieira H. L.; Cohen I.; Haouzi D.; Metivier D.; Marchetti P.; Brenner C.; Kroemer G. Oncogene 2001, 20, 7579. [DOI] [PubMed] [Google Scholar]
- Larochette N.; Decaudin D.; Jacotot E.; Brenner C.; Marzo I.; Susin S. A.; Zamzami N.; Xie Z.; Reed J.; Kroemer G. Exp. Cell Res. 1999, 249, 413. [DOI] [PubMed] [Google Scholar]
- Zheng Y.; Shi Y.; Tian C.; Jiang C.; Jin H.; Chen J.; Almasan A.; Tang H.; Chen Q. Oncogene 2003, 23, 1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong H. L.; Liebler D. C. Chem. Res. Toxicol. 2008, 21, 796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carré M.; André N.; Carles G.; Borghi H.; Brichese L.; Briand C.; Braguer D. J. Biol. Chem. 2002, 277, 33664. [DOI] [PubMed] [Google Scholar]
- Psarra A.; Hermann S.; Panayotou G.; Spyrou G. Biochem. J. 2009, 422, 521. [DOI] [PubMed] [Google Scholar]
- Miranda-Vizuete A.; Damdimopoulos A. E.; Spyrou G. Antioxid. Redox Signaling 2000, 2, 801. [DOI] [PubMed] [Google Scholar]
- Requejo R.; Chouchani E. T.; James A. M.; Prime T. A.; Lilley K. S.; Fearnley I. M.; Murphy M. P. Arch. Biochem. Biophys. 2010, 504, 228. [DOI] [PubMed] [Google Scholar]
- Marchetti P.; Decaudin D.; Macho A.; Zamzami N.; Hirsch T.; Susin S. A.; Kroemer G. Eur. J. Immunol. 1997, 27, 289. [DOI] [PubMed] [Google Scholar]
- McStay G. Biochem. J. 2002, 367, 541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sionov R. V.; Cohen O.; Kfir S.; Zilberman Y.; Yefenof E. J. Exp. Med. 2006, 203, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman R. D.; Lane M. D. J. Biol. Chem. 1992, 267, 14005. [PubMed] [Google Scholar]
- Li Y. M.; Broome J. D. Cancer Res. 1999, 59, 776. [PubMed] [Google Scholar]
- Menzel D. B.; Hamadeh H. K.; Lee E.; Meacher D. M.; Said V.; Rasmussen R. E.; Greene H.; Roth R. N. Toxicol. Lett. 1999, 105, 89. [DOI] [PubMed] [Google Scholar]
- Spuches A. M.; Wilcox D. E. J. Am. Chem. Soc. 2008, 130, 8148. [DOI] [PubMed] [Google Scholar]
- Simons S. S.; Chakraborti P. K.; Cavanaugh A. H. J. Biol. Chem. 1990, 265, 1938. [PubMed] [Google Scholar]
- Stoica A.; Pentecost E.; Martin M. B. Endocrinology 2000, 141, 3595. [DOI] [PubMed] [Google Scholar]
- He X.; Ma Q. Mol. Pharmacol. 2009, 76, 1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miseta A.; Csutora P. Mol. Biol. Evol. 2000, 17, 1232. [DOI] [PubMed] [Google Scholar]
- Kitchin K. T.; Wallace K. J. Inorg. Biochem. 2008, 102, 532. [DOI] [PubMed] [Google Scholar]
- Reimer K. J.; Koch I.; Cullen W. R. Met. Ions Life Sci. 2010, 7, 165. [DOI] [PubMed] [Google Scholar]
- Charoensuk V.; Gati W. P.; Weinfeld M.; Le X. C. Toxicol. Appl. Pharmacol. 2009, 239, 64. [DOI] [PubMed] [Google Scholar]
- Naranmandura H.; Suzuki K. T. Chem. Res. Toxicol. 2008, 21, 678. [DOI] [PubMed] [Google Scholar]
- Styblo M.; Thomas D. J. Toxicol. Appl. Pharmacol. 1997, 147, 1. [DOI] [PubMed] [Google Scholar]
- Delnomdedieu M.; Styblo M.; Thomas D. J. Chem. Biol. Interact. 1995, 98, 69. [DOI] [PubMed] [Google Scholar]
- De Kimpe J.; Cornelis R.; Mees L.; Vanholder R. Toxicol. Sci. 1996, 34, 240. [DOI] [PubMed] [Google Scholar]
- Marafante E.; Vahter M.; Envall J. Chem. Biol. Interact. 1985, 56, 225. [DOI] [PubMed] [Google Scholar]
- Vahter M.; Marafante E. Chem. Biol. Interact. 1983, 47, 29. [DOI] [PubMed] [Google Scholar]
- Styblo M.; Delnomdedieu M.; Thomas D. J. Chem. Biol. Interact. 1996, 99, 147. [DOI] [PubMed] [Google Scholar]
- Cornelis R.; Borguet F.; De Kimpe J. Anal. Chim. Acta 1993, 283, 183. [Google Scholar]
