

Abstract
Photoisomerization of biliverdin (BV) chromophore triggers the photoresponse in native Agp1 bacteriophytochrome. We discuss heterogeneity in phytochrome Pr form to account for the shape of the absorption profile. We investigated different regions of the absorption profile by angle balanced polarization resolved femtosecond VIS pump–IR probe spectroscopy. We studied the Pr form of Agp1 with its natural chromophore and with a sterically locked 18Et-BV (locked Agp1). We followed the dynamics and orientations of the carbonyl stretching vibrations of ring D and ring A in their ground and electronically excited states. Photoisomerization of ring D is reflected by strong signals of the ring D carbonyl vibration. In contrast, orientational data on ring A show no rotation of ring A upon photoexcitation. Orientational data allow excluding a ZZZasa geometry and corroborates a nontwisted ZZZssa geometry of the chromophore. We found no proof for heterogeneity but identified a new, to our knowledge, electronic transition in the absorption profile at 644 nm (S0→S2). Excitation of the S0→S2 transition will introduce a more complex photodynamics compared with S0→S1 transition. Our approach provides fundamental information on disentanglement of absorption profiles, identification of chromophore structures, and determination of molecular groups involved in the photoisomerization process of photoreceptors.
Introduction
Phytochromes are a family of photoreceptors with a bilin chromophore that have been found in plants, bacteria, and fungi. Typically, a phytochrome is switched between the two stable forms termed Pr (red-absorbing form) and Pfr (far-red absorbing form) by light. Photoconversion is initiated by an ultrafast isomerization of the C15 = C16 methine bridge between ring C and ring D accompanied by rotation of ring D of the bilin chromophore (1). Phytochrome proteins comprise at least a PAS, GAF, and PHY domain in the N-terminus, and a C-terminal output region with a variable domain arrangement. The C terminus of bacterial phytochromes or bacteriophytochromes and of fungal phytochromes most often contains a histidine-kinase domain, whereas plant phytochromes have two PAS domains and a histidine-kinase–like domain in their C terminus. The N-terminal PAS, GAF, and PHY domains are the sites for chromophore insertion and photoconversion. The GAF domain forms most of the chromophore contacts, and a number of cyanobacterial proteins that have bilin-binding GAF domains have been found that are also photoactive. These cyanobacteriochromes can absorb light of the entire visible spectrum (2). In phytochromes with the tripartite N-terminal domain arrangement, PAS, GAF, and PHY domains interact with each other in a characteristic manner; PAS and GAF domains form an unusual knotted structure. Within the large group of phytochromes, variations of biochemical and spectral properties are found that are often characteristic for specific phylogenetic groups (3). The chromophore can either be phytochromobilin with absorbance maxima of 660 nm for Pr and 730 nm for Pfr, or phycocyanobilin (PCB), with 655 nm and 700 nm absorbance maxima for Pr and Pfr, respectively, or biliverdin (BV), with 700 nm and 750 nm absorbance maxima for Pr and Pfr, respectively. Phytochromobilin and PCB chromophores are covalently attached to a Cys in the GAF domain; the binding site for BV is a Cys residue at the N-terminus of the PAS domain (4). Whereas most phytochromes have a Pr ground state, the so-called bathy phytochromes exist as Pfr in darkness. In this study we deal with bacteriophytochrome Agp1 from the soil bacterium Agrobacterium tumefaciens, which has been used as model phytochrome in biochemical studies (5). Agp1 incorporates BV as a chromophore and has a “normal” Pr ground state and Pr to Pfr photoconversion, but a low quantum yield for Pfr to Pr photoconversion. NMR and crystal structure data have shown that phytochromes exhibit a ZZZssa and ZZEssa chromophore geometry in their Pr and Pfr forms, respectively (6–9). The same stereochemistry has been confirmed for Agp1 by studies using locked chromophores (see Fig. 1 for the stereochemistry of a 15Za chromophore) (10).
Figure 1.
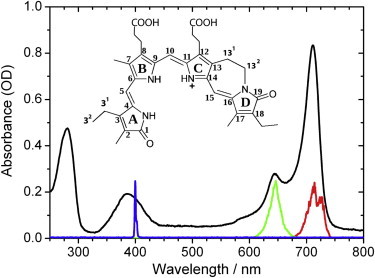
Absorption spectrum of Agp1 bacteriophytochrome adduct (black line) with 15Za-18Et-BV chromophore (see inset) in its ZZZssa geometry. Femtosecond excitation pulses at 400 nm (blue line), 644 nm (green line), and 713 nm (red line).
Heterogeneity of the chromophore ground state geometry can induce a broadening of absorption lines in the visible absorption spectrum and new, to our knowledge, spectral features in the vibrational pattern due to diverse interactions between the chromophore and the protein binding pocket, e.g., hydrogen bonds (11). For the Pr form, it is still under debate as of this writing whether the shoulder in the absorption spectrum around 620 nm and the biexponential decay with time constants of ∼3 ps and 30 ps of the isomerization dynamics originate from heterogeneity of the chromophore (12–14). With our method we are able to identify chromophore heterogeneity and separate contributions from different electronic transitions.
It has been demonstrated that the Pr absorption spectrum of the cyanobacterial phytochrome Cph1 can be explained by vibrational progressions of a homogeneous chromophore ensemble (15), but another study resolved two different ZZZssa chromophore geometries in the Pr form of Cph1 (6).
For native Agp1 single molecule spectroscopy demonstrated structurally heterogeneous population of bacteriophytochromes at low temperatures (13), and resonance Raman studies found at least two chromophore conformers in Agp1 (16).
In this study, we compare structural and dynamic information on locked Agp1 with measurements on the Pr form of native Agp1. We demonstrate the existence of two different electronic transitions in the Pr form of 15Za-18Et-BV Agp1 (locked Agp1) of the same chromophore geometry at the main peak at 713 nm and at the shoulder at 644 nm (Fig. 1) by polarization resolved femtosecond VIS pump–IR probe experiments.
Experimental and Theoretical Methods
Materials and Methods
We synthesized 15Za-18Et-BV according to the reported procedure in Hammam et al. (17). Recombinant Agp1 with a C-terminal poly-His tag was purified by affinity chromatography as described in other studies (4,18,19). After assembly with 15Za-18Et-BV or BV (10), we dissolved the Agp1 protein samples in D2O and set to pH 7.4 with 20 mM Tris.
We performed angle balanced polarization resolved femtosecond VIS pump–IR probe experiments at room temperature (23°C), using a Coherent Legend USP Ti:Sa laser/amplifier system (55 fs pulses, centered at 800 nm) with 1 kHz repetition rate. Mid-IR probe pulses were generated in a spectral range between 5.6 and 6.3 μm.
