

Abstract
Amphitropic proteins, such as the virulence factor phosphatidylinositol-specific phospholipase C (PI-PLC) from Bacillus thuringiensis, often depend on lipid-specific recognition of target membranes. However, the recognition mechanisms for zwitterionic lipids such as phosphatidylcholine, which is enriched in the outer leaflet of eukaryotic cells, are not well understood. A 500 nanosecond long molecular dynamics simulation of PI-PLC at the surface of a lipid bilayer revealed a strikingly high number of interactions between tyrosines at the interfacial binding site and lipid choline groups with structures characteristic of cation-π interactions. Membrane affinities of PI-PLC tyrosine variants mostly tracked the simulation results, falling into two classes: (i) those with minor losses in affinity, Kd(mutant)/Kd(wildtype)≤5, and (ii) those where the apparent Kd was 50-200 times higher than wildtype. Estimating ΔΔG for these Tyr/PC interactions from the apparent Kd values reveals that the free energy associated with class I is ~1 kcal/mol, comparable to the value predicted by the Wimley-White hydrophobicity scale. In contrast, removal of class II tyrosines has a higher energy cost: ~2.5 kcal/mol towards pure PC vesicles. These higher energies correlate well with the occupancy of the cation-π adducts throughout the MD simulation. Together, these results strongly indicate that PI-PLC interacts with PC headgroups via cation-π interactions with tyrosine residues, and suggest that cation-π interactions at the interface may be a mechanism for specific lipid recognition by amphitropic and membrane proteins.
Keywords: amphitropic membrane proteins, cation-π, molecular dynamics simulations, fluorescence correlation spectroscopy, phosphatidylinositol-specific phospholipase C (PI-PLC)
INTRODUCTION
Cation-π interactions are ubiquitous noncovalent interactions that can be energetically equivalent or stronger than hydrogen bonds.1,2 In proteins cation-π interactions are a driving force for structure formation and stability as well as function,1,3 and are typically observed between aromatic amino acids (phenylalanine, tryptophan, histidine or tyrosine) and ammonium or guanidinium groups from lysines and arginines, respectively.4 Cation-π interactions are also important for substrate and ligand binding, particularly for molecules containing a choline group5-9 as observed, for example, in the X-ray structure of the human phosphatidylcholine transfer protein (PC-TP)5 where the choline moiety of the lipid substrate is involved in cation-π interactions within a cage of three tyrosine residues.
Cation-π interactions with lipids in cell membranes have been suggested to play a role in the structural stability and lipid specificity of integral membrane proteins. In molecular dynamics (MD) simulations of the gramicidin (gA) dimer as a model for membrane-spanning proteins containing interfacial Trp residues, Petersen and coworkers show that phosphatidylethanolamine (PE), and, to a lesser extent, phosphatidylcholine (PC), interact with interfacial tryptophans via cation-π interactions.10 Trp involvement in cation-π interactions with the membrane has also been observed in other simulations, for short peptides11,12 and for an amphitropic enzyme.13 Combined NMR and modeling studies on small model systems describe the interactions between choline-containing lipid headgroups and indole groups as complex and involving cation-π interactions, hydrogen bonds and carbonyl-cation interactions.10,14,15 The available literature thus strongly suggests that cation-π interactions mediated by protein indole groups can play an important role in protein-lipid interfacial binding and lipid specificity. Although tyrosine residues can in principle engage in cation-π interactions with choline-containing lipids,15 much less is known about these contributions to membrane binding by proteins.
Unlike integral membrane proteins, amphitropic proteins do not span the lipid bilayer and their affinity for cell membranes is mostly accounted for by their interactions with the lipid headgroups. They bind at the surface of cell or organelle membranes in order to perform their function. Examples include cytochrome P450, annexins as well as phospholipases, and many of these protein-membrane interactions facilitate cellular reactions to environmental changes.16 Amphitropic proteins bind reversibly to lipid vesicles with equilibrium association constants typically between 103 M−1 to 107 M−1.16 Electrostatic interactions drive their positioning and orientation at the membrane surface thus facilitating the intercalation of a few hydrophobic groups. It is generally acknowledged that the association of amphitropic proteins with lipid bilayers is fast while the dissociation is slow; the dissociation rate constant is thus the main determinant of the binding strength. As a consequence, in simple systems where the protein does not undergo conformational changes and does not interact with other proteins, the affinity for the membrane is mostly accounted for by interactions between the protein interfacial binding site and lipids. The energy and specificity of these interactions is generally estimated by including contributions from electrostatics, ~−1.4 kcal/mol for each positively charged amino acid interacting with the lipid headgroups,17 and hydrophobic interactions with the bilayer core, ~−0.8 kcal/mol per acyl chain CH2 group interacting with the protein.18 However, as we have seen above, electrostatic interactions with lipid headgroups may not necessarily be restricted to charged amino acids and hydrogen bonds. It is thus possible that interfacial cation-π interactions between aromatic residues and lipids have been over-looked for amphitropic proteins.
To investigate the possible role of cation-π interactions in the binding of amphitropic proteins to membranes, we used a combination of molecular dynamics (MD) simulations, including explicit lipid bilayers, and experimental measurements of membrane binding using fluorescence correlation spectroscopy (FCS). Bacillus thuringiensis phosphatidylinositol specific phospholipase C (PI-PLC) was chosen as a model system because it is activated by binding to membranes containing PC or sphingomyelin and a number of Trp and Tyr residues have been implicated in PC binding.19-22 This system also allows facile testing of the simulation results by generating mutant PI-PLCs lacking one or more Tyr residue.
In what follows we present a combined in silico and in vitro study of the binding to PC-rich bilayers of the wild type (WT) enzyme and eleven tyrosine mutants (seven single and four double mutants). We first describe the analysis of a 500 ns MD simulation of the wild type (WT) enzyme in a bilayer contining 256 dimyristoylphosphatidylcholine (DMPC) lipids using the Charmm36 force field.23 The trajectory obtained is analyzed to inventory protein-lipid interactions at the interfacial binding site and in particular the interactions involving interfacial aromatic amino acids. Next we present the results of experiments prompted by the results of the MD simulations. The binding of the wildtype enzyme and tyrosine mutants to small unilamellar vesicules (SUVs) was measured using FCS and the severity of binding defects was evaluated by comparison to WT PI-PLC. With only one exception, Tyr residues with long-lived cation-pi interactions in the simulations showed the most severe binding defects. The results also indicate that two Tyr residues may cooperatively form adducts with the same lipid headgroup. We finally present a semi-quantitative analysis of how tyrosine-choline cation-π interactions contribute to PI-PLC membrane binding affinity.
RESULTS
PI-PLC specifically cleaves the sn-3 phosphodiester bond in phosphatidylinositol (PI). While eukaryotic PI-PLCs are usually multidomain proteins containing both membrane binding domains (PH and C2 domains) and catalytic domains, the bacterial enzymes combine membrane binding and catalytic activity in a single αβ barrel. The well-studied Bacillus thuringiensis PI-PLC is a 34.8 kDa secreted protein that targets eukaryotic cells and, like many other bacterial PI-PLC enzymes, likely plays a role in bacterial virulence. It folds to a distorted (αβ)8-barrel24 structure and anchors to lipid bilayers via a small α-helix (helix B) as well as neighboring loops and two longer α-helices, F and G,19-22 regions that contain at least eight tyrosines (Tyr) and two tryptophans (Trp) (Figure 1). These aromatic amino acids are associated with tighter binding of B. thuringiensis PI-PLC to PC containing membranes and the activation of substrate cleavage by membranes containing 0.1 to 0.5 mole fraction PC (XPC) suggests that this Bacillus PI-PLC specifically recognizes PC headgroups.19,21,22
FIGURE 1.

PI-PLC membrane binding orientations from MD simulations. A. IMM1 orientation of PI-PLC used to initiate the simulations with explicit lipids. Helices B, D, F and G are magenta, blue, green and orange, respectively. The active site is represented by a red fuzzy dot. B. Positioning of PI-PLC in the explicit bilayer model. Lipids are represented with thin sticks colored according to the atomic elements (N: blue, O: red, P: orange, C and H: grey). Electrostatic potential of PI-PLC; the equipotential contours at −/+ 1kT (red/blue) calculated using APBS.25 The potential between −1kT and +1kT is mapped on the molecular surface (negative: red, positive: blue).
