

Abstract
The 4H syndrome (hypomyelination, hypodontia, hypogonadotropic hypogonadism) is a newly recognized leukodystrophy. The classical form is characterized by the association of hypomyelination, abnormal dentition, and hypogonadotropic hypogonadism, but the recent identification of 2 genes responsible for the syndrome demonstrates that these 3 main characteristics can be variably combined among “Pol-III related leukodystrophies”. The pathophysiology of this group of diseases is still to be elucidated and there are no neuropathological descriptions of brain tissue. We report the clinical, neuroradiological and neuropathological findings of a patient affected by 4H syndrome with confirmed POLR3A mutations. We found a marked loss of oligodendrocytes varying in severity in different brains regions accompanied by severe loss of myelin, moderately severe loss of axons and patchy perivascular regions of better-preserved white matter. There was relatively mild white matter astrogliosis and microgliosis. A macrophage reaction involving viable, normal-appearing oligodendroglia suggests the possibility of an immunologic process in this disorder. Cortical laminar astrogliosis and mineralization of layers I and II in particular was present. Thus, despite the uniformly hypomyelinating pattern seen on magnetic resonance imaging, neuropathological examination reveals a complex heterogeneous leukodystrophy with prominent neuro-axonal and glial involvement in this disorder.
Keywords: Hypodontia, Hypogonadotropic hypogonadism, Hypomyelination, Leukodystrophy, Pol-III
INTRODUCTION
Pol-III related leukodystrophies are a recently recognized group of leukodystrophies that include a series of overlapping clinical disorders (1). These include 4H syndrome (hypomyelination, hypodontia, hypogonadotropic hypogonadism), first described in 2006 by Timmons et al in 4 patients (2), but also earlier phenotypes of oligodontia with leukodystrophy (3), as well as later described cases of this association (4, 5). The classical form is characterized by the association of hypomyelination, abnormal dentition, and hypogonadotropic hypogonadism. The recent identification of 2 genes responsible for this autosomal recessive syndrome (POLR3A and POLR3B) demonstrates that these 3 main characteristics are variably present in patients with the so-called “Pol-III-related leukodystrophies” (1, 6, 7). The age of onset may be from early infancy to late childhood; the neurological picture is characterized by prominent cerebellar signs combined with a variable degree of pyramidal signs and may seem to be almost stable over several years (2, 4, 5, 8–14). A high level myopia has also been reported in several patients, as well as mild intellectual disability and/or slowly progressive cognitive difficulties (4, 5, 10, 11). Neuroradiological images show diffuse hypomyelination with cerebellar atrophy (2, 4, 5, 8–14). Published data of peripheral nerve histopathology show numerous intermediate and large myelinated axons with dark granular rings of material within the myelin sheath on cross section (2), but to our knowledge, no descriptions of brain neuropathology in clinically or genetically defined 4H syndrome have been published. Here we report the clinical, neuroradiological and neuropathological findings of a patient affected by mutation defined 4H syndrome.
MATERIALS AND METHODS
Case Report
The patient was a male already briefly reported (patient 2 in [2]) and included in the description of Pol-III mutations (1). He was born after an uneventful pregnancy and delivery and his early childhood was unremarkable except for the presence of delayed and missing teeth. At the age of 13 years, he was investigated because of the absence of spontaneous puberty and hypogonadotropic hypogonadism was diagnosed. Decline in school performance became evident during late childhood. Progression of cognitive dysfunction and onset of ataxia resulted in a first magnetic resonance imaging (MRI) performed at the age of 26 years, which revealed diffuse white matter hypomyelination sparing brainstem white matter and marked thinning of corpus callosum with mild cerebellar atrophy. He was referred for diagnostic work-up at the age of 28 years. When first examined by the authors, he presented with high-level myopia, mild dysphagia for solids, and gait dysfunction. Neurological examination showed upbeat nystagmus, hypometric saccades, fragmented smooth pursuit, dysarthria, spastic paraparesis, ataxia, hyporeflexia, dysmetria, and dysdiadochokinesia. Neuropsychiatric evaluation showed a total IQ of 62, with a verbal IQ of 72 and a performance IQ of 57. A follow-up MRI showed no significant changes (Fig. 1). Sural nerve biopsy was performed showing findings consistent with a demyelinating neuropathy (2); electromyogram and nerve conduction velocities were normal. Extensive laboratory and metabolic and genetic investigations were normal except for strongly positive antinuclear antibodies (4.2 EU) of still unexplained significance. Based on the clinical and neuroradiological data the diagnosis of 4H syndrome was made.
Figure 1.

Magnetic resonance (MR) imagines of patient with 4H syndrome. (A) MR T1-weighted axial scan demonstrates iso- or hyperintensity of the cerebral white matter. (B) MR T2-weighted axial scan demonstrates irregular hyperintensity of the cerebral white matter with hypointensity of the optic radiations (white arrow), lateral thalamus (thick black arrow), and dentate nucleus (thin black arrow).
The patient was followed until the age of 36 years. The clinical and neuroimaging findings (Fig. 1) remained globally stable except for the development of vertical gaze palsy, horizontal restrictive gaze and absence of smooth pursuit. The patient died unexpectedly at 36 years of age because of a foreign body obstruction in the supraglottic larynx and hypopharynx. Death occurred within minutes of accidental choking. There was no intercurrent illness, medications or nutritional deficiency reported at the time of death. The autopsy was performed within 12 hours of death.