Three VIS pump pulses with frequencies of 400 nm, 644 nm, and 713 nm were generated using SHG (Second Harmonic Generation) and multistage OPG (Optical Parametric Generation) processes for excitation of locked Agp1. More details on pump pulse generation can be found in Theisen et al. (20) Fig. 1 presents the spectral profiles. The obtained tunable mid-IR probe pulses typically had 160 fs pulse duration, 100 cm−1 spectral width, and 0.6 μJ energy. After passing the sample, both probe pulses are dispersed with an imaging spectrograph (grating 150 lines/mm, f = 320 mm) and recorded with a 2 × 32 element MCT array detector (resolution ∼1.5 cm−1). The absolute frequency position was calibrated by water vapor absorption line spectrum (see Fig. 8 a). Excitation of native Agp1 in the Pr form was performed at 680 nm under continuous background illumination at wavelength longer than 760 nm. The VIS pump–IR probe time resolution was 0.3 to 0.4 ps, and mainly limited by group velocity mismatch. Pump pulses were attenuated to excitation efficiencies below 6% of the native Agp1 molecules. The probe beam is split into two beams with polarizations perpendicular to each other and overlapped with the pump beam within the sample, for measuring signals for both parallel and perpendicular pump-probe polarizations simultaneously. The spatial arrangement of the pump and probe beams were set to enable angle balanced measurements (see Fig. 2 and the Supporting Material).
Figure 8.
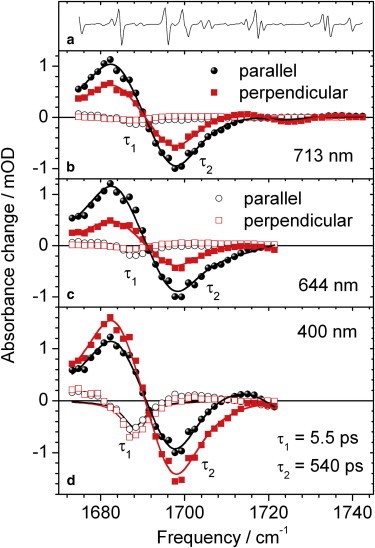
Decay associated spectra (DAS) for time constant τ1 = 5.5 ps (open circles and open squares) and time constant τ2 = 540 ps (solid circles and solid squares) for polarizations parallel and perpendicular to pump pulse polarization. (a) Derivative of water vapor spectrum inducing small distortions on spectral shape. We used the distortions to test our spectral alignment. (b) Excitation at 713 nm. (c) Excitation at 644 nm. (d) Excitation at 400 nm.
Figure 2.
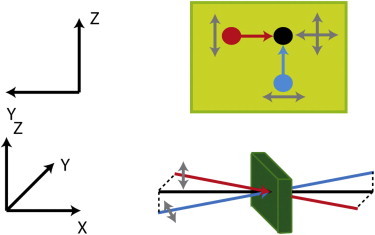
Spatial arrangement of the pump pulse (black) and the two probe pulses. The probe pulse 1 (blue) is polarized along the y axis, the probe pulse 2 (red) is polarized along the z axis. Polarization direction of the pump pulse is switched between y and z axis. Yellow box: Polarization directions (gray arrows) of the probe beams (blue and red dots) and of the pump beam (black dot) in the plane of the sample; red and blue arrows indicate the projections of the probe beams as they approach the pump beam overlapping in the sample volume.
DFT calculations
All quantum-chemical calculations were done with Gaussian 09 (21). Geometry optimization of the 15Za-18Et-BV chromophore, vibrational frequency calculations, and determination of the vibrational tdm orientations in the molecular scaffold were performed with density functional theory (DFT) (22) using the B3LYP exchange-correlation functional and the 6-31G∗∗ basis set. We calculated the vibrational frequencies for the different 15Za-18Et-BV chromophore geometries ZZZssa and ZZZasa, the different helicities at ring D, and the different ring A geometries. The Pr and Pfr chromophores of (bacterio-)phytochromes including Agp1 are usually protonated (23–25). For this reason, we introduced a chlorine counterion to stabilize the 15Za-18Et-BV chromophore geometry before geometry optimization (see the Supporting Material).
Results and Discussion
Dynamics of locked Agp1
Excitation at 713 nm
Ultrafast transient dynamics of Agp1 assembled with 15Za-18Et-BV as chromophore were investigated upon S0→S1 excitation at 713 nm (Fig. 1, red curve), the Pr absorption maximum. The 15Za-18Et-BV chromophore exhibits a covalent linkage between ring C and ring D inhibiting Z-to-E isomerization around C15 = C16 methine bridge. No lumi-R photoproduct formation was observed up to 1 ns. Fig. 3 presents transients of the chromophore’s carbonyl stretching vibrations in their electronically excited state (positive signals) and ground state (negative signals). As reported in Schumann et al., the C19 = O stretching vibration of native Agp1 in H2O absorbs at ∼1700 cm−1 and at ∼1710 cm−1 in its electronically excited state and in its Pr ground state, respectively (26).
Figure 3.
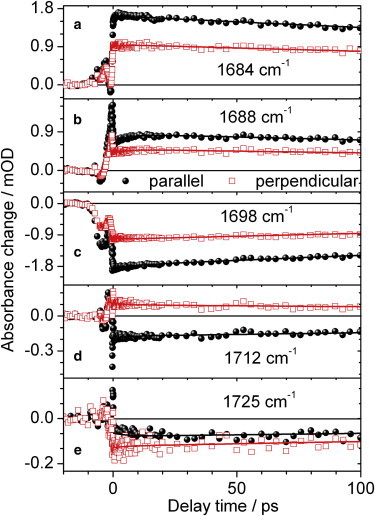
Polarization resolved vibrational transients of locked Agp1 upon excitation at 713 nm. Signals measured for probe polarization parallel (black circles) or perpendicular (red open squares) to pump polarization. C19 = O stretching vibration in the excited state at 1684 cm−1 and 1688 cm−1 (a), (b),; bleach signal of the C19 = O stretching vibration at 1698 cm−1 (c); C1 = O stretching vibration in the excited state at 1712 cm−1 (d); bleach signal of the C1 = O stretching vibration at 1725 cm−1 (e). Biexponential simulations (black and red lines) with time constants of 5.5 ps and 540 ps.
Positive and negative signals around 1684 cm−1, 1689 cm−1, 1712 cm−1, and 1725 cm−1 show constant signals for parallel and perpendicular probe polarizations, thus no anisotropy decay on a time scale of 100 ps. Therefore, these signals are caused by chromophore vibrations that are fixed in orientation within the protein, in contrast to amino acid side chains that are much more flexible. For the 15Za-18Et-BV locked Agp1, the C19 = O stretching vibration absorbs maximally around 1684 cm−1 in its electronically excited state, and the C19 = O stretching vibration in the ground state is found at 1698 cm−1. These vibrations exhibit similar features, but there is a shift of ∼10 cm−1 to lower frequencies, compared with the native BV-adduct of Agp1 in H2O (27). However, the absorption of the C1 = O stretching vibration in the electronically excited state is displayed by the positive signal at 1712 cm−1 for perpendicular polarization. The bleaching signal of the C1 = O stretching vibration in its electronic ground state is located at 1725 cm−1, which is in agreement with previous assignments of this vibration in Agp1 (27).
