

Abstract
We explore noninvasive clinical applications of multiple disease-specific fusion markers recently discovered in prostate cancer to predict the risk of cancer occurrence and aggressiveness at diagnosis. A total of 92 men who were prostate-specific antigen (PSA) screened and scheduled for diagnostic biopsy were enrolled for this study. Prospectively collected urine was blind coded for laboratory tests. RNA from urine sediments was analyzed using a panel of 6 TMPRSS2:ETS fusion markers with a sensitive quantitative PCR platform. The pathology reported 39 biopsy-positive cases from 92 patients (42.4%). In urine test, 10 unique combinations of fusion types were detected in 32 of 92 (34.8%) prebiopsy samples. A novel combination of fusion markers, termed Fx (III, IV, ETS), was identified with a sensitivity of 51.3% and an odds ratio of 10.1 in detecting cancer on biopsy. Incorporating a categorical variable of Fx (III, IV, ETS) with urine PCA3 and serum PSA, a regression model was developed to predict biopsy outcomes with an overall accuracy of 77%. Moreover, the overexpression of Fx (III, IV, or ETS) was shown to be an independent predictor to the high-grade cancer, with a predictive accuracy of 80% when coupled with PSA density. The individualized risk scores further stratified a high-risk group that is composed of 92% high-grade cancers and a low-risk group that harbors mainly clinically insignificant cancers. In conclusion, we have identified a novel combination of fusion types very specific to the clinically significant prostate cancer and developed effective regression models to predict biopsy outcomes and aggressive cancers at diagnosis.
Keywords: Biopsy cohort, diagnosis and prognosis, ETS fusions, prostate cancer, TMPRSS2, urine test
Introduction
The serum prostate-specific antigen (PSA) test coupled with needle biopsy is the standard clinical practice in prostate cancer diagnosis, but it is limited by the outcomes of excessive negative biopsies, overdiagnosis of clinically insignificant cancers and significant false-negative biopsy rate [1–4]. There is currently no reliable method for early detection of the life-threatening form of cancer. As a seminal discovery [5], many prostate tumors contain a specific genetic change that involves the fusion of an androgen-regulated gene with an oncogene. The most common fusion involves androgen-regulated TMPRSS2 gene with an oncogenic ETS family transcription factor ERG gene, which is reported in 50% of surgical prostate tumors [5–12]. Moreover, diverse TMPRSS2:ERG fusion subtypes have been uncovered, ranging from chromosomal rearrangements to fusion transcripts [10, 12–15]. This fusion gene and its many subtypes not only allow stratification of clinically aggressive forms of cancer [10,13], but also provide redundant and cancer-specific transcript markers for noninvasive cancer detection in bodily fluids. Indeed, the TMPRSS2:ERG fusion RNA is shown to be detectable in the urine of men with prostate cancer [16–18]. A common subtype of this fusion, in combination with urine PCA3, enhanced the predictive performance of serum PSA for prostate cancer risk and clinically relevant cancer on biopsy in a recent clinical study [19]. However, the single subtype-based test excludes multiple alternative fusion markers for informed clinical tests.
In addition to the common fusion with ERG gene, TMPRSS2 is also fused to several other ETS transcription factor genes, such as ETV1, ETV4, and ETV5, in approximately 10% of prostate tumors [5–8, 20]. Intriguingly, many of the low-prevalent TMPRSS2:ETS fusion events are associated with aggressive and metastatic cancers [21,22]. Moreover, ETS genes have also been found to fuse with an increasing number of androgen-regulated genes, all at low prevalence. Among these genes, ETV1 emerges as a highly connected ETS gene, fused to more than 10 different androgen-regulated genes [22–25]. These fusion genes along with others have expanded into an interconnected fusion network, consisting of a dominant fusion and many low-prevalent fusion genes that may or may not be mutually exclusive in clinical tumors. Together, these fusion events provide not only a common mechanism for androgen-regulated overexpression of ETS transcription factor genes, but also novel molecular markers that are only detectable in prostate cancer [26–28]. However, the clinical significance of the increasing number of novel fusion genes is poorly studied due to their low prevalence as individual events and the lack of effective tools. Therefore, the potential clinical application of low-prevalent fusion gene markers in prostate cancer detection has yet to be reported. As multiple genomic alterations in prostate tumors have been recently shown to define a group of patients with high-risk cancer [29], a panel of multiple fusion gene markers may provide a new perspective for urine-based detection of the genetic instability or heterogeneity, and for better stratification of the clinically significant prostate cancer.
We are among the first to use a panel of TMPRSS2:ERG subtype markers for urine-based prostate cancer detection with high specificity and sensitivity [30]. In this study, we hypothesize that multiple TMPRSS2:ERG fusion subtypes and additional low-prevalent TMPRSS2:ETS fusion genes are collectively more informative than any single marker alone in the noninvasive detection and stratification of clinically significant prostate cancer. To test this hypothesis, a new panel of TMPRSS2:ETS fusion gene markers were investigated in the prospectively collected urine from PSA-screened men scheduled for diagnostic biopsy. We demonstrated for the first time, the feasibility of incorporating fusion subtypes and low-prevalent fusion genes into clinical practice. Moreover, we identified several alternative fusion markers very specific to clinically significant cancers and developed effective regression models to predict the risk of both cancer occurrence and aggressiveness prospectively.
Materials and Methods
Human subjects
Patients who were screened by serum PSA and scheduled for diagnostic needle biopsy were recruited at the prostate cancer clinics at the McGill University Health Center (MUHC). The research protocol was approved by the MUHC institutional review board and written informed consent was obtained from every participant. A total of 97 patients were recruited from April to November 2010 to form a prebiopsy cohort, among which 92 patients generated informative samples for molecular analysis (Fig. S1). Urine was collected post attentive digital rectal exam (DRE) and prior to needle biopsy. The prebiopsy urine specimens were coded for anonymity. Laboratory investigators were blinded to sample allocation for prospective molecular diagnosis using a panel of molecular markers. The results from this panel were then compared to needle biopsy results in a double-blinded protocol. The pathology of each biopsy with 10 needle cores was reviewed by a single genitor-urinary pathologist at MUHC. All included slides were assigned a grade according to the modified Gleason grading system. Men with previous treatment for prostate cancer or with repeat biopsy were excluded from this study. The baseline clinical–pathological information of the study cohort and 1-year follow-up are provided in Table 1.
Table 1.
Baseline clinical and pathological characteristics of biopsy cases
| Characteristic | Biopsy positive (39) | Biopsy negative (53) |
| Age | ||
| Mean1 | 67.46 ± 7.85 | 68.55 ± 8.12 |
| Median2 | 68 (50–86) | 70 (52–87) |
| n | 39 | 53 |
| Pre-Bx PSA (ng/mL) | ||
| Mean | 15.67 ± 42.99 | 6.75 ± 4.07 |
| Median | 6.71 (1.18–271.57) | 5.87 (0.74–25.25) |
| n | 39 | 53 |
| F/U PSA (ng/mL) | ||
| Mean | 31.29 ± 99.42 | 7.55 ± 5.65 |
| Median | 8.26 (1.19–484.89) | 6.35 (9–145) |
| n | 23 | 23 |
| Prostate size (cc) | ||
| Mean | 40.46 ± 19.93 | 48.73 ± 23.65 |
| Median | 35.00 (16–110)* | 46 (3–60) |
| n | 39 | 51 |
| PSA density | ||
| Mean | 0.37 ± 0.71 | 0.17 ± 0.14 |
| Median | 0.2 (0.05–4.24)** | 0.12 (0.03–0.63) |
| n | 39 | 51 |
| Gleason score | ||
| Mean | 6.74 ± 1.02 | NA |
| Median | 6 (6–9) | NA |
| n | 39 | 53 |
| HG PIN3 | ||
| n | 17 | 16 |
| No. of cores positive | ||
| Mean | 3.90 ± 2.99 | NA |
| Median | 3 (1–10) | NA |
| n | 39 | 53 |
| Maximum percentage cancer involvement in any core | ||
| Mean | 44.23 ± 33.77 | NA |
| Median | 40 (5–100) | NA |
| n | 39 | 53 |
| Treatment | ||
| RP4 (n) | 9 | 0 |
PIN, prostatic intraepithelial neoplasia; PSA, prostate-specific antigen.