Anatomical Pathology
The patient had signed consent under an IRB approved protocol at the National Institutes of Health, and the family provided consent for a research autopsy. The brain was extracted in its entirety by the usual bimastoidal incision and was bisected to separate left and right hemispheres. The right hemisphere was serially sectioned and frozen for research. After fixation in 10% formalin for 2 weeks, the left hemisphere, brainstem and cerebellum were serially sectioned and examined. Thalamus, midbrain-substantia nigra, pons-locus coeruleus-hypoglossal, vagus nucleus, floor of the 4th ventricle, medulla-olivary-pyramidal tracts, medulla-obex, mamillary body, globus pallidus, and septum-fornix were not available for review.
Hematoxylin and eosin (H&E), Luxol-fast blue/periodic acid-Schiff, and von Kossa stains and Bielschowsky impregnation were performed according to standard protocols: Luxol-fast blue (0.5% in 95% alcohol with acetic acid [Rowley Biochemical, Danvers, MA]); Bielschowsky stain kit was from American MasterTech, Lodi, CA (#KTBIE). Counterstaining was performed with PAS; von Kossa staining was performed with the American MasterTech kit # KTVKOPT.
Immunohistochemical sections of brain tissue were prepared from 4-μm-thick formalin-fixed paraffin sections, dried for 20 to 30 minutes in a 65° to 70°C oven. Deparaffinization and rehydration were carried out on the Ventana BenchMark XT or Ventana Ultra automated immunostainer (Ventana Medical Systems, Tucson, AZ) with Ventana EZ Prep and working Reaction Buffer per pre-programmed steps. Antigen retrieval was accomplished, as needed, with Ventana Cell Conditioning 1 (CC1, Tris-EDTA) or Ventana Cell Conditioning 2 (CC2, Citrate). Sections were incubated with either rabbit polyclonal, mouse monoclonal or rabbit monoclonal, as appropriate for the antibody (Ventana Medical Systems or other vendor) at 37°C. Detection and visualization were accomplished with Ventana’s I-View DAB kit with avidin/biotin blocking added, counterstained with Ventana Hematoxylin for 4 minutes and blued with Ventana Bluing Reagent for 4 minutes. Slides were removed from the autostainer, washed in warm soapy water, rinsed well with warm tap water, dehydrated through graded alcohols, cleared in xylene and mounted with resinous mounting medium. The Olig2 immunohistochemistry used OLIG2 Clone EPR2673, Rabbit monoclonal at 1:100, dilution (#ab109186, Abcam, Cambridge, MA); CC1 for 60 minutes, and incubation in primary antibody for 48 minutes. The anti-glial fibrillary acidic protein (GFAP) antibody used was Clone EP672Y, Mouse monoclonal, prediluted (Ventana #760-4345), exposed to CC1 for 60 minutes, incubation in primary for 4 minutes. The anti-CD68 antibody, Clone KP-1, Mouse monoclonal, was a predilute Ventana #790-2931, placed CC1 for 30 minutes, incubation in primary for 36 minutes. The anti-CD3 antibody was Clone 2GV6, rabbit monoclonal, predilute Ventana #790-4341, placed in CC1 for 60 minutes, incubation in primary for 32 minutes. The anti-CD20 was Clone L26, Mouse monoclonal, prediluted Ventana #760-2531 placed in CC1 for 30 minutes, incubation in primary for 40 minutes. Avidin/biotin block timing, detection reagents and DAB were programmed by Ventana according to the instrument and not alterable.
High-powered fields (HPFs) for cell counting were selected manually for the absence of staining artifact and avoiding areas with perivascular preservation. Cell counts per HPF were calculated as averages of manual cell counts of 10 HPFs at 40x objective 400 magnification using the 40x objective (Nikon Labophot-2 adapted to use Leica Fluocar Lenses). Relative numbers of oligodendroglia on H&E stain were compared to cell counts of Olig2-immunoreactive oligodendroglia per HPF in the cerebral white matter, centrum semiovale, and corpus callosum of the 4H patient vs. a control. The control was a 37-year-old male who died of a sudden pulmonary embolus. Additionally, random cell counts of 25 HPFs at 40x objective (400 magnification using the 40x objective) of Olig2 staining oligodendrocytes were performed in the centrum semiovale, corpus callosum and cerebral cortex at the cingulate gyrus in the 4H and control patients. These field sets were analyzed with a descriptive methodology because the sample size was too small for predictive statistics. Cell counts were assessed for mean and range, after discarding greatest and least values.
RESULTS
Neuroimaging
Cerebral MRI findings in this patient were consistent with previously reported abnormalities in 4H syndrome. There was increased intensity on T2 images and abnormal iso-or the normal hyperintense white matter signal on T1 imaging, consistent with a radiologic diagnosis of hypomyelination (15). There was also relative T2 hypointensity of the optic radiation, lateral nucleus of the thalamus and dentate nucleus (16). Cerebellar atrophy, an often-predominant feature, was not marked (2, 5).
Neuropathological Description
The brain weighed 1140 g. Macroscopic examination demonstrated a thin corpus callosum and dilated ventricles (Fig. 2A). Coronal sectioning of the left cerebral hemisphere revealed areas suggestive of hypo/demyelination of white matter, including the internal capsule (Fig. 2B, C). There were areas within the cerebral white matter that appeared lighter on gross examination (Fig. 2A–C). Serial sectioning of the brainstem and cerebellum also revealed areas suggestive of white matter demyelination. The brain was otherwise structurally intact.
Figure 2.

Macroscopic appearance of the brain. (A) Left hemisphere, sagittal view shows a thin corpus callosum and enlarged lateral ventricle. (B, C) Coronal sections reveal variegated areas of white matter granulation/discoloration suggestive of hypo/demyelination.