All transients are best fitted with a biexponential function. The time constants are determined to be τ1 = 5.5 ps and τ2 = 540 ps. The 1σ error margins, determined by exhaustive search analysis (28), range from 4 to 8 ps for the shorter time constant and from 510 to 570 ps for the longer time constant. The shorter time constant τ1 is in agreement with the fast photoreaction time constant of the reported wild type Pr form of the BV-adduct (26) but has an amplitude of only 10%. In contrast, the corresponding 3.3 ps component is a major component in native Agp1 (26). Since no photoisomerization occurs in locked Agp1, we assign the fast time constant τ1 to relaxation processes and the slow time constant τ2 to fluorescence decay (29). A comparison with nonisomerizing Cph1-PEB cyanobacterial phytochrome reconstituted with phycoerythrobilin (PEB) shows that the 5.5 ps found in locked Agp1 is absent in Cph1-PEB (12). Furthermore, the slow time constant τ2 in locked Agp1 is faster than for Cph1-PEB with a time constant in the range of 0.7 to 2.0 ns (12), which is in agreement with the lower fluorescence quantum yield of 15Za-18Et-BV locked Agp1 as reported by Zienicke et al. (29).
Polarization resolved VIS pump–IR probe data provide information on the relative angle between the electronic transition dipole moment (tdm) vector μel and the tdm vector of the investigated vibration μvib. The ratio of the transient amplitudes for parallel and perpendicular polarization defines the dichroic ratio DR (DR = A‖/A⊥). From the dichroic ratio, the angle θ between μel and μvib can be calculated as θ = acos[(2 DR − 1)/(DR + 2)]1/2.
Fig. 3 displays transients at selected frequencies for probe pulse polarizations parallel (black circles) and perpendicular (red squares) with respect to pump pulse polarization. Fig. 3 a displays the transients of the C19 = O stretching vibration in its electronically excited state, whereas the Fig. 3 c and e show the bleaching signals of the C19 = O stretching vibration, and C1 = O stretching vibration, respectively. Using the biexponential fit function we calculated the spectrum at time zero for both polarizations. We modeled the vibrational bands with Lorentzian line shapes (see Fig. 8 b) and from their amplitude values we derived the dichroic ratio. Thus, contributions from adjacent vibrations are minimized. The 1σ error ranges were gained from exhaustive search analysis.
The angle between the electronic tdm and C19 = O stretching vibration tdm in the electronically excited state is calculated from the dichroic ratio to be μC19=O∗ = (36 ± 2)°. The C19 = O ground state vibration at 1698 cm−1 shows a similar relative angle of μC19=O = (40 ± 2)°. In contrast, we found distinct angles for the C1 = O vibrations. The C1 = O bleaching vibration has a relative angle of 90° with an 1σ error ranging from 71° to 90°. In the electronically excited state we found a similar angle of 73° (1σ range of 65° to 90°). Consequently, we ascertain distinct relative orientations of the C19 = O and C1 = O tdm vectors with respect to the electronic S0→S1 transition. On the basis of these angles, we then compared the ZZZssa and ZZZasa chromophore geometries.
Excitation at 644 nm
The absorption spectrum in Fig. 1 displays an absorption peak at 713 nm and a pronounced shoulder at 644 nm. The existence of a shoulder is common for Pr spectra of most phytochromes. Whether the shoulder reflects a strong vibronic progression (15), another chromophore geometry, or a second electronic transition is not resolved as of this writing (12). We excited the locked Agp1 sample at the shoulder at 644 nm and determined the relative angles between the C19 = O stretching vibrations and the electronic tdm. If the shoulder is caused by vibronic progression, the relative angle μC19=O of the tdm of the C19 = O bleaching band with the electronic tdm would be the same for excitation at 713 nm and 644 nm. This results from the fact that tdm orientations are identical in the harmonic approximation for the lower vibrational levels and the electronic tdm vector is the same for a given transition. If the relative angle μC19=O determined upon excitation at 644 nm deviates from the angle upon excitation at 713 nm, a different electronic tdm is involved.
Fig. 4 depicts polarization resolved transients upon excitation at 644 nm. The overall signals are more than twofold smaller compared with excitation at 713 nm due to lower absorption; however, the shape of the transients are very similar.
Figure 4.
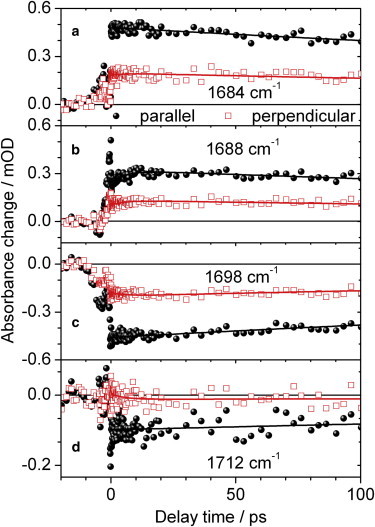
Polarization resolved vibrational transients of locked Agp1 upon excitation at 644 nm. Signals measured for probe polarization parallel (black circles) or perpendicular (red open squares) to pump polarization. Excited state C19 = O stretching vibration at 1684 cm−1 and 1688 cm−1 (a), (b); bleach signal of the C19 = O stretching vibration at 1698 cm−1 (c); C1 = O stretching vibration in the excited state at 1712 cm−1 (d). Biexponential simulations (black and red lines) with time constants of 5.5 ps and 540 ps.
The transients are well fitted by the two time constants τ1 and τ2 of 5.5 and 540 ps, respectively.
In the investigated spectral range from 1670 cm−1 to 1722 cm−1, the amplitude of the fast time constant τ1 is with 14% larger than for excitation at 713 nm, but still small. The small increase of the amplitude can account for energy redistribution of the higher excitation energy. The angle μC19=O between the tdm of the C19 = O bleaching vibration at 1698 cm−1 and the electronic tdm is 29° (1σ range of 24° to 32°), which is significantly different from the value determined upon exciting the S0→S1 transition at 713 nm. The same holds for the angle μC19=O∗ in the electronically excited state at 1684 cm−1 that is determined to (20 ± 5)°.
This result demonstrates vibronic progression cannot explain the absorption peak at 644 nm alone. Another source of absorption around 644 nm has to be taken into account—either another electronic transition (S0→S2) or the existence of a different chromophore geometry. In the investigated spectral window, the C1 = O stretching vibration is not fully resolved and thus not further investigated.
Excitation at 400 nm
The Soret band of the 15Za-18Et-BV chromophore is located around 400 nm. Excitation at 400 nm results in high electronic transitions S0→Sx (x > 2). Thus, we expect a stronger signal contribution from energy redistribution pathways. Fig. 5 presents selected transients in the spectral range of 1670 cm−1 to 1722 cm−1.
Figure 5.
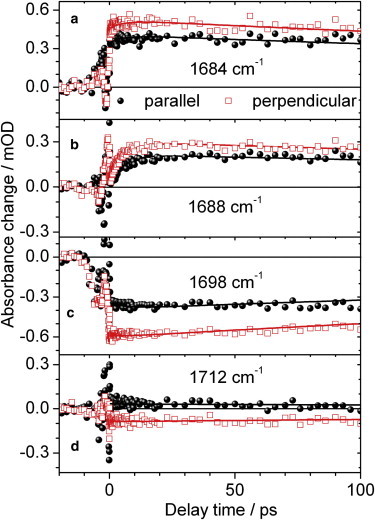
Polarization resolved vibrational transients of locked Agp1 upon excitation at 400 nm. Signals measured for probe polarization parallel (black circles) or perpendicular (red open squares) to pump polarization. C19 = O stretching vibration in the excited state at 1684 cm−1 and 1688 cm−1; bleach signal of the C19 = O stretching vibration at 1698 cm−1; C1 = O stretching vibration in the excited state at 1712 cm−1. Biexponential simulations (black and red lines) with time constants of 5.5 ps and 540 ps. Note, the best fit is found with time constants of 5.5 ps and 1 ns.