Mean with standard deviation, Student's t-test (*P <0.05, **P <0.01).
Median with min–max values, Mann–Whitney test (*P <0.05, **P <0.01).
High-grade PIN.
Radical prostatectomy.
Urine collection and whole-transcriptome cDNA library preparation
From each subject, 10–40 mL of the first voided urine post DRE was collected in a sterile collection cup containing RNA/DNA preservatives (Sierra Molecular, Incline Village, NV) and processed within 4 h of sampling. Urine sediments were collected by low-speed centrifugation at 4°C and resuspended in TRIzol Reagent (Invitrogen, Carlsbad, CA) for immediate RNA extraction or stored at −80°C until use. Total RNA was extracted from urine sediments using a miRNAeasy Mini kit (Qiagen, Germantown, MD) according to the manufacturer's instructions with minor modifications. A whole-transcriptome cDNA library was generated for each sample using a TransPlex WTA2 kit (Sigma-Aldrich, St. Louis, MO) [30].
Detection of multiple molecular markers
Real-time quantitative PCR (qPCR) was used to detect a panel of fusion markers consisting of three TMPRSS2:ERG fusion subtypes (I, III, and IV) and three TMPRSS2:ETS fusion genes (ETV1, ETV4, and ETV5) using the previously established protocol [30]. Additional molecular markers were also quantified, including two ERG markers targeting exons 5–6 and 6–7, PCA3, PSA, and the housekeeping gene GAPDH. The probe sequences, amplification features, and gene locations are listed in Table S1 and Figure S2. Briefly, 9 ng of cDNA was amplified in a 20-μL reaction containing 1× SYBR Green Supermix (Bio-Rad, Hercules, CA) and 300 nmol/L of each forward and reverse primers, using a two-step amplification program. The qPCR program consisted of initial denaturing at 95°C for 1.5 min, followed by 50 cycles of a two-step reaction at 95°C for 15 sec, and 67–70°C (varying for marker pairs) for 30 sec. The qPCR was performed using the MyiQ real-time PCR system (Bio-Rad). The relative expression of each target gene was normalized to GADPH for nonfusion markers, or both GAPDH and PSA for fusion markers using the comparative Ct method (Applied Biosystems User Bulletin 2, Foster City, CA). A control sample was included in all amplifications to serve as a common calibrator for relative expression.
Data analysis
All statistical analyses except for the DeLong's test were performed using IBM SPSS statistics 19, version 19.0.0 (IBM Corporation, Armonk, NY). The DeLong's test was performed using R, version 2.13.1 (R Project for Statistical Computing, http://www.R-project.org). Two-sided tests were used for all comparisons and P values <0.05 were considered statistically significant. The REMARK guidelines were followed in data analysis [31].
Associations between biopsy outcomes, molecular subgroups, and clinical–pathological variables were assessed with Student's t-test (parametric), Mann–Whitney test (nonparametric), Fisher's exact test or chi-square test (categorical), or Spearman's ρ. Diagnostic values of biomarkers were quantified with sensitivity, specificity, predictive accuracy, odds ratio, and area under the curve (AUC) in the receiver operating characteristic (ROC) curve. The significance in AUCs between different markers was examined with the DeLong's test [32]. The probability of different combination of biomarkers to predict risk of cancer occurrence (i.e., biopsy outcome) or risk of high-grade cancer (i.e., Gleason score ≥7) was assessed using multivariate logistic regression models.
For cancer risk assessment, individual molecular scores were computed from regression models for each subject and used directly to stratify men into three risk groups (i.e., high, intermediate, and low risk) for either biopsy outcome in all biopsy cases or high-grade cancer in biopsy-positive cases. The relative risk (RR) between different risk groups was calculated to measure the probability of biopsy outcome, high-grade cancer, high risk of recurrence (Gleason ≥8, pre-bx PSA ≥ 20 ng/mL or stage T3a) defined by the National Comprehensive Cancer Network (NCCN) guidelines (http://www.nccn.org), features indicative of clinical significance (i.e., prostate-specific antigen density [PSAD] ≥ 0.15 ng/mL or positive cores ≥3 or maximum cancer involvement in a single core ≥50%), and clinically insignificant cancers (i.e., Gleason ≤6, 1–2 positive cores, <50% cancer involvement in any core and PSAD < 0.15 ng/mL) defined by the Epstein criteria [33,34].
Results
Fusion type and clinical sensitivity of a panel of TMPRSS2:ETS fusion markers
The common fusion transcript between TMPRSS2-exon1 and ERG-exon4 (TMP-e1:ERG-e4), subtype I in this study, was the primary marker used in urine-based detection of prostate cancer [16–19]. We previously reported a panel of TMPRSS2:ERG fusion-subtype markers for urine-based cancer detection with high specificity and sensitivity [30]. In this study, we expanded a new panel of TMPRSS2:ETS fusion markers (Table S1 and Fig. S2) and demonstrated reproducible detection of individual fusion markers in a dynamic range from 1.8 million to 18 copies using the qPCR platform (Fig. S3). Using this expanded fusion panel in a urine test, we detected 10 unique combinations of fusion genes and/or subtypes that we termed as “fusion types,” distributed with various frequencies in 32 cases (34.8%) from a prebiopsy cohort of 92 patients (Table 2). Six of the fusion types contained the common subtype I. The remaining four fusion types contained no subtype I and were detected in five of 32 (15.6%) fusion-positive cases. Significantly, we showed for the first time the detection of three low-prevalent TMPRSS2:ETS fusion genes in four urine specimens, among which two of the specimens also had at least one TMPRSS2:ERG subtype. The pathology, on the other hand, identified 39/92 (42.4%) biopsy-positive cases (Table 1). We reasoned that the diverse fusion types provided not only redundant fusion markers for improved sensitivity in cancer detection but also a molecular basis for stratification of cancer risks. Indeed, we demonstrated that different fusion types exhibited different predictive values on biopsy outcomes; the most informative fusion markers composed of TMPRSS2:ERG subtype III, IV or any TMPRSS2:ETS (ETV1, ETV4, ETV5) fusions, a special combination we termed Fx (III, IV, ETS). When used as a categorical variable, this combination of fusion markers had a sensitivity of 51.3% and a specificity of 90.6% with an odds ratio of 10.1 in detecting prostate cancer on biopsy (Table 3). Significantly, this novel combination of fusion markers outperformed fusion types containing the common subtype I in prospective cancer detection. Indeed, six of seven cases detected with the common subtype alone were biopsy negative. Taken together, fusion-typing of a novel panel of TMPRSS2:ETS fusion markers allows detection of diverse fusion types in urine; multiple alternative fusion markers other than the common subtype I provide improved detection of prostate cancer in prebiopsy patients. To rule out potential contamination in fusion detection, fusion-positive results were independently validated in a second aliquot of original RNA from all 32 fusion-positive cases (Fig. S1 and Table S2), in corresponding prostatectomy cancer tissues from three available fusion-positive cases (Table S3) and by DNA sequencing (Fig. S4).
Table 2.
