

Abstract
Cytogenetic and molecular cytogenetic studies demonstrate association between congenital diaphragmatic hernia (CDH) and chromosome 1q41q42 deletions. In this study, we screened a large CDH cohort (N=179) for microdeletions in this interval by the multiplex ligation-dependent probe amplification (MLPA) technique, and also sequenced two candidate genes located therein, dispatched 1 (DISP1) and homo sapiens H2.0-like homeobox (HLX). MLPA analysis verified deletions of this region in two cases, an unreported patient with a 46,XY,del(1)(q41q42.13) karyotype and a previously reported patient with a Fryns syndrome phenotype [Kantarci et al., 2006]. HLX sequencing showed a novel but maternally inherited single nucleotide variant (c.27C>G) in a patient with isolated CDH, while DISP1 sequencing revealed a mosaic de novo heterozygous substitution (c.4412C>G; p.Ala1471Gly) in a male with a left-sided Bochdalek hernia plus multiple other anomalies. Pyrosequencing demonstrated the mutant allele was present in 43%, 12%, and 4.5% of the patient’s lymphoblastoid, peripheral blood lymphocytes, and saliva cells, respectively. We examined Disp1 expression at day E11.5 of mouse diaphragm formation and confirmed its presence in the pleuroperitoneal fold, as well as the nearby lung which also expresses Sonic hedgehog (Shh).
Our report describes the first de novo DISP1 point mutation in a patient with complex CDH. Combining this finding with Disp1 embryonic mouse diaphragm and lung tissue expression, as well as previously reported human chromosome 1q41q42 aberrations in patients with CDH, suggests that DISP1 may warrant further consideration as a CDH candidate gene.
Keywords: congenital diaphragmatic hernia (CDH), chromosome 1q41q42 deletion, microdeletion, MLPA, pyrosequencing, Sonic Hedgehog (SHH) pathway, pleuroperitoneal fold (PPF)
INTRODUCTION
The etiology of the common birth defect, congenital diaphragmatic hernia (CDH), in the majority of cases is unknown though growing evidence suggests genetic aberrations can cause or contribute to CDH. Recurrent deletions, involving chromosomes 15q26.1 [OMIM 142340, DIH1] and 8p23.1 [OMIM 222400, DIH2], are one such class of defects and constitute CDH “hotspots” or critical regions. Their further characterization may ultimately pinpoint genes responsible for CDH [Holder et al., 2007; Kantarci et al., 2006; Klaassens et al., 2005; Scott et al., 2007; Shaffer et al., 2007].
Our previous report of a patient with a Fryns syndrome phenotype was the first observation of a de novo 1q41q42.12 microdeletion in a patient with complex CDH (i.e., CDH plus additional anomalies) [Kantarci et al., 2006]. Rearrangements involving this locus detected by standard chromosome analyses and array-based comparative genomic hybridization (aCGH) have been reported in other patients with CDH [Rogers et al., 1995; Shaffer et al., 2007; Slavotinek et al., 2006; Youssoufian et al., 1988]. A compelling candidate gene mapping to this interval is Dispatched 1 (DISP1) [OMIM 607502] given its role in organogenesis [Ma et al., 2002; Tian et al., 2004].
We used several approaches to determine the importance to diaphragmatic hernia of the human chromosome region 1q41q42.12 and selected genes therein. We applied the multiplex ligation-dependent probe amplification (MLPA) technique to a large cohort of CDH patients to determine the prevalence of microdeletions and microduplications; we also screened a subset of these patients for exon copy number changes in the candidate gene, DISP1. We sequenced the DISP1 and HLX coding regions in a large cohort of chromosomally normal patients with CDH. Finally, we examined the developing mouse diaphragm, lung, and other adjacent tissues for expression of Disp1 and several related genes, since such studies can provide evidence that putative gene candidates contribute to diaphragm organogenesis or the often lethal pulmonary hypoplasia and pulmonary hypertension that co-exists with CDH [Lage et al., 2008; Naxerova et al., 2008]. Rodents are the most accepted model system for the study of diaphragm development, and in mice, the diaphragm is completely formed and muscularized by day 15-16 of gestation (of 19.5 days average gestation). The primordial diaphragm, first visible at day E11-12 of gestation in the mouse, consists of mesenchymal cells that migrate from the lateral dermatomyotome cephalad to their final position near the liver forming a structure referred to as the pleuroperitoneal fold (PPF). A defect in the integrity of the PPF has been demonstrated in association with diaphragmatic hernia in mice [Allan and Greer, 1997; Clugston et al., 2006] and genes known to be important for diaphragm development have been identified in these tissues.
MATERIAL AND METHODS
Study participants
We analyzed 179 CDH patients recruited from several centers into our ongoing NICHD funded study “Gene Mutations and Rescue in Human Congenital Diaphragmatic Hernia.” The families enrolled in this study gave their informed consent. CDH patients were phenotyped by the same geneticist (BRP) as having either isolated CDH (the absence of additional major malformations) or complex CDH (the presence of other birth defects, chromosome abnormalities or recognized genetic syndromes) based on information obtained from a parental questionnaire, medical record review, family pedigree, physical examination, and clinical or research diagnostic studies such as routine karyotyping, array Comparative Genomic Hybridization (aCGH), and imaging of the brain or heart. Two-thirds of this cohort was classified as having isolated CDH, while the remainder were classified as having complex CDH.
Blood, skin, cheek swabs, saliva, or amniotic fluid samples were obtained on probands as a prerequisite for study participation. Patients who died from complications of CDH had DNA extracted from formalin-fixed paraffin-embedded or frozen tissues. The Institutional Review Boards at MassGeneral Hospital for Children and Children’s Hospital, Boston provided annual review and approval of our study protocol.
Three patients in our cohort initiated these studies.
Patient 1
A previously unreported 6-year-old male patient was the full-term 3.3 kg first born product of healthy unrelated Caucasian parents. Family history was unremarkable for congenital anomalies except for a maternal uncle with cleft lip/palate who died at three months of age. Prenatal monitoring revealed fetal heart abnormalities (ventricular septal defect and abnormal aorta), left-sided Bochdalek congenital diaphragmatic hernia, and a left-sided cleft lip with bilateral cleft palate. Amniocentesis was not performed. Multiple abnormalities were confirmed at delivery. He was hypotonic and a G-tube was required for feeding. Postnatal ultrasound examination of the brain was normal. Chromosome analysis and subtelomeric FISH studies on peripheral blood samples were normal. Surgical repairs of his cardiac, diaphragm, and craniofacial anomalies were performed, as was surgical release of his postnatally detected spinal cord tethering. A short neck, torticollis, scoliosis, bilateral single transverse palmar creases, and wide thumbs were evident. His latest neurodevelopmental assessment at almost 5 years of age was normal save for hypotonia. He currently attends a regular classroom and no longer needs educational supports such as speech and language therapy. He had no history of seizures. Growth parameters at 210/12 were as follows: weight = 4 kg (50-75th centile), height =89.5 cm (10th centile), and head circumference = 48 cm (10-25th centile), while at 8 years of age all growth parameters were between the 3rd-10th centile.