The transients are well fitted by two time constants τ1 and τ2 of 5.5 and 540 ps, respectively. Here, the 1σ error margins, determined by exhaustive search analysis (28), range from 4 to 8 ps for the shorter time constant and from 500 ps to 2 ns for the longer time constant. The amplitude of the fast time constant τ1 = 5.5 ps is significant with 33% and directly visible in the signal increase at 1688 cm−1 in Fig. 5 b. Remarkably, the perpendicular polarized signals are larger than the parallel polarized signals for the C19 = O stretching vibration in its electronically excited state and ground state at 1684 cm−1, and 1698 cm−1, respectively. This demonstrates a major angle change for μC19=O and μC19=O∗ to angles above 54.7°.
Since the same vibrational transitions are observed, we conclude that the orientation of electronic tdm has changed significantly going from S0→S1 to S0→Sx transitions. The angle μC19=O between the electronic tdm of the S0→Sx transition and the C19 = O stretching vibration tdm in the electronic ground state is μC19=O = (63 ± 3)°. In the electronically excited state the angle μC19=O∗ is found to be (67 ± 3)°. The C1 = O stretching vibration dominates the signals at 1712 cm−1 in Fig. 5 d. The polarized signals are interchanged compared with Fig. 3 d upon excitation at 713 nm. From this result we conclude that μC1=O∗ < 54°.
Comparison of different excitation frequencies
Locked Agp1 exhibits similar photoreactions of the C19 = O and C1 = O stretching vibration upon excitation at 713 nm, 644 nm, and 400 nm. All transients are well fitted by a biexponential function with time constants of τ1 = 5.5 ps and τ2 = 540 ps and no photoisomerization occurs. Nevertheless, the relative amplitudes of the time constants differ, and the angles between the vibrational tdm and the excited electronic tdm show distinct values. By direct comparison of the C19 = O bleaching transients for parallel and perpendicular polarization, the existence of distinct dichroic ratios and hence, distinct angles becomes clear. Fig. 6 presents the C19 = O bleaching signals at 1698 cm−1 for different excitation frequencies, scaled on their signal strength for parallel polarization. For perpendicular polarization, the transient at 644 nm excitation has a significantly lower signal level compared with excitation at 713 nm. The signal strengths of both transients do not overlap demonstrating the existence of distinct angles.
Figure 6.
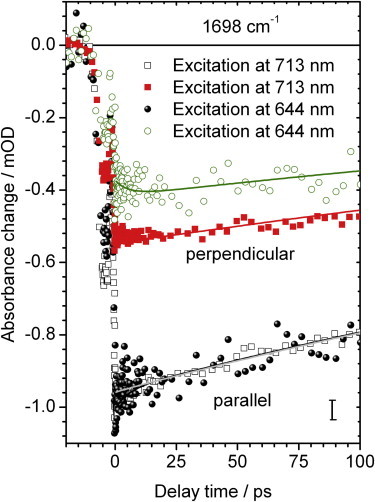
Polarization resolved vibrational transients of the C19 = O bleaching signal upon excitation of locked Agp1 at 713 nm and 644 nm. Signals for parallel polarization (black dots and gray open squares) are scaled to follow the same dynamics for long delay times. The signals for perpendicular polarization (green open circles and red squares) are clearly separated in amplitude. Biexponential simulations (solid lines) with time constants of 5.5 ps and 540 ps.
Fig. 7 presents the difference spectra detected at various delay times. The overall shape is similar for all excitation frequencies with absorption of the C19 = O stretching vibration in the electronically excited state at 1682 cm−1 (positive signal), and absorption of the C19 = O stretching vibration bleach signal at 1698 cm−1 (negative signal). The spectral shape is rather smooth with exception of four small dips located at 1676 cm−1, 1685 cm−1, 1696 cm−1, and 1701 cm−1, caused by water vapor absorption (Figs. 7 and 8 a). However, a distinct spectral shift is observed for excitation at 400 nm, indicated by an arrow in Fig. 7 c. At early delay times before 5 ps the C19 = O stretching vibration in the electronically excited state ν(C19 = O)∗ exhibits a slightly red shifted absorption. The absorption below 1680 cm−1 on the low energy side of the ν(C19 = O)∗ band decreases, whereas the absorption around 1688 cm−1 on the high energy side of the ν(C19 = O)∗ band increases. This is a typical signature of vibrational cooling. This becomes also evident from the polarization resolved decay associated spectra (DAS) plotted in Fig. 8 b–d for different excitation frequencies. Although the DAS of time constant τ2 shows positive absorption of ν(C19 = O)∗ at 1682 cm−1, negative bleaching signals of ν(C19 = O) at 1698 cm−1, positive absorption of ν(C1=O)∗ at 1716 cm−1, and negative bleaching signals of ν(C1 = O) at 1725 cm−1, the DAS of time constant τ1 shows significant contributions only below 1694 cm−1.
Figure 7.
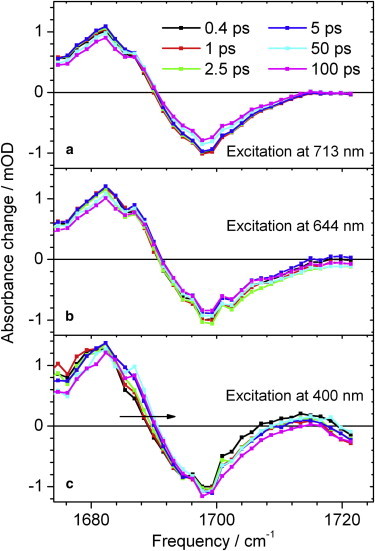
Isotropic absorbance difference spectra in the spectral range of the C19 = O stretching vibration and C1 = O stretching vibration. Spectra at different delay times as indicated; dots display the individually measured signal. Normalized C19 = O bleaching signal at 0.4 ps. Excitation at 713 nm (a), at 644 nm (b), and at 400 nm (c).
Most prominent contributions from time constant τ1 are provided by the DAS upon excitation at 400 nm (Fig. 8 d). The positive DAS values below 1680 cm−1 indicate a signal decrease, whereas the negative DAS values between 1684 cm−1 and 1694 cm−1 indicate a signal increase. This is in complete agreement with vibrational cooling of the C19 = O stretching vibration in the electronically excited state, and thus a transient blue shift of the absorption band. Additionally, the dichroic ratio of the polarization resolved DAS of time constant τ1 is the same as for the ν(C19 = O)∗ band, supporting the same origin of the signal.
Small contributions of time constant τ1 are also visible in the spectral range from 1700 cm−1 to 1716 cm−1 in Figs. 7 c and 8 d. These signals could be due to vibrational cooling of the C1 = O stretching vibration in the electronically excited state.