Fusion types: combination of fusion genes and subtypes found in individual biopsy patients
| No. of patients (n) | TMP:ERG I (T-e1:E-e4) | TMP:ERG III (T-e1:E-e2) | TMP:ERG IV (T-e1:E-e5) | TMP:ETV1 (T-e1:V1-e6) | TMP:ETV4 (T-e1:V4-e5) | TMP:ETV5 (T-e1:V5-e2) | Fusion types1 |
| 7 | + | TMP:ERG I | |||||
| 7 | + | + | TMP:ERG (I + III) | ||||
| 4 | + | + | TMP:ERG (I + IV) | ||||
| 1 | + | + | TMP:ERG I + TMP:ETV5 | ||||
| 2 | + | TMP:ERG III | |||||
| 1 | + | TMP:ERG IV | |||||
| 7 | + | + | + | TMP:ERG (I + III + IV) | |||
| 1 | + | + | + | TMP:ERG (I + IV) + TMP:ETV1 | |||
| 1 | + | TMP:ETV4 | |||||
| 1 | + | TMP:ETV5 |
Combination of fusion markers detected in the same patient.
Table 3.
Sensitivity and specificity of models based on different TMPRSS2:ETS fusion types to predict biopsy outcome
| Features | Pre-bx PSA1 | TMP:ERG I2 | Fx (III, IV, ETS)3 | All fusions4 |
| Frequency (%) | – | 27/92 (29.35) | 25/92 (27.17) | 32/92 (34.78) |
| Sensitivity (%) | 9/30 (23.1) | 17/39 (43.6) | 20/39 (51.3) | 21/39 (53.8) |
| Specificity (%) | 45/53 (84.9) | 43/53 (81.1) | 48/53 (90.6) | 42/53 (79.2) |
| Overall percentage | 58.7 | 65.2 | 73.9 | 68.5 |
| Odds ratio (OR) (95% CI) | 1.629 (0.960–2.763) | 3.323 (1.305–8.463) | 10.105 (3.315–30.808) | 4.455 (1.784–11.121) |
| P | 0.070 | 0.012 | <0.001 | 0.001 |
PSA, prostate-specific antigen.
Prebiopsy serum PSA (quantitative variable).
Any fusion-type event containing TMP:ERG subtype I fusion (categorical variable).
Any fusion-type event containing TMP:ERG subtype III or IV or other TMP:ETS (ETV1, ETV4, ETV5) (categorical variable).
Any fusion-type event containing TMP:ERG subtype I or III or IV or other TMP:ETS (ETV1, ETV4, ETV5) (categorical variable).
Distinct molecular subgroups stratified by biopsy outcome and fusion status
Biopsy outcomes stratified by fusion status (all fusion types) identified four distinctive molecular subgroups exhibiting different molecular (Fig. 1) and clinical–pathological (Table 4) features. For example, a biopsy-positive/fusion-positive [Bx(+)/Fx(+)] subgroup exhibited a very significant increase in urine expression of cancer-specific markers ERG(5-6), ERG(6-7), and PCA3 when compared with a double-negative subgroup [Bx(−)/Fx(−)]. This result was consistent with our previous observation for a strong correlation between TMPRSS2:ERG fusion and ERG expression levels in urine of prostate cancer patients [30]. In contrast, a biopsy-positive/fusion-negative subgroup [Bx(+)/Fx(−)] exhibited a residual level of ERG markers, but a significantly elevated PCA3 expression. Thus, the PCA3 marker may be informative to identify prostate cancer in the Bx(+)/Fx(−) subgroup. It is useful to note that the overall urine PSA level was similar between [Bx(+)/Fx(+)] and [Bx(+)/Fx(−)] subgroups (Fig. 1D). Thus, the Fx(−) status in Bx(+) cases could not simply be attributed to the biased sampling. On the other hand, a small subgroup of biopsy-negative/fusion-positive [Bx(−)/Fx(+)] cases (n = 11) was also identified in this study, which had a molecular profile similar to the Bx(+)/Fx(+) subgroup. Interestingly, most cases detected with the common subtype I alone belonged to this subgroup. However, the Bx(−)/Fx(+) subgroup was shown to be associated with older age (P < 0.001) and increased incidence of high-grade prostatic intraepithelial neoplasia (PIN) lesions (P < 0.05) as compared with the double-negative subgroup (Table 4). These features raise the possibility that the Bx(−)/Fx(+) subgroup has an elevated risk for either “false” biopsy-negative events or future development of cancer in at least some of the cases (e.g., cases with both fusion-positive and with HG PIN lesions). Finally, the double-negative subgroup [Bx(−)/Fx(−)] was shown to exhibit baseline levels of molecular and clinical features as compared with the other three subgroups.
Figure 1.
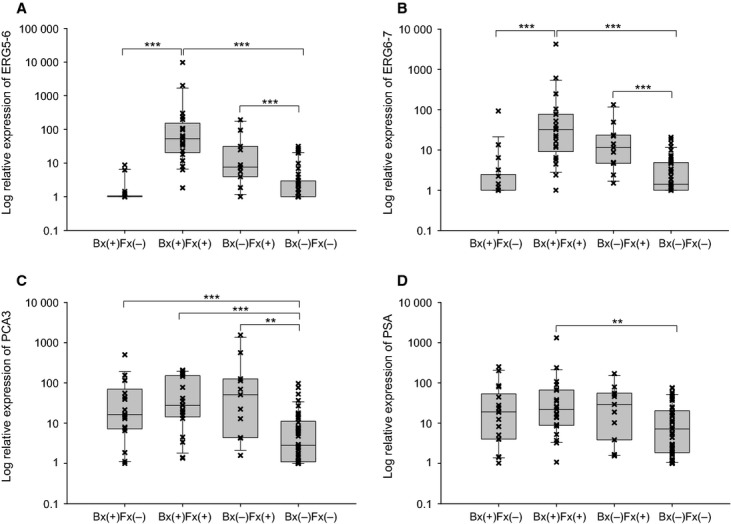
Distinctive molecular subgroups stratified by biopsy outcomes and fusion status in urine. A total of 92 biopsy patients were stratified into the four subgroups based on biopsy outcomes (Bx) and fusion status (Fx): Bx(+)/Fx(−) (n = 18), Bx(+)/Fx(+) (n = 21), Bx(−)/Fx(+) (n = 11), and Bx(−)/Fx(−) (n = 42). (A, B), the expression levels of two ERG markers (ERG [5–6] and [6–7], respectively) in urine were stratified by Bx and Fx status. (C, D), the expression levels of PCA3 and PSA in urine were stratified by Bx and Fx status. The relative expression was calculated using ∆∆ threshold cycle method and transformed into log(x + 1) values. Mann–Whitney test was performed (**P < 0.01, and ***P < 0.001).
Table 4.
Clinical and pathological characteristics of four distinct molecular subgroups stratified by biopsy (Bx) and fusion (Fx) status
| Clinical parameters1 | Bx(+)/Fx(−) | Bx(+)/Fx(+) | Bx(−)/Fx(+) | Bx(−)/Fx(−) |
| No. of cases (n) | 18 | 21 | 11 | 42 |
| Age | 69.50 (54–86) | 68 (50–81) | 75.00 (65–83)*** | 66 (52–87) |
| Pre-Bx prostate-specific antigen (ng/mL) | 6.60 (2.33–271.57) | 6.71 (1.18–54.46) | 7.51 (1.29–25.25) | 5.83 (0.74–14.31) |
| Prostate Size (cc) | 35.00 (19–110) | 35 (16–88) | 46 (19–80) | 46.50 (9–145) |
| Prostate-specific antigen density | 0.21 (0.05–4.24)** | 0.18 (0.05–1.88)* | 0.15 (0.05–0.63) | 0.12 (0.03–0.56) |
| HG PIN (n)2 | 8 | 9 | 6* | 10 |
| HG PIN core no. >2 (n)2 | 4 | 7 | 4* | 6 |
| Gleason score | 6 (6–9) | 6 (6–9) | – | – |
| No. of cores with cancer | 3 (1–10) | 3 (1–10) | – | – |
| Max% inv. of single core | 40 (5–100) | 40 (5–100) | – | – |
| Treatment (n)3 | 4 RP3 | 5 RP | – | – |
PIN, prostatic intraepithelial neoplasia.
Expressed as median with min–max values unless indicated differently, Mann–Whitney test (vs. Bx(−)/Fx(−) where applicable; *P <0.05, **P <0.01, ***P <0.001).