Histologic analysis demonstrated variable myelin rarefaction and loss accompanied by a significant, but less marked, reduction in axons. There was loss of myelin in the deep white matter and the optic radiations (Fig. 3A). Areas seen macroscopically to have a white appearance within the white matter were selected for analysis; the subcortical white matter of the insular cortex demonstrated irregular patches of relative myelin preservation and paler regions of marked myelin loss (Fig. 3B). Similarly, in the centrum semiovale there were areas of severe myelin loss (Fig. 3C) and axonal loss (Fig. 3D) along with areas of relative perivascular preservation.
Figure 3.

Brain histology. Regions that appear whiter on gross examination of the brain sections are shown in panels A–D. (A) Insula, cortical grey matter (CORTEX) and white matter (WM) have a reverse staining density pattern on Luxol fast-blue/periodic acid Schiff (LFB-PAS) stain. In a normal subject, the cortex is light blue and the WM is darker blue. A WM tract from a portion of the optic radiations has a marked reduction of myelin (arrows). Subcortical WM of the insular cortex has irregular patches of relative myelin preservation and paler regions of severe myelin loss (LFB-PAS). WM of centrum semiovale with severe myelin loss as demonstrated by staining with LFB-PAS (C). Myelin focally shows relatively better preservation in some perivascular areas. In a section adjacent to C, centrum semiovale WM shows marked axonal loss with focal perivascular preservation in a Bielschowsky preparation (D). (E) Staining of the cerebellar dentate nucleus and surrounding cerebellar deep white matter with LFB-PAS demonstrates am appropriate darker blue of the white matter and lighter blue of the dentate nucleus (DENTATE). There are large irregular regions of pallor and marked loss of myelin to the left of the dentate nucleus region. (F) In a Bielschowsky preparation on an adjacent section to E, the region immediately surrounding the dentate has relatively well preserved axons as well of the cerebellar dentate nucleus and surrounding cerebellar deep white matter. (G) The white matter of the cerebellar folia stained with LFB-PAS has a marked loss of myelin. In comparison, cerebellar cortical molecular (MOL), granular (GRAN) and Purkinje (arrowhead) cell layers are relatively well preserved. Atrophy of the cerebellar folia is mostly due to loss of WM myelin and axons. H: WM of the cerebellar folia with Bielschowsky preparation has a marked loss of axons. In comparison, cerebellar molecular (MOL), granular (GRAN) and Purkinje (arrowhead) cell layers are relatively well preserved. Atrophy of the cerebellar folia is mostly due to loss of myelin and axons of the white matter. Original magnification: A, B, 100x; C–H, 25x.
The cerebellar dentate nucleus and the immediate surrounding cerebellar deep white matter were well preserved although there was greater loss of myelin and axons within the cerebellar white matter further from the dentate (Fig. 3E, F). The cerebellar folia showed loss of myelin and axons with relative preservation of the granular layer (Fig. 3G, H). Atrophy of the cerebellar folia was mostly due to loss of white matter myelin and axons. White matter rarefaction, demyelination and astrogliosis extends to the distal gyral (folial) white matter. Rosenthal fibers were notably absent.
The cerebellar molecular layer had low cellularity with minimal astrogliosis. The pyriform layer had the normal complement of Purkinje cells without inclusions, vacuolation, degeneration, or axonal bulbs; Bergmann glia were minimally reactive. Granular cell layer neurons appeared to be mildly to moderately decreased in cellularity, corresponding to the loss to the extent seen in patients with longstanding chronic diseases. There was a small 5-mm cerebellar heterotopia in the cerebellar cortex and some displacement of the Purkinje cells into the molecular layer and granular cell layers. The rare displaced Purkinje cell bodies did not have the characteristics of axonal bulbs on Bielschowsky and appeared as neuron cell bodies with Bielschowsky and Luxol fast blue/PAS stain. There were no other abnormalities of the cerebellar cortex.
Oligodendroglial loss was most severe in the centrum semiovale, but there were better preserved areas with less rarefaction and preserved oligodendroglia in the white matter surrounding small capillaries, venules and arterioles (Fig. 4A–C, G). Axonal loss was moderate in the centrum semiovale (Fig. 4I), with areas of better-preserved axons near blood vessels (Fig. 4H). The striatal subcortical white matter tracts and internal capsule showed patchy rarefaction and myelin loss (Fig. 4D). U-fibers were not spared, i.e. there was rarefaction, myelin loss and loss of oligodendroglia underlying most gyri.
Figure 4.

Characterization of regional white matter (WM). (A) There is marked rarefaction (pale areas) indicative of myelin loss of the subcortical WM and throughout the centrum semiovale (lower half of figure). Darker areas indicate relative preservation of WM myelin. The relatively preserved areas tend to be around blood vessels (H&E). (B) Centrum semiovale showing marked rarefaction and myelin loss. The white matter in close proximity to blood vessels is better preserved or is less affected by the disease process (H&E). (C) Cerebral WM around small blood vessels and capillaries is less rarified and has more numerous oligodendroglia; the surrounding rarefied WM is less cellular with sparse glia (H&E). (D) Internal capsule WM is mostly intact but there are scattered patches of rarefaction (H&E). (E) WM tracts in the striatum are relatively preserved (H&E). (F) Centrum semiovale is rarified with marked myelin and oligodendrocyte loss (H&E). (G) Luxol-fast blue/Periodic acid Schiff (LFB/PAS) stain reveals rarefied pale-blue myelin in centrum semiovale and perivascular areas of better-preserved WM myelin. (H) Axons near blood vessels are also better preserved (Bielschowsky preparation). (I) Axonal loss is greater in regions with more prominent WM rarefaction and myelin loss (Bielschowsky). (J) Glial fibrillary acidic protein (GFAP)-immunoreactive fibrillary astrocytes are present in cortical layers V and VI. (K) In contrast, there is a lack of prominent astrogliosis in most regions of the subcortical WM and centrum semiovale, including most of the U fibers (GFAP). (L) Laminar, fibrillary astrogliosis in cortical layers I, V and VI. Laminar astrogliosis of layer III, which often accompanies gliosis in layer V, is not present. (GFAP). (M) Laminar mineralization of cortical layers I and II corresponds to the laminar astrogliosis (von Kossa stain for calcium). Original magnification: A, J, L, M, 2x; B–E, G, K, 4x; C, F, 10x; H, I, 20x.