This patient’s DNA was extracted from several tissue sources including an EBV transformed lymphoblastoid cell line, peripheral blood lymphocytes, and saliva. Parental DNA was obtained from both transformed cell lines and saliva, while DNA from the proband’s brother without CDH was obtained only from saliva (given that he was being evaluated for Factor IX deficiency). Sequencing, restriction endonuclease, and pyrosequencing analyses (see below) were carried out using the available sources of patient, parent, and sibling DNA.
Patient 2
A previously unreported full-term male infant with left medial diaphragmatic eventration, pulmonary hypoplasia and lobation abnormalities, bilateral talipes equinovarus, undescended testes, and dysmorphic facial features died at one month of age. Neuropathology was normal except for subdural hematomas of unknown origin. Chromosome analysis revealed a 46,XY,del(1)(q41q42.13) karyotype. The parents were a nonconsanguineous couple with one prior healthy child, but were not available for cytogenetic study. Patient DNA was isolated from formalin-fixed paraffin-embedded tissue.
Patient 3
A term female infant with findings of Fryns syndrome including a large left posterior medial hernia, a right medial diaphragmatic eventration, bilateral pulmonary hypoplasia, coarse facial features, hypertelorism, enlarged fontanelles, midline cleft of the soft palate, and moderate hypoplasia of the nails was previously reported [Kantarci et al., 2006; Shaffer et al., 2007]. Neuropathologic examination revealed no abnormalities. Prenatal and postnatal routine chromosome analyses were normal but a Spectral Genomechip V1.2 array demonstrated a ~5 million base pair deletion including RP11-124J24 (218,853,835-219,017,536 at 1q41) and RP5-1090A23 (223,724,297-223,981,854 at 1q42.12) BAC probes.
DNA extraction
Genomic DNA was isolated from blood, frozen tissue, and amniotic fluid samples using the Puregene DNA Purification Kit (Gentra Systems, Minneapolis, MN) according to the manufacturer’s recommendations, while a standard phenol/chloroform extraction protocol was used for transformed lymphoblastoid and cultured fibroblast lines. The QIAamp DNA Mini Kit (Qiagen Inc., Valencia, CA) was used to extract DNA from saliva and formalin-fixed paraffin embedded autopsy tissue. Ethnically matched control DNA samples were provided by the laboratory of Drs. John and Christine Seidman. The amount of DNA contained in each sample was quantified with the NanoDrop ND-Spectrophotometer (NanoDrop Technologies, Wilmington, DE).
MLPA
The MLPA kit reagent EK5 was obtained from MRC-Holland (Amsterdam, the Netherlands) and all reactions were carried out as recommended (http://www.mrcholland.com/mlpa_dna_protocol.htm). This kit contains the necessary reagents except the MLPA probe mixture. The target probes for the 1q41q42.12 region and the DISP1 exons (see below) were adapted from a method previously described [Stern et al., 2004]. Control probes were chosen from the chromosome 15q26 region. Probes were localized according to the USCS Genome browser [the March 2006 (hg18) human reference sequence (NCBI Build 36.1)] The MLPA probe sequences are available upon request. To verify both the position and the uniqueness (especially across the ligation site) of each probe, we used BLAT (UCSC;http://genome.ucsc.edu/cgi-bin/hgBlat) and BLAST (NCBI; http://www.ncbi.nlm.nih.gov/BLAST/) programs. The probes were ordered from Sigma Genosys (http://www.sigmaaldrich.com/life-science/custom-oligos.html) and were desalted without further purification. Product separation was performed using an ABI 3730XL DNA Analyzer (Applied Biosystems). Following analysis of peaks by GeneMapper v3.5 (ABI), the peak height data were exported to an Excel file. An Excel program was adapted [Kozlowski et al., 2007] and used to normalize peak height data, in which control samples were assigned a value of 1 after normalization. As a summary, all peak heights belonging to each probe were divided by the average signal from two control probes located on different chromosomes.
A) 1q41q42.12 Region
All CDH patients were screened for copy number changes (i.e., microdeletions and microduplications) of the 1q41q42.12 region by MLPA. Target probes were selected from eleven genes located in this interval: PTPN14 (212,768,242-212,791,265 at 1q41), USH2A (214,413,915-214,663,361 at 1q41), EPRS (218,227,107-218,286,623 at 1q41), BPNT1 (218,297,696-218,329,814 at 1q41), DUSP10 (219,941,389-219,982,084 at 1q41), CAPN2 (222,011,949-222,030,343 at 1q41), LBR (223,660,887-223,682,407 at 1q41.12), ACBD3 (224,399,003-224,441,046 at 1q41.12), ITPKB (224,961,518-224,993,499 at 1q41.12), PSEN2, (225,129,689-225,150,427 at 1q41.12 at 1q41.13) and TAF5L (227,801,564-227,828,417 at 1q42.13), respectively. These probes were spaced ~1.5 Mb apart throughout this interval.
B) DISP1 exons
We designed seven MLPA probes targeting DISP1 exons 2-8 to screen for copy number changes in the coding regions. We applied this probe panel to two dozen complex CDH patients whose anomalies were most compatible with the broad expression pattern of SHH.
Sequencing of Gene Candidates
The coding exons and flanking intronic base pairs of DISP1 and a second candidate gene mapping to the chromosome 1q41q42.12 interval, homo sapiens H2.0-like homeobox (HLX) [OMIM 142995], were sequenced in both directions in all 179 patients in our cohort. Sequencing was carried out by fluorescent dye-terminator chemistry using ABI Prism 3730 XL. Sequences were analyzed using Phred 2003 base-calling software [Stein, 2003].
Three previously unreported DISP1 variants and one HLX variant were genotyped in probands and available parents. The primers were designed using Primer3 (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi) and were synthesized by Sigma-Genosys (The Woodlands, TX). Genotypes were analyzed using Sequencher™ DNA sequencing software (Gene Codes Corporation, Ann Arbor, MI). Primer sequences for sequencing and genotyping are available upon request. Sequence changes were evaluated by PolyPhen (http://genetics.bwh.harvard.edu/pph) and SIfT (http://SIfT.jcvi.org/) programs to predict if changes were damaging.
Restriction Endonuclease Analysis
We performed restriction endonuclease analysis to confirm the de novo c.4412C>G DISP1 mutation (p.Ala1471Gly) identified in Patient 1. The PCR products using forward primer 5′-GGAAAGTGGAGCTGAGCTTG-3′ and reverse primer 5′-CCATGTTTGGCATTTGACAG-3′ were digested by the restriction endonuclease, CviKI-1 enzyme (New England Biolabs, Ipswich, MA) which specifically recognizes and cleaves the nucleotide sequence 5′-RG↓CY-3′. The 10μl of PCR products (195bp) were incubated with CviKI-1 (10U) overnight at 37°C. The digestion products were separated by gel electrophoresis on a 3% Nusieve-agarose gel (FMC Corporation Philadelphia, PA) and photographed under UV light.