Since the chromophore is not able to relax energy via photoisomerization, increasing excitation energy results in higher excess energy that is redistributed within the chromophore vibrations and strongly coupled vibrations of the protein in a first step.
The energy redistribution process is modeled with a time constant τ1 of 5.5 ps (1σ range of 4 to 8 ps). The energy redistribution pathway strongly involves the ν(C19 = O)∗ ring D vibrations in agreement with fluorescence studies on bacteriophytochrome adducts with synthetic locked chromophores (29). Furthermore, we observed no spectral shifts in addition to the vibrational cooling or new emerging vibrational bands. Hence, in Agp1 with 15Za-18Et-BV chromophore, we do not see any evidence for excited state proton transfer in the spectral range from 1670 cm−1 to 1740 cm−1 (30).
The distinct angles measured at 713 and 644 nm excitation indicate a new electronic transition at 644 nm or a different chromophore geometry absorbing at 644 nm. For a different chromophore geometry, this significant energy difference would be reflected in the fluorescence spectrum. The carbonyl vibration of ring D is strongly involved in energy relaxation process, but the carbonyl vibration of ring A shows negligible coupling to the electronic excitation and its involvement in the photoreaction process is insignificant.
Ground state geometry of locked Agp1
Excitation of the S0→S1 transition at 713 nm with linearly polarized light induces photoselection within the bacteriophytochrome molecules in solution. Polarization resolved femtosecond VIS pump–IR probe spectroscopy allows for measuring the relative angles between the electronic tdm of the S0→S1 transition and the carbonyl tdms of ring A and ring D. The three-dimensional (3D) orientation of the vibrational C19 = O carbonyl tdm is fixed for a given geometry within the molecule. The single angle μC19=O indicates that the electronic tdm lies somewhere on a cone with an angle μC19=O round the vibrational tdm. A second cone, based on the angle μC1=O that another vibrational tdm forms with the electronic tdm, has at most two intersections with the first cone. The intersections fixing the electronic tdm orientation create a solution volume in the 3D space. If no solution volume is found, the geometry does not fit to the measured data.
Figs. 9 and 10 present the 15Za-18Et-BV chromophore with ZZZssa and ZZZasa geometry, respectively. The blue arrows display the orientation of the calculated ν(C19 = O) vibrational tdm, and the red arrows show the orientation of the calculated ν(C1 = O) vibrational tdm. The relative angles μC19=O and μC1=O are used to plot the cones around the vibrational tdms of ν(C19 = O) and ν(C1 = O), respectively. For each relative angle two cones representing the 1σ error ranges are plotted. The blue and cyan cone corresponding to angles μC19=O of 38° and 42°. The red and the pink cone corresponding to angles μC1=O of 71° and 90°.
Figure 9.
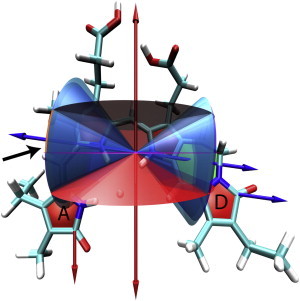
Showing 15Za-18Et-BV chromophore with ZZZssa geometry. Rings A, B, C, and D (from left to right) are marked by red areas; the ring formed by the covalent linkage between rings C and D is marked by a green area. Blue arrows: ν(C19 = O) tdm, red arrows: ν(C1 = O) tdm. Blue and cyan cones show 1σ range of angle μC19 = O around vibrational tdm of ν(C19 = O); red and pink cones show 1σ range of angle μC1 = O around vibrational tdm of ν(C1 = O). Electronic tdm orientation is given by the intersection area of the cones, indicated by orange rectangular and the black arrow. Note that each cone of angle θ has also a cone of angle 180°-θ.
Figure 10.
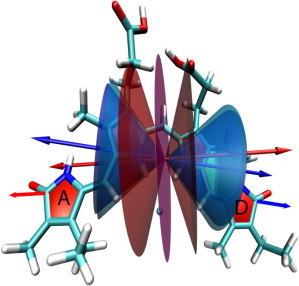
Showing 15Za-18Et-BV chromophore with ZZZasa geometry (as in Fig. 9). Blue arrows: ν(C19 = O) tdm, red arrows: ν(C1 = O) tdm. Blue and cyan cones show 1σ range of angle μC19 = O around vibrational tdm of ν(C19 = O); red and pink cones show 1σ range of angle μC1 = O around vibrational tdm of ν(C1 = O).
The orientation of the excited electronic tdm at 713 nm is fixed by the intersection of cones around the vibrational tdms of ν(C19 = O) and ν(C1 = O). Since, the measured relative angles have error ranges, we have a set of intersections of cones (31). In Fig. 9 the possible 3D orientations of the electronic tdm are given by the area between the blue and cyan cone around vibrational tdms of ν(C19=O) and between the red and pink cone around vibrational tdms of ν(C1 = O). We found eight areas of solutions of which one is indicated by an orange rectangular in Fig. 9. Hence, the ZZZssa geometry agrees with our experimental data.
In contrast to the ZZZssa geometry, the orientation of the vibrational tdms of ν(C19 = O) and ν(C1 = O) are similar for the ZZZasa geometry (Fig. 10). Consequently, the cones around the vibrational tdms show a different pattern as compared with Fig. 9. The ZZZasa geometry show no intersection of the cones in the 1σ error ranges and no solution volume at all. Hence, the ZZZasa geometry conflicts with our experimental data.
The frequency position of the carbonyl vibrations at ring A is strongly influenced by an adjacent double bond between C3 and C31 or between C2 and C3. A double bond between C3 and C31 decreases conjugation at the C1 = O group, and thereby increasing its stretching frequency. Experimentally we found a frequency difference of 27 cm−1 between ν(C19 = O) and ν(C1 = O) vibrations in agreement with calculations for a planar ring A geometry with a double bond between C2 and C3 in locked Agp1 (see Supporting Material) (16).
By combining DFT calculations and measured orientational data, we can exclude a ZZZasa and confirm a ZZZssa chromophore geometry. Moreover, our data favor a planar ring A geometry in contrast to a nonplanar ring A geometry with a double bond between C3 and C31.
Origin of the new electronic transition
The absorption spectrum of 15Za-18Et-BV Agp1 displayed in Fig. 1 exhibits two pronounced peaks at 713 nm and 644 nm. Excitation at 713 nm and 644 nm result in two different dichroic ratios of the ν(C19 = O) ground state bleaching band at 1698 cm−1 of DR713 = 1.52 and of DR644 = 2.07, respectively. Consequently, two different electronic transitions with distinct electronic tdm orientations contribute to the absorption spectrum. However, excitation in the shoulder of the absorption spectrum at 644 nm could be influenced by contributions from the main peak at 713 nm, e.g., vibronic progressions. Therefore, we estimate the contribution of the main peak at 713 nm to the signals measured upon excitation at 644 nm. Assuming two components contributing to the measured signal upon excitation at 644 nm with probabilities p713 and p644 (and p713 + p644 = 1) and pure dichroic ratios D713 and D644. The measured dichroic ratio DR644 is given by DR644 = p713D713 + p644D644. Since contributions from the electronic transition at 644 nm are negligible at 713 nm excitation (DR713 = D713), we can determine p713 using (p644 = 1− p713) to be p713 = (DR644 − D644)/(D713 − D644). The pure dichroic ratio D644 can range from 2.07 to 3, because DR644 is greater than DR713 and contributions of p713 could only have decreased DR644. Consequently, p713 varies from 0 to 0.63. This corresponds to an angle less than or equal to 29°. Thus, the contribution of the second electronic transition to the signals measured upon excitation at 644 nm is at least 37% and maximally 100%.