Dense confluent laminar astrogliosis was most prominent in cortical layer VI, followed by cortical layers I and V (Fig. 4J, L). Laminar mineralization of cortical layers I and II corresponded to the laminar astrogliosis (Fig. 4M). Mineralization of layers I followed by layer II was most consistent and was diffuse. Small foci and patchy mineralization of deeper cortical layers V and VI were also seen. Similar areas of mineralization were seen within the granular layer of the cerebellum (not shown). Areas of the brain that had a chalky white appearance within the affected white matter on gross examination (Fig. 2) were analyzed using von Kossa staining for calcification, but were negative. In addition, areas with hypointense signal on MRI, such as the dentate, were assessed using von Kossa staining but were also negative. Astrogliosis was present in the white matter of the corpus callosum and centrum semiovale, but was relatively sparse. Regularly scattered, fibrillary astrogliosis was seen in the deeper layers of the cortex and in the immediate subcortical white matter, but dissipated within a few millimeters (Fig. 4K). The astrogliosis in the white matter was mostly perivascular with a few scattered fibrillary astrocytes in the more remote parenchyma.
Myelin loss in the centrum semiovale was accompanied by decreased numbers of oligodendroglia, as determined by H&E morphology and decreased numbers of Olig2-immunoreactive cells (Figs. 5, 6A–L). In the centrum semiovale, H&E counts were greater in the control (mean: 96; range: 82–100) vs. the 4H patient (mean: 35; range: 22–49); Olig2-reactive cell counts in the control (mean: 95; range: 90–97) vs. 4H patient (mean: 4.6; range: 0–7) indicated a 63% and 95% loss of oligodendrocytes, respectively, in the 4H patient. In the corpus callosum, H&E counts in the control (mean: 159; range: 120–178) were also greater than in the 4H patient (mean: 87; range: 84–91); Olig2-reactive cell counts in the control (mean: 105; range: 96–142) vs. the 4H patient (mean: 31; range: 27–42) indicated a 47% (H&E) and 70% (Olig2) loss of oligodendrocytes in the 4H patient. In the cerebral cortex H&E counts in the control (mean: 40; range: 32–50) were greater than in the 4H patient (mean: 6.6; range: 0–8); Olig2-reactive cell counts in the control (mean: 34.9; range: 27–44) vs. the 4H patient (mean: 5.6; range: 0–8) indicated a 83% (H&E) and 83% (Olig2) loss of oligodendrocytes on in the 4H patient. The disparity between the H&E counts and Olig2 counts in the centrum semiovale may be due to difficulty in counting the oligodendrocytes based on morphologic criteria alone, but may also indicate a preferential loss of Olig2-reactive oligodendrocytes.
Figure 5.

Quantification of oligodendroglia. Oligodendrocytes per high-powered field (HPF) were counted in the corpus callosum, centrum semiovale and cerebral cortex in the 4H patient and a control using both H&E and Olig2 staining. There are decreased numbers of oligodendrocytes in the 4H patient vs. the control. Average counts and ranges are shown.
Figure 6.
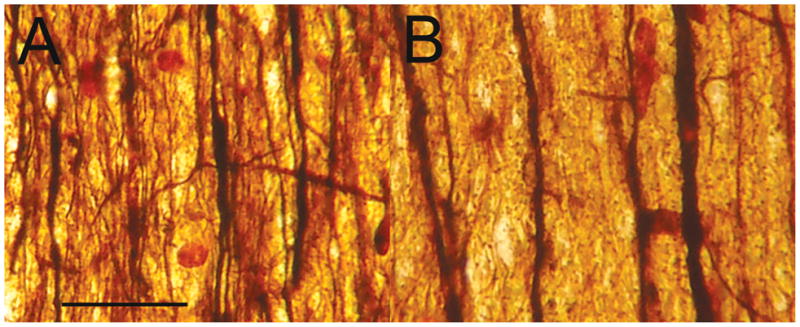
Details, distribution and characteristics of various cell types. (A) Centrum semiovale oligodendrocytes, some with a clear perinuclear halo, in a control sample (H&E). (B) Oligodendrocytes with nuclear reactivity for Olig2 in the control centrum semiovale. (C) Centrum semiovale the 4H patient with rarefaction of myelin, increased microglia, astrocytes and decreased numbers of oligodendrocytes (H&E). (D) Centrum semiovale in the 4H patient with markedly decreased numbers of Olig2-positive oligodendrocytes; only 4 to 5 oligodendrocytes are seen in this high power field. (E, F) Corpus callosum oligodendrocytes in a control sample (H&E, E; Olig2, F). (G) Corpus callosum in 4H patient with rarefaction, increased microglia and astrocytes and decreased numbers of oligodendroglia (H&E). (H) Corpus callosum in 4H patient oligodendrocytes with nuclear reactivity for olig2 showing decreased numbers of labeled oligodendrocytes. (I) Control sample cerebral cortex; there are 3 to 6 perineuronal oligodendrocytes per neuron in this plane of section (H&E). (J) Control sample cerebral cortex showing perineural and perivascular oligodendrocytes immunoreactive to Olig2. (K) Cerebral cortex in 4H patient with 0 to 1 perineural oligodendrocyte1 per neuron (H&E). (L) Cerebral cortex in 4H patient with a few Olig2-reactive oligodendrocytes; compare with (J). (M) Centrum semiovale in 4H patient. CD68-immunopositive macrophages/microglia are interspersed in the white matter. (N) Centrum semiovale blood vessel in 4H patient, with CD68-positive macrophages adherent or invested in perivascular adventitia or basement membrane. There are a few macrophages in the Virchow-Robin space. (O) CD68-immunoreactive macrophages in 4H patient within the endothelial layer or basement membrane layer of a microvessel. (P1–P4) In the 4H patient, CD68-positive macrophages extend toward (P1), encroach upon (P3) and partially (P2) encircle or completely encircle (P4) oligodendrocytes with clear perinuclear cytoplasm. (Q) CD68-positive macrophages with abundant cytoplasm are closely apposed to 2 oligodendrocytes in the 4H patient (original magnification 40x, CD68). (R) The vast majority of CD68 reactivity in the 4H patient is in macrophages associated with blood vessels. Only rare CD68 reactivity outlines oligodendrocytes (cell with clear cytoplasm) indicating current or prior interaction with macrophages or microglia. These features were not seen in the control. Original magnifications: A, C–E, G, M, N, 10x; B, F, H, I–L, O, 20x; P–R, 40x.