- Cornelis R.; Kimpe J. J. Anal. At. Spectrom. 1994, 9, 945. [Google Scholar]
- Schmidt A.; Steier S.; Otto M. Talanta 2009, 77, 1830. [DOI] [PubMed] [Google Scholar]
- Bogdan G. M.; Sampayo-Reyes A.; Aposhian H. V. Toxicology 1994, 93, 175. [DOI] [PubMed] [Google Scholar]
- Schmidt A.; Steier S.; Otto M. Talanta 2009, 77, 1830. [DOI] [PubMed] [Google Scholar]
- Vahter M.; Marafante E. Biol. Trace Elem. Res. 1989, 21, 233. [DOI] [PubMed] [Google Scholar]
- Buchet J. P.; Lauwerys R. Arch. Toxicol. 1985, 57, 125. [DOI] [PubMed] [Google Scholar]
- Oladimeji A. A. Ecotoxicol. Environ. Saf. 1985, 9, 1. [DOI] [PubMed] [Google Scholar]
- Piotrowski J. K.; Szymańska J. A. J. Toxicol. Environ. Health, Part A 1976, 1, 991. [DOI] [PubMed] [Google Scholar]
- Arpadjan S.; E̅elik G.; Taskesen S.; Güe̅er S. Food Chem. Toxicol. 2008, 46, 2871. [DOI] [PubMed] [Google Scholar]
- Hanlon D. P.; Ferm V. H. Environ. Res. 1986, 40, 380. [DOI] [PubMed] [Google Scholar]
- Styblo M.; Yamauchi H.; Thomas D. J. Toxicol. Appl. Pharmacol. 1995, 135, 172. [DOI] [PubMed] [Google Scholar]
- Styblo M.; Hughes M. F.; Thomas D. J. J. Chromatogr. B: Biomed. Appl. 1996, 677, 161. [DOI] [PubMed] [Google Scholar]
- Schmidt A. C.; Mattusch J.; Wennrich R. Microchim. Acta 2005, 151, 167. [Google Scholar]
- Reyes L. H.; Mar J. L. G.; Rahman G. M.; Seybert B.; Fahrenholz T.; Kingston H. M. Talanta 2009, 78, 983. [DOI] [PubMed] [Google Scholar]
- Naranmandura H.; Suzuki N.; Suzuki K. T. Chem. Res. Toxicol. 2006, 19, 1010. [DOI] [PubMed] [Google Scholar]
- Chen B.; Cao F.; Yuan C.; Lu X.; Shen S.; Zhou J.; Le X. C. Anal. Bioanal.Chem. 2013, 405, 1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B.; Arnold L. L.; Cohen S. M.; Thomas D. J.; Le X. C. Toxicol. Sci. 2011, 124, 320. [DOI] [PubMed] [Google Scholar]
- De Kimpe J.; Cornelis R. Toxicol. Environ. Chem. 1999, 71, 279. [Google Scholar]
- Reinke J.; Uthe J. F.; Freeman H. C.; Johnston J. R. Environ. Lett. 1975, 8, 371. [DOI] [PubMed] [Google Scholar]
- Lu M.; Wang H.; Li X.; Lu X.; Le X. C. J. Chromatogr. A 2009, 1216, 3985. [DOI] [PubMed] [Google Scholar]
- Raab A.; Wright S. H.; Jaspars M.; Meharg A. A.; Feldmann J. Angew. Chem., Int. Ed. 2007, 46, 2594. [DOI] [PubMed] [Google Scholar]
- Gong Z.; Lu X.; Ma M.; Watt C.; Le X. C. Talanta 2002, 58, 77. [DOI] [PubMed] [Google Scholar]
- Francesconi K. A.; Kuehnelt D. Analyst 2004, 129, 373. [DOI] [PubMed] [Google Scholar]
- Dietz C.; Sanz J.; Sanz E.; Muñoz-Olivas R.; Cámara C. J. Chromatogr. A 2007, 1153, 114. [DOI] [PubMed] [Google Scholar]
- Ma R.; McLeod C. W.; Tomlinson K.; Poole R. K. Electrophoresis 2004, 25, 2469. [DOI] [PubMed] [Google Scholar]
- Feldmann J. TrAC Trends Anal. Chem. 2005, 24, 228. [Google Scholar]
- Mounicou S.; Szpunar J.; Lobinski R. Chem. Soc. Rev. 2009, 38, 1119. [DOI] [PubMed] [Google Scholar]
- Szpunar J.; Lobinski R.; Prange A. Appl. Spectrosc. 2003, 57, 102. [DOI] [PubMed] [Google Scholar]
- Harrington C. F.; Clough R.; Hansen H. R.; Hill S. J.; Pergantis S. A.; Tyson J. F. J. Anal. At. Spectrom. 2009, 24, 999. [Google Scholar]
- B’hymer C.; Caruso J. J. Chromatogr. A 2004, 1045, 1. [DOI] [PubMed] [Google Scholar]
- Patel P. C.; Goulhen F.; Boothman C.; Gault A. G.; Charnock J. M.; Kalia K.; Lloyd J. R. Arch. Microbiol. 2007, 187, 171. [DOI] [PubMed] [Google Scholar]
- Feldmann J.; Devalla S.; Raab A.; Hansen H. R. In Organic Metal and Metalloid Species in the Environment: Analysis, Distribution, Processes and Toxicological Evaluation; Hirner A. V., Emons H., Eds.; Springer Verlag: Berlin, 2004; p 41. [Google Scholar]