Number of cases, chi-square test (P <0.05).
Radical prostatectomy.
Individualized risk model to predict cancer occurrence at prostate biopsy
The combined detectability of the informative fusion markers [i.e., Fx (III, IV, ETS)] had an improved predictive value on cancer occurrence. By incorporating the categorical variable of fusion types Fx (III, IV, ETS) with log-transformed continuous variables of PCA3 and serum PSA, a logistic regression model was developed to calculate an individualized molecular score for optimal prediction of biopsy outcomes (Table 5). As demonstrated in the ROC curve analysis, the Fx (III, IV, ETS) + PCA3 + serum PSA (FPP) score had a significantly increased AUC (0.80) as compared with serum PSA (0.58, P < 0.001), TMPRSS2:ERG subtype I (0.65, P < 0.01), or Fx (III, IV, ETS) fusion type (0.71, P < 0.05) in the prebiopsy cohort of 92 patients (Fig. 2A). For clinical-friendly applications, the FPP molecular scores were used to stratify prebiopsy patients prospectively into three distinctive risk groups: the high-risk group (FPP scores [1–0.47], n = 31), the intermediate-risk group (scores [0.47–0.25], n = 30), and the low-risk group (scores [0.25–0], n = 31) (Fig. 2B). As such, the positive predictive value (PPV) for prostate cancer detection was 81% in the high-risk group, but only 30% and 16% in the intermediate- and low-risk groups (Table 6). On the other hand, a small fraction of biopsy-negative cases were identified in the high-risk group; such cases may be prioritized for repeat biopsies to identify potentially false-negative biopsies. Meanwhile, all except two biopsy-negative cases in the low-risk group were also negative to any fusion markers and could be truly low-risk subjects to prostate cancer. Interestingly, three Bx(−) patients from the low-risk group were biopsied again within 12 months and remained negative in the follow-up biopsy. However, the FPP model was not effective to predict significant cancers when only the biopsy-positive cases were considered (Table 6, but also Table S4).
Table 5.
A logistic regression model based on informative fusion markers [Fx (III, IV, ETS)], PCA3, and prebiopsy serum PSA to predict cancer risk on biopsy
| Univariable logistic regression models | Multivariable logistic regression model1 | |||||||
| Biopsy cohort (n) | Dependent variable | Diagnostic Variable | OR (95% CI) | P | Overall accuracy (%) | OR (95% CI) | P | Overall accuracy (%)2 |
| Fx (III, IV, ETS)3 | 10.11 (3.32–30.81) | <0.0001 | 73.9 | 7.10 (2.20–22.89) | 0.001 | |||
| 92 | Biopsy outcome | PCA34 | 1.34 (1.11–1.60) | 0.002 | 65.2 | 1.22 (1.00–1.49) | 0.045 | 77.25 |
| Serum PSA4 | 1.63 (0.96–2.76) | 0.07 | 58.7 | 1.58 (0.89–2.77) | 0.116 | |||
PSA, prostate-specific antigen.
Hosmer–Lemeshow Goodness-of-Fit of logistic regression model: P = 0.307.
Defined as (true positives + true negatives)/all.
TMP:ERG subtype III or IV, or TMP:ETS (ETV 1, 4, or 5) (binary categorical variable).
PCA3 and serum PSA were log-transformed continuous variables.
61.5% sensitivity and 88.7% specificity at 50% cut-off value.
Figure 2.
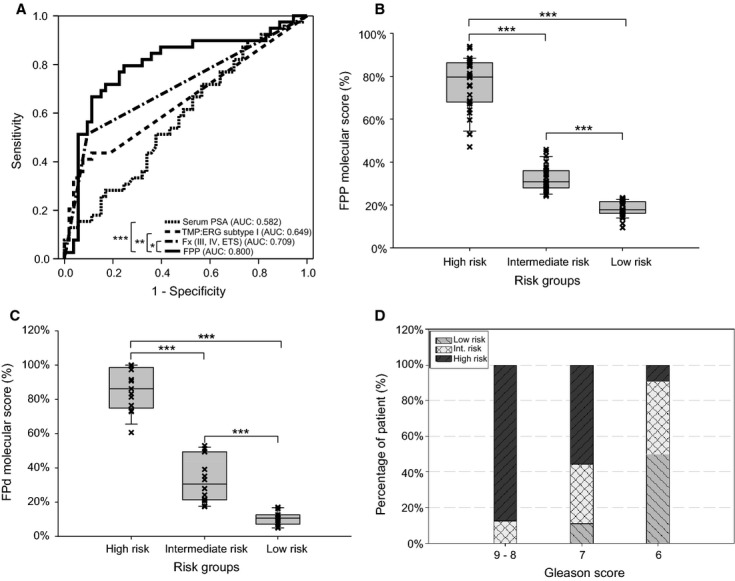
Molecular risk groups to predict either biopsy outcomes (A, B) or high-grade prostate cancers (C, D) in a prebiopsy cohort. (A) Receiver operating characteristic (ROC) curves for pre-Bx serum prostate-specific antigen (PSA) (blue), TMPRSS2:ERG (TMP:ERG) fusion subtype I (green), informative fusion types Fx (III, IV, ETS) (brown), and the FPP scores (purple). The FPP score [Fx (III, IV, ETS) + PCA3 + Pre-Bx PSA] was used for the prediction of biopsy outcomes in the prebiopsy cohort (n = 92). (B) The FPP scores were used to stratify prebiopsy patients into high- (n = 31), intermediate- (n = 30), and low (n = 31)-risk groups. (C) The FPd scores [Fx (III, IV, ETS) + prostate-specific antigen density] were used to stratify biopsy-positive patients (n = 39) into high- (n = 13), intermediate- (n = 14), and low (n = 12)-risk groups to high-grade cancers (Gleason >6). (D) Distribution of high-, intermediate-, and low-risk groups (%) in patients with total Gleason scores of 9–8 (n = 8), 7 (n = 9), and 6 (n = 22). The area under the curve of the ROC curves were compared with DeLong's test (*P <0.05, **P <0.01, and ***P <0.001). The Mann–Whitney test was performed between each risk group (***P < 0.001).
Table 6.
Risk groups to cancer occurrence stratified by the FPP molecular scores
| High | Intermediate | Low | |
| Risk score1 | |||
| n (%) | 31 (34) | 30 (33) | 31 (34) |
| Median (min–max) | 0.7967 (0.4707–0.9391) | 0.3077 (0.2415–0.4582) | 0.1772 (0.0943–0.2335) |
| Cancer | |||
| n (%)2 | 25 (81) | 9 (30) | 5 (16) |
| RR (H vs. L), P3 | 5.000 (2.199–11.370), P <0.0001 | ||
| RR (H vs. [I + L]), P4 | 3.514 (2.150–5.743), P <0.0001 | ||
| Gleason score ≥7 | |||
| n (%)5 | 14 (56) | 3 (33) | 0 (0) |
| RR (H vs. L), P | Undefined, P =0.0447 | ||
| RR (H vs. [I + L]), P | 2.613 (0.9038–7.557), P =0.0490 | ||
| High risk of recurrence by NCCN6 | |||
| n (%)5 | 7 (28) | 2 (22) | 0 (0) |
| RR (H vs. L), P | Undefined, P =0.3041 | ||
| RR (H vs. [I + L]), P | 1.960 (0.469–8.183), P =0.4448 | ||
| No. of cores positive ≥3 | |||
| n (%)5 | 16 (64) | 6 (67) | 2 (40) |
| RR (H vs. L), P | 1.600 (0.526–4.871), P =0.3644 | ||
| RR (H vs. [I + L]), P | 0.960 (0.584–1.577), P =1 | ||
| Max.% cancer inv. in any core ≥50% | |||
| n (%)5 | 12 (48) | 2 (22) | 0 (0) |
| RR (H vs. L), P | Undefined, P =0.0657 | ||
| RR (H vs. [I + L]), P | 3.360 (0.874–12.920), P =0.0444 | ||
NCCN, National Comprehensive Cancer Network; PSA, prostate-specific antigen; RR, relative risk.