Pyrosequencing
To quantify the tissue levels of the DISP1 mutation identified in Patient 1, an assay was designed using Pyrosequencing Design Software (Biotage, Uppsala, Sweden). We specifically interrogated the base underlined in the following sequence: GSTGGTTGTA GGTCTTGCCC AAATAAT The optimal primers chosen were: Forward primer 5′-ACGGATGCAAGTGTGAACTCAGA-3′; and 5′ biotinylated reverse primer 5′-TTTGTGAATTATTTGGGCAAGACC-3′.
PCR was performed with these primers according to standard protocols and generated a product 109bp in length, with a biotin attached to the bottom strand. This strand was isolated by immobilization onto streptavidin-coated beads, and annealed to the sequencing primer (5′-TCATTTAATGGGGGAG-3′), whose 3′ end is one base away from the base of interest at nucleotide position 4412 of DISP1. Pyrosequencing [Fakhrai-Rad et al., 2002] was carried out using the PSQ96 Reagent Kit according to the manufacturer’s recommendation, on a Pyromark MD machine (Pyrosequencing AB, Uppsala, Sweden). Percentages of the variant base were calculated using the built in AQ (Allele Quantification) software module (Biotage, Uppsala, Sweden).
Gene Expression Studies
In situ hybridization was used to detect expression of Disp1, Sonic Hedgehog (Shh), Indian Hedgehog (Ihh), and Patched1 (Ptch1) during diaphragm development. Embryos from timed pregnant wildtype FVB/J or A/J mice were collected on day E11.5 with morning of plug defined as day 0.5. All procedures were performed in accordance with University Committee on Animal Resources guidelines. Embryos were fixed in 4% fresh paraformaldehyde (PFA) overnight, processed with standard techniques, and paraffin embedded. Non-radioactive in situ hybridization was performed with standard techniques (additional methods provided in Supplement 1 available online).
Fate Mapping Shh Expressing Cells to the Diaphragm
Mice with EGFP-Cre recombinase knocked in to the Shh locus have been previously described [Harfe et al., 2004]. These mice, Shh tm1(EGFP/cre)Cjt were obtained from Jackson Laboratories (JAX #005622) and are abbreviated as Shh GFP-Cre. Mice carrying a reporter (B6.129S4-Gt(ROSA)26Sortm1Sor/J) (“R26R”, JAX #003474) were also obtained from Jackson. Timed pregnancies were set between Shh GFP-Cre male heterozygotes and R26R female mice, and embryos were collected either at E11.5 or just after birth. E11.5 embryos were fixed with glutaraldehyde fixative for X-Gal staining as previously described [Ackerman et al., 2005]. Whole stained embryos were processed, embedded in paraffin, sectioned, and counterstained with Vector® Nuclear Fast Red counterstain (Vector H-3403). Newborn pups were euthanized in accordance with and after approval from the University Committee on Animal Resources. Whole diaphragms were dissected and fixed overnight prior to X-Gal staining as described. Diaphragms were post-fixed in 4% paraformaldehyde.
RESULTS
MLPA
A) 1q41q42.12 Region
A total of 179 patients CDH were screened by the MLPA technique to detect copy number changes in the 1q41q42.12 region. Analysis was successful on 168 patients; the remaining 11 samples failed, all of which were extracted either from formalin-fixed paraffin-embedded autopsy tissue or from frozen tissue.
In Patient 2, previously unreported, the cytogenetically visible deletion [46,XY,del(1)(q41q42.13)] was confirmed by MLPA to extend from BPNT1 (218,297,696-218,329,814 at 1q41) to PSEN2 (225,129,689-225,150,422 at 1q41.13) (data not shown). In Patient 3, reported as having a Fryns syndrome phenotype by our group [Kantarci et al., 2006], MLPA further defined the minimally deleted interval detected by aCGH, demonstrating it extended from EPRS (218,227,107-218,286,623 at 1q41) to ACBD3 (224,399,003-224,441,046 at 1q41.12) (Fig 1).
Figure 1.
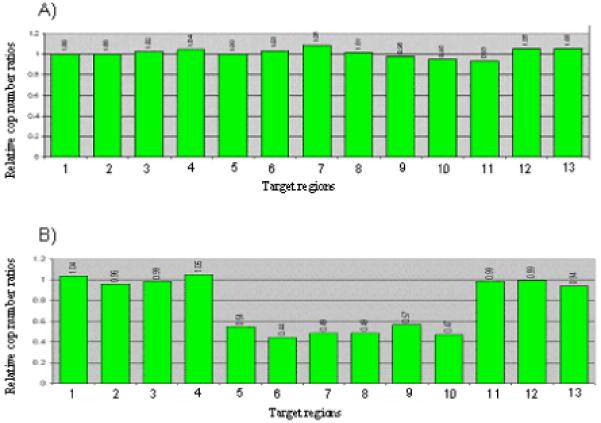
MLPA results for 1q41q42.12 microdeletion screening. Normalized peak height graphs are shown for two CDH patients. (A) a CDH patient without a 1q41q42.12 microdeletion (B) Patient 3, the previously reported Fryns phenotype patient [Kantarci et al., 2006]. Each bar represents the normalized peak height for the probe focusing on a gene on the X-axis, and is arranged in genomic order. Bars 1-2: Contol regions on 15q26. Bar 3: PTPN14 (212,768,242-212,791,265 at 1q41); Bar 4: USH2A (214,413,915-214,663,361 at 1q41); Bar 5 EPRS (218,227,107-218,286,623 at 1q41); Bar 6: BPNT1 (218,297,696-218,329,814 at 1q41); Bar 7: DUSP10 (219,941,389-219,982,084 at 1q41); Bar 8: CAPN2 (222,011,949-222,030,343 at 1q41); Bar 9: LBR (223,660,887-223,682,407at 1q41.12); Bar 10: ACBD3 (224,399,003-224,441,046 at 1q41.12); Bar 11: ITKPKB (224,961,518-224,993,499 at 1q41.12); Bar 12: PSEN2 (225,129,689-225,150,427 at 1q41.12 at 1q41.13); and Bar 13: TAF5L (227,801,564-227,828,417 at 1q42.13).
Note that the deletion extends from EPRS to ACBD3 in (B) Patient 3.
The remaining 166 patients demonstrated no submicroscopic copy number changes in this interval.
B) DISP1 exons
Twenty-four patients selected because they had complex CDH underwent MLPA analysis targeting DISP1 exons. Similar to the 1q41q42.12 region results, only the two patients mentioned above demonstrated DISP1 exon deletions (data not shown).
Sequencing, endonuclease, and pyrosequencing analyses
All patients in our CDH cohort (N=179) were sequenced for DISP1 and HLX. The 8-exon DISP1 demonstrated three previously unreported variants (p.Met1096Thr, p.Asn1281Ile, and p.Ala1471Gly). One putatively deleterious exon 8 variant, the heterozygous C-G transversion (c.4412C>G), was found in Patient 1 on DNA extracted from EBV transformed lymphoblastoid cells; this mutation was predicted to change an alanine to glycine at position 1471 (p.Ala1471Gly). Genotyping of the parents and the healthy brother indicated the transversion arose de novo in the proband. Following confirmation of paternity (data not shown), we genotyped 96 control DNA samples from ethnically-matched normal individuals without observing this point mutation. The p.Ala1471Gly variant was predicted to be ‘not tolerated’ by SIfT based on sequence homology and the physical properties of amino acids, but ‘benign’ by PolyPhen based on multiple alignments.