Docking PI-PLC to an anionic implicit membrane model
Experimental data21 indicate that PI-PLC interacts with phospholipid bilayers via helix B and surrounding loops. Yet, there is no direct structural data showing PI-PLC bound to lipid layers. To initiate all-atoms molecular dynamics simulations we therefore had to generate a model of the membrane bound form of PI-PLC. Electrostatic interactions between PI-PLC and membranes are relatively weak, as mutagenesis of a single Lys residue to Ala (K44A) is sufficient to increase the apparent Kd towards PC containing vesicles by at least two orders of magnitude21. We therefore did not expect to be able to observe spontaneous binding of PI-PLC to the membrane using all atoms simulations within tractable time scales (unlike what has been other observed for other proteins with strong electrostatic binding26). We used simulations with an implicit membrane model (IMM1-GC27,28) to determine the initial orientation of PI-PLC relative to the membrane, allowing a more cost-effective exploration of potential interface binding sites on the protein surface.
Simulations using anionic implicit membranes were initiated using six PI-PLC orientations relative to the membrane. Each of these orientations corresponds to one face of a cube containing the enzyme. Half (3/6) of these orientations led to an anchored PI-PLC at the model membrane. In the corresponding 9 simulations (3 for each initial orientation), the anchorage is always achieved via helix B (Cf. Figure 1) in agreement with published experimental data.21
The simulation yielding the lowest IMM1-GC effective energy was selected for further analysis. The average binding energy (ΔW) calculated for the last 1.5 ns of this simulation is −5.6±1.0 kcal/mol. Decomposition of the binding energy shows that the largest contributions arise from hydrophobic interactions with residues in helix B. Interestingly, despite the proximity of interfacial Tyr residues to the membrane plane, these residues show no significant energetically favorable interactions of Trp or Tyr residues with the implicit membrane (a detailed list of all favorable contributions is provided as Supporting Information). This is similar to what we observed for another amphitropic protein, proteinase 3, where a tryptophan residue (Trp218) did not significantly contribute to binding in IMM1 simulations29 but all-atom simulations13 revealed its involvement in hydrogen bonds and cation-π interactions with the carbonyl and choline moieties of dimyristoylphosphatidylcholine (DMPC) lipids. Although the implicit membrane model is useful for predicting the correct orientation of PI-PLC at the membrane, in our experience it underestimates the interfacial interactions of aromatic amino acids.
Simulation of PI-PLC interactions with DMPC bilayers
The orientation of PI-PLC predicted by the implicit membrane simulations was used to initiate simulations of the protein with an all-atom lipid bilayer model. Although the binding of PI-PLC is strongest in PC/PG mixtures, we chose to use pure DMPC bilayers to avoid artifacts due to the initial positioning of neutral lipids with respect to the protein.
PI-PLC exhibits very little structural change during the 500 ns MD simulation. The root mean square deviation (RMSD) of the backbone atoms is on average low (1.4 Å) and stays stable throughout the simulation. A plot of the RMSD time evolution is provided as Supporting Information. We evaluated the amino acid anchorage depth at the interfacial binding site (IBS) by calculating the distance between the residue centers of mass and the average plane defined by the position of the phosphorus atoms. This distance was calculated for every frame in the trajectory and averaged. Results are reported in Table 1, where the list of amino acids is sorted by structural elements, and in the Supporting Information. The most deeply buried amino-acids are located on helix B (Ile43, Pro42, Asn41) and on the β7-αG loop (Trp242, Thr240, Ala241 and Gly239). These residues penetrate 2 to 4 Å, on average, inside the plane defined by the phosphorus atoms. Other amino acids of the IBS, such as tyrosines, are situated on the other side of the phosphate plane.
Table 1. Anchorage of PI-PLC in DMPC lipids: depth and inventory of interactions.
| SSEa | aa # | depthb (Å) |
hydro- phobicc |
Hbondsd (%) |
cations-πe (%) |
|---|---|---|---|---|---|
| αB | Q40 | 0.6 ±2.3 | 2.1 | 98.1/34.0 | |
| N41 | 3.3 ±1.8 | 1.4 | 97.0/55.1 | ||
| P42 | 4.2 ±1.9 | 5.7 | |||
| I43 | 4.3 ±1.7 | 7.7 | |||
| K44 | 0.4 ±1.7 | 2.5 | 99.0 | ||
| Q45 | −0.6 ±1.9 | 32.8 | |||
| V46 | 0.4 ±1.9 | 4.7 | |||
| W47 | −0.8 ±1.9 | 3.0 | |||
|
| |||||
| β2 | R71 | −10.1 ±2.0 | 50.3 | ||
|
| |||||
| β2-αD | P84 | −3.2 ±1.9 | 2.9 | ||
| L85 | −3.6 ±1.6 | 1.9 | |||
| Y86 | −5.5 ±1.9 | 1.0 | 30.1 | 22.5 | |
| Y88 | −4.9 ±2.0 | 58.1 | 95.6 | ||
|
| |||||
| β3-αE | Y118 | −8.7 ±2.1 | 47.9 | 23.8 | |
| K122 | −6.1 ±2.9 | 54.7 | |||
|
| |||||
| β6-αF | Y200 | −10.9 ±2.2 | 64.4 | ||
| K201 | −7.6 ±2.6 | 53.4 | |||
|
| |||||
| αF | Y204 | −7.3 ±2.6 | 25.2 | ||
|
| |||||
| β7-αG | S236 | −6.6 ±2.0 | 51.9 | ||
| S237 | −3.0 ±1.9 | 1.3 | |||
| G238 | 0.1 ±2.0 | 1.1 | |||
| G239 | 2.2 ±1.9 | 3.4 | |||
| T240 | 3.3 ±2.0 | 2.6 | |||
| A241 | 2.6 ±2.2 | 3.0 | |||
| W242 | 3.9 ±2.4 | 3.0 | |||
| N243 | −0.3 ±2.3 | 39.0 | |||
| S244 | −2.3 ±2.1 | 75.2 | |||
|
| |||||
| αG | Y246 | −4.5 ±2.4 | 38.7 | 77.3 | |
| Y247 | −2.3 ±2.5 | 44.6 | 8.9 | ||
| S250 | −7.2 ±2.7 | 24.1 | |||
| Y251 | −6.7 ±3.2 | 25.8 | 36.7 | ||
| K279 | −8.1 ±3.1 | ||||
Secondary structure elements; α: helix, β: strand, βi-αX: loop between strand i and helix X.
Mean values and standard deviations. Positive values indicate that the center of mass of the amino acid is buried in the bilayer, beyond the plane defined by the phosphate groups.
Average number of hydrophobic contacts per frame (listed if above 1).
Occupancies of hydrogen bonds in % (if > 20; bold numbers for backbone hydrogen bonds).
Occupancy of cation-π adducts (if > 10%).
Interactions between PI-PLC residues and membrane lipids were monitored during the MD simulation and the most frequent ones are reported in Table 1. For each amino acid, we report the average number of hydrophobic contacts per frame, occupancies of hydrogen bonds and cation-π interactions. The criteria for detection of the interacting pairs are given in the Methods section. The geometry of the PI-PLC binding region after 300 ns of simulation is shown in Figure 2 where we highlight the amino acid side chains involved in hydrophobic contacts with the lipid tails (for those with an average number of contacts per frame greater than 1). Ile43 in helix B has the highest number of hydrophobic contacts, consistent with it being the most deeply buried residue in PI-PLC. In addition, each of the helix B hydrophobic side chains (Pro42, Ile43, Val46) is in contact with more than one lipid hydrophobic group and these contacts remain throughout most of the simulation.
FIGURE 2.

PI-PLC amino acids involved in hydrogen bonds and hydrophobic contacts with DMPC lipids. The structure of PI-PLC after 300ns is represented with cartoons. The side chains of amino acids involved in long-lasting hydrogen bonds (occupancies > 75%) are represented with a pink mesh while those involved in hydrophobic contacts (if average number of contacts per frame above 2) are represented with sticks, and colored following a cyan (equal to 2) to red (equal to 8) gradient.