- Whaley-Martin K.; Koch I.; Moriarty M.; Reimer K. Environ. Sci. Technol. 2012, 46, 3110. [DOI] [PubMed] [Google Scholar]
- Caumette G.; Koch I.; Moriarty M.; Reimer K. J. Sci. Total Environ. 2012, 432, 243. [DOI] [PubMed] [Google Scholar]
- Sun X.; Tsang C.; Sun H. Metallomics 2009, 1, 25. [Google Scholar]
- Munro K. L.; Mariana A.; Klavins A. I.; Foster A. J.; Lai B.; Vogt S.; Cai Z.; Harris H. H.; Dillon C. T. Chem. Res. Toxicol. 2008, 21, 1760. [DOI] [PubMed] [Google Scholar]
- Miot J.; Morin G.; Skouri-Panet F.; Férard C.; Aubry E.; Briand J.; Wang Y.; Ona-Nguema G.; Guyot F.; Brown G. E. Environ. Sci. Technol. 2008, 42, 5342. [DOI] [PubMed] [Google Scholar]
- Mustra D. J.; Warren A. J.; Wilcox D. E.; Hamilton J. W. Chem. Biol. Interact. 2007, 168, 159. [DOI] [PubMed] [Google Scholar]
- Matzapetakis M.; Farrer B. T.; Weng T.; Hemmingsen L.; Penner-Hahn J. E.; Pecoraro V. L. J. Am. Chem. Soc. 2002, 124, 8042. [DOI] [PubMed] [Google Scholar]
- Chong S.; Dill K.; McGown E. J. Biochem. Toxicol. 1989, 4, 39. [DOI] [PubMed] [Google Scholar]
- Vahter M.; Marafante E.; Lindgren A.; Dencker L. Arch. Toxicol. 1982, 51, 65. [Google Scholar]
- Pott W. A.; Benjamin S. A.; Yang R. S. H. Rev. Environ. Contam. Toxicol. 2001, 169, 165. [DOI] [PubMed] [Google Scholar]
- Das A. K.; Chakraborty R.; Luisa Cervera M.; de la Guardia M. Microchim. Acta 1996, 122, 209. [Google Scholar]
- Wang T.; Yan P.; Squier T. C.; Mayer M. U. ChemBioChem 2007, 8, 1937. [DOI] [PubMed] [Google Scholar]
- Griffin B. A.; Adams S. R.; Tsien R. Y. Science 1998, 281, 269. [DOI] [PubMed] [Google Scholar]
- Adams S. R.; Campbell R. E.; Gross L. A.; Martin B. R.; Walkup G. K.; Yao Y.; Llopis J.; Tsien R. Y. J. Am. Chem. Soc. 2002, 124, 6063. [DOI] [PubMed] [Google Scholar]
- Stroffekova K.; Proenza C.; Beam K. G. Pflügers Arch.-Eur. J. Physiol. 2001, 442, 859. [DOI] [PubMed] [Google Scholar]
- Hearps A. C.; Pryor M. J.; Kuusisto H. V.; Rawlinson S. M.; Piller S. C.; Jans D. A. J. Fluoresc. 2007, 17, 593. [DOI] [PubMed] [Google Scholar]
- Huang C.; Yin Q.; Zhu W.; Yang Y.; Wang X.; Qian X.; Xu Y. Angew. Chem., Int. Ed. 2011, 50, 7551. [DOI] [PubMed] [Google Scholar]
- Hannestad U.; Lundqvist P.; Sörbo B. Anal. Biochem. 1982, 126, 200. [DOI] [PubMed] [Google Scholar]
- Zhou G.; Jauhiainen M.; Stevenson K.; Dolphin P. J. J. Chromatogr. B: Biomed. Sci. Appl. 1991, 568, 69. [PubMed] [Google Scholar]
- Kalef E.; Walfish P. G.; Gitler C. Anal. Biochem. 1993, 212, 325. [DOI] [PubMed] [Google Scholar]
- Berleth E. S.; Kasperek E. M.; Grill S. P.; Braunscheidel J. A.; Graziani L. A.; Pickart C. M. J. Biol. Chem. 1992, 267, 16403. [PubMed] [Google Scholar]
- Boerman M. H. E. M.; Napoli J. L. Arch. Biochem. Biophys. 1995, 321, 434. [DOI] [PubMed] [Google Scholar]
- Winski S. L.; Carter D. E. J. Toxicol. Environ. Health, Part A 1995, 46, 379. [DOI] [PubMed] [Google Scholar]
- Mizumura A.; Watanabe T.; Kobayashi Y.; Hirano S. Toxicol. Appl. Pharmacol. 2010, 242, 119. [DOI] [PubMed] [Google Scholar]
- Jin Y. J.; Yu C. L.; Burakoff S. J. J. Biol. Chem. 1999, 274, 28301. [DOI] [PubMed] [Google Scholar]
- Gitler C.; Zarmi B.; Kalef E. Anal. Biochem. 1997, 252, 48. [DOI] [PubMed] [Google Scholar]
- Foley T. D.; Stredny C. M.; Coppa T. M.; Gubbiotti M. A. Neurochem. Res. 2010, 35, 306. [DOI] [PubMed] [Google Scholar]
- Foley T. D.; Melideo S. L.; Healey A. E.; Lucas E. J.; Koval J. A. Neurochem. Res. 2011, 36, 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bognumil R.; Ullrich V. Methods. Enzymol. 2002, 348, 271. [DOI] [PubMed] [Google Scholar]