FPP molecular score: FPP regression model = −2.890 + 1.960*Fx(III, IV, ETS) + 0.201*log 2 (PCA3 + 1) + 0.454*log 2 (serum PSA + 1), Risk score = 1/(1 + e(−FPP)), where e is the number of ∼2.71828.
Percentage of each risk group or positive predictive value in a total of 92 prebiopsy patients.
RR between high (H)- and low (L)-risk groups with 95% CI, P value of Fisher exact test.
RR between high- and intermediate + low (I + L)-risk groups with 95% CI, P value of Fisher exact test.
Percentage of each risk group or positive predictive value in 39 biopsy-positive patients.
Gleason score ≥8, PSA ≥20 ng/mL or T3a as defined by the NCCN guidelines.
Individualized risk model to predict the risk of aggressive cancer
One of the most challenging tasks of early cancer detection is to identify the aggressive forms of cancer at diagnosis. We explored risk models to predict high-grade cancers (Gleason ≥7) in the context of biopsy-positive cases. Unlike the detectability (i.e., categorical variable) of Fx (III, IV, or ETS) used in predicting biopsy outcomes, the overexpression profiles of these fusion markers were more informative to identify clinically significant cancers. Using a common calibrating sample sparked with fusion molecules, we devised a strategy that ranked the relative quantity of different informative fusion markers as a new continuous variable. Instead of using each fusion marker separately with reduced statistic power, we proposed to calculate the quantitative feature of combined fusion markers based on the sum of quantity values (or the highest relative quantity) of fusion markers in each sample. Coupling this new continuous variable of Fx (III, IV, or ETS) with the continuous variable of PSA density (PSAD) that we termed as Fx (III, IV, ETS) + PSA density (FPd), a logistic regression model was developed to predict the risk of high-grade cancer (Gleason ≥7) with an overall accuracy of 80% (Table 7). The essentially same scores were obtained when the combined fusion markers were re-ranked based on the highest quantity in each sample (data not shown). The FPd scores were then used to stratify biopsy-positive cases equally into high-, intermediate-, and low-risk groups for clinically significant cancers (Fig. 2C and D). We demonstrated that the high-risk group not only identified 92.3% (12/13) Gleason ≥7 cancers (P < 0.0001) but also associated with the high-risk-of-recurrence cases (P < 0.001) as defined by the NCCN guidelines (Table 8). Indeed, the FPd molecular scores were highly correlated with Gleason scores (r = 0.65, P < 0.0001), the number of positive cores (r = 0.47, P < 0.01), and the percentage of cancer involvement in a single core (r = 0.61, P < 0.001) when analyzed by the Spearman's ρ (Table 9). Conversely, a low-risk group for cancer progression was also identified using the same score system, which consisted of 91.6% (11/12) cases with Gleason 6 cancers. Coincidentally, six clinically insignificant cancers that were identified among the 39 Bx(+) cases by the Epstein criteria all belonged to this low-risk group. The significant association (P < 0.01) of the low-risk cancers based on molecular scores with the insignificant cancers based on pathology may suggest an indolent property for such cancer patients. Indeed, three Bx(+) cases (Gleason 6) from the low-risk group exhibited no progression in 12-month follow-up biopsies, while two Bx(+) cases (Gleason 6) from the intermediate-risk group failed to detected cancer cells in repeat biopsy (data not shown). However, long-term follow-up is required to determine the clinical outcomes that are beyond the scope of this study.
Table 7.
A logistic regression model based on informative fusion types [Fx (III, IV, ETS)] and PSAD to predict risk of aggressive cancer on biopsy
| Univariable logistic regression models | Multivariable logistic regression model1 | |||||||
| Biopsy cohort (n) | Dependent variable | Diagnostic variable | OR (95% CI) | P | Overall accuracy (%) | OR (95% CI) | P | Overall accuracy (%)2 |
| 39 | Gleason score (≥7 or 6) | Fx (III, IV, ETS)3 | 2.06 (1.11–3.81) | 0.022 | 69.2 | 2.23 (1.09–4.59) | 0.029 | 79.54 |
| PSAD5 | 21942.60 (7.80–6.18 × 107) | 0.014 | 71.8 | 51723.691 (11.54–2.31 × 108) | 0.011 | |||
PSAD, prostate-specific antigen density.
Hosmer–Lemeshow Goodness-of-Fit of logistic regression model: P = 0.910.
Defined as (true positives + true negatives)/all.
TMP:ERG subtype III, IV, or TMP:ETS (ETV 1, 4, or 5) (log-transformed continuous variable).
70.6% sensitivity and 86.4% specificity at 50% cut-off value.
PSAD continuous variable.
Table 8.
Risk groups to clinically significant cancers stratified by the FPd molecular scores
| High | Intermediate | Low | |
| Risk score1 | |||
| n (%) | 13 (33) | 14 (36) | 12 (31) |
| Median (min–max) | (0.6064–1.0000) | (0.1735–0.5297) | (0.0489–0.1706) |
| Gleason score ≥7 | |||
| n (%)2 | 12 (92) | 4 (29) | 1 (8) |
| RR (H vs. L), P3 | 10.150 (1.556–66.260), P < 0.0001 | ||
| RR (H vs. [I + L]), P4 | 4.800 (2.149–10.720), P < 0.0001 | ||
| High risk of recurrence by NCCN5 | |||
| n (%)2 | 8 (62) | 1 (7) | 0 (0) |
| RR (H vs. L), P | Undefined, P = 0.0016 | ||
| RR (H vs. [I + L]), P | 16.000 (2.232–114.700), P = 0.0002 | ||
| No. of cores ≥3 | |||
| n (%)2 | 11 (85) | 8 (57) | 5 (42) |
| RR (H vs. L), P | 2.031 (1.000–4.125), P = 0.0414 | ||
| RR (H vs. [I + L]), P | 1.692 (1.080–2.651), P = 0.0449 | ||
| Max.% cancer inv. in any core ≥50% | |||
| n (%)2 | 9 (69) | 5 (36) | 0 (0) |
| RR (H vs. L), P | Undefined, P = 0.0005 | ||
| RR (H vs. [I + L]), P | 3.600 (1.512–8.570), P = 0.0041 | ||
| Insignificant cancer by the Epstein criteria6 | |||
| n (%)2 | 0 (0) | 0 (0) | 6 (50) |
| RR (L vs. H), P | Undefined, P = 0.0052 | ||
| RR (L vs. [H + I]), P | Undefined, P = 0.0003 | ||
NCCN, National Comprehensive Cancer Network; PSAD, prostate-specific antigen density; RR, relative risk.
FPd molecular score: FPd = −3.481 + 0.803*log 2 Fx(III, IV, ETS) + 10.854*PSAD, High-grade cancer risk score = 1/(1 + e(−FPd)).
% Of each risk group or positive predictive value in 39 biopsy positive patients.
RR between high- and low-risk group with 95% CI, P value of Fisher exact test.
RR between high- and intermediate + low-risk group with 95% CI, P value of Fisher exact test.
Gleason score ≥8, PSA ≥ 20 ng/mL or T3a as defined by the NCCN guidelines.
Gleason score ≤6, no. of cores ≤2, percentage cancer involvement ≤50%, and PSAD ≤ 0.15 ng/mL met at the same time. RR between low- and high-, low- and high + intermediate (H + I)-risk group with 95% CI, P value of Fisher exact test.
Table 9.