For the purposes of mutation (p.Ala1471Gly) confirmation and subsequent genetic counseling, DNA from Patient 1 was extracted from two other sources, peripheral blood lymphocytes and saliva. Genotyping did not detect the mutation in saliva derived DNA, but did indicate low-level mosaicism in peripheral blood lymphocyte derived DNA (Fig 2). To resolve these tissue differences, we developed an endonuclease restriction assay to take advantage of the fact that the c.4412C>G mutation abolishes a CviKI-1 restriction site. This assay demonstrated the c.4412C>G transversion in Patient 1’s lymphoblast cell line derived DNA, the presence of low level mutation fragments in peripheral blood derived DNA, but absence of mutation fragments in saliva derived DNA. Neither parent, nor the healthy sibling, showed the mutation by this assay using available DNA (Fig 3).
Figure 2.
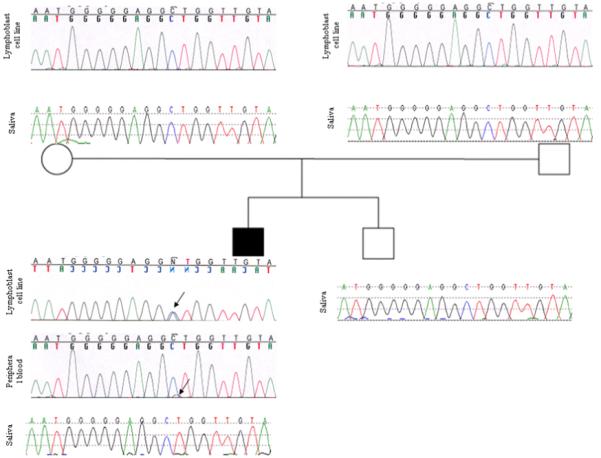
The family pedigree and sequencing chromatograms of the DISP1-exon 8 showing a de novo heterozygous mutation [c.4412C>G (p.Ala1471Gly)] on lymphoblastoid cell line derived DNA of Patient 1. The peripheral blood derived DNA of the patient demonstrates a low level of the mutant allele, while there is no detectable mutant allele in his saliva derived DNA, indicative of tissue specific mosaicism. Arrows show the heterozygous C to G substitution in the patient.
Figure 3.
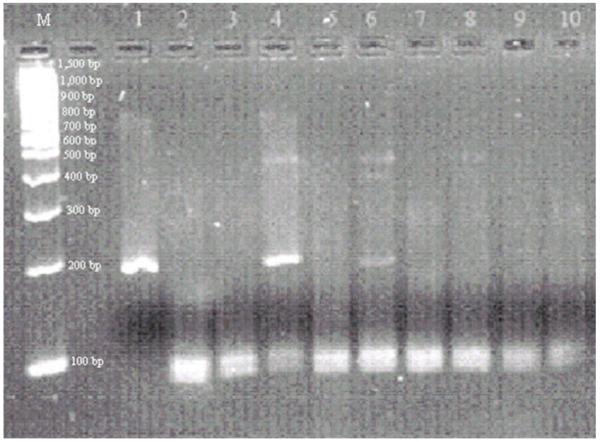
Restriction enzyme digestion of a 195-bp DISP1 exon 8 product with CviKI-1. Wild type product with CviKI-1 recognition site (GG ↓CT) is digested into fragments of 103-bp and 92-bp. The mutation [c.4412C>G (p.Ala1471Gly)] abolishes a CviKI-1 site (GG GT). The presence of the fragments of 195-bp, 103-bp and 92-bp indicates a heterozygous mutation. Samples are as follows: Promega 100-bp DNA ladder (M); undigested PCR product (lane 1); Patient 1’s lymphoblastoid cell line (lane 4), peripheral blood (lane 6), and saliva (lane 9); mother’s lymphoblastoid cell line (lane 2) and saliva (lane 7); father’s lymphoblastoid cell line (lane 3) and saliva (lane 8); healthy brother’s saliva DNA (lane 10); and wild type control (lane 5). Both lane 4 (lymphoblastoid cell line DNA) and lane 6 (peripheral blood DNA) show a heterozygous mutation in Patient 1. Note that the 195-bp mutant fragment is less intense in lane 6 compared to lane 4. There is no observed mutant fragment in the patient’s saliva DNA (lane 9).
Pyrosequencing analysis quantified the percent of the mutant allele in Patient 3’s DNA from different tissues as follows: 43% in transformed lymphoblastoid cells; 12% in peripheral blood lymphocytes; and 4.5% in saliva, compatible with tissue mosaicism for this mutation (Fig 4). Alanine 1471 is highly conserved across mammalian species (Fig 5).
Figure 4.
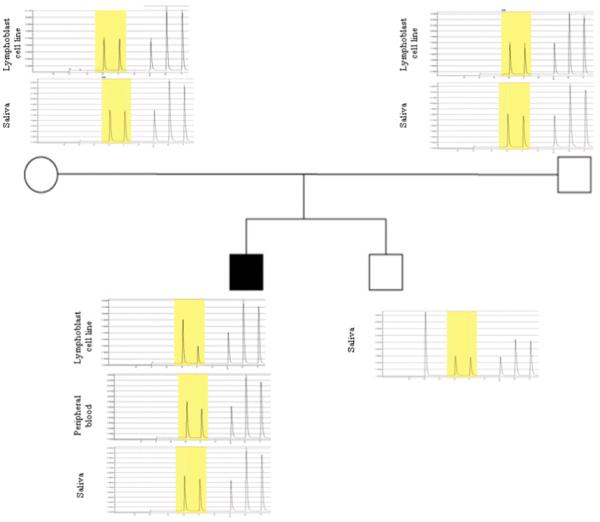
Pyrosequencing analysis of the DISP1 mosaic mutation [c.4412C>G (p.Ala1471Gly)] in Patient 1 and his family members. The mutated base (C>G) is highlighted in yellow. Quantitative analysis identifies that the mutation is present in 43%, 12%, 4.5% of the patient’s samples from lymphoblastoid cell line, peripheral blood cells, and saliva, respectively.
Figure 5.

A) Schematic representation of DISP1 protein and the position of p.Ala1471Gly mutation. B) The protein sequence alignment by ClustalW2 (http://www.ebi.ac.uk/Tools/clustalw2/index.html) in different species shows that wild type alanine (A) at position 1471 is evolutionarily conserved. Conserved amino acids compared to humans are indicated in red.
The additional DISP1 variants, p.Met1096Thr and p.Asn1281Ile, were found in three and two patients, respectively. Each variant, predicted to be “benign” by PolyPhen and to be “tolerated” by SIfT, was inherited from a healthy parent in the three families with available parental samples.
The 4-exon HLX was successfully sequenced in all patients. A previously unreported single nucleotide variation (c.27C>G) resulting in a change from phenylalanine to leucine at amino acid position 9 (p.Phe9Leu) was identified in a patient with isolated CDH. Parental genotyping demonstrated this was inherited from the phenotypically normal mother. The p.Phe9Leu variant was predicted to be “probably damaging” by PolyPhen and “tolerated” by SIfT databases, respectively.