- Yan H.; Wang N.; Weinfeld M.; Cullen W. R.; Le X. C. Anal. Chem. 2009, 81, 4144. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Yang F.; Shim J. Y.; Kirk K. L.; Anderson D. E.; Chen X. Cancer Lett. 2007, 255, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heredia-Moya J.; Kirk K. L. Bioorg. Med. Chem. 2008, 16, 5743–5746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donoghue N.; Yam P. T. W.; Jiang X. M.; Hogg P. J. Protein Sci. 2000, 9, 2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donoghue N.; Hogg P. J. Meth. Enzymol. 2002, 348, 76. [DOI] [PubMed] [Google Scholar]
- Dilda P. J.; Decollogne S.; Rossiter-Thornton M.; Hogg P. J. Cancer Res. 2005, 65, 11729. [DOI] [PubMed] [Google Scholar]
- Park D.; Don A. S.; Massamiri T.; Karwa A.; Warner B.; MacDonald J.; Hemenway C.; Naik A.; Kuan K. T.; Dilda P. J. J. Am. Chem. Soc. 2011, 133, 2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moaddel R.; Sharma A.; Huseni T.; Jones G. S. Jr.; Hanson R. N.; Loring R. H. Bioconjugate Chem. 1999, 10, 629. [DOI] [PubMed] [Google Scholar]
- Bennett T. A.; Edwards B. S.; Sklar L. A.; Rogelj S. J. Immunol. 2000, 164, 4120. [DOI] [PubMed] [Google Scholar]
- Foley T. D.; Clark A. R.; Stredny E. S.; Wierbowski B. M. Cell. Mol. Neurobiol. 2011, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S.; Chen Y.; Rosen B. P. Mol. Microbiol. 2001, 41, 687. [DOI] [PubMed] [Google Scholar]
- Gong Z.; Jiang G.; Cullen W. R.; Aposhian H. V.; Le X. C. Chem. Res. Toxicol. 2002, 15, 1318. [DOI] [PubMed] [Google Scholar]
- Mounicou S.; Lobinski R. Pure Appl. Chem. 2008, 80, 2565. [Google Scholar]
- Lobinski R.; Becker J. S.; Haraguchi H.; Sarkar B. Pure Appl. Chem. 2010, 82, 493. [Google Scholar]
- Garcia J. S.; Magalhães C. S.; Arruda M. A. Z. Talanta 2006, 69, 1. [DOI] [PubMed] [Google Scholar]
- Ge R.; Sun H. Sci. China Ser. B: Chem. 2009, 52, 2055. [Google Scholar]
- Sekhon B. Proc. Natl. Acad. Sci. India Section B-Biol. Sci. 2008, 78, 299. [Google Scholar]
- Haraguchi H. J. Anal. At. Spectrom. 2003, 19, 5. [Google Scholar]
- Jakubowski N.; Lobinski R.; Moens L. J. Anal. At. Spectrom. 2003, 19, 1. [Google Scholar]
- Aebersold R.; Mann M. Nature 2003, 422, 198. [DOI] [PubMed] [Google Scholar]
- Lin D.; Tabb D. L.; Yates J. R. Biochim. Biophys. Acta, Proteins Proteomics 2003, 1646, 1. [DOI] [PubMed] [Google Scholar]
- Thilakaraj R.; Raghunathan K.; Anishetty S.; Pennathur G. Bioinformatics 2007, 23, 267. [DOI] [PubMed] [Google Scholar]
- Fomenko D. E.; Marino S. M.; Gladyshev V. N. Mol. Cell 2008, 26, 228. [PMC free article] [PubMed] [Google Scholar]
- Delnomdedieu M.; Basti M. M.; Styblo M.; Otvos J. D.; Thomas D. J. Chem. Res. Toxicol. 1994, 7, 621. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee H.; Rosen B. P. J. Biol. Chem. 1996, 271, 24465. [DOI] [PubMed] [Google Scholar]
- Qin J.; Fu H. L.; Ye J.; Bencze K. Z.; Stemmler T. L.; Rawlings D. E.; Rosen B. P. J. Biol. Chem. 2007, 282, 34346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T.; Radaev S.; Rosen B. P.; Gatti D. L. EMBO J. 2000, 19, 4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob C.; Giles G. I.; Giles N. M.; Sies H. Angew. Chem., Int. Ed. 2003, 42, 4742. [DOI] [PubMed] [Google Scholar]
- Torrance J. W.The Geometry and Evolution of Catalytic Sites and Metal Binding Sites. Ph.D. Thesis, European Bioinformatics Institute, Robinson College, Cambridge, U.K., 2008.