Similar results in Olig2 reactive oligodendrocyte counts were obtained on random cell counts in the centrum semiovale. Olig2-reactive counts in the control (mean: 117.6; range: 85–163) vs. the 4H patient (mean: 1.2; range: 0–9) indicated a 99% loss of oligodendrocytes in the 4H patient; in the corpus callosum Olig2 counts in the control (mean: 90.5; range: 54–138) vs. 4H patient (mean: 1.8; range: 0–8) indicated a 98% loss of oligodendrocytes in the 4H patient; in the cerebral cortex at the cingulate gyrus, Olig2 reactive oligodendroglia counts in the control (mean: 19,2; range: 9–31) vs. the 4H patient (mean: 3.5; range: 1–7) indicated an 82% loss of oligodendrocytes in the latter. Interneuron oligodendroglia were present in numbers equal to the perineuronal oligodendrocytes, although in normal circumstances the perineuronal oligodendrocytes outnumber the interneuronal oligodendrocytes. Because of the small sample sizes, the fact that only 1 4H patient vs. 1 control were studied, and because the range of cell counts had no overlap between the patient and control, statistical comparison of cell counting was not performed.
Macrophages and microglial activation was mild to moderate (Fig. 6M), and mostly involved the adventitia or basement membranes of capillaries, small venules and arterioles (Fig. 6N, O). Macrophages in the Virchow-Robin spaces, usually a common feature of many disease conditions, were not easily found. Rare CD68 reactive cells corresponding to macrophages were closely juxtaposed to oligodendroglia (Fig. 6P3, Q) and appeared to extend cell membrane pseudopodia (Fig. 6P1–3) toward or around oligodendroglia (Fig. 6P4, R). The membranes of a few rare oligodendrocytes were circumferentially highlighted with CD68 antigen (Fig. 6P4, R). There was a loss of axons in the cortex and white matter that was demonstrated in a comparison of the rarified centrum semiovale region and adjacent, relatively preserved perivascular region. Ten fields, each consisting of 20 μm transverse sections across axons, were counted. The perivascular regions had a total of 142 axons (14.2 axons/section) and 126 small caliber axons (12.6/section). The adjacent, demyelinated, centrum semiovale region had a total of 29 axons (2.9/section) and only 7 small caliber axons (Fig. 7). Together, this suggests significant axonal loss, although there were no regions with axonal swelling or other evidence of axonal disruption.
Figure 7.

Axonal component of 4H leukodystrophy. (A, B) The transverse sectional density of axons in the better preserved or less affected perivascular regions of the centrum semiovale (A) is compared to the greater rarefaction, myelin and axonal loss less than 1 mm away from the perivascular region in (B). Both intermediate and small caliber axons are present in greater numbers in (A); small caliber axons are particularly depleted in (B). Bielschowsky stain. Original magnification: 400x; Horizontal bar = 20 μm for both panels.
The cerebellar molecular layer had low cellularity with minimal astrogliosis. The pyriform layer had the normal complement of Purkinje cells without inclusions, vacuolation, degeneration, or axonal bulbs; Bergmann glia were minimally reactive. Granular cell layer neurons appeared to be mildly to moderately decreased in cellularity, corresponding to the loss to the extent seen in patients with longstanding chronic diseases. A single 0.5-cm cerebellar cortical heterotopia was noted. There were rare displaced Purkinje cells; Purkinje cells with neuron cell bodies completely in the molecular or granular cell layer in an otherwise clean cross section of cerebellum.
The hippocampus and related structures are mostly intact. White matter rarefaction was confined to scattered foci in the fimbria, along the alveus and into the corona radiate (gyri) hippocampi. The hippocampal fissure and lamina medullaris superficialis lacked calcifications and excessive astrogliosis and polyglucosan bodies. The CA4 proper, outer rim of the CA4 (third layer of the dentate cortex), dentate gyrus, outer layer of dentate cortex, CA3, CA2, proximal CA1 transitioning to Sommer’s sector, and distal CA1 were without significant loss of pyramidal cell neurons or astrogliosis. No degenerative changes were seen in the hippocampal CA1-4 regions. The regions of relative preservation of myelin and axons were in the perivascular regions directly abutting and surrounding some of the capillaries, small arterioles and small venules. These vessels had a thin wall composed of normal appearing endothelial cells. A few had a thin connective tissue basement membrane or at most a single smooth muscle layer. The vascular lumens were widely patent. Hyalinization, or thickening of the vascular walls were not seen. A few hemosiderin-laden macrophages were present in the perivascular spaces. Inflammation, reduplication of the basement membrane and perivascular astrogliosis were absent.