Expression patterns of Disp1 and Shh in developing diaphragm and lung
Expression of Disp1 was evaluated by in situ hybridization in the developing lung and primordial PPF diaphragm tissue of wild type mouse embryos at E11.5. Disp1 was specifically expressed in the PPF, the developing lung mesothelium, and in the developing lung epithelium and mesenchyme (Fig 6 A-F). There was no Disp1 expression in the heart.
Figure 6.
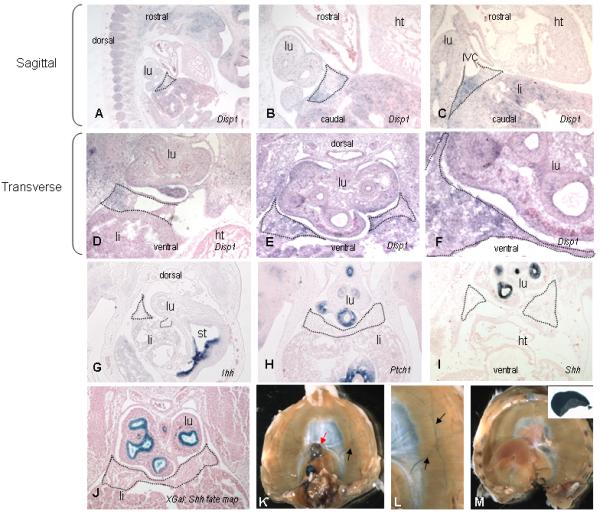
Disp1 and Hedgehog pathway genes in the diaphragm. In situ hybridization on E11.5 mouse embryos (A-I) and X-gal staining (J at E11.5 and K-M mature diaphram) during diaphragm development. Developing diaphragmatic tissue including PPF is outlined with dotted lines in A-J. Disp1 has a generally diffuse pattern of expression but is not expressed in the heart (shown in sections A-D as negative control). Disp1 is highly expressed in the pleuroperitoneal folds (PPFs), especially in the region adjacent to the inferior vena cava (C, D). The expression pattern in the lung includes cells from the mesothelium, mesenchyme, and epithelium (A-F). Indian hedgehog (Ihh) is not expressed in the PPF but is highly expressed in the stomach (G). Patched1 is expressed in the esophagus, lung, and stomach but not in the diaphragm (H). Shh is expressed in lung epithelium but not in the PPF (I). Shh expressing cells (Shh GFP-Cre) do not contribute to the developing diaphragm tissue but do contribute to the lung and esophagus (J). X-gal staining of mature diaphragms from a Shh-Cre fate mapping experiment show that Shh expressing cells contribute to the esophagus (red arrow) and phrenic nerve (black arrows) but not to the mature diaphragmatic parenchyma (K, L). A control littermate (stained but not Cre expressing) shows no staining of the lung, diaphragm, or esophagus (M). The insert shows a piece of lung as a positive control (M, inset). (lu-lung, ht-heart, IVC-inferior vena cava, li-liver, st-stomach)
Since Disp1 is required for Shh function, we also evaluated expression of Shh, Indian hedgehog, and the Hedgehog target gene, Patched1 (Ptch1). [Note that Desert hedgehog (Dhh) was not studied as review of publically available expression databases in mouse embryos revealed no evidence of expression close to the region of diaphragm development]. Shh, Ihh, and Ptch1 were not expressed in the PPF/primordial diaphragm tissue, though expression was strong in other regions of the embryo including lung and esophagus, notochord and ventral plate of the neural tube (Fig 6 G,H,I).
We examined the contribution of cells to the diaphragm that expressed Shh at any time during development by fate mapping, since Shh, a secreted factor, could be restricted to a time or a place not currently recognized as being important for diaphragm development. Fate mapping was conducted using Shh GFP-Cre and Rosa26 reporter (B-galactosidase reporter) mice. Tissues were evaluated in E11.5 mouse embryos (in the tissue that was found to express Disp1) and in the mature diaphragm in newborn pups. There was no contribution of Shh expressing cells to either the primordial or to the mature posterior diaphragm tissue. Shh expressing cells did, however, contribute to the lungs and esophagus, as would be expected based on a known role in lung and esophageal development [Pepicelli et al., 1998], as well as to the phrenic nerve of the diaphragm (Fig 6 K, L).
DISCUSSION
Using standard cytogenetic techniques, chromosomal aneuploidy is found in ~10% of patients with CDH, primarily those with additional anomalies (as reviewed in [Pober, 2008]). Recent advances using higher resolution technologies, such as aCGH and MLPA, reveal that recurrent submicroscopic aneuploidies also cause or contribute to CDH. Identification of these loci is important not only for clinical management and genetic counseling, but also can provide a powerful tool for discovery and prioritization of CDH candidate genes in the deleted regions, such as the previously reported COUP-TFII (NR2F2) in 15q26.2 [Klaassens et al., 2005; Scott et al., 2007] and Gata4 in 8p23.1 [Jay et al., 2007]. In light of reports of 1q41q42 rearrangements in patients with CDH [Rogers et al., 1995; Shaffer et al., 2007; Slavotinek et al., 2006; Youssoufian et al., 1988], we performed a comprehensive molecular screening of the 1q41q42.12 interval in a large cohort of CDH patients, and also studied mouse expression of a candidate gene and its pathway partners.
Of the 168 CDH patients successfully screened by MLPA analysis we identified only two 1q41a42.12 novel deletions, each having been previously detected by other methods (in Patient 2 by standard cytogenetic analysis and in Patient 3 by aCGH [Kantarci et al., 2006]). The remaining 166 patients screened by MLPA did not have copy number aberrations in this interval, though the probe panel resolution was ~1.5 Mb, so copy number changes located between probes could escape detection.
Following examination of the known functions of the 28 genes located in the 1q41q42.12 interval we selected the DISP1 gene for sequencing and expression analyses, given the possible role of DISP1 in diaphragm and lung development [Ma et al., 2002], and its known role in SHH signaling [Burke et al., 1999]. Shh is critical for normal lung development and not surprisingly aberrant Hh activity causes either lethality or a number of other congenital abnormalities [Garcia-Barcelo et al., 2008; Heussler and Suri, 2003; Hu and Helms, 1999; Roessler et al., 2009]. Extrapolation from the VACTERL association [VACTERL = V-vertebral anomalies, A-anal atresia, C-cardiovascular anomalies, T-tracheoesophageal fistula, E-esophageal atresia, R-renal and/or radial anomalies, L-limb anomalies] suggests that Shh or the Hedgehog pathway may also play an important role in diaphragm development [Kim et al., 2001]. First, CDH and tracheoesophageal fistula have been observed in the same patient, with the latter anomaly being a well-described VACTERL, possibly Shh induced, defect [Al-Salem and Alkhuwaher, 2008]. Very recently, VACTERL syndrome was directly associated with the Shh pathway, as deletion in a downstream target, HOXD13, was identified in an affected patient [Garcia-Barcelo et al., 2008]. Independent evidence comes from the teratogenic model of rodent congenital diaphragmatic hernia, the nitrofen model, in which Shh expression abnormalities have been observed in the lungs of exposed animals [Kim et al., 2001; Unger et al., 2003]. Unfortunately, the use of mouse models to examine the effect of reduced Hh pathway dosage has been limited by early embryonic lethality due to mutations in these genes, though mice with mutations of Gli, the downstream target of SHH, were reported to have diaphragmatic defects (reviewed in [Rottier and Tibboel, 2005]).