- Ramadan D.; Rancy P. C.; Nagarkar R. P.; Schneider J. P.; Thorpe C. Biochemistry 2008, 48, 424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touw D. S.; Nordman C. E.; Stuckey J. A.; Pecoraro V. L. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 11969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramadan D.; Cline D. J.; Bai S.; Thorpe C.; Schneider J. P. J. Am. Chem. Soc. 2007, 129, 2981. [DOI] [PubMed] [Google Scholar]
- Cruse W. B. T.; James M. N. G. Acta Crystallogr. Sect. B: Struct. Crystallogr. Cryst. Chem. 1972, 28, 1325. [Google Scholar]
- Brenna O.; Perrella M.; Pace M.; Pietta P. G. Biochem. J. 1975, 151, 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X.; Larsen N. A.; Basran A.; Bruce N. C.; Wilson I. A. J. Biol. Chem. 2003, 278, 2008. [DOI] [PubMed] [Google Scholar]
- Glazer A. N. J. Biol. Chem. 1968, 243, 3693. [PubMed] [Google Scholar]
- Jain M. K. In Handbook of Enzyme Inhibitors; Wiley: New York, 1982. [Google Scholar]
- Zollner H.Handbook of Enzyme Inhibitors; VCH: New York, 1993; p 1065 [Google Scholar]
- Tsao D. H. H.; Maki A. H. Biochemistry 1991, 30, 4565. [DOI] [PubMed] [Google Scholar]
- Thompson A. A.; Peersen O. B. EMBO J. 2004, 23, 3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maignan S.; Guilloteau J. P.; Zhou-Liu Q.; Clément-Mella C.; Mikol V. J. Mol. Biol. 1998, 282, 359. [DOI] [PubMed] [Google Scholar]
- Tęte-Favier F.; Cobessi D.; Boschi-Muller S.; Azza S.; Branlant G.; Aubry A. Structure 2000, 8, 1167. [DOI] [PubMed] [Google Scholar]
- Raman C. S.; Li H.; Martásek P.; Babu B. R.; Griffith O. W.; Masters B. S. S.; Poulos T. L. J. Biol. Chem. 2001, 276, 26486. [DOI] [PubMed] [Google Scholar]
- Ye J.; Ajees A. A.; Yang J.; Rosen B. P. Biochemistry 2010, 49, 5206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J.; He Y.; Skalicky J.; Rosen B. P.; Stemmler T. L. Biomol. NMR Assign. 2011, 5, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y. F.; Yang J.; Rosen B. P. J. Biol. Chem. 2007, 282, 16783. [DOI] [PubMed] [Google Scholar]
- Gettins P.; Coleman J. E. J. Biol. Chem. 1984, 259, 4987. [PubMed] [Google Scholar]
- Sowadski J. M.; Handschumacher M. D.; Krishna Murthy H. M.; Foster B. A.; Wyckoff H. W. J. Mol. Biol. 1985, 186, 417. [DOI] [PubMed] [Google Scholar]
- Coutinho E. Pharma Times 2006, 38, 10. [Google Scholar]
- Forster M. J. Micron 2002, 33, 365. [DOI] [PubMed] [Google Scholar]
- Schwede T.; Sali A.; Eswar N.; Peitsch M. C.. Protein Structure Modeling. In Computational Structural Biology; World Scientific Publishing: Singapore, 2008. [Google Scholar]
- Fiser A. Expert Rev. Proteom. 2004, 1, 97. [DOI] [PubMed] [Google Scholar]
- Tsai M. D.; Yan H. Biochemistry 1991, 30, 6806. [DOI] [PubMed] [Google Scholar]
- Ruan X.; Bhattacharjee H.; Rosen B. P. J. Biol. Chem. 2006, 281, 9925. [DOI] [PubMed] [Google Scholar]
- Konermann L.; Stocks B. B.; Pan Y.; Tong X. Mass Spectrom. Rev. 2010, 29, 651. [DOI] [PubMed] [Google Scholar]
- Lim J.; Vachet R. W. Anal. Chem. 2003, 75, 1164. [DOI] [PubMed] [Google Scholar]
- Suzuki K. T.; Tomita T.; Ogra Y.; Ohmichi M. Chem. Res. Toxicol. 2001, 14, 1604. [DOI] [PubMed] [Google Scholar]