If the second electronic transition at 644 nm presented the S0→S1 transition of an unbound chromophore or a different chromophore geometry in the protein sample (e.g., ZZZasa), it would result in differences between emission excitation spectra and absorption spectra. However, the emission excitation spectra and the absorption spectra display the same shape between 625 and 730 (29). Hence, we conclude that this electronic transition is the S0→S2 transition of locked Agp1.
Dynamics of native Agp1
The absorption spectrum of native Agp1 in its Pr form is very similar to the locked Agp1 with a less-pronounced shoulder at 644 nm. Upon excitation at 680 nm the infrared absorbance difference spectra, presented in Fig. 11 a, are dominated by positive signals from 1670 cm−1 to 1695 cm−1, and negative signals from 1695 cm−1 to 1713 cm−1.
Figure 11.
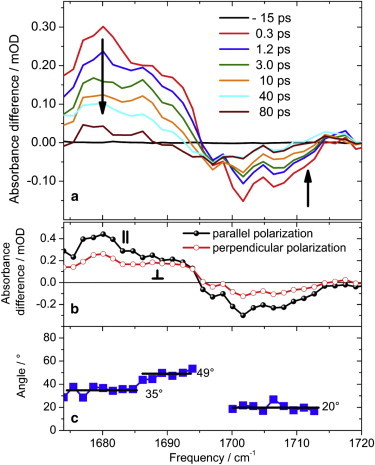
(a) Absorbance difference of native Agp1 upon excitation at 680 nm for various pump-probe delay times under isotropic conditions. Bleaching signals around 1705 cm−1 and above 1718 cm−1 are due to ν(C19 = O) and ν(C1 = O) absorption. (b) Decay associated infrared absorption spectra calculated to time zero for parallel polarization (A‖, black solid circles) and perpendicular polarization (A⊥, red open circles). (c) Angle between electronic and vibrational tdm calculated by the dichroic ratio (DR = A‖/A⊥). The solid lines indicate regions of constant angle.
The positive signal peaking at 1680 cm−1 represents mainly the ν(C19 = O)∗ vibration of ring D in its electronically excited state, whereas the ν(C19 = O) vibration of the ground state bleaching band peaks at 1702 cm−1 for short delay times and at 1697 cm−1 for long delay times. The zero crossing at 1695 cm−1 is shifted by 5 cm−1 to higher frequencies and longer delay times in native Agp1. This is caused by a faster decay of the positive signal at 1690 cm−1 compared with 1680 cm−1. In contrast to the difference spectra of locked Agp1, small positive signals are clearly visible around 1716 cm−1 assigned to ν(C1 = O)∗ ring A carbonyl vibration in the electronic excited state and ring D carbonyl vibration of the photoproduct Lumi-R (32). The signal strength of the ν(C1 = O)∗ and ν(C1 = O) carbonyl vibrations of ring A are more than 10 times smaller than the ν(C19 = O)∗ and ν(C19 = O) carbonyl vibrations of ring D.
Taking the whole time range into account, the ν(C19 = O) bleaching band exhibits a maximal FWHM (full width at half maximum) of ∼16 cm−1 comparable with the FWHM of 14 cm−1 in locked Agp1. The observed bleaching band is broad enough to consist of two bleaching bands, representing two different ground state geometries. However, the difference spectra show no further indications supporting such heterogeneity.
Our transient data are well globally fitted by two exponentials with time constants of τ1 = 1.0 ± 0.4 ps and τ2 = 20 ± 3 ps, as well as by three exponentials with time constants of τ1 = 1.0 ± 0.6 ps, τ2 = 9 ± 3 ps, and τ3 = 40 ± 10 ps. Since introduction of the third time constant improved the χ2 only by 1.7%, the significance of a third time constant is negligible for our data set. However, the time constants agree well with findings of Schumann et al. (26). They found three time constants of τ1 = 2.0 ps, τ2 = 4.3 ps, and τ3 = 46.8 ps and vibrations that were slightly shifted to higher energies, probably due to H/D (Hydrogen / Deuterium) exchange.
In Fig. 11 b DAS calculated to time zero are presented for parallel and perpendicular polarization with respect to pump pulse polarization. From the dichroic ratio of both spectra, we determined the relative angle of vibrational to electronic tdm. In Fig. 11 c the relative angles are depicted for the spectral range from 1670 cm−1 to 1714 cm−1. Around 1680 cm−1, the ν(C19 = O)∗ ring D vibration exhibits an angle of (35 ± 8)°, and the ν(C19 = O) ring D bleaching band has an angle of (20 ± 8)°. These findings are in agreement with a ZZZssa chromophore geometry (32). Since the DAS in Fig. 11 b shows very similar features from 1710 cm−1 to 1720 cm−1 as the DAS for locked Agp1 (Fig. 8 b), we also expect an angle of > 54° for the ν(C1 = O) carbonyl vibration in native Agp1.
In native Agp1, an additional vibrational contribution with an angle of (49 ± 8)° is found in the spectral range from 1685 cm−1 to 1694 cm−1 (Fig. 11 c). We tentatively assign this contribution to an amino acid or a high energy amide I vibration.
Fig. S11 in the Supporting Material presents transients and simulations with time constants of 1 ps and 20 ps for parallel and perpendicular polarizations at 1680 cm−1 and 1702 cm−1, respectively. The data show clearly a fast component of ∼1 ps, first reported by Schumann et al. (26).
Tracking the dichroic ratio of ν(C19 = O)∗ in time, we were not able to identify orientational changes for delay times longer than 1.5 ps. For early delay times, we see indications for an angle decrease of ∼4°, but the signal-to-noise ratio is not good enough to provide reliable statements.
Excitation of the Soret band at 400 nm shows similar time constants and similar isotropic absorbance change differences (Fig. S13). Polarization resolved measurements provide distinct angles for the carbonyl vibrations at 400 nm excitation than found upon 680 nm excitation (Fig. S12 c). The data can be explained by an angle between the electronic tdms of S0→S1 and S0→Sx of 104°, and by electronic and vibrational tdms all lying roughly in the pyrrole plane (Fig. S13).
Orientational differences of ring D’s ν(C19 = O) tdm compared with locked Agp1 are due to stronger protein-chromophore interaction at ring D in native Agp1.