Other regions of the enzyme are also in contact with hydrophobic lipid tails although they are less deeply buried than Ile43. This is the case for residues in the loop between the second strand of the beta barrel (β2) and helix D. This long (>15 aa) loop contains the catalytic residue His82 and is partially structured in two short strands. Pro84, Leu85 and Tyr86 from this loop are in contact with the hydrophobic lipid tails during most of the simulation time. The contacts from Pro84 and Leu85 have occupancies above 75%, while Tyr86 has an occupancy of 30%. Only one other structural element, the loop preceding helix G (Ser237 to Trp242) is involved in hydrophobic contacts with the lipid tails. Hydrophobic intercalation is thus restricted to three regions of B. thuringiensis PI-PLC: helix B, the β2-helix D loop and the β7-helix G loop.
Most hydrogen bonds between the protein and membrane involve phosphate groups. From Table 1 one can see that a few amino acid residues in helix B and the β7-helix G loop achieve long-lasting hydrogen bonds with occupancy above 70% while the other structural elements have shorter-lived interactions. Interestingly Gln40 and Asn41 interact through their backbone atoms. The side chains of Lys44, Ser244 and Asn41 also mediate long lasting hydrogen bonds. Tyr86 is the other residue involved in a backbone mediated hydrogen bond, but to a lesser extent. β2 is involved via Arg71 and the subsequent loop carrying Tyr88. Two other loops, β3-helix E and β6-helix F interact with phosphate groups via Lys122 and Lys201, respectively.
The frequency of aromatic amino acids, especially tyrosines, at the interface between PI-PLC and the membrane is strikingly high. We screened the trajectory frames to identify potential cation-π interactions between choline and aromatic (Trp indole or Tyr phenol) groups, using a geometric criteria (see Methods section). Figure 3A shows the time evolution of these interactions while snapshots of such choline-tyrosine interactions are highlighted in Figures 3B-D. It is interesting to note that some tyrosine side chains seem to undergo significant changes of their orientation compared to the starting structure due to interaction with lipids. Our analysis reveals three tyrosines that are involved in very long-lasting cation-π interactions (>50% lifetime): Tyr88, Tyr200, Tyr246.
FIGURE 3.

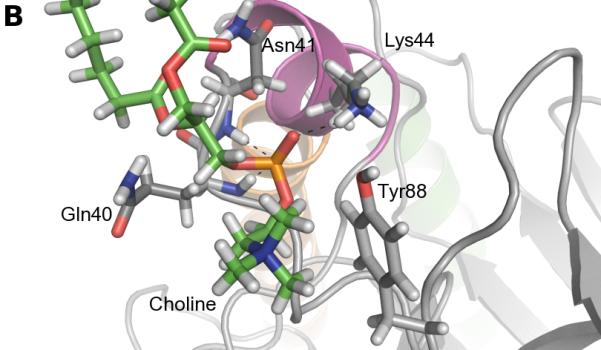
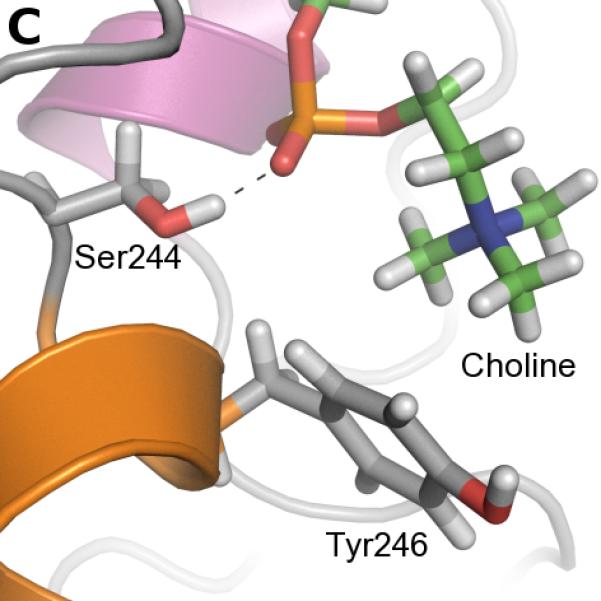
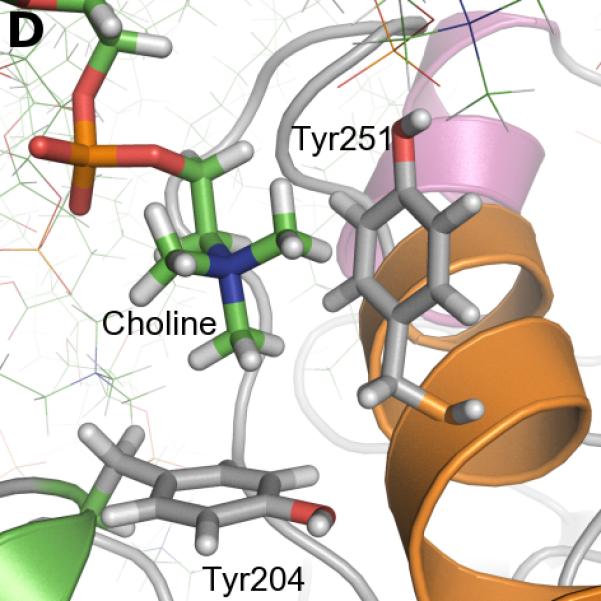
Cation-π interactions identified during the simulation. A. Occurrences of cation-π interactions for aromatic residues versus simulation time (occupancies>5%). B. C. and D. Snapshots of cation-π interactions collected along the simulation (representative frames taken between 400 and 500 ns). Color code for cartoons as in Figure 1A.
Interestingly, cation-π interactions of Tyr88 and Tyr246 engage the same lipids as the longest-lasting hydrogen bonds. The phospholipid, the head of which is involved in a cation-π interaction with Tyr88, is also hydrogen-bonded via its phosphate to the Lys44 sidechain and the backbone of Gln40 and Asn41 (Figure 3B). Tyr246 (cation-π) and Ser244 (hydrogen bond) also share the same partner approximately 60% of the time. The corresponding arrangement of the two amino acids and a lipid is illustrated on Figure 3C. It is worth noting that the hydrogen bonds described are not present in the starting structure, and are formed during the first 50 ns of simulation. Unlike Tyr88 and Tyr246, the cation-π interactions involving Tyr200 are not interfacial. Tyr200 is the most distant of the three from the membrane and it forms an interaction with the choline group of a lipid that sticks out of the bilayer half-way into the active site.
Four other tyrosines are involved in less stable cation-π interactions (occupancy from 20 to 40%) that nevertheless occur regularly throughout the simulation: Tyr86, Tyr118, Tyr204, Tyr251. A few other aromatic amino acids are observed mediating cation-π interactions with choline groups but these are less frequent events: Tyr53 (2.5%), Tyr247 (8.9%), Tyr248 (1.9%). In our simulations, all significant cation-π interactions with cholines are thus mediated by tyrosines while tryptophans interact with the lipid tails (compare the inventory of hydrophobic contacts in Table 1).
Among the four tyrosines (246, 247, 248 and 251) in helix G, three are involved in cation-π interactions and two of them (246 and 251) rather strongly. Because of their consecutive location on an α-helix, the side chains of tyrosines 246, 247 and 248 point in different directions and interact with different lipids.
In contrast, the sidechains of Tyr247 and Tyr251 are on the same side of the helix, separated by one turn, and these residues can interact with the same lipid choline group. Similarly, the orientation of Tyr204 on helix F and Tyr251 on helix G allow simultaneous interactions with the same lipid. Tyr247 is involved in cation-π interactions with choline headgroups only 8.9% of the time, but for about half of these interactions (ca 5% of the simulation time) Tyr251 is also interacting with this headgroup (Figure 3A). More frequently, 14.3% of the time, Tyr204 and Tyr251 are involved in cation-π interactions with the same choline headgroup (Figure 3D), and this lipid is distinct from that interacting with Tyr247. This correlation between Tyr204 and Tyr251 lipid binding is reflected in Figure 3A and is in agreement with a study using a combination of high-resolution field-cycling 31P-NMR and molecular docking that implicated both 204 and 251 in a tight PC binding site in B. thuringiensis PI-PLC.30 In our simulations, we never observe more than two tyrosines simultaneously interacting with the same lipid. Yet homologues to Y204, Y247 and Y251 are shown to be involved in choline binding in a recent X-ray crystal structure of a Staphylococcus aureus PI-PLC variant with two introduced Tyr residues.31
Unlike the other Tyr residues at the top of helix G, Tyr248 is almost never involved in interactions with lipids (cation-π occupancy: 1.9%, hydrogen bonds < 10%). It instead engages in long-lived intramolecular hydrophobic interactions with Leu235 and Pro245 which appear to maintain the structure and orientation of the rim loop between β7 and helix G. The hydrogen bond between the backbone nitrogen of Asn243 and the backbone oxygen of Gly238 also helps stabilize the shape of this loop. Figure 4 shows the difference between structures of Tyr248Ala mutants and WT obtained from MD simulations in water (Figure 4). In the mutant simulations the rim loop is more flexible and the distance to helix B increases. Accordingly there is a significant increase in the atomic fluctuations of both the rim loop and helix B (root mean square fluctuations are provided as Supporting Information). The simulations thus indicate that the role of Tyr248 in membrane binding is to restrict PI-PLC conformational space rather than making direct contacts with the lipids.