In Patient 1, we identified a de novo heterozygous DISP1 missense mutation [c.4412C>G], predicted to alter a residue (p.Ala1471Gly)] near the C terminus of the DISP1 protein (Fig 5). This 1471 residue resides in a linker domain not currently known to have functional significance but which is highly conserved when present (e.g., across all mammals tested and in the chick). The significance of lack of sequence conservation in lower species in this case is unknown and does not necessarily indicate absence of functional significance, as lower species lack lungs and a diaphragm [Monroe, 2009]. Ideally, mutations such as the one we detected could be tested in a functional assay, however attempts by others to show the functional significance of human DISP1 mutations in vitro has been unsuccessful likely due to redundancy and incomplete understanding of factors involved in the mechanism by which DISP1 influences development [Roessler et al., 2009].
The p.Ala1471Gly mutation was initially detected by sequencing in DNA extracted from EBV transformed lymphoblastoid cell lines. Interestingly, both sequencing and restriction endonuclease analysis detected only low levels of the mutant allele in DNA extracted from peripheral blood lymphocytes, but no mutant allele in DNA extracted from saliva. Pyrosequencing, a real-time DNA sequencing method based on the sequencing-by-synthesis principle, provided allele specific quantification and resolved these seemingly contradictory results by confirming the presence of tissue mosaicism in this patient; mutation levels ranged from a high of 43% in the lymphoblastoid cell line to a low of 4.5% in DNA extracted from saliva. We hypothesize the process of EBV transformation randomly stimulated growth of B cells harboring the mutant allele, thereby optimizing this tissue source for mutation recovery. Though the level of mutant allele was extremely low in DNA extracted from saliva, samples from other cases have shown a high mutant load in DNA extracted from buccal cells; accordingly, mosaicism does not always result in a consistent pattern or tissue distribution [Gripp et al., 2006; Wuyts et al., 2005].
Disp1, first identified in Drosophilia, encodes a transmembrane Disp protein that regulates the release of cholesterol and palmitoyl modified hedgehog proteins. Full length Sonic and other Hh proteins must undergo an autocatalytic cleavage reaction to remove a C-terminal domain and attach a cholesterol molecule to the N-terminal signaling fragment (Shh-N). Following autoproteolysis, a cholesterol molecule is covalently bound to the C terminus of Shh-N and a palmitoyl group is added to the N terminus of Shh-N (Shh-Np). The loss of Disp function results in accumulation of the Shh-Np in Hh-producing cells [Kawakami et al., 2002; Ma et al., 2002]. Disp in Drosophilia has two murine homologs, with Disp1 being important for Hh-signaling in early development [Caspary et al., 2002]. It is difficult to investigate the consequences of loss of Disp1 since mice with complete or severe disruption of Disp1 (Disp1Δ8 [Kawakami et al., 2002; Ma et al., 2002] and Disp1C289F [Tian et al., 2005; Tian et al., 2004] die in embryogenesis at E9.5. Mice with the hypomorphic allele, Disp1Δ2, survive until birth and exhibit facial midline patterning defects which also phenocopy Shh mutants [Tian et al., 2005; Tian et al., 2004]. It is unknown whether these mutants have diaphragmatic defects. However, a role in diaphragm development should be considered as we found strong Disp1 expression in the mesenchyme (PPF) of the developing diaphragm. Though neither Shh, Ihh, nor the receptor Patched1 were expressed in the PPF at E11.5, we chose to perform fate mapping of Shh expressing cells to determine if at any point in time they contributed to diaphragmatic formation. Shh expressing cells did not contribute directly to the cells of the mature diaphragm musculature or the non-muscular (eg, membranous) portion of the diaphragm, though did contribute to phrenic nerve branches as well as structures in proximity to the diaphragm, including the lung and esophagus. The phrenic nerve is critical for diaphragmatic formation with migration of the nerve branches closely following the migration of myotubes onto the membranous diaphragm [Babiuk et al., 2003]. However, it is uncertain whether expression of genes in the phrenic nerve further plays a role in the maturation of the diaphragm during this critical time in organogenesis.
Though Disp1 is required in SHH expressing cells for normal hedgehog function, the expression pattern of Disp1 is known to be different from that of Shh and include distinct regions potentially influenced by hedgehog protein gradients [Kawakami et al., 2002]. We hypothesize that hedgehog signaling could still influence the Disp1 expressing PPF and subsequent diaphragm development through distant inductive effects. However, the lack of a reported diaphragmatic phenotype in Shh null mice makes this less likely, although most Hh pathway mouse mutants have not adequately been studied. It also cannot be excluded that DISP1 is important for normal diaphragm development, but via an independent uncharacterized Hh pathway which should be the object of future study.
We also studied, by exonic sequencing, a second candidate gene, HLX because the human gene maps to our target interval 1q41q42.12, the mouse Hlx gene product is normally expressed during diaphragmatic development, and a knockout model dies at E15 with an undermuscularized diaphragm [Bates et al., 2000; Hentsch et al., 1996] [Bates et al., 2005]. Sequencing of HLX showed a previously unreported single nucleotide variation (c.27C>G) resulting in a phenylalanine to leucine change at amino acid position 9 (p.Phe9Leu) in a patient with isolated CDH. Parental genotyping indicated this was inherited from a phenotypically normal mother and was predicted to be ‘probably damaging’ by PolyPhen and ‘tolerated’ by SIfT algorithms (data not shown).
HLX sequencing in another human CDH cohort revealed four different previously unreported SNPs with predicted amino acid changes in highly conserved regions of the protein, though parental samples were not available to determine whether these variants were de novo or familial [Slavotinek et al., 2009]. None of the SNPs reported to date, including the one detected in our cohort (p.Phe9Leu) are contained within the predicted DNA binding domain according to UniProtKB/Swiss-Prot [accession # Q14774 (HLX_HUMAN)].
CONCLUSIONS
Our study of a large CDH cohort (N=168) were screened for copy number aberrations in the 1q41q42.12 CDH interval using the MLPA technique. We also performed MLPA analysis targeting DISP1 exons in a selected 24 patient subset with complex CDH. These platforms confirmed and refined deletions in two patients in our cohort. Sequencing analysis of 179 CDH patients revealed the first de novo point mutation in a candidate gene located in this region, DISP1, in a patient with CDH plus additional malformations. Given initial failure to confirm the mutation in saliva derived DNA, we employed several molecular techniques, including pyrosequencing, to confirm tissue mosaicism with high levels of the mutant allele in lymphoblastoid extracted DNA, intermediate levels in peripheral blood extracted DNA, but low levels in saliva extracted DNA. We demonstrated that Disp1 is highly expressed in the developing mesenchyme of the diaphragm as well as in the early embryonic lung, while its molecular partner, Shh, was expressed in the adjacent lung and contributes to phrenic nerve branches of the mature diaphragm. It will be important in the future to understand further the role of Disp1 during diaphragm development.