- Shiobara Y.; Ogra Y.; Suzuki K. T. Chem. Res. Toxicol. 2001, 14, 1446. [DOI] [PubMed] [Google Scholar]
- Schlichting I.X-ray Crystallography of Protein–Ligand Interactions. In Protein–Ligand Interactions: Methods and Applications; Nienhaus G. U., Ed.; Humana Press: Totowa, NJ, 2005; p 155. [DOI] [PubMed] [Google Scholar]
- Hu P.; Ye Q. Z.; Loo J. A. Anal. Chem. 1994, 66, 4190. [DOI] [PubMed] [Google Scholar]
- Hille R.; Stewart R. C.; Fee J. A.; Massey V. J. Biol. Chem. 1983, 258, 4849. [PubMed] [Google Scholar]
- Cline D. J.; Thorpe C.; Schneider J. P. J. Am. Chem. Soc. 2003, 125, 2923. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Rosen B. P. J. Biol. Chem. 1997, 272, 14257. [DOI] [PubMed] [Google Scholar]
- Jiang H.; Ding J.; Chang P.; Chen Z.; Sun G. Chromatographia 2010, 71, 1075. [Google Scholar]
- Lazdunski C.; Petitclerc C.; Chappelet D.; Lazdunski M. Biochem. Biophys. Res. Commun. 1969, 37, 744. [DOI] [PubMed] [Google Scholar]
- Petitclerc C.; Lazdunski C.; Chappelet D.; Moulin A.; Lazdunski M. Eur. J. Biochem. 1970, 14, 301. [DOI] [PubMed] [Google Scholar]
- Chen B.; Cao H.; Yan P.; Mayer M. U.; Squier T. C. Bioconjugate Chem. 2007, 18, 1259. [DOI] [PubMed] [Google Scholar]
- Wilcox D. E. Inorg. Chim. Acta 2008, 361, 857. [Google Scholar]
- Spuches A. M.; Kruszyna H. G.; Rich A. M.; Wilcox D. E. Inorg. Chem. 2005, 44, 2964. [DOI] [PubMed] [Google Scholar]
- Rey N. A.; Howarth O. W.; Pereira-Maia E. C. J. Inorg. Biochem. 2004, 98, 1151. [DOI] [PubMed] [Google Scholar]
- Schmidt A. C.; Neustadt M.; Otto M. J. Mass Spectrom. 2007, 42, 771. [DOI] [PubMed] [Google Scholar]
- Schmidt A. C.; Steier S. J. Mass Spectrom. 2010, 45, 870. [DOI] [PubMed] [Google Scholar]
- Schmidt A. C.; Mickein K. J. Mass Spectrom. 2012, 47, 949. [DOI] [PubMed] [Google Scholar]
- Raji M.; Frycak P.; Beall M.; Sakrout M.; Ahn J. M.; Bao Y.; Armstrong D.; Schug K. Int. J. Mass Spectrom. 2007, 262, 232. [Google Scholar]
- Gabelica V.; Galic N.; Rosu F.; Houssier C.; De Pauw E. J. Mass Spectrom. 2003, 38, 491. [DOI] [PubMed] [Google Scholar]
- Pizauro J. M.; Ciancaglini P.; Leone F. A. Mol. Cell. Biochem. 1995, 152, 121. [DOI] [PubMed] [Google Scholar]
- Crans D. C.; Simone C. M.; Holz R. C.; Que L. Jr. Biochemistry (N. Y.) 1992, 31, 11731. [DOI] [PubMed] [Google Scholar]
- Hirano K.; Iiizumi Y.; Mori Y.; Toyoshi K.; Sugiura M.; Iino S. J. Biochem. 1985, 97, 1461. [DOI] [PubMed] [Google Scholar]
- Saha A. K.; Crans D. C.; Pope M. T.; Simone C. M.; Glew R. H. J. Biol. Chem. 1991, 266, 3511. [PubMed] [Google Scholar]
- Pappas P. W.; Leiby D. A. J. Cell. Biochem. 1989, 40, 239. [DOI] [PubMed] [Google Scholar]
- Cyboron G. W.; Wuthier R. E. J. Biol. Chem. 1981, 256, 7262. [PubMed] [Google Scholar]
- Fernandes S. S.; Furriel R. P. M.; Petenusci S. O.; Leone F. A. Biochem. Pharmacol. 1999, 58, 841. [DOI] [PubMed] [Google Scholar]
- Sugiura Y.; Kawabe H.; Tanaka H.; Fujimoto S.; Ohara A. J. Biol. Chem. 1981, 256, 10664. [PubMed] [Google Scholar]
- Rezende A. A.; Pizauro J. M.; Ciancaglini P.; Leone F. A. Biochem. J. 1994, 301, 517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton R. H.; Moss D. W. Biochem. J. 1967, 102, 917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messens J.; Martins J. C.; Brosens E.; Van Belle K.; Jacobs D. M.; Willem R.; Wyns L. J. Biol. Inorg. Chem. 2002, 7, 146. [DOI] [PubMed] [Google Scholar]