Conclusions
There is a long-standing debate whether the Pr form of phytochrome is heterogeneous or not. Schmidt et al. showed the existence of an isosbestic point between the main absorption peak and the shoulder upon temperature change in oat phytochrome (33). These findings were in contrast to a single electronic transition and were interpreted as heterogeneity of the Pr form as reported by Sineshchekov et al. (14). Single molecule fluorescence studies on native Agp1 bacteriophytochrome showed clear evidence for heterogeneity at very low temperatures (13). Additionally, von Stetten et al. found the coexistence of at least two similar conformers in Agp1 solution (16). Spillane et al. demonstrated that the Pr form linear absorption spectrum of Cph1 cyanobacterial phytochrome can be completely simulated by vibronic transitions of a single Pr conformer of Cph1 (15).
Since the absorption spectra of Cph1 and Agp1 are similar, we assume the contribution of vibronic transitions to the Agp1 absorption spectrum to be relevant. However we investigated whether another contribution to the absorption spectrum exists.
By angle balanced polarization resolved femtosecond VIS pump–IR probe spectroscopy we were able to assign the shoulder in the absorption spectrum at 644 nm to a S0→S2 electronic transition of locked Agp1. The transition has a contribution of at least 37% at this spectral position in addition to vibronic transitions. These findings may shed new light on the interpretation of the absorption spectrum of the Pr form and could change the interpretation of transient data on the Pr form of phytochromes, by including additional relaxation processes from S2 to S1.
Furthermore, in our study we found no conclusive evidence for heterogeneity in the ground state geometry of ring D. Nevertheless, we also cannot exclude heterogeneity, since the line width of the ν(C19 = O) ring D bleaching band of ∼15 cm−1 is broad enough to consist of two different vibrational absorption bands in Agp1.
We acquired orientational information on the electronic tdms and the vibrational tdms of the carbonyl groups of ring A and ring D. By comparing these data with DFT calculations we were able to support the ZZZssa geometry of the chromophore in locked 15Za-18Et-BV Agp1 and exclude a ZZZasa geometry. Moreover, our data agrees well with a ZZZssa geometry of the chromophore in native Agp1.
For locked Agp1, time constants of 5.5 and 540 ps were determined. The fast time constant was assigned to vibrational cooling of the hot ν(C19 = O)∗ ring D stretching vibration in its electronically excited state. The orientation of the hot ν(C19 = O)∗ ring D vibration is the same as the cold ν(C19 = O)∗ ring D vibration in the electronic excited state. The 540 ps time constant was assigned to fluorescence decay.
Energy redistribution pathway strongly involves the ν(C19 = O)∗ vibration of ring D, whereas contributions of the ν(C1 = O)∗ vibration of ring A were negligible. The carbonyl bleaching signal of ring A has only negligible contribution in locked Agp1 and native Agp1. Photoisomerization dynamics of native Agp1 showed time constants of 1 and 20 ps upon excitation in the main absorption band and in the Soret band. We identified Lumi-R formation and determined the orientations of ring D and ring A carbonyl vibrations.
Our polarization resolved data show no transient change of orientation of ring A, and no photoproduct was observed in locked Agp1 up to 1 ns. Hence, we can rule out that there is a light-induced rotation of the ring A on the photoisomerization time scale as proposed by Ulijasz et al. (34). In addition, the measured relative angles between vibrational and electronic tdms are in conflict with the twisted ring A conformation found in cyanobacterium Synechococcus OSB’ (34). Therefore, the findings on cyanobacterium Synechococcus OSB’ cannot be transferred to the bacteriophytochrome Agp1.
We showed that with our angle balanced polarization resolved femtosecond VIS pump–IR probe setup orientations of individual carbonyl groups within a protein can be detected and followed in time. From these data structural and dynamical information on conformations, heterogeneity, molecular groups involved in the photoreaction process, and individual electronic and vibrational states can be studied to understand the underlying dynamical processes in photoreceptors.
Acknowledgments
We thank the Computing Centers of MPI-Halle and FUB (ZEDAT) for their services.
This work was supported by the Deutsche Forschungsgemeinschaft (SFB 1078 TP B3).
Footnotes
This is an Open Access article distributed under the terms of the Creative Commons-Attribution Noncommercial License (http://creativecommons.org/licenses/by-nc/2.0/), which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Supporting Material
References
- 1.Rockwell N.C., Su Y.S., Lagarias J.C. Phytochrome structure and signaling mechanisms. Annu. Rev. Plant Biol. 2006;57:837–858. doi: 10.1146/annurev.arplant.56.032604.144208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Narikawa R., Ishizuka T., Ikeuchi M. Structures of cyanobacteriochromes from phototaxis regulators AnPixJ and TePixJ reveal general and specific photoconversion mechanism. Proc. Natl. Acad. Sci. USA. 2013;110:918–923. doi: 10.1073/pnas.1212098110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ulijasz A.T., Vierstra R.D. Phytochrome structure and photochemistry: Recent advances toward a complete molecular picture. Curr. Opin. Plant Biol. 2011;14:498–506. doi: 10.1016/j.pbi.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 4.Lamparter T., Michael N., Esteban B. Phytochrome from Agrobacterium tumefaciens has unusual spectral properties and reveals an N-terminal chromophore attachment site. Proc. Natl. Acad. Sci. USA. 2002;99:11628–11633. doi: 10.1073/pnas.152263999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scheerer P., Michael N., Lamparter T. Light-induced conformational changes of the chromophore and the protein in phytochromes: Bacterial phytochromes as model systems. ChemPhysChem. 2010;11:1090–1105. doi: 10.1002/cphc.200900913. [DOI] [PubMed] [Google Scholar]
- 6.Song C., Psakis G., Matysik J. Two ground state isoforms and a chromophore D-ring photoflip triggering extensive intramolecular changes in a canonical phytochrome. Proc. Natl. Acad. Sci. USA. 2011;108:3842–3847. doi: 10.1073/pnas.1013377108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang X.J., Ren Z., Moffat K. Temperature-scan cryocrystallography reveals reaction intermediates in bacteriophytochrome. Nature. 2011;479:428–432. doi: 10.1038/nature10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wagner J.R., Brunzelle J.S., Vierstra R.D. A light-sensing knot revealed by the structure of the chromophore-binding domain of phytochrome. Nature. 2005;438:325–331. doi: 10.1038/nature04118. [DOI] [PubMed] [Google Scholar]