Supplementary Material
ACKNOWLEDGMENTS
We thank the many CDH families who have participated in this research study. We are greatly thankful for the family of Patient 3 who allowed us share findings from their son with others. We thank the Laboratory for Molecular Medicine (LMM), a CLIA-certified clinical diagnostic laboratory at Harvard Medical School for confirming the mutation found in DISP1. We thank the Seidman Laboratory at Harvard Medical School for supplying control DNA samples. This work was generously supported by National Institutes of Health R01 HD55150-01, the Broad Institute SPARC grant, and the MGH Austen and McBride Funds. We would like to thank A. McMahon for generously supplying materials for Ihh and Disp1 probes.
REFERENCES
- Ackerman KG, Herron BJ, Vargas SO, Huang H, Tevosian SG, Kochilas L, Rao C, Pober BR, Babiuk RP, Epstein JA, Greer JJ, Beier DR. Fog2 is required for normal diaphragm and lung development in mice and humans. PLoS Genet. 2005;1:58–65. doi: 10.1371/journal.pgen.0010010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Salem AH, Alkhuwaher H. Coexisting congenital diaphragmatic hernia, esophageal atresia, and tracheoesophageal fistula: a case report and review of the literature. Int Surg. 2008;93:141–144. [PubMed] [Google Scholar]
- Allan DW, Greer JJ. Pathogenesis of nitrofen-induced congenital diaphragmatic hernia in fetal rats. J Appl Physiol. 1997;83:338–347. doi: 10.1152/jappl.1997.83.2.338. [DOI] [PubMed] [Google Scholar]
- Babiuk RP, Zhang W, Clugston R, Allan DW, Greer JJ. Embryological origins and development of the rat diaphragm. J Comp Neurol. 2003;455:477–487. doi: 10.1002/cne.10503. [DOI] [PubMed] [Google Scholar]
- Bates MD, Schatzman LC, Lints T, Hamlin PE, Harvey RP, Potter SS. Structural and functional characterization of the mouse Hlx homeobox gene. Mamm Genome. 2000;11:836–842. doi: 10.1007/s003350010179. [DOI] [PubMed] [Google Scholar]
- Bates MD, Wells JM, Venkatesh B. Comparative genomics of the Hlx homeobox gene and protein: conservation of structure and expression from fish to mammals. Gene. 2005;352:45–56. doi: 10.1016/j.gene.2005.03.021. [DOI] [PubMed] [Google Scholar]
- Burke R, Nellen D, Bellotto M, Hafen E, Senti KA, Dickson BJ, Basler K. Dispatched, a novel sterol-sensing domain protein dedicated to the release of cholesterol-modified hedgehog from signaling cells. Cell. 1999;99:803–815. doi: 10.1016/s0092-8674(00)81677-3. [DOI] [PubMed] [Google Scholar]
- Caspary T, Garcia-Garcia MJ, Huangfu D, Eggenschwiler JT, Wyler MR, Rakeman AS, Alcorn HL, Anderson KV. Mouse Dispatched homolog1 is required for long-range, but not juxtacrine, Hh signaling. Curr Biol. 2002;12:1628–1632. doi: 10.1016/s0960-9822(02)01147-8. [DOI] [PubMed] [Google Scholar]
- Clugston RD, Klattig J, Englert C, Clagett-Dame M, Martinovic J, Benachi A, Greer JJ. Teratogen-induced, dietary and genetic models of congenital diaphragmatic hernia share a common mechanism of pathogenesis. Am J Pathol. 2006;169:1541–1549. doi: 10.2353/ajpath.2006.060445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakhrai-Rad H, Pourmand N, Ronaghi M. Pyrosequencing: an accurate detection platform for single nucleotide polymorphisms. Hum Mutat. 2002;19:479–485. doi: 10.1002/humu.10078. [DOI] [PubMed] [Google Scholar]
- Garcia-Barcelo MM, Wong KK, Lui VC, Yuan ZW, So MT, Ngan ES, Miao XP, Chung PH, Khong PL, Tam PK. Identification of a HOXD13 mutation in a VACTERL patient. Am J Med Genet A. 2008;146A:3181–3185. doi: 10.1002/ajmg.a.32426. [DOI] [PubMed] [Google Scholar]
- Gripp KW, Stabley DL, Nicholson L, Hoffman JD, Sol-Church K. Somatic mosaicism for an HRAS mutation causes Costello syndrome. Am J Med Genet A. 2006;140:2163–2169. doi: 10.1002/ajmg.a.31456. [DOI] [PubMed] [Google Scholar]
- Harfe BD, Scherz PJ, Nissim S, Tian H, McMahon AP, Tabin CJ. Evidence for an expansion-based temporal Shh gradient in specifying vertebrate digit identities. Cell. 2004;118:517–528. doi: 10.1016/j.cell.2004.07.024. [DOI] [PubMed] [Google Scholar]
- Hentsch B, Lyons I, Li R, Hartley L, Lints TJ, Adams JM, Harvey RP. Hlx homeo box gene is essential for an inductive tissue interaction that drives expansion of embryonic liver and gut. Genes Dev. 1996;10:70–79. doi: 10.1101/gad.10.1.70. [DOI] [PubMed] [Google Scholar]
- Heussler HS, Suri M. Sonic hedgehog. Mol Pathol. 2003;56:129–131. doi: 10.1136/mp.56.3.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holder AM, Klaassens M, Tibboel D, de Klein A, Lee B, Scott DA. Genetic factors in congenital diaphragmatic hernia. Am J Hum Genet. 2007;80:825–845. doi: 10.1086/513442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D, Helms JA. The role of sonic hedgehog in normal and abnormal craniofacial morphogenesis. Development. 1999;126:4873–4884. doi: 10.1242/dev.126.21.4873. [DOI] [PubMed] [Google Scholar]
- Jay PY, Bielinska M, Erlich JM, Mannisto S, Pu WT, Heikinheimo M, Wilson DB. Impaired mesenchymal cell function in Gata4 mutant mice leads to diaphragmatic hernias and primary lung defects. Dev Biol. 2007;301:602–614. doi: 10.1016/j.ydbio.2006.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarci S, Casavant D, Prada C, Russell M, Byrne J, Haug LW, Jennings R, Manning S, Blaise F, Boyd TK, Fryns JP, Holmes LB, Donahoe PK, Lee C, Kimonis V, Pober BR. Findings from aCGH in patients with congenital diaphragmatic hernia (CDH): a possible locus for Fryns syndrome. Am J Med Genet A. 2006;140:17–23. doi: 10.1002/ajmg.a.31025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami T, Kawcak T, Li YJ, Zhang W, Hu Y, Chuang PT. Mouse dispatched mutants fail to distribute hedgehog proteins and are defective in hedgehog signaling. Development. 2002;129:5753–5765. doi: 10.1242/dev.00178. [DOI] [PubMed] [Google Scholar]
- Kim PC, Mo R, Hui Cc C. Murine models of VACTERL syndrome: Role of sonic hedgehog signaling pathway. J Pediatr Surg. 2001;36:381–384. doi: 10.1053/jpsu.2001.20722. [DOI] [PubMed] [Google Scholar]