- Messens J.; Hayburn G.; Desmyter A.; Laus G.; Wyns L. Biochemistry 1999, 38, 16857. [DOI] [PubMed] [Google Scholar]
- Zakharyan R. A.; Ayala-Fierro F.; Cullen W. R.; Carter D. M.; Aposhian H. V. Toxicol. Appl. Pharmacol. 1999, 158, 9. [DOI] [PubMed] [Google Scholar]
- Cheng Y.; Prusoff W. H. Biochem. Pharmacol. 1973, 22, 3099. [DOI] [PubMed] [Google Scholar]
- Wildfang E.; Zakharyan R. A.; Aposhian H. V. Toxicol. Appl. Pharmacol. 1998, 152, 366. [DOI] [PubMed] [Google Scholar]
- Ji G.; Garber E. A. E.; Armes L. G.; Chen C. M.; Fuchs J. A.; Silver S. Biochemistry 1994, 33, 7294. [DOI] [PubMed] [Google Scholar]
- Kedderis G. L.; Elmore A. R.; Crecelius E. A.; Yager J. W.; Goldsworthy T. L. Chem. Biol. Interact. 2006, 161, 139. [DOI] [PubMed] [Google Scholar]
- Hayakawa T.; Kobayashi Y.; Cui X.; Hirano S. Arch. Toxicol. 2005, 79, 183. [DOI] [PubMed] [Google Scholar]
- Helmreich E.; Cori C. F. Proc. Natl. Acad. Sci. U. S. A. 1964, 51, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmreich E.; Cori C. F. T. Proc. Natl. Acad. Sci. U. S. A. 1964, 52, 647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Verdier C. H.; Gould B. J. Biochim. Biophys. Acta 1963, 68, 333. [DOI] [PubMed] [Google Scholar]
- Eis C.; Nidetzky B. Biochem. J. 2002, 363, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nidetzky B.; Eis C.; Albert M. Biochem. J. 2000, 351, 649. [PMC free article] [PubMed] [Google Scholar]
- Schinzel R.; Drueckes P. FEBS Lett. 1991, 286, 125. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee H.; Sheng J.; Ajees A. A.; Mukhopadhyay R.; Rosen B. P. Biochemistry 2010, 49, 802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keng Y. F.; Wu L.; Zhang Z. Y. Eur. J. Biochem. 1999, 259, 809. [DOI] [PubMed] [Google Scholar]
- Zhang Y. L.; Zhang Z. Y. Anal. Biochem. 1998, 261, 139. [DOI] [PubMed] [Google Scholar]
- Szajn H.; Csopak H.; Folsch G. Biochim. Biophys. Acta, Enzymol. 1977, 480, 154. [DOI] [PubMed] [Google Scholar]
- Whisnant A. R.; Gilman S. D. Anal. Biochem. 2002, 307, 226. [DOI] [PubMed] [Google Scholar]
- VanEtten R. L.; Waymack P. P.; Rehkop D. M. J. Am. Chem. Soc. 1974, 96, 6782. [DOI] [PubMed] [Google Scholar]
- Zhang Z. Y.; Van Etten R. L. Arch. Biochem. Biophys. 1990, 282, 39. [DOI] [PubMed] [Google Scholar]
- Chawla S.; Mutenda E. K.; Dixon H. B.; Freeman S.; Smith A. W. Biochem. J. 1995, 308, 931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutenda E. K.; Sparkes M. J.; Dixon H. B. Biochem. J. 1995, 310, 983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams S. R.; Sparkes M. J.; Dixon H. B. Biochem. J. 1984, 221, 829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla S.; Dixon H. B. F. J. Enzyme Inhib. Med. Chem. 1995, 8, 255. [DOI] [PubMed] [Google Scholar]
- Visedo-Gonzalez E.; Dixon H. B. Biochem. J. 1989, 260, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambeir A. M.; Opperdoes F. R.; Wierenga R. K. Eur. J. Biochem. 1987, 168, 69. [DOI] [PubMed] [Google Scholar]
- Krietsch W. K. G.; Pentchev P. G.; Klingenbürg H.; Hofstätter T.; Bücher T. Eur. J. Biochem. 1970, 14, 289. [DOI] [PubMed] [Google Scholar]
