

Abstract
Sphingosine-1-phosphate (S1P) regulates a wide array of biological functions. However, the role of S1P signaling in tumorigenesis remains to be elucidated. In this study, we show that S1P receptor subtype 3 (S1P3) is markedly up-regulated in a subset of lung adenocarcinoma cells compared to normal lung epithelial cells. Specific knockdown of S1P3 receptors inhibits proliferation and anchorage-independent growth of lung adenocarcinoma cells. Mechanistically, we demonstrate that S1P3 signaling increases epidermal growth factor receptor (EGFR) expression via the Rho kinase (ROCK) pathway in lung adenocarcinoma cells. Nuclear run-off analysis indicates that S1P/S1P3 signaling transcriptionally increases EGFR expression. Knockdown of S1P3 receptors diminishes the S1P-stimulated EGFR expression in lung adenocarcinoma cells. Moreover, S1P treatment greatly enhances EGF-stimulated colony formation, proliferation and invasion of lung adenocarcinoma cells. Together, these results suggest that the enhanced S1P3-EGFR signaling axis may contribute to the tumorigenesis or progression of lung adenocarcinomas.
Keywords: sphingosine-1-phosphate, sphingosine-1-phosphate receptor subtype 3, epidermal growth factor, epidermal growth factor receptor, S1P3, lung carcinoma
Introduction
Spingosine-1-phosphate (S1P), a serum-borne bioactive lipid mediator, regulates an array of biological activities in various cell types (1–4). Most, if not all, of S1P-regulated functions are mediated by the S1P family of G protein-coupled receptors (GPCRs) (5–7). There are five identified members of the S1P receptor family: S1P1, S1P2, S1P3, S1P4, and S1P5 (previous nomenclature: EDG-1, −5, −3, −6, −8, respectively) (8). S1P receptor subtypes couple to different Gα polypeptides to regulate distinct signaling pathways (9–11). The S1P receptor subtypes are expressed in distinct combinations in different cell types to produce appropriate biological effects. For example, S1P1 and S1P3 receptors are expressed in endothelial cells (12). The signaling pathways regulated by the S1P1 and S1P3 receptors are required for the chemotaxis of endothelial cells, adherens junction assembly, endothelial morphogenesis, and angiogenic responses (5,12,13). Moreover, the balance of S1P1 and S1P2 signaling contributes to vascular integrity in vivo (14). Disturbance of this balance, i.e., by up-regulation of S1P2 signaling, may have functional implications in vascular dysfunction, e