Correlation of clinical–pathological and molecular characteristics with the FPd molecular scores for aggressiveness
| Parameter | No. of patients (n) | FPd score1 (Spearman rs) | P |
| Age | 39 | 0.167 | 0.311 |
| Prebiopsy PSA | 39 | 0.536 | <0.0001 |
| Prostate size | 39 | −0.467 | 0.003 |
| PSA density | 39 | 0.845 | <0.0001 |
| Gleason score | 39 | 0.659 | <0.001 |
| No. of cores with cancer | 39 | 0.423 | 0.007 |
| Max. % cancer inv. in any core | 39 | 0.564 | <0.0001 |
| Urine PSA | 39 | 0.185 | 0.258 |
| Urine PCA3 | 39 | 0.083 | 0.616 |
| Urine ERG(5-6) | 39 | 0.227 | 0.164 |
PSA, prostate-specific antigen.
Spearman's rank correlation test to measure correlation of the FPd score with each parameter.
Discussion
In this study, we report a prospective study for molecular diagnosis of PSA-screened patients who are scheduled for diagnostic biopsy (prebiopsy cohort) using a novel panel of both common TMPRSS2:ERG subtypes and low-prevalent TMPRSS2:ETS (ETV1, ETV4, and ETV5) fusion markers. We demonstrated for the first time the clinical utility of multiple low-prevalent fusion markers and diverse fusion types in urine-based cancer detection. Moreover, we identified multiple alternative fusion genes and subtypes very specific to clinically significant prostate cancers than the common subtype I, and developed effective regression models to predict both the risk of cancer occurrence and the risk of aggressive cancer prospectively.
Prostate cancer is characterized by its extensive clinical heterogeneity; early stratification of aggressive disease from a majority of indolent cancers at diagnosis is a critical clinical task in cancer management and treatment. Our work builds on an increasing number of disease-specific fusion genes recently discovered in prostate cancer [26–28]. We argue that a panel of multiple fusion markers is superior to any single one alone in the noninvasive detection and stratification of clinically significant tumors. First, both high- and low-prevalent fusion genes/subtypes together provide improved clinical sensitivity through redundant transcript markers and mutually exclusive nature of some fusion genes [35]. Second, the panel approach offers rich genetic diversity for better stratification of heterogeneous tumors through unique genetic and molecular profiles associated with each individual patient. Some fusion subtypes are shown to have prognostic values [10], while many low-prevalent fusion events are associated with aggressive cancers [21,22]. Third, the panel approach provides new insights into the complexity and the extent of genetic heterogeneity or instability associated with multiple independent fusion events detectable in the same patients [35]. Such information may be very useful for noninvasive identification of a group of highly aggressive prostate cancer defined by extensive genomic alterations [29]. To explore this new perspective, we have developed a qPCR platform for urine-based detection of multiple fusion markers. This platform is shown to have a high specificity (>99%) in a negative control group and a sensitivity of detecting a single fusion-positive cancer cell in at least 3000 normal cells in men's urine sediments [30]. Reproducible detection of fusion-subtype molecules are also demonstrated in the dynamic range from 1.8 million to 18 copies (Fig. S3) and from independent preparation of the same samples (Fig. S1). Using this sensitive platform to screen a panel of six TMPRSS2:ETS fusion markers, we not only detected diverse fusion types in urine of prebiopsy patients but also identified several alternative fusion markers very specific to biopsy outcomes. The informative fusion markers, Fx (III, IV, or ETS), are collectively more sensitive than the dominant fusion subtype I in prospective prostate cancer detection. Moreover, by incorporating the categorical variable of Fx (III, IV, or ETS) with urine PCA3 and serum PSA, the multivariate FPP model not only has an overall predictive accuracy of 77% to overall prostate cancer detection, but also allows individualized stratification of prebiopsy patients into distinctive risk groups. As such, the PPV on the biopsy outcome is 81% in a high-risk group, but only 16% in a low-risk group. On the other hand, the combined quantity of Fx (III, IV, or ETS) in urine are shown to be an independent predictor to the high-grade cancers (P < 0.022). When coupled with PSAD, the FPd model detects the aggressive prostate cancer with an overall predictive accuracy of 80% in biopsy-positive patients. The resulting FPd scores further stratify not only a high-risk group that is composed of 92% high-grade cancer, but also a low-risk group that harbors mainly clinically insignificant cancers. However, it remains to be elucidated on the basis of different clinical implications between a single versus a panel of markers as well as between detectability and overexpression among the same set of informative fusion markers. Interestingly, the less common subtypes (III and IV) are frequently accompanied by the common subtype I in Bx(+) cases, but the common subtype alone is overrepresented in the [Bx(−)/Fx(+)] subgroup. Thus, the genetic complexity of fusion types and the active expression of less common fusion markers in urine may be associated with an active state of cancer proliferation and hence serve as better predictive markers to the biopsy outcomes and the risk of aggressive cancer. This hypothesis is further supported by the identification of multiple novel fusion subtypes associated with the high-risk cancers when the same urine specimens were screened by a DNA-chip-based high-throughput method (P.-N Nguyen and J. Z. Chen, unpubl. data). Therefore, the panel approach is better suited to identify clinical significant cancers over a single marker-based test. It is feasible to further improve the clinical sensitivity by including additional fusion markers (e.g., SLC45A3, KLK2, and so on) into the same platform, and to simplify the multimarker analyses into a single test using a qPCR-array format. However, it is necessary to point out that the assay based the new fusion panel was a laboratory-developed test and that our prospective study is limited by a relatively small sample size and short follow-up time. Independent validation studies are necessary and current ongoing to address these limitations by the authors.
In conclusion, we propose a urine-based clinical test for early detection of clinically significant prostate cancer using a panel of multiple TMPRSS2:ETS fusion markers. The extensive diversity of fusion types identified in urine of PSA-screened men provides better genetic and molecular bases to stratify the clinical heterogeneity of prostate cancer. Clinically applicable risk models are developed to generate individualized molecular scores to identify distinctive risk groups. As such, patients identified with a high molecular score will likely lead to the detection of aggressive forms of cancer in biopsy-positive cases or “false” diagnosis in biopsy-negative cases who should be prioritized for repeat biopsies. On the other hand, patients characterized with a low-risk score may represent indolent cancer in biopsy-positive cases or truly low-risk subjects in biopsy-negative cases. The former may be benefitted by conservative management such as active surveillance, while the latter may be excluded from further screening to reduce the biopsy burden. It is logical to expect that the conceptual advances and simultaneous analysis of all existing fusion markers using high-throughput technologies will provide a new paradigm for personalized molecular diagnosis of the clinically significant prostate cancer.
Acknowledgments
The authors acknowledge M. Chevrette for comments on the early version of the manuscript and D. Ayele for statistical assistance. This work was supported by grants from the Canadian Institute of Health Research (NGH99087) and the Canada Foundation for Innovation (#11623) to J. Z. Chen.
Conflict of Interest
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
A flowchart for the procurement of urine specimens, processing of biological materials, fusion marker analysis and validation. Urine specimens were procured from 97 PSA‐screened men scheduled for diagnostic biopsy. A total of 92 informative specimens were used for molecular diagnosis, while five specimens were excluded from analyses due to failed RNA extraction or WTA amplification. The dashed line indicates independent confirmation of fusion positive specimens using a second aliquot of original RNA from all 32 fusion‐positive cases.
Figure S2. A diagram to show the index gene isoforms and the locations of PCR primers used to amplify fusion subtypes. Forward (F) chimeric primers from TMPRSS2‐exon 1 were paired with reversed (R) primers from ERG‐exons 4, 2, and 5 to amplify TMPRSS2:ERG subtypes I, III, and IV. The forward primer from TMPRSS2‐exon 1 (TMP‐e1F) was paired with reverse primers from ETV1 (ETV1‐e6R), ETV4 (ETV4‐e5R), and ETV5 (ETV5‐e2R) to amplify corresponding TMPRSS2:ETS fusion genes. The fusion partner genes were drawn in proportion to actual structures.