FIGURE 4.
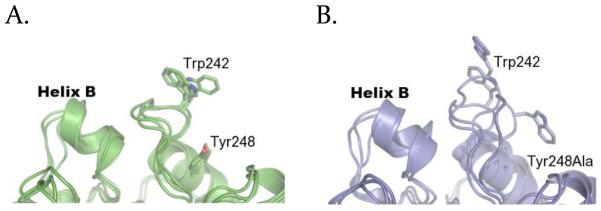
Effect of the Y248A mutation on PI-PLC. Structure of the interfacial binding sites of WT PI-PLC (A) and Y248A (B) after 20 ns MD simulations in water, from three independent simulations in each case.
Effects of Tyr mutations on PI-PLC binding to small unilamellar vesicles (SUVs)
The MD simulations with an explicit DMPC membrane identify Tyr residues in the loop between β2 and helix D (86, 88), in helix F (204) and in helix G (246-247, 251) as important for cation-π interactions with PC headgroups. Tyr248, also at the interfacial binding site, seems to have a slightly different role. These predictions were tested using single or double Tyr mutants of PI-PLC, and measuring protein binding to SUVs using FCS. Because B. thuringiensis PI-PLC binding to SUVs is synergistic, with lower affinity for SUVs composed only of anionic lipids or pure PC SUVs,21,32 binding was measured as a function of mole fraction PC (XPC) for phosphatidylglycerol (PG) SUVs. The anionic lipid PG was used as a substrate analog because the production of diacylglycerol from phosphatidylinositol (PI) would lead to vesicle fusion compromising FCS binding experiments. Apparent Kds and apparent Kds relative to wild type are plotted in Figure 5 as a function of XPC. Values of apparent Kds at XPC=1 are given in Table 2.
FIGURE 5.

Binding of PI-PLC Tyr variants to SUVs. Apparent Kd (A) and apparent Kd relative to WT* (B, C, and D) as a function of XPC. The error bars are the largest uncertainties generated during the FCS data analysis, either the standard deviations from two independent repeats or the uncertainty generated by error propagation for the two independent data sets.
Table 2.
Apparent Kd values of mutants of B. thuringiensis PI-PLC variants towards pure PC SUVs (XPC=1)
| Mutant | Apparent Kd (mM)a |
|---|---|
| WT* | 0.016 ± 0.003 |
| Y86A* | 0.072 ± 0.005 |
| Y88A* | 1.2 ± 0.1 |
| Y204S* | 0.87 ± 0.58 |
| Y246A* | 1.2 ± 0.3 |
| Y247A* | 0.082 ± 0.007 |
| Y248A* | 2.5 ± 0.5 |
| Y251A* | 0.81 ± 0.41 |
|
| |
| Y86A/Y88A* | 1.1 ± 0.4 |
| Y246A/Y247A | 2.0 ± 0.1 |
| Y248A/Y204S | 11 ± 3 |
| Y251A/Y204A | 2.8 ± 2.1 |
The uncertainties are the largest uncertainties generated during the FCS data analysis. Either the standard deviations from two independent repeats or the uncertainty generated by error propagation for the two independent data sets.
None of the single Tyr mutations significantly perturb the PI-PLC structure as judged by far-UV circular dichroism, and all of the variants exhibit significant activity towards PI presented in small unilamellar vesicles (the preferred substrate) that increases as PC is added to the vesicle (see Supporting Information). In general, WT* PI-PLC and the Tyr* variants, (where the asterisk indicates the Asn168Cys mutation required for Cys-mediated fluorescent labeling of PI-PLC) have similar affinities for anionic SUVs containing only PG (Figure 5), and Tyr* defects in binding are greatest for SUVs with high XPC. This is consistent with the simulations where most of the Tyr residues mediate interactions with choline headgroups.
For the loop between β2 and helix D, mutagenesis of Tyr88 to Ala (Y88A*) increases the apparent Kd by at least one order of magnitude for XPC≥0.5 while defects in binding of Y86A* are barely significant (Figure 5A). As might be expected, decreases in membrane affinity for the Y86A/Y88A* double mutant are dominated by the effect of mutating Tyr88. These results are consistent with the simulations where, compared to Tyr86, occupancies of cation-π interactions between Tyr88 and choline headgroups are much higher and are stabilized by additional interactions mediated by the phosphate group of the same lipid.
The Tyr residues in the helix F/helix G region can be broken into two groups: (i) The adjacent Tyr residues 246 and 247 on helix G, and (ii) Tyr204 on helix F as well as Tyr residues 248 and 251 on helix G. The Y246A* variant shows significant binding defects even at 0.2 XPC (Figure 5C). Mutagenesis of Tyr247 is much less perturbing, and Y247A* has apparent Kds that are only 5 times higher than WT* at XPC≥0.8 where Y246A* has apparent Kds more than 60 times larger than WT*. As expected from these results, the double mutant, Y246A/Y247A*, has reduced binding affinity similar to that seen for Y246A*. Again, this mirrors the simulations where Tyr246 cation-π interactions have high occupancies and are supported by a stable hydrogen bond mediated by S244 while Y247 mediates fewer cation-π interactions.
The results for Tyr residues 204, 248 and 251 are more complicated. The Tyr204 and Tyr251 variants behave similarly (Figure 5D). Mutagenesis of either Tyr residue has no significant effect on binding at XPC= 0 or 0.2, but increases the apparent Kd by 5-fold for 0.5 XPC and at least an order of magnitude for vesicles with higher PC content. Additionally, the double mutant behaves similarly to either single mutant suggesting that the effects of these two mutations are not additive. In both the S. aureus N254Y/H258Y variant structure 31 (where S. aureus PI-PLC residues 254 and 258 correspond to B. thuringiensis PI-PLC residues 247 and 251, respectively), and in the simulations with DMPC, Tyr residues corresponding to B. thuringiensis 204 and 251 interact with the same choline ion diffused into the crystal. Thus, the mutagenesis data with the Bacillus PI-PLC suggest that disrupting one of these interactions may be sufficient to disrupt the other.
Mutating Tyr residues 88, 246 or 248 has the largest effects on binding (Figure 5). However, unlike 88 and 246, the simulations predict that Tyr248 does not interact with the membrane, but rather restricts conformations of the PI-PLC rim loop by interactions with other amino acids. The serious losses in binding affinity for the Y248A* variant when XPC>0 (Figure 5D) suggest that either the simulation underestimates the extent of interactions between 248 and lipids, or that the change in rim loop dynamics observed in the protein-only simulations for Y248A reduce binding affinity particularly for PC-rich vesicles.
DISCUSSION
This combination of simulations and experiments details the molecular mechanisms PI-PLC uses to bind PC-rich membranes. Simulations with an implicit membrane model yield an orientation of B. thuringiensis PI-PLC in good agreement with experimental data, even though the model significantly underestimates the energetic contribution of aromatic amino acids and possibly the overall binding energy. The all-atom simulation with a DMPC bilayer predicts specific interactions between individual PI-PLC residues and lipids. Two 500 ns simulations were also performed using a DMPC/DMPG 4:1 lipid bilayer and starting with different lipid distribution around the protein but almost identical PI-PLC orientations relative to the membrane. The occupancies and interactions between PC lipids and PI-PLC residues (data not shown) are not significantly different from those obtained with a pure PC bilayer. These results support the significance of the data obtained from the 500 ns-long DMPC simulation and suggest that these interactions are likely to occur for a variety of PC-rich bilayers.