DISCUSSION
This first neuropathological description of 4H syndrome in a POLR3A-mutated patient demonstrates a more complex picture than the diffuse appearance of hypomyelination that characterizes the neuroimaging of patients with this disorder. Histopathologic analysis demonstrates white matter rarefaction, marked myelin and oligodendrocyte loss with moderate axonal loss. Patchy regions around small blood vessels have relatively better-preserved white matter with less rarefaction, myelin, and cellular loss. There was no clear involvement of blood vessels, which have normal appearing walls and lumens.
Myelin loss was also variable in different brain regions. Oligodendrocyte loss was most prominent in the centrum semiovale and cerebral cortex. The corpus callosum had a significant, but lesser degree of oligodendrocyte loss and the internal capsule appeared to be better preserved. Focal patchy rarefaction and demyelination was seen in the striatum, but in sporadic pattern, i.e. not a regional swath of demyelination as seen in osmotic demyelination syndrome in the striatum (17). Similarly, the area around the cerebellar dentate nucleus was relatively preserved. This suggests that areas of hypointensity on MRI may correspond to areas of preserved myelination and more normal signal intensity relative to the remainder of the abnormal white matter. Von Kossa staining showed no evidence of mineralization of these regions but it did demonstrate unexpected mineralization of a layer of cells within the cortical and cerebellar mantle.
There was a lesser degree of astrogliosis and microgliosis than might be expected in view of the severity of the other findings. Astrogliosis was present in the cortical grey matter, most prominent in layers I, V and VI, and most abundantly around small blood vessels. Astrocytes seemed to be decreased in number (especially in the white matter) in the 4H patient, but cell counts were not attempted. Fewer white matter astrocytes and scarce white matter astrogliosis on GFAP staining might reflect decreased number of astrocytes, decreased astrocytic reactivity, or suppressed astrocytic function or activity.
Macrophages and activated microglia were, for the most part, situated in the adventitia and basement membrane of small blood vessels. Fewer than expected were in the Virchow-Robin spaces. Only a few CD68-positive macrophages were present in the brain parenchyma and, even more rarely, they were associated with oligodendrocytes. Rare CD68-positive macrophages extended cytoplasmic and membranous pseudopodia around viable-appearing oligodendrocytes, a finding more consistent with an immunologic reaction than a phagocytic, “clean-up” function (Fig. 6P–R). A few rare CD3-positive T cells were seen in close approximation, juxtaposed against the cell membrane of an oligodendrocyte (not shown). Similarly, rare macrophages projected pseudopodia that partially encircled an astrocytic cell (not shown). This might represent a chance association, immune surveillance or an immunologic reaction. Of note, Pol-III detects cytosolic DNA and induces type I interferons through the RIG-I pathway, and thus an alteration of innate immune mechanisms might be involved (18).
Although axonal loss was seen in areas of demyelination, there was no apparent inherent abnormality of axons. The hippocampal CA2-CA4 pathology pattern associated with both ceroid lipofuscinosis and Gaucher disease, i.e. neuronal degeneration, neuron loss, microgliosis, astrogliosis with sparing of CA1 (19, 20), was not seen. We presume that axonal loss is secondary to myelin abnormalities and not a primary manifestation of this disorder, in particular because axonal loss was less prominent in perivascular areas of relatively spared myelination.
The underlying pathophysiologic mechanisms of Pol-III-related leukodystrophy are largely unknown. Pol III-related leukodystrophies are hypomyelinating autosomal recessive disorders caused by mutations in POLR3A and POLR3B (6–8, 21, 22). POLR3A and POLR3B encodes for the 2 largest subunits of the RNA polymerase III (Pol III). Together, they form the catalytic core of the polymerase. There are 3 different types of polymerases: Pol I, II and III. They are responsible for the transcription of DNA into RNA. Pol III is responsible for the transcription of small RNA such as all tRNA, the RNA 5S RNA, 2 elementary types of RNA that participate in the translation of mRNAs into proteins (24). In addition, Pol III transcribes the U6 RNA as well as the RNA components of RNAses P and MRP. These RNAses participate in the post-transcriptional processing of specific precursor RNAs. Furthermore, Pol III transcribes the 7SL RNA, which is a component of the signal recognition particle, and the 7SK, Alu and B2 RNAs, which are regulatory factors of Pol II transcription (24–26). The pathophysiology of these diseases could involve the reduction of certain specific RNAs, such as the tRNA, as has been suggested in other diseases with white matter involvement (27–29).
It is completely unknown, however, how this proposed mechanism would result in the perivascular sparing seen prominently in this case. Of note, there is no known clinical vasculopathy associated with disorder and, except for a few hemosiderin-laden macrophages in the perivascular spaces, the vessels and perivascular area were unremarkable in this patient. Breakdown of the blood-brain barrier often elicits a reaction from the endothelial cells, basement membrane and perivascular astrocytes resulting in reduplication of the basement membrane and pronounced perivascular astrogliosis (30). There was no evidence of other vasculopathy, inflammation or evidence of blood-brain barrier abnormality as seen in other conditions (31, 32). The relatively preserved cells and axons of the perivascular region might reflect increased availability or access to some protective factor in the perivascular zone. Possibilities include access to diffusible nutrients and oxygen, or greater clearance of compounds or metabolites either through diffusion or improved clearance by perivascular macrophages (33–35).