Figure S3. Dynamic ranges of TMPRSS2:ERG subtype markers in serial dilution experiments. Three DNA fragments containing TMPRSS2:ERG subtype I, III, and IV were purified and used to generate 10× serial dilutions ranging from 1.8 million to 18 copies for each DNA fragment. Each fusion‐subtype marker was used to amplify the serial dilutions plus a reference urine sample (indicated by red dots and arrows) and a negative control containing no fusion DNA using the protocols described in the Materials and Methods. The qPCR standard curve was constructed for fusion subtypes I (A), III (B) and IV (C) by the MyIQ System Software (v 1.0.410). The Ct (threshold cycle) was plotted on the y‐axis and the log10 transformation of the starting material (copy number) on the x‐axis. The standard curve was described as: Ct value = slope × log10 (copy number) + y‐intercept. The qPCR efficiencies for subtypes I, III, and IV were 94.9%, 91.4%, and 94.1%, respectively. Consistent amplifications were generated in the large dynamic range for each subtype marker, while no signal was generated in the negative control in a 50‐cycle reaction.
Figure S4. The junction sequences of TMPRSS2:ERG subtypes (I, III, and IV) and TMPRSS2:ETS fusion genes (ETV1, ETV4 and ETV5) validated by DNA sequencing. The arrows indicate the fusion transcript junctions between TMPRSS2 and ERG (NM_004449.3), ETV1 (NM_004956.3), ETV4 (NM_001986.1), or ETV5 (NM_004454.1). The sequence traces were generated using Applied Biosystems 3730xl DNA Analyzer at the McGill University Genome Centre.
Table S1. Primer sequences of TMPRSS2:ETS fusion markers and additional molecular markers.
Table S2. Combination of fusion markers (or fusion‐types) and raw Ct values of each fusion marker in urine of 32 fusion‐positive men.
Table S3. Confirmation of fusion‐positive urine specimens in three corresponding prostatectomy cancer tissues.
Table S4. Risk groups to cancer occurrence stratified by the FPP molecular scores.
References
- 1.Schroder F. H., Hugosson J., Roobol M. J., Tammela T. L., Ciatto S., Nelen V. Screening and prostate-cancer mortality in a randomized European study. N. Engl. J. Med. 2009;360:1320–1328. doi: 10.1056/NEJMoa0810084. [DOI] [PubMed] [Google Scholar]
- 2.Andriole G. L., Crawford E. D., Grubb R. L., 3rd, Buys S. S., Chia D., Church T. R. Mortality results from a randomized prostate-cancer screening trial. N. Engl. J. Med. 2009;360:1310–1319. doi: 10.1056/NEJMoa0810696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Strope S. A., Andriole G. L. Prostate cancer screening: current status and future perspectives. Nat. Rev. Urol. 2010;7:487–493. doi: 10.1038/nrurol.2010.120. [DOI] [PubMed] [Google Scholar]
- 4.Sandblom G., Varenhorst E., Rosell J., Lofman O., Carlsson P. Randomised prostate cancer screening trial: 20 year follow-up. BMJ. 2011;342:d1539. doi: 10.1136/bmj.d1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tomlins S. A., Rhodes D. R., Perner S., Dhanasekaran S. M., Mehra R., Sun X. W. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 6.Tomlins S. A., Mehra R., Rhodes D. R., Smith L. R., Roulston D., Helgeson B. E. TMPRSS2:ETV4 gene fusions define a third molecular subtype of prostate cancer. Cancer Res. 2006;66:3396–3400. doi: 10.1158/0008-5472.CAN-06-0168. [DOI] [PubMed] [Google Scholar]
- 7.Hermans K. G., van Marion R., van Dekken H., Jenster G., van Weerden W. M., Trapman J. TMPRSS2:ERG fusion by translocation or interstitial deletion is highly relevant in androgen-dependent prostate cancer, but is bypassed in late-stage androgen receptor-negative prostate cancer. Cancer Res. 2006;66:10658–10663. doi: 10.1158/0008-5472.CAN-06-1871. [DOI] [PubMed] [Google Scholar]
- 8.Perner S., Demichelis F., Beroukhim R., Schmidt F. H., Mosquera J. M., Setlur S. TMPRSS2:ERG fusion-associated deletions provide insight into the heterogeneity of prostate cancer. Cancer Res. 2006;66:8337–8341. doi: 10.1158/0008-5472.CAN-06-1482. [DOI] [PubMed] [Google Scholar]
- 9.Iljin K., Wolf M., Edgren H., Gupta S., Kilpinen S., Skotheim R. I. TMPRSS2 fusions with oncogenic ETS factors in prostate cancer involve unbalanced genomic rearrangements and are associated with HDAC1 and epigenetic reprogramming. Cancer Res. 2006;66:10242–10246. doi: 10.1158/0008-5472.CAN-06-1986. [DOI] [PubMed] [Google Scholar]
- 10.Wang J., Cai Y., Ren C., Ittmann M. Expression of variant TMPRSS2/ERG fusion messenger RNAs is associated with aggressive prostate cancer. Cancer Res. 2006;66:8347–8351. doi: 10.1158/0008-5472.CAN-06-1966. [DOI] [PubMed] [Google Scholar]
- 11.Yoshimoto M., Joshua A. M., Chilton-Macneill S., Bayani J., Selvarajah S., Evans A. J. Three-color FISH analysis of TMPRSS2/ERG fusions in prostate cancer indicates that genomic microdeletion of chromosome 21 is associated with rearrangement. Neoplasia. 2006;8:465–469. doi: 10.1593/neo.06283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clark J., Merson S., Jhavar S., Flohr P., Edwards S., Foster C. S. Diversity of TMPRSS2-ERG fusion transcripts in the human prostate. Oncogene. 2007;26:2667–2673. doi: 10.1038/sj.onc.1210070. [DOI] [PubMed] [Google Scholar]
- 13.Attard G., Clark J., Ambroisine L., Fisher G., Kovacs G., Flohr P. Duplication of the fusion of TMPRSS2 to ERG sequences identifies fatal human prostate cancer. Oncogene. 2008;27:253–263. doi: 10.1038/sj.onc.1210640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Demichelis F., Fall K., Perner S., Andren O., Schmidt F., Setlur S. R. TMPRSS2:ERG gene fusion associated with lethal prostate cancer in a watchful waiting cohort. Oncogene. 2007;26:4596–4599. doi: 10.1038/sj.onc.1210237. [DOI] [PubMed] [Google Scholar]
- 15.Nam R. K., Sugar L., Wang Z., Yang W., Kitching R., Klotz L. H. Expression of TMPRSS2:ERG gene fusion in prostate cancer cells is an important prognostic factor for cancer progression. Cancer Biol. Ther. 2007;6:40–45. doi: 10.4161/cbt.6.1.3489. [DOI] [PubMed] [Google Scholar]
- 16.Laxman B., Morris D. S., Yu J., Siddiqui J., Cao J., Mehra R. A first-generation multiplex biomarker analysis of urine for the early detection of prostate cancer. Cancer Res. 2008;68:645–649. doi: 10.1158/0008-5472.CAN-07-3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laxman B., Tomlins S. A., Mehra R., Morris D. S., Wang L., Helgeson B. E. Noninvasive detection of TMPRSS2:ERG fusion transcripts in the urine of men with prostate cancer. Neoplasia. 2006;8:885–888. doi: 10.1593/neo.06625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hessels D., Smit F. P., Verhaegh G. W., Witjes J. A., Cornel E. B., Schalken J. A. Detection of TMPRSS2-ERG fusion transcripts and prostate cancer antigen 3 in urinary sediments may improve diagnosis of prostate cancer. Clin. Cancer Res. 2007;13:5103–5108. doi: 10.1158/1078-0432.CCR-07-0700. [DOI] [PubMed] [Google Scholar]