- Nickbarg E. B.; Knowles J. R. Biochemistry 1988, 27, 5939. [DOI] [PubMed] [Google Scholar]
- Legrain C.; Stalon V. Eur. J. Biochem. 1976, 63, 289. [DOI] [PubMed] [Google Scholar]
- Wargnies B.; Legrain C.; Stalon V. Eur. J. Biochem. 1978, 89, 203. [DOI] [PubMed] [Google Scholar]
- Glazer A. N. Proc. Natl. Acad. Sci. U. S. A. 1968, 59, 996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galardy R. E.; Kortylewicz Z. P. Biochem. J. 1985, 226, 447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi N.; Matsumoto H. Comp. Biochem. Physiol., Part B: Comp. Biochem. 1985, 81, 587. [DOI] [PubMed] [Google Scholar]
- Kovach I. M. J Enzyme Inhib. Med. Chem. 1988, 2, 199. [DOI] [PubMed] [Google Scholar]
- Johnson J. L.; Cusack B.; Hughes T. F.; McCullough E. H.; Fauq A.; Romanovskis P.; Spatola A. F.; Rosenberry T. L. J. Biol. Chem. 2003, 278, 38948. [DOI] [PubMed] [Google Scholar]
- Auletta J. T.; Johnson J. L.; Rosenberry T. L. Chem. Biol. Interact. 2010, 187, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberry T. L. J. Mol. Neurosci. 2010, 40, 32. [DOI] [PubMed] [Google Scholar]
- Ripoll D. R.; Faerman C. H.; Axelsen P. H.; Silman I.; Sussman J. L. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson I. B.; Silman I. Biochemistry 1977, 16, 2701. [DOI] [PubMed] [Google Scholar]
- Fulladosa E.; Debord J.; Villaescusa I.; Bollinger J. C.; Murat J. C. Environ. Chem. Lett. 2007, 5, 115. [Google Scholar]
- Nafisi S.; Sobhanmanesh A.; Alimoghaddam K.; Ghavamzadeh A.; Tajmir-Riahi H. A. DNA Cell Biol. 2005, 24, 634. [DOI] [PubMed] [Google Scholar]
- Mandal S.; Ghosh A.; Pati B.; Das A. Toxicol. Environ. Chem. 2009, 91, 219. [Google Scholar]
- Kim M.; Um H. J.; Bang S. B.; Lee S. H.; Oh S. J.; Han J. H.; Kim K. W.; Min J.; Kim Y. H. Environ. Sci. Technol. 2009, 43, 9335. [DOI] [PubMed] [Google Scholar]
- Phatak P.; Dai F.; Butler M.; Nandakumar M.; Gutierrez P. L.; Edelman M. J.; Hendriks H.; Burger A. M. Clin. Cancer Res. 2008, 14, 4593. [DOI] [PubMed] [Google Scholar]
- Stewart R. C.; Hille R.; Massey V. J. Biol. Chem. 1984, 259, 14426. [PubMed] [Google Scholar]
- George G. N.; Bray R. C. Biochemistry 1983, 22, 1013. [DOI] [PubMed] [Google Scholar]
- Zechel D. L.; Konermann L.; Withers S. G.; Douglas D. Biochemistry 1998, 37, 7664. [DOI] [PubMed] [Google Scholar]
- Konermann L.; Pan J.; Wilson D. J. Compr. Anal. Chem. 2008, 52, 103. [Google Scholar]
- McMullen J. P.; Jensen K. F. Org. Process Res. Dev. 2011, 15, 398. [Google Scholar]
- Shin S. H.; Luchian T.; Cheley S.; Braha O.; Bayley H. Angew. Chem. 2002, 114, 3859. [DOI] [PubMed] [Google Scholar]
- Shin S. H.; Steffensen M. B.; Claridge T. D. W.; Bayley H. Angew. Chem., Int. Ed. 2007, 46, 7412. [DOI] [PubMed] [Google Scholar]
- Dilda P. J.; Hogg P. J. Cancer Treat. Rev. 2007, 33, 542. [DOI] [PubMed] [Google Scholar]
- Dixon H. B. F. Adv. Inorg. Chem. 1996, 44, 191. [Google Scholar]
- Rossman T.; Stone D.; Molina M.; Troll W. Environ. Mutagen. 1980, 2, 371. [DOI] [PubMed] [Google Scholar]
- Jacobson-Kram D.; Montalbano D. Environ. Mutagen. 1985, 7, 787. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.