The neuropathology of 4H contrasts with demyelinating disorders (e.g. multiple sclerosis) that have prominent perivascular demyelination (36, 37), and with other conditions that have perivascular patterns of injury around small venules and arterioles, such as hypertension, ischemia, radiation injury, edema, and autoimmune, and chronic inflammatory conditions with increased lymphocytes or macrophages (32).
In summary, the brain pathology of the present 4H patient demonstrates variable myelin deficits in different brain regions consistent with hypomyelination, accompanied by less severe loss of axons and patchy perivascular regions of better-preserved white matter. Certain larger regions of preserved myelination, including that seen at the dentate nucleus, may correspond to previously described areas of hypointense signaling on brain MRI in these patients. Other unexpected findings, including macrophage reaction involving viable, normal-appearing oligodendroglia and laminar cortical mineralization require validation in additional cases. Together, these features underline the complexity of pathological processes that may appear on neuroimaging as diffuse and fairly uniform hypomyelination.
Acknowledgments
This work was supported by the intramural program at the National Institutes of Health, National Human Genome Research Institute and the National Institute of Neurologic Disorders and Stroke, and by the Myelin Disorders Bioregistry Project at Children’s National Medical Center. The contributions of GB were supported by the “Fondation sur les Leucodystrophies”, the “Fondation Go”, the Montreal Children’s Hospital and Montreal University Health Center Research Institutes and FRSQ (Fonds de Recherche en Santé du Québec).
References
- 1.Bernard G, Chouery E, Putorti ML, et al. Mutations of POLR3A encoding a catalytic subunit of RNA Polymerase Pol III cause a recessive Hhypomyelinating leukodystrophy. Am J Hum Genet. 2011;89:415–23. doi: 10.1016/j.ajhg.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Timmons M, Tsokos M, Asab MA, et al. Peripheral and central hypomyelination with hypogonadotropic hypogonadism and hypodontia. Neurology. 2006;67:2066–9. doi: 10.1212/01.wnl.0000247666.28904.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Atrouni S, Darazé A, Tamraz J, et al. Leukodystrophy associated with oligodontia in a large inbred family: fortuitous association or new entity? Am J Med Genet A. 2003;118:76–81. doi: 10.1002/ajmg.a.10019. [DOI] [PubMed] [Google Scholar]
- 4.Sato I, Onuma A, Goto N, et al. A case with central and peripheral hypomyelination with hypogonadotropic hypogonadism and hypodontia (4H syndrome) plus cataract. J Neurol Sci. 2011;300:179–81. doi: 10.1016/j.jns.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 5.Wolf NI, Harting I, Innes AM, et al. Ataxia, delayed dentition and hypomyelination: a novel leukoencephalopathy. Neuropediatrics. 2007;38:64–70. doi: 10.1055/s-2007-985137. [DOI] [PubMed] [Google Scholar]
- 6.Tetreault M, Choquet K, Orcesi S, et al. Recessive mutations in POLR3B, encoding the second largest subunit of Pol III, cause a rare hypomyelinating leukodystrophy. Am J Hum Genet. 2011;89:652–5. doi: 10.1016/j.ajhg.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bernard G, et al. Mutations of POLR3A encoding a catalytic subunit of RNA polymerase Pol III cause a recessive hypomyelinating leukodystrophy. Am J Hum Genet. 2011;89:415–23. doi: 10.1016/j.ajhg.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saitsu H, Osaka H, Sasaki M, et al. Mutations in POLR3A and POLR3B encoding RNA Polymerase III subunits cause an autosomal-recessive hypomyelinating leukoencephalopathy. Am J Hum Genet. 2011;89:644–51. doi: 10.1016/j.ajhg.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wolff A, Koch MJ, Benzinger S, et al. Rare dental peculiarities associated with the hypomyelinating leukoencephalopathy 4H syndrome/ADDH. Pediatr Dent. 2010;32:386–92. [PubMed] [Google Scholar]
- 10.Vazquez-Lopez M, Ruiz-Martín Y, de Castro-Castro P, et al. Central hypomyelination, hypogonadotrophic hypogonadism and hypodontia: a new leukodystrophy. Rev Neurol. 2008;47:204–8. [PubMed] [Google Scholar]
- 11.Outteryck O, Devos D, Jissendi P, et al. 4H syndrome: a rare cause of leukodystrophy. J Neurol. 2010;257:1759–61. doi: 10.1007/s00415-010-5598-0. [DOI] [PubMed] [Google Scholar]
- 12.Orcesi S, Tonduti D, Uggetti C, et al. New case of 4H syndrome and a review of the literature. Pediatr Neurol. 2010;42:359–64. doi: 10.1016/j.pediatrneurol.2010.01.015. [DOI] [PubMed] [Google Scholar]
- 13.Bekiesinska-Figatowska M, Mierzewska H, Kuczynska-Zardzewialy A, et al. Hypomyelination, hypogonadotropic hypogonadism, hypodontia - First Polish patient. Brain Dev. 2010;32:574–8. doi: 10.1016/j.braindev.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 14.Chouery E, Delague V, Jalkh N, et al. A whole-genome scan in a large family with leukodystrophy and oligodontia reveals linkage to 10q22. Neurogenetics. 2010;12:73–78. doi: 10.1007/s10048-010-0256-3. [DOI] [PubMed] [Google Scholar]