Mutagenesis of the amino acids involved in the specific interactions observed during the simulation generally reduces PI-PLC affinity for lipid vesicles, validating the simulation results. In particular the importance of helix B seen in the simulations is in agreement with PI-PLC binding data towards SUVs.20 The I43A mutant has an apparent Kd ca. 5.5 times greater than the WT protein. Mutating Lys44 to alanine increases Kd by more than one order of magnitude (Cf. Supp. Inf.). In addition to helix B, containing the amino acids that are most deeply anchored in the bilayer, two other regions carry amino acids that interact with the lipid tails via hydrophobic interactions: the loop between β2 and helix D, and the loop between β7 and helix G (rim loop region). Among all the interactions identified along the simulation, we observe a strikingly high number of interactions involving aromatic residues and in particular conformations of choline groups and tyrosine side chains characteristic of cation-π interactions. Again, mutagenesis of these Tyr residues reduces membrane affinity, particularly for PC-rich membranes.
The fact that FCS experiments show the greatest binding defects for vesicles with high PC/PG ratio confirms that tyrosines play an important role in specific PC, or at least choline, binding. Mutagenesis of tyrosines 88, 204, 246 and 251 increases the apparent Kd by at least an order of magnitude compared to wild type PI-PLC. These are also the tyrosines for which we observe the highest number of cation-π occurrences during the MD simulation, suggesting that the cation-π interactions observed in the simulation are relevant for binding.
In contrast, mutagenesis of Tyr248 also reduces the apparent Kd by two orders of magnitude at high XPC (Table 2), but barely any cation-π adduct is formed during the 500 ns simulation. While we cannot exclude that the simulations underestimate lipid-Tyr interactions, the intramolecular hydrophobic interactions of Tyr248 in the PI-PLC/DMPC simulation and the simulation of the Y248A mutant in water indicate that Tyr248 may help maintain the structure and rigidity of the rim loop region. In previous MD simulations on PI-PLC in solution, this rim loop (residues 238-244) was observed to open and close over the active site in concert with the helix B region.33 Mutagenesis of Pro245 at the top of helix G, resulted in a loss of this correlated motion in MD simulations and Pro245 mutants show both reduced enzymatic activity and reduced binding affinity particularly towards PC-rich SUVs.33 Taken all together the current and previous simulations along with helix G mutations suggest that the dynamics and orientation of the rim loop are important for PI-PLC activity and binding in PC-rich environments such as the outer membrane of eukaryotic cells.
The energetics of Tyr/PC cation-π interactions are reflected in the occupancies observed during the simulations (Figure 3A) and may also be estimated by calculating ΔΔG (=ΔGmutant−ΔGWT) from the apparent Kds for the Tyr mutants. Both of these methods only estimate the energetics because cation-π interactions may be underestimated in MD simulations (see discussion below), and the apparent Kds are calculated based on the total lipid concentrations rather than the lipid concentration in the SUV outer leaflet. (We chose to use the total lipid concentration because the SUVs used for the binding experiments are polydisperse21 complicating esimates of the lipid concentration in the outer leaflet.) Despite these caveats, the cation-π occupancies and ΔΔGs calculated from the apparent Kds are reasonably well-correlated for the 500 ns simulation (Figure 6). With the exception of Tyr248, the most perturbing mutations (high ΔΔG) are associated with Tyr residues exhibiting long cation-π occupancies indicating that the simulation does a good, qualitative job of modeling interactions between PC and Tyr residues.
FIGURE 6.

Comparison between occupancies of predicted cation-π interactions from the simulation and evaluation of ΔΔG (kcal/mol) from FCS measurements for PI-PLC WT* and single tyrosine mutants.
Comparing the ΔΔG calculated from the apparent Kd values can also provide qualitative information about the type of interaction between tyrosines and PC. The free energy change associated with Tyr86 and Tyr247 is approximately −1 kcal/mol. This value is comparable to that predicted by the Wimley-White scale (0.94±0.06 kcal/mol for the transfer of tyrosines34 from water to POPC) and in the range of a hydrogen bonded interaction.35 It is also close to the Wimley-White scale for transfer to octanol (−0.71±0.11 kcal/mol) which reflects hydrophobic interactions. In contrast, removal of tyrosines 88, 204, 246 or 251 is linked to a higher energy cost: 2.5-3 kcal/mol towards pure PC vesicles. These higher energies may indicate cation-π interactions. Gallivan and Dougherty report an estimated upper limit of −3.6 kcal/mol for a methylammonium-benzene cation-π interaction.35 Altogether, these results strongly indicate the presence of these cation-π interactions.
Some of the most important cation-π interactions are also correlated with strong Hbonds (Lys44, Ser244), suggesting a network of interactions at the interfacial binding site involving the cation-π interactions. However, longer MD simulations would be needed to confirm the stability of this network.
To the best of our knowledge, investigations of cation-π interactions between membrane proteins and lipids have so far mostly focused on tryptophans. While the two tryptophan residues present at the PI-PLC binding interface are deeply anchored, and interact mostly with the hydrophobic lipid chains, we observe cation-π interactions mediated almost exclusively by tyrosines. This is in agreement with protein structures5-9 and studies on simpler systems, both experimental and theoretical, that provide evidence for the existence of cation-π adducts between tetramethylammonium or choline moieties and phenolic groups of tyrosine.
Cation-π interactions result from electrostatic interactions between pi electrons of an aromatic system, in proteins typically Phe, Tyr or Trp, and a cation (e.g. −NH3+, −N(CH3)3+). The origin of the dominating component (electrostatic interaction with the quadrupole, polarization of the aromatic system, charge-transfer, dispersion effects) has been debated over the years. Although several studies argue for a dominant electrostatic contribution 1,36-38 other studies have concluded that the other contributions cannot be overlooked.39,40 Whichever scenario we envisage there are good reasons to doubt the ability of additive pairwise potentials from molecular mechanics force field to accurately reproduce the energetics of cation-π interactions.10,41 Yet molecular mechanics force fields have been shown to capture geometries of cation-π pairs in proteins fairly well.41,42 Our work expands previous results and suggests that the Charmm force field provides a qualitatively reasonable picture of the cation-π interactions between PI-PLC and phosphatidylcholine lipids. However, we cannot exclude that our simulation misses or underestimates some interactions, such as the formation of cages of Tyr residues as recently observed for S. aureus PI-PLC.31,43 This could be caused by the fact that positive charges approaching along the plane of the aromatic ring are not well described by Charmm.37 Yet the qualitatively good agreement between the simulation and FCS experiments reported here indicate that MD simulations with a classical mechanics force field have strong predictive power for protein-lipid interfacial interactions, even when they involve “challenging” adducts such as the cation-π systems.
The binding affinity between membrane lipids and amphitropic proteins is typically the result of interfacial interactions, most often referred to as the sum of electrostatics (hydrogen bonds) between basic amino acids (Lys, Arg) and the interface (phospholipid heads), and hydrophobic interactions of aliphatic amino acids with the lipid tails. The role of interfacial aromatic amino acids is seldom mentioned despite the importance of these residues. Our results suggest that tyrosine residues are involved in interfacial cation-π interactions that are potentially energetically equivalent to or stronger than hydrophobic interactions or hydrogen bonds. It has been suggested that cation-π interactions are less sensitive to the solvent dielectric constant than salt bridges35 making them well-adapted for the interface between a low dielectric environment and a high dielectric environment. Moreover other phospholipids have amino groups capable of interacting with aromatic systems. Phosphatidylethanolamine (PE) is abundant in bacteria and phosphatidylserine (PS) distribution between the inner and outer leaflet of eukaryotic plasma membranes is modified in apoptotic cells. Our results suggest that overlooked cation-π interactions between membranes and aromatic amino acids of amphitropic proteins may play an important role not only in membrane binding but also in lipid specificity.
CONCLUSION
Experimental mutagenesis of PI-PLC Tyr residues implicated in cation-π interactions with PC revealed an excellent correlation between the occupancy of cation-π interactions in the MD simulation and the severity of binding defects in the Tyr variants. This relationship did not hold for Tyr248 which is instead implicated in constraining protein intrinsic dynamics that are critical for membrane binding as suggested recently.23 This study also presents a semi-quantitative analysis of the energetics of choline-tyrosine cation-π interactions in protein-membrane binding suggesting that strong cation-π interactions may provide up to 3 kcal/mol of binding energy at the interface. It demonstrates the utility of relatively long protein plus membrane simulations using molecular mechanics force fields for insight into the molecular mechanisms used for membrane binding by amphitropic proteins including both amino acid-lipid interactions and possible roles for protein dynamics. Cation-π interactions between aromatic residues, including tyrosines, and the interfacial membrane region may be an overlooked, important mechanism utilized by other amphitropic proteins with aromatic rich regions.