- 9.Mailliet J., Psakis G., Hughes J. Spectroscopy and a high-resolution crystal structure of Tyr263 mutants of cyanobacterial phytochrome Cph1. J. Mol. Biol. 2011;413:115–127. doi: 10.1016/j.jmb.2011.08.023. [DOI] [PubMed] [Google Scholar]
- 10.Inomata K., Hammam M.A.S., Lamparter T. Sterically locked synthetic bilin derivatives and phytochrome Agp1 from Agrobacterium tumefaciens form photoinsensitive Pr- and Pfr-like adducts. J. Biol. Chem. 2005;280:24491–24497. doi: 10.1074/jbc.M504710200. [DOI] [PubMed] [Google Scholar]
- 11.Nibbering E.T.J., Tschirschwitz F., Elsaesser T. Femtochemistry of hydrogen bonded complexes after electronic excitation in the liquid phase: The case of coumarin 102. J. Phys. Chem. A. 2000;104:4236–4246. [Google Scholar]
- 12.Heyne K., Herbst J., Diller R. Ultrafast dynamics of phytochrome from the cyanobacterium synechocystis, reconstituted with phycocyanobilin and phycoerythrobilin. Biophys. J. 2002;82:1004–1016. doi: 10.1016/S0006-3495(02)75460-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nieder J.B., Brecht M., Bittl R. Dynamic intracomplex heterogeneity of phytochrome. J. Am. Chem. Soc. 2009;131:69–71. doi: 10.1021/ja8058292. [DOI] [PubMed] [Google Scholar]
- 14.Sineshchekov V., Loskovich A., Takano M. Two native pools of phytochrome A in monocots: Evidence from fluorescence investigations of phytochrome mutants of rice. Photochem. Photobiol. 2006;82:1116–1122. doi: 10.1562/2005-12-10-ra-749. [DOI] [PubMed] [Google Scholar]
- 15.Spillane K.M., Dasgupta J., Mathies R.A. Homogeneity of phytochrome Cph1 vibronic absorption revealed by resonance Raman intensity analysis. J. Am. Chem. Soc. 2009;131:13946–13948. doi: 10.1021/ja905822m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.von Stetten D., Günther M., Hildebrandt P. Chromophore heterogeneity and photoconversion in phytochrome crystals and solution studied by resonance Raman spectroscopy. Angew. Chem. Int. Ed. Engl. 2008;47:4753–4755. doi: 10.1002/anie.200705716. [DOI] [PubMed] [Google Scholar]
- 17.Hammam M.A.S., Nakamura H., Inomata K. Syntheses of biliverdin derivatives sterically locked at the CD-ring components. Bull. Chem. Soc. Jpn. 2006;79:1561–1572. [Google Scholar]
- 18.Inomata K., Khawn H., Lamparter T. Assembly of Agrobacterium phytochromes Agp1 and Agp2 with doubly locked bilin chromophores. Biochemistry. 2009;48:2817–2827. doi: 10.1021/bi802334u. [DOI] [PubMed] [Google Scholar]
- 19.Scheerer P., Michael N., Krauss N. Crystallization and preliminary X-ray crystallographic analysis of the N-terminal photosensory module of phytochrome Agp1, a biliverdin-binding photoreceptor from Agrobacterium tumefaciens. J. Struct. Biol. 2006;153:97–102. doi: 10.1016/j.jsb.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 20.Theisen M., Linke M., Heyne K. Femtosecond polarization resolved spectroscopy: A tool for determination of the three-dimensional orientation of electronic transition dipole moments and identification of configurational isomers. J. Chem. Phys. 2009;131:124511–124519. doi: 10.1063/1.3236804. [DOI] [PubMed] [Google Scholar]
- 21.Frisch M.J., Trucks G.W., Fox D.J. Gaussian, Inc.; Wallingford, CT: 2009. Gaussian 09, Revision A.02. [Google Scholar]
- 22.Kohn W., Sham L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965;140:A1133–A1138. [Google Scholar]
- 23.Borucki B., von Stetten D., Hildebrandt P. Light-induced proton release of phytochrome is coupled to the transient deprotonation of the tetrapyrrole chromophore. J. Biol. Chem. 2005;280:34358–34364. doi: 10.1074/jbc.M505493200. [DOI] [PubMed] [Google Scholar]
- 24.von Stetten D., Seibeck S., Lamparter T. Highly conserved residues Asp-197 and His-250 in Agp1 phytochrome control the proton affinity of the chromophore and Pfr formation. J. Biol. Chem. 2007;282:2116–2123. doi: 10.1074/jbc.M608878200. [DOI] [PubMed] [Google Scholar]
- 25.Seibeck S., Borucki B., Heyn M.P. Locked 5Zs-biliverdin blocks the Meta-RA to Meta-RC transition in the functional cycle of bacteriophytochrome Agp1. FEBS Lett. 2007;581:5425–5429. doi: 10.1016/j.febslet.2007.10.043. [DOI] [PubMed] [Google Scholar]
- 26.Schumann C., Gross R., Diller R. Sub-picosecond mid-infrared spectroscopy of phytochrome Agp1 from Agrobacterium tumefaciens. ChemPhysChem. 2007;8:1657–1663. doi: 10.1002/cphc.200700210. [DOI] [PubMed] [Google Scholar]
- 27.Piwowarski P., Ritter E., Bartl F. Light-induced activation of bacterial phytochrome Agp1 monitored by static and time-resolved FTIR spectroscopy. ChemPhysChem. 2010;11:1207–1214. doi: 10.1002/cphc.200901008. [DOI] [PubMed] [Google Scholar]
- 28.Roelofs T.A., Lee C.H., Holzwarth A.R. Global target analysis of picosecond chlorophyll fluorescence kinetics from pea chloroplasts. Biophys. J. 1992;61:1147–1163. doi: 10.1016/s0006-3495(92)81924-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zienicke B., Chen L.Y., Lamparter T. Fluorescence of phytochrome adducts with synthetic locked chromophores. J. Biol. Chem. 2011;286:1103–1113. doi: 10.1074/jbc.M110.155143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Toh K.C., Stojkovic E.A., Kennis J.T.M. Proton-transfer and hydrogen-bond interactions determine fluorescence quantum yield and photochemical efficiency of bacteriophytochrome. Proc. Natl. Acad. Sci. USA. 2010;107:9170–9175. doi: 10.1073/pnas.0911535107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Linke M., Lauer A., Heyne K. Three-dimensional orientation of the Qy electronic transition dipole moment within the chlorophyll a molecule determined by femtosecond polarization resolved VIS pump-IR probe spectroscopy. J. Am. Chem. Soc. 2008;130:14904–14905. doi: 10.1021/ja804096s. [DOI] [PubMed] [Google Scholar]
- 32.Yang Y., Linke M., Heyne K. Real-time tracking of phytochrome’s orientational changes during Pr photoisomerization. J. Am. Chem. Soc. 2012;134:1408–1411. doi: 10.1021/ja209413d. [DOI] [PubMed] [Google Scholar]
- 33.Schmidt P., Gensch T., Schaffner K. The complexity of the P-r to P-fr phototransformation kinetics is an intrinsic property of native phytochrome. Photochem. Photobiol. 1998;68:754–761. [Google Scholar]
- 34.Ulijasz A.T., Cornilescu G., Vierstra R.D. Structural basis for the photoconversion of a phytochrome to the activated Pfr form. Nature. 2010;463:250–254. doi: 10.1038/nature08671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wagner J.R., Zhang J.R., Forest K.T. High resolution structure of Deinococcus bacteriophytochrome yields new insights into phytochrome architecture and evolution. J. Biol. Chem. 2007;282:12298–12309. doi: 10.1074/jbc.M611824200. [DOI] [PubMed] [Google Scholar]
- 36.Brueggemeier R.W., Snider C.E., Counsell R.E. Substituted C-19 steroid analogs as inhibitors of aromatase. Cancer Res. 1982;42:3334–3337. [PubMed] [Google Scholar]
- 37.Oswald A.A., Griesbaum K., Hudson B.E. Organic sulfur compounds. 8. Addition of thiols to conjugated diolefins. J. Am. Chem. Soc. 1962;84:3897–3904. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.