- 19.Tomlins S. A., Aubin S. M., Siddiqui J., Lonigro R. J., Sefton-Miller L. Urine TMPRSS2:ERG fusion transcript stratifies prostate cancer risk in men with elevated serum PSA. Sci. Transl. Med. 2011;3:94ra72. doi: 10.1126/scitranslmed.3001970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Helgeson B. E., Tomlins S. A., Shah N., Laxman B., Cao Q., Prensner J. R. Characterization of TMPRSS2:ETV5 and SLC45A3:ETV5 gene fusions in prostate cancer. Cancer Res. 2008;68:73–80. doi: 10.1158/0008-5472.CAN-07-5352. [DOI] [PubMed] [Google Scholar]
- 21.Clark J. P., Cooper C. S. ETS gene fusions in prostate cancer. Nat. Rev. Urol. 2009;6:429–439. doi: 10.1038/nrurol.2009.127. [DOI] [PubMed] [Google Scholar]
- 22.Tomlins S. A., Laxman B., Dhanasekaran S. M., Helgeson B. E., Cao X., Morris D. S. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007;448:595–599. doi: 10.1038/nature06024. [DOI] [PubMed] [Google Scholar]
- 23.Hermans K. G., van der Korput H. A., van Marion R., van de Wijngaart D. J. Ziel-van der Made A, Dits NF, Boormans JL, van der Kwast TH, van Dekken H, Bangma CH, Korsten H, Kraaij R, et al. Truncated ETV1, fused to novel tissue-specific genes, and full-length ETV1 in prostate cancer. Cancer Res. 2008;68:7541–7549. doi: 10.1158/0008-5472.CAN-07-5930. [DOI] [PubMed] [Google Scholar]
- 24.Han B., Mehra R., Dhanasekaran S. M., Yu J., Menon A., Lonigro R. J. A fluorescence in situ hybridization screen for E26 transformation-specific aberrations: identification of DDX5-ETV4 fusion protein in prostate cancer. Cancer Res. 2008;68:7629–7637. doi: 10.1158/0008-5472.CAN-08-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hermans K. G., Bressers A. A., van der Korput H. A., Dits N. F., Jenster G., Trapman J. Two unique novel prostate-specific and androgen-regulated fusion partners of ETV4 in prostate cancer. Cancer Res. 2008;68:3094–3098. doi: 10.1158/0008-5472.CAN-08-0198. [DOI] [PubMed] [Google Scholar]
- 26.Kumar-Sinha C., Tomlins S. A., Chinnaiyan A. M. Recurrent gene fusions in prostate cancer. Nat. Rev. Cancer. 2008;8:497–511. doi: 10.1038/nrc2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tomlins S. A., Bjartell A., Chinnaiyan A. M., Jenster G., Nam R. K., Rubin M. A. ETS gene fusions in prostate cancer: from discovery to daily clinical practice. Eur. Urol. 2009;56:275–286. doi: 10.1016/j.eururo.2009.04.036. [DOI] [PubMed] [Google Scholar]
- 28.Pflueger D., Terry S., Sboner A., Habegger L., Esgueva R., Lin P. C. Discovery of non-ETS gene fusions in human prostate cancer using next-generation RNA sequencing. Genome Res. 2011;21:56–67. doi: 10.1101/gr.110684.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taylor B. S., Schultz N., Hieronymus H., Gopalan A., Xiao Y., Carver B. S. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nguyen P. N., Violette P., Chan S., Tanguay S., Kassouf W., Aprikian A. A panel of TMPRSS2:ERG fusion transcript markers for urine-based prostate cancer detection with high specificity and sensitivity. Eur. Urol. 2011;59:407–414. doi: 10.1016/j.eururo.2010.11.026. [DOI] [PubMed] [Google Scholar]
- 31.McShane L. M., Altman D. G., Sauerbrei W., Taube S. E., Gion M., Clark G. M. Reporting recommendations for tumor marker prognostic studies (REMARK) J. Natl. Cancer Inst. 2005;97:1180–1184. doi: 10.1093/jnci/dji237. [DOI] [PubMed] [Google Scholar]
- 32.DeLong E. R., DeLong D. M., Clarke-Pearson D. L. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics. 1988;44:837–845. [PubMed] [Google Scholar]
- 33.Bastian P. J., Mangold L. A., Epstein J. I., Partin A. W. Characteristics of insignificant clinical T1c prostate tumors. A contemporary analysis. Cancer. 2004;101:2001–2005. doi: 10.1002/cncr.20586. [DOI] [PubMed] [Google Scholar]
- 34.Epstein J. I., Walsh P. C., Carmichael M., Brendler C. B. Pathologic and clinical findings to predict tumor extent of nonpalpable (stage T1c) prostate cancer. JAMA. 1994;271:368–374. [PubMed] [Google Scholar]
- 35.Svensson M. A., LaFargue C. J., MacDonald T. Y., Pflueger D., Kitabayashi N., Santa-Cruz A. M. Testing mutual exclusivity of ETS rearranged prostate cancer. Lab. Invest. 2010;91:404–412. doi: 10.1038/labinvest.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A flowchart for the procurement of urine specimens, processing of biological materials, fusion marker analysis and validation. Urine specimens were procured from 97 PSA‐screened men scheduled for diagnostic biopsy. A total of 92 informative specimens were used for molecular diagnosis, while five specimens were excluded from analyses due to failed RNA extraction or WTA amplification. The dashed line indicates independent confirmation of fusion positive specimens using a second aliquot of original RNA from all 32 fusion‐positive cases.
Figure S2. A diagram to show the index gene isoforms and the locations of PCR primers used to amplify fusion subtypes. Forward (F) chimeric primers from TMPRSS2‐exon 1 were paired with reversed (R) primers from ERG‐exons 4, 2, and 5 to amplify TMPRSS2:ERG subtypes I, III, and IV. The forward primer from TMPRSS2‐exon 1 (TMP‐e1F) was paired with reverse primers from ETV1 (ETV1‐e6R), ETV4 (ETV4‐e5R), and ETV5 (ETV5‐e2R) to amplify corresponding TMPRSS2:ETS fusion genes. The fusion partner genes were drawn in proportion to actual structures.
Figure S3. Dynamic ranges of TMPRSS2:ERG subtype markers in serial dilution experiments. Three DNA fragments containing TMPRSS2:ERG subtype I, III, and IV were purified and used to generate 10× serial dilutions ranging from 1.8 million to 18 copies for each DNA fragment. Each fusion‐subtype marker was used to amplify the serial dilutions plus a reference urine sample (indicated by red dots and arrows) and a negative control containing no fusion DNA using the protocols described in the Materials and Methods. The qPCR standard curve was constructed for fusion subtypes I (A), III (B) and IV (C) by the MyIQ System Software (v 1.0.410). The Ct (threshold cycle) was plotted on the y‐axis and the log10 transformation of the starting material (copy number) on the x‐axis. The standard curve was described as: Ct value = slope × log10 (copy number) + y‐intercept. The qPCR efficiencies for subtypes I, III, and IV were 94.9%, 91.4%, and 94.1%, respectively. Consistent amplifications were generated in the large dynamic range for each subtype marker, while no signal was generated in the negative control in a 50‐cycle reaction.
Figure S4. The junction sequences of TMPRSS2:ERG subtypes (I, III, and IV) and TMPRSS2:ETS fusion genes (ETV1, ETV4 and ETV5) validated by DNA sequencing. The arrows indicate the fusion transcript junctions between TMPRSS2 and ERG (NM_004449.3), ETV1 (NM_004956.3), ETV4 (NM_001986.1), or ETV5 (NM_004454.1). The sequence traces were generated using Applied Biosystems 3730xl DNA Analyzer at the McGill University Genome Centre.
Table S1. Primer sequences of TMPRSS2:ETS fusion markers and additional molecular markers.
Table S2. Combination of fusion markers (or fusion‐types) and raw Ct values of each fusion marker in urine of 32 fusion‐positive men.
Table S3. Confirmation of fusion‐positive urine specimens in three corresponding prostatectomy cancer tissues.
Table S4. Risk groups to cancer occurrence stratified by the FPP molecular scores.