METHODS
Molecular dynamics simulations
Simulations with an implicit membrane model
There is no X-ray structure of wild type B. thuringiensis PI-PLC. We thus used the X-ray structure of the B. thuringiensis Y247S/Y251S22 mutant (PDB ID: 3EA1) for which we constructed the missing tyrosine side chains using the x-ray structure of the W47A/W242A44 mutant (PDB ID: 2OR2). The obtained structure was then subjected to an energy minimization using harmonic restraints (50 kcal/mol/Å2 on the backbone and 15 kcal/mol/Å2 on sidechains) using Charmm (v33b1)45. Simulations with the IMM1-GC implicit membrane model27,28 were also performed with Charmm (v33b1),45 using a salt concentration of 0.1 M, a membrane thickness of 23 Å, an area per lipid of 70 Å2 and a 50% ratio of anionic “lipid” of valence 1. The plane of smeared charge was positioned 3 Å above the membrane plane (i.e. at z = ± 14.5 Å). The Charmm polar hydrogen force field46 was used to describe the protein. The simulations were run for 4 ns and, as described previously,29 starting from six different orientations of the proteins with respect to the membrane. Each orientation corresponds to one face of a cube containing the protein and was used as a starting point for three independent simulations using different distributions of the initial velocities. The conformations were stored every picosecond. For each stored conformation we calculated the effective energy of the enzyme in water, using the EEF1.1 force field.27 The binding energy of PI-PLC to membranes could thus be calculated every picosecond as the difference between the effective energy with the membrane model IMM1-GC and the effective energy in water (EEF1.1) (as described in detail in Ref.29). The structural and energetic analyses were done on the simulation that led to the lowest average effective energy. The trajectory between 2.5 and 4 ns was used for analysis as the anchorage depth and binding energy were stable on this time scale. Note that we also ran the same simulations with a neutral implicit bilayer but these did not lead to significant anchorage of PI-PLC (data not shown). IMM1 generally underestimates the binding of PI-PLC, both with zwitterionic and anionic membranes presumably because it underestimates the non-hydrophobic contribution of aromatic amino acids. The contributions of each PI-PLC amino acid to the solvation free energy was calculated for every frame in the last 1.5 ns of the simulation and then averaged. We then calculated the difference with the corresponding terms in water to evaluate the contribution of each atom to the membrane binding energy. The atomic contributions were summed to obtain residue contributions.
Simulation with an explicit bilayer
The last 3 ns of the three IMM1 simulations leading to membrane anchorage were merged to evaluate the backbone angle and center of gravity. We then selected the conformation that had the orientation with the lowest deviations relative to the average binding energy values, angle as well as center of gravity, and used it to initiate the all-atom simulation. Only the orientation and not the protein structure was used since long implicit solvent simulations can result in high protein flexibility and unrealistic deformations. Instead a combination of x-ray structures 2OR2 and 3EA1 was used (see above) and oriented following the IMM1 results. The bilayer contains 256 dimyristoylphosphatidylcholine (DMPC) lipids. It was taken from Broemstrup et al47 and further equilibrated for 100 ns with the Charmm36 force field23(average value of surface area per lipid was 60.9 +/− 0.9 Å2). In order to reduce steric clashes, four lipids were removed after insertion of the protein into the leaflet. Membrane and protein were then submitted to an energy minimization using the following harmonic restraints: 150 kcal/mol/Å2 for protein backbone and water molecules; 100 kcal/mol/Å2 for membrane atoms located further than 5 Å from the protein and protein sidechain atoms located further than 5 Å from the membrane; 50 kcal/mol/Å2 for membrane atoms located within 5 Å of the protein; 15kcal/mol/Å2 for protein sidechains atoms located 5 Å or less from the membrane. The minimization consisted of 20 consecutive cycles of 600 minimization steps (500 steepest descent, 100 conjugated gradients) with restraints being scaled by 0.65 after each cycle. TIP3P water molecules were then added to the minimized structure using VMD48 which resulted in a system with dimension 80 × 90 × 115 Å. Finally, we replaced seven water molecules, chosen randomly by sodium ions to neutralize the system.
The program NAMD(v2.9)49 was used to perform the simulations at 310 K with an integration timestep of 2 fs, in the NPT ensemble, without surface tension in the isobaric-isothermal ensemble using the Langevin piston method50 (target pressure: 1 atm, oscillation period: 200 fs, damping time scale: 50 fs) and Langevin dynamics to control the temperature (temperature damping coefficient: 1.0). We used the Charmm force field51 (c27 including the CMAP corrections52) with the force field update for lipids (Charmm36)23. The equations of motion were integrated using a multiple time step algorithm;53 bonded interactions and short-range nonbonded forces were evaluated every step, and long-range electrostatics every second step. Force-based switching was used for both electrostatic and van der Waals interactions, with a switch distance of 11 Å and a cutoff of 12 Å. Particle-Mesh Ewald54 was used for long-range electrostatic interactions. SHAKE55 was applied to constrain all bonds between hydrogen and heavy atoms. We first performed two short initial equilibrations of 400 ps each in the NVT ensemble, velocities being reassigned every 100 fs and 1 ps, respectively, with a constrained protein backbone. Constraints were then removed and the system was further equilibrated for 2 ns with periodic reassignment of the velocities every 1ps. The production phase was finally run for 500 ns. Backbone RMSD compared to the minimized X-ray structure was monitored and reveal an overall stable structure of PI-PLC.
Simulations in water
The wild type enzyme and the Y248A mutant were simulated in TIP3P water. The starting structure was the same as above and the procedure similar except for the heating/equilibration procedure. The structures were first submitted to four successive heating stages of 2 ps each at 10 K, 100 K, 200 K and 300 K, respectively. The equilibration procedure was 200 ps long with a velocity reassignment every picosecond. A timestep of 1 fs and a simulation temperature of 300 K were used. The systems were solvated in boxes of 90 × 90 × 90 Å.
Trajectory analysis
Analyses presented here were performed on the whole trajectory minus the first fifty nanoseconds (450 ns). The only exception is the calculation of depth of anchorage which is done over the whole simulation.
Hydrophobic contacts are considered to exist if two unbound candidate atoms are within 3 Å for at least 10 ps. Candidate atoms are atoms in aliphatic groups of amino acids side chains (Charmm force field nomenclature: ca; cb; cg1; cg2; ha*; hb*; hg; hg2*; type cg except the one of hsd, hse, asn, asp; type hg1 except for cys, thr, ser; type cd except for arg, gln glu; type cd1; type cd2 except for hsd, hse; type ce1, ce2, cz and associated hydrogens of phe, tyr; type cd1, cd2, ce2, ce3, cz2 cz3 and associated hydrogens of trp; type cay and type hy*). The criteria for hydrogen bonds are the following: acceptor (A) to hydrogen distance equal or below 2.4 Å, and angle D-H-A (D: hydrogen bond donor) equal or greater than 130°. These two criteria must be met for at least 10 ps. The donor and acceptor definitions are from the Charmm 51 force field. Cation-π interactions between the aromatic rings of tyrosines and tryptophans were considered to exist when all distances between the aromatic ring atoms and the choline nitrogen were below 7 Å. Additionally these distances should not differ by more than 1.5 Å.10,42 We report occupancies (number of trajectory conformations where the interaction is present / total number of conformations) for cation-π interactions and hydrogen bonds. We chose to report the average number of contacts per frame for hydrophobic interactions because a given residue can mediate several interactions at a time, and reporting occupancies would not reflect the number of interacting partners. Hydrogen bonds and cation-π interactions on the other hand rarely involve more than one simultaneous partner. Due to lipid diffusion, a given residue can interact with different DMPC molecules along the simulation but we consider the membrane as a whole and lipids as interchangeable. Time-evolution plots of cation-pi interactions (Figure 3A), hydrogen bonds and hydrophobic contacts (Figure 1 in Supporting Information) provide insight into the stability of the main interactions. These plots are generated as follows: the simulation is divided into 1ns-long windows, and one dot is plotted if the interaction/contact is observed on at least half of the conformations of the window centered around that point.