- Klaassens M, van Dooren M, Eussen HJ, Douben H, den Dekker AT, Lee C, Donahoe PK, Galjaard RJ, Goemaere N, de Krijger RR, Wouters C, Wauters J, Oostra BA, Tibboel D, de Klein A. Congenital diaphragmatic hernia and chromosome 15q26: determination of a candidate region by use of fluorescent in situ hybridization and array-based comparative genomic hybridization. Am J Hum Genet. 2005;76:877–882. doi: 10.1086/429842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlowski P, Roberts P, Dabora S, Franz D, Bissler J, Northrup H, Au KS, Lazarus R, Domanska-Pakiela D, Kotulska K, Jozwiak S, Kwiatkowski DJ. Identification of 54 large deletions/duplications in TSC1 and TSC2 using MLPA, and genotype-phenotype correlations. Hum Genet. 2007;121:389–400. doi: 10.1007/s00439-006-0308-9. [DOI] [PubMed] [Google Scholar]
- Lage K, Hansen NT, Karlberg EO, Eklund AC, Roque FS, Donahoe PK, Szallasi Z, Jensen TS, Brunak S. A large-scale analysis of tissue-specific pathology and gene expression of human disease genes and complexes. Proc Natl Acad Sci U S A. 2008;105:20870–20875. doi: 10.1073/pnas.0810772105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Erkner A, Gong R, Yao S, Taipale J, Basler K, Beachy PA. Hedgehog-mediated patterning of the mammalian embryo requires transporter-like function of dispatched. Cell. 2002;111:63–75. doi: 10.1016/s0092-8674(02)00977-7. [DOI] [PubMed] [Google Scholar]
- Monroe D. Genomic Clues to DNA Treasure Sometimes Lead Nowhere. Science. 2009;35:142–143. doi: 10.1126/science.325_142. [DOI] [PubMed] [Google Scholar]
- Naxerova K, Bult CJ, Peaston A, Fancher K, Knowles BB, Kasif S, Kohane IS. Analysis of gene expression in a developmental context emphasizes distinct biological leitmotifs in human cancers. Genome Biol. 2008;9:R108. doi: 10.1186/gb-2008-9-7-r108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepicelli CV, Lewis PM, McMahon AP. Sonic hedgehog regulates branching morphogenesis in the mammalian lung. Curr Biol. 1998;8:1083–1086. doi: 10.1016/s0960-9822(98)70446-4. [DOI] [PubMed] [Google Scholar]
- Pober BR. Genetic aspects of human congenital diaphragmatic hernia. Clin Genet. 2008;74:1–15. doi: 10.1111/j.1399-0004.2008.01031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler E, Ma Y, Ouspenskaia MV, Lacbawan F, Bendavid C, Dubourg C, Beachy PA, Muenke M. Truncating loss-of-function mutations of DISP1 contribute to holoprosencephaly-like microform features in humans. Hum Genet. 2009;125:393–400. doi: 10.1007/s00439-009-0628-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers JC, Harris DJ, Pasztor LM. Interstitial deletion of the long arm of chromosome 1: del(1)(pter!42.11::q42.3!qter) Am J Hum Genet. 1995:57. [Google Scholar]
- Rottier R, Tibboel D. Fetal lung and diaphragm development in congenital diaphragmatic hernia. Semin Perinatol. 2005;29:86–93. doi: 10.1053/j.semperi.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Scott DA, Klaassens M, Holder AM, Lally KP, Fernandes CJ, Galjaard RJ, Tibboel D, de Klein A, Lee B. Genome-Wide Oligonucleotide-Based Array Comparative Genome Hybridization Analysis of Non-Isolated Congenital Diaphragmatic Hernia. Hum Mol Genet. 2007;16:424–430. doi: 10.1093/hmg/ddl475. [DOI] [PubMed] [Google Scholar]
- Shaffer LG, Theisen A, Bejjani BA, Ballif BC, Aylsworth AS, Lim C, McDonald M, Ellison JW, Kostiner D, Saitta S, Shaikh T. The discovery of microdeletion syndromes in the post-genomic era: review of the methodology and characterization of a new 1q41q42 microdeletion syndrome. Genet Med. 2007;9:607–616. doi: 10.1097/gim.0b013e3181484b49. [DOI] [PubMed] [Google Scholar]
- Slavotinek AM, Moshrefi A, Davis R, Leeth E, Schaeffer GB, Burchard GE, Shaw GM, James B, Ptacek L, Pennacchio LA. Array comparative genomic hybridization in patients with congenital diaphragmatic hernia: mapping of four CDH-critical regions and sequencing of candidate genes at 15q26.1-15q26.2. Eur J Hum Genet. 2006 doi: 10.1038/sj.ejhg.5201652. [DOI] [PubMed] [Google Scholar]
- Slavotinek AM, Moshrefi A, Lopez Jiminez N, Chao R, Mendell A, Shaw GM, Pennacchio LA, Bates MD. Sequence variants in the HLX gene at chromosome 1q41-1q42 in patients with diaphragmatic hernia. Clin Genet. 2009;75:429–439. doi: 10.1111/j.1399-0004.2009.01182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein L. Large scale sequencing. 2003. Chapter 11: Unit 11-11. [DOI] [PubMed] [Google Scholar]
- Stern RF, Roberts RG, Mann K, Yau SC, Berg J, Ogilvie CM. Multiplex ligation-dependent probe amplification using a completely synthetic probe set. Biotechniques. 2004;37:399–405. doi: 10.2144/04373ST04. [DOI] [PubMed] [Google Scholar]
- Tian H, Jeong J, Harfe BD, Tabin CJ, McMahon AP. Mouse Disp1 is required in sonic hedgehog-expressing cells for paracrine activity of the cholesterol-modified ligand. Development. 2005;132:133–142. doi: 10.1242/dev.01563. [DOI] [PubMed] [Google Scholar]
- Tian H, Tenzen T, McMahon AP. Dose dependency of Disp1 and genetic interaction between Disp1 and other hedgehog signaling components in the mouse. Development. 2004;131:4021–4033. doi: 10.1242/dev.01257. [DOI] [PubMed] [Google Scholar]
- Unger S, Copland I, Tibboel D, Post M. Down-regulation of sonic hedgehog expression in pulmonary hypoplasia is associated with congenital diaphragmatic hernia. Am J Pathol. 2003;162:547–555. doi: 10.1016/S0002-9440(10)63848-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuyts W, Biervliet M, Reyniers E, D’Apice MR, Novelli G, Storm K. Somatic and gonadal mosaicism in Hutchinson-Gilford progeria. Am J Med Genet A. 2005;135:66–68. doi: 10.1002/ajmg.a.30663. [DOI] [PubMed] [Google Scholar]
- Youssoufian H, Chance P, Tuck-Muller CM, Jabs EW. Association of a new chromosomal deletion [del(1)(q32q42)] with diaphragmatic hernia: assignment of a human ferritin gene. Hum Genet. 1988;78:267–270. doi: 10.1007/BF00291674. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.