- 15.Bernard G, Thiffault I, Tetreault M, et al. Tremor–ataxia with central hypomyelination (TACH) leukodystrophy maps to chromosome 10q22.3–10q23. 31. Neurogenetics. 2010;11:457–464. doi: 10.1007/s10048-010-0251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schiffmann R, van der Knaap MS. Invited article: an MRI-based approach to the diagnosis of white matter disorders. Neurology. 2009;72:750–9. doi: 10.1212/01.wnl.0000343049.00540.c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steenweg ME, Vanderver A, Blaser S, et al. Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain. 2010;133:2971–82. doi: 10.1093/brain/awq257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Souza A, Desai PK. More often striatal myelinolysis than pontine? A consecutive series of patients with osmotic demyelination syndrome. Neurol Res. 2012;34:262–71. doi: 10.1179/1743132812Y.0000000009. [DOI] [PubMed] [Google Scholar]
- 19.Chiu YH, Macmillan JB, Chen ZJ. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell. 2009;138:576–91. doi: 10.1016/j.cell.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tyynelä J, Cooper JD, Khan MN, et al. Hippocampal pathology in the human neuronal ceroid-lipofuscinoses: distinct patterns of storage deposition, neurodegeneration and glial activation. Brain Pathol. 2004;14:349–57. doi: 10.1111/j.1750-3639.2004.tb00077.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong K, Sidransky E, Verma A, et al. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol Genet Metab. 2004;82:192–207. doi: 10.1016/j.ymgme.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 22.Potic A, Brais B, Choquet K, et al. 4H Syndrome With Late-Onset Growth Hormone Deficiency Caused by POLR3A Mutations. Arch Neurol. 2012;69:920–3. doi: 10.1001/archneurol.2011.1963. [DOI] [PubMed] [Google Scholar]
- 23.Terao Y, Saitsu H, Segawa M, et al. Diffuse central hypomyelination presenting as 4H syndrome caused by compound heterozygous mutations in POLR3A encoding the catalytic subunit of polymerase III. J Neurol Sci. 2012;320:102–5. doi: 10.1016/j.jns.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 24.Szymanski M, Barciszewska MZ, Erdmann VA, et al. 5 S rRNA: structure and interactions. Biochem J. 2003;371:641–51. doi: 10.1042/BJ20020872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dieci G, Fiorino G, Castelnuovo M, et al. The expanding RNA polymerase III transcriptome. Trends Genet. 2007;23:614–22. doi: 10.1016/j.tig.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 26.Jeronimo C, Forget D, Bouchard A, et al. Systematic analysis of the protein interaction network for the human transcription machinery reveals the identity of the 7SK capping enzyme. Mol Cell. 2007;27:262–74. doi: 10.1016/j.molcel.2007.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krueger BJ, Jeronimo C, Roy BB, et al. LARP7 is a stable component of the 7SK snRNP while P-TEFb, HEXIM1 and hnRNP A1 are reversibly associated. Nucleic Acids Res. 2008;36:2219–29. doi: 10.1093/nar/gkn061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feinstein M, Markus B, Noyman I, et al. Pelizaeus-Merzbacher-like disease caused by AIMP1/p43 homozygous mutation. Am J Hum Genet. 2010;87:820–8. doi: 10.1016/j.ajhg.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Edvardson S, Shaag A, Kolesnikova O, et al. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am J Hum Genet. 2007;81:857–62. doi: 10.1086/521227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scheper GC, van der Klok T, van Andel RJ, et al. Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat Genet. 2007;39:534–9. doi: 10.1038/ng2013. [DOI] [PubMed] [Google Scholar]
- 31.Abbott NJ. Astrocyte-endothelial interactions and blood-brain barrier permeability. J Anat. 2002;200:629–38. doi: 10.1046/j.1469-7580.2002.00064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Errede M, Girolamo F, Ferrara G, et al. Blood-brain barrier alterations in the cerebral cortex in experimental autoimmune encephalomyelitis. J Neuropathol Exp Neurol. 2012;71:840–54. doi: 10.1097/NEN.0b013e31826ac110. [DOI] [PubMed] [Google Scholar]
- 33.Nico B, Ribatti D. Morphofunctional aspects of the blood-brain barrier. Curr Drug Metab. 2012;13:50–60. doi: 10.2174/138920012798356970. [DOI] [PubMed] [Google Scholar]
- 34.Denis J, Delorme ML, Boschat M, et al. Respective roles of ammonia, amino acids, and medium-sized molecules in the pathogenesis of experimentally induced acute hepatic encephalopathy. J Neurochem. 1983;40:10–9. doi: 10.1111/j.1471-4159.1983.tb12646.x. [DOI] [PubMed] [Google Scholar]
- 35.Deguchi T, Isozaki K, Yousuke K, et al. Involvement of organic anion transporters in the efflux of uremic toxins across the blood-brain barrier. J Neurochem. 2006;96:1051–9. doi: 10.1111/j.1471-4159.2005.03550.x. [DOI] [PubMed] [Google Scholar]
- 36.Pardridge WM. Drug transport across the blood-brain barrier. J Cereb Blood Flow Metab. 2012 doi: 10.1038/jcbfm.2012.126. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ge Y, Law M, Herbert J, et al. Prominent perivenular spaces in multiple sclerosis as a sign of perivascular inflammation in primary demyelination. AJNR Am J Neuroradiol. 2005;26:2316–9. [PMC free article] [PubMed] [Google Scholar]
- 38.Alvarez JI, Dodelet-Devillers A, Kebir H, et al. The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science. 2011;334:1727–31. doi: 10.1126/science.1206936. [DOI] [PubMed] [Google Scholar]