To evaluate the depth of anchorage, we used the average z coordinate of the phosphorus atoms as a reference (the plane of the membrane is perpendicular to the z axis). For each frame, the z coordinate of the center of mass of each residue was calculated, and its distance to the reference was measured. All coordinates statistics were done using the corman module of the Charmm program. Values reported are averages of the distances calculated over the whole simulation.
Plots along simulation time of RMSD, occupancies of hydrogen bonds, number of hydrophobic contacts, and depth of anchorage are provided as Supporting Information.
Enzyme expression and purification
All PI-PLC mutants were constructed in the N168C B. thuringiesis PI-PLC background using QuikChange methodology with a site-directed mutagenesis kit (Agilent Technologies). The salt-free purified mutagenic primers were purchased from Operon. All mutated genes were sequenced at Genewiz to confirm that the desired mutation was introduced. A plasmid containing the mutant B. thuringiensis gene was transformed into Escherichia coli BL21- Codonplus (DE3)-RIL cells. Overexpression and purification of these mutants followed procedures described previously.19 These mutants expressed well in E. coli, and >90% purity of isolated protein, as monitored by SDS-PAGE, was achieved by chromatography on a Q-Sepharose fast flow column followed by a phenyl-Sepharose column as described for the recombinant B. thuringiensis PI-PLC (rPI-PLC).19 Protein solutions were concentrated using Millipore Central plus 10 filters, and concentrations were estimated by absorption at 280 nm using the extinction coefficient calculated using ProtParam.56 Secondary structure as estimated from far UV circular dichroism (CD) data using an AVIV 202 CD spectrophotometer, were essentially the same for all the single tyrosine mutants indicating no significant change in secondary structure. Small changes were seen for some of the double mutants, but these were close to the error in the deconvolution of the spectra, performed using the CDNN program 57(see Supporting Information). Thermal stability of PI-PLC variants was assessed by monitoring the ellipticity at 222 nm while increasing the sample temperature 0.5 degrees per minute.20,33 Activities for all the variants (see Supporting Information), determined towards 4 mM PI in small vesicles with various amounts of PC, were determined by 31P NMR spectroscopy as described previously.21,30
Preparation of small unilamellar vesicles (SUVs)
Stock solutions of 1-palmitoyl-2-oleoyl-phosphatidylcholine (POPC) and dioleoylphosphatidylglycerol (DOPG) in choloroform were purchased from Avanti Polar Lipids. SUVs with increasing mole fractions of POPC were prepared by sonication in phosphate buffered saline, pH 7.4 (PBS) as previously described.32
Measuring PI-PLC binding to SUVs with FCS
The FCS based SUV binding experiments take advantage of the fact that protein binding to vesicles slows translational diffusion. FCS experiments were performed using PI-PLC variants labeled at N168C with Alexa Fluor 488 maleimide and a home-built confocal setup based on an IX-70 inverted microscope (Olympus) as previously described.32 FCS experiments were carried out at 22°C on 300 μL samples in PBS, pH 7.4, plus 1 mg/ml bovine serum albumin (BSA) to stabilize PI-PLC, in chambered coverglass wells (LabTek), coated with 10 mg/ml BSA and rinsed with PBS prior to use to prevent protein adhesion to the sides of the wells. 10 nM labeled PI-PLC was titrated with unlabeled SUVs, and the fraction of protein bound to vesicles was determined from two species fits to the autocorrelations (obtained in crosscorrelation mode), G(τ) 33,58-60
| (1) |
Where p and v denote free protein and SUVs that are fluorescent due to PI-PLC binding, respectively, and Aj is the amplitude of species j. The correlation function for species j, gj(τ), accounts for diffusion of the molecules through the observation volume which depends on the radius and extent of the observation volume, determined from fits to rhodamine 110 calibration data using D=280 m2 s−1 at 22°C61, and Dj, the diffusion coefficient for each species.58,62 Dp for the free protein was experimentally determined in the absence of vesicles while Dv for the SUVs was determined from global fits to all of the titration experiments for a particular XPC using Origin (OriginLab). The apparent fraction of protein bound to the SUVs, f, can be determined from Ap and the time-averaged number of proteins in the observation volume in the absence of vesicles,59,60 <No>:
| (2) |
where Ap,o=1/<No> is the autocorrelation amplitude for free PI-PLC prior to titration corrected for volume changes. The apparent dissociation constant, Kd, representing PI-PLC partitioning to the vesicle surface, and a cooperativity coefficient, n, was determined from fits to a Hill equation:
| (3) |
where f, is determined for different total lipid concentrations, [PL], at fixed XPC, and fmax is the apparent maximum fraction bound. Fitting the correlation curves to equation 1 assumes that all of the SUVs have a single well-defined radius with a single diffusion coefficient. However the SUVs are not a single population, but rather have a distribution of sizes. Fitting a polydisperse population to a single size, using equation 1, has no significant effects on the value for the apparent Kd, but does reduce the value of fmax (see the supplemental material for references 21 and 63). Reductions in the apparent fraction bound means that even in the case of 100% binding, the value of fmax determined from fits to equations 3 is less than one. FCS experiments were repeated twice using different vesicle and protein preparations. Assuming that uncertainties are normally distributed, we consider a change of 3 times the standard deviation in apparent Kd to be significant. Based on this assumption and the standard deviation values from all of the FCS measurements, a 2-3 times increase in apparent Kd relative to N168C PI-PLC (WT*) is considered significant.
Supplementary Material
ACKNOWLEDGMENT
Parallab (High Performance Computing Laboratory at the University of Bergen) and NOTUR (Norwegian metacenter for computational science) are thankfully acknowledged for provision of CPU time.
Funding Sources
This work was supported by grants from the Bergen Research Foundation to N.R. and C.G., and by the National Institute of General Medical Sciences of the National Institutes of Health under award number R01GM060418 to MFR.
ABBREVIATIONS
- DMPC
dimyristoylphosphatidylcholine
- DMPG
dimyristoylphosphitdylglycerol
- FCS
fluorescence correlation spectroscopy
- MD
molecular dynamics
- PI-PLC
phosphatidylinositol specific phospholipase C
- PC
phosphatidylcholine
Footnotes
ASSOCIATED CONTENT
Supporting Information. Implicit membrane energy decomposition per amino acid (Table 1); RMSD, depth of anchorage, hydrogen bonds and hydrophobic contacts along simulation time (Figure 1); RMSF from simulations of WT and Y248A in water (Figure 2); activity (Figure 3) and far UV CD data (Table 2) for WT* (N168C) and double Tyr variants of PI-PLC are also provided. Binding affinities of the K44A* variant to PG/PC SUVs as a function of mole fraction of PC (Figure 4). “This material is available free of charge via the Internet at http://pubs.acs.org.”
REFERENCES
- (1).Ma JC, Dougherty DA. Chem Rev. 1997;97:1303–1324. doi: 10.1021/cr9603744. [DOI] [PubMed] [Google Scholar]
- (2).Mahadevi AS, Sastry GN. Chem. Rev. 2013;113:2100–38. doi: 10.1021/cr300222d. [DOI] [PubMed] [Google Scholar]
- (3).Xiu X, Puskar NL, Shanata JA, Lester HA, Dougherty DA. Nature. 2009;458:534–7. doi: 10.1038/nature07768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Gallivan JP, Dougherty DA. Proc Natl Acad Sci U S A. 1999;96:9459–64. doi: 10.1073/pnas.96.17.9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Roderick SL, Chan WW, Agate DS, Olsen LR, Vetting MW, Rajashankar KR, Cohen DE. Nat Struct Biol. 2002;9:507–511. doi: 10.1038/nsb812. [DOI] [PubMed] [Google Scholar]
- (6).Beene DL, Brandt GS, Zhong W, Zacharias NM, Lester HA, Dougherty DA. Biochemistry. 2002;41:10262–9. doi: 10.1021/bi020266d. [DOI] [PubMed] [Google Scholar]
- (7).Celie PHN, van Rossum-Fikkert SE, van Dijk WJ, Brejc K, Smit AB, Sixma TK. Neuron. 2004;41:907–914. doi: 10.1016/s0896-6273(04)00115-1. [DOI] [PubMed] [Google Scholar]
- (8).Pittelkow M, Tschapek B, Smits SH, Schmitt L, Bremer E. J Mol Biol. 2011;411:53–67. doi: 10.1016/j.jmb.2011.05.037. [DOI] [PubMed] [Google Scholar]
- (9).Tschapek B, Pittelkow M, Sohn-Bosser L, Holtmann G, Smits SH, Gohlke H, Bremer E, Schmitt L. J Mol Biol. 2011;411:36–52. doi: 10.1016/j.jmb.2011.05.039. [DOI] [PubMed] [Google Scholar]
- (10).Petersen FNR, Jensen MO, Nielsen CH. Biophys J. 2005;89:3985–3996. doi: 10.1529/biophysj.105.061804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Aliste MP, MacCallum JL, Tieleman DP. Biochemistry. 2003;42:8976–8987. doi: 10.1021/bi027001j. [DOI] [PubMed] [Google Scholar]
- (12).de Jesus AJ, Allen TW. Biochim Biophys Acta. 2013;1828:864–76. doi: 10.1016/j.bbamem.2012.09.009. [DOI] [PubMed] [Google Scholar]
- (13).Broemstrup T, Reuter N. Phys Chem Chem Phys. 2010;12:7487–96. doi: 10.1039/b924117e. [DOI] [PubMed] [Google Scholar]
- (14).Sanderson JM. Org Biomol Chem. 2007;5:3276–3286. doi: 10.1039/b707502b. [DOI] [PubMed] [Google Scholar]
- (15).Sanderson JM, Whelan EJ. Phys Chem Chem Phys. 2004;6:1012–1017. [Google Scholar]
- (16).Johnson JE, Cornell RB. Mol Membr Biol. 1999;16:217–235. doi: 10.1080/096876899294544. [DOI] [PubMed] [Google Scholar]
- (17).Kim JY, Mosior M, Chung LA, Wu H, Mclaughlin S. Biophys J. 1991;60:135–148. doi: 10.1016/S0006-3495(91)82037-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Luckey M. Membrane Structural Biology: With Biochemical and Biophysical Foundations. Cambridge University Press; 2008. [Google Scholar]
- (19).Feng J, Wehbi H, Roberts MF. J Biol Chem. 2002;277:19867–75. doi: 10.1074/jbc.M200938200. [DOI] [PubMed] [Google Scholar]
- (20).Guo S, Zhang X, Seaton BA, Roberts MF. Biochemistry. 2008;47:4201–10. doi: 10.1021/bi702269u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Pu M, Roberts MF, Gershenson A. Biochemistry. 2009;48:6835–45. doi: 10.1021/bi900633p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Shi X, Shao C, Zhang X, Zambonelli C, Redfield AG, Head JF, Seaton BA, Roberts MF. J Biol Chem. 2009;284:15607–18. doi: 10.1074/jbc.M901601200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, Jr., Pastor RW. J Phys Chem B. 2010;114:7830–43. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Heinz DW, Ryan M, Bullock TL, Griffith OH. EMBO J. 1995;14:3855–63. doi: 10.1002/j.1460-2075.1995.tb00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Proc Natl Acad Sci USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Rogaski B, Klauda JB. J Mol Biol. 2012;423:847–61. doi: 10.1016/j.jmb.2012.08.015. [DOI] [PubMed] [Google Scholar]
- (27).Lazaridis T. Proteins. 2003;52:176–92. doi: 10.1002/prot.10410. [DOI] [PubMed] [Google Scholar]
- (28).Lazaridis T. Proteins. 2005;58:518–27. doi: 10.1002/prot.20358. [DOI] [PubMed] [Google Scholar]
- (29).Hajjar E, Mihajlovic M, Witko-Sarsat V, Lazaridis T, Reuter N. Proteins. 2008;71:1655–1669. doi: 10.1002/prot.21853. [DOI] [PubMed] [Google Scholar]
- (30).Pu M, Orr A, Redfield AG, Roberts MF. J Biol Chem. 2010;285:26916–22. doi: 10.1074/jbc.M110.123083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Goldstein RI. PhD dissertation. Boston College; 2012. [Google Scholar]
- (32).Pu M, Fang X, Redfield AG, Gershenson A, Roberts MF. J Biol Chem. 2009;284:16099–107. doi: 10.1074/jbc.M809600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Cheng J, Karri S, Grauffel C, Wang F, Reuter N, Roberts MF, Wintrode PL, Gershenson A. Biophys J. 2013;104:1–11. doi: 10.1016/j.bpj.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Wimley WC, Creamer TP, White SH. Biochemistry. 1996;35:5109–5124. doi: 10.1021/bi9600153. [DOI] [PubMed] [Google Scholar]
- (35).Gallivan JP, Dougherty DA. J Am Chem Soc. 2000;122:870–874. [Google Scholar]
- (36).Dougherty DA. Science. 1996;271:163–8. doi: 10.1126/science.271.5246.163. [DOI] [PubMed] [Google Scholar]
- (37).Kumpf RA, Dougherty DA. Science. 1993;261:1708–10. doi: 10.1126/science.8378771. [DOI] [PubMed] [Google Scholar]
- (38).Luhmer M, Bartik K, Dejaegere A, Bovy P, Reisse J. B Soc Chim Fr. 1994;131:603–606. [Google Scholar]
- (39).Caldwell JW, Kollman PA. Journal of the American Chemical Society. 1995;117:4177–4178. [Google Scholar]
- (40).Yau WM, Wimley WC, Gawrisch K, White SH. Biochemistry. 1998;37:14713–8. doi: 10.1021/bi980809c. [DOI] [PubMed] [Google Scholar]
- (41).Woolf TB, Grossfield A, Pearson JG. Int J of Quant Chem. 1999;75:197–206. [Google Scholar]
- (42).Minoux H, Chipot C. J Am Chem Soc. 1999;121:10366–10372. [Google Scholar]
- (43).Goldstein R, Cheng J, Stec B, Roberts MF. Biochemistry. 2012;51:2579–87. doi: 10.1021/bi300057q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Shao C, Shi X, Wehbi H, Zambonelli C, Head JF, Seaton BA, Roberts MF. J Biol Chem. 2007;282:9228–35. doi: 10.1074/jbc.M610918200. [DOI] [PubMed] [Google Scholar]
- (45).Brooks BR, Brooks CL, 3rd, Mackerell AD, Jr., Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. J Comput Chem. 2009;30:1545–614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Neria E, Fischer S, Karplus M. J Chem Phys. 1996;105:1902–1921. [Google Scholar]
- (47).Broemstrup T, Reuter N. Biophys J. 2010;99:825–33. doi: 10.1016/j.bpj.2010.04.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Humphrey W, Dalke A, Schulten K. J Molec Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- (49).Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. J Comput Chem. 2005;26:1781–802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Feller SE, Zhang YH, Pastor RW, Brooks BR. J Chem Phys. 1995;103:4613–4621. [Google Scholar]
- (51).MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. Journal of Physical Chemistry B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- (52).Mackerell AD, Feig M, Brooks CL. Journal of Computational Chemistry. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- (53).Izaguirre JA, Reich S, Skeel RD. J Chem Phys. 1999;110:9853–9864. [Google Scholar]
- (54).Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- (55).Andersen HC. J Comput Phys. 1983;52:24–34. [Google Scholar]
- (56).Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. In: The Proteomics Protocols Handbook. Walker JM, editor. Humana Press; New York: 2005. pp. 571–607. [Google Scholar]
- (57).Bohm G, Muhr R, Jaenicke R. Protein Eng. 1992;5:191–5. doi: 10.1093/protein/5.3.191. [DOI] [PubMed] [Google Scholar]
- (58).Thompson NL. In: Topics in Fluorescence Spectroscopy. Lakowicz JR, editor. Plenum Press; New York: 1991. pp. 337–378. [Google Scholar]
- (59).Rusu L, Gambhir A, McLaughlin S, Radler J. Biophys J. 2004;87:1044–1053. doi: 10.1529/biophysj.104.039958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Middleton ER, Rhoades E. Biophys J. 2010;99:2279–2288. doi: 10.1016/j.bpj.2010.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Magde D, Elson EL, Webb WW. Biopolymers. 1974;13:29–61. doi: 10.1002/bip.1974.360130103. [DOI] [PubMed] [Google Scholar]
- (62).Elson EL, Magde D. Biopolymers. 1974;13:1–27. doi: 10.1002/bip.1974.360130103. [DOI] [PubMed] [Google Scholar]
- (63).Cheng J, Karri S, Grauffel C, Wang F, Reuter N, Roberts MF, Wintrode PL, Gershenson A. Biophys J. 2013;104:185–95. doi: 10.1016/j.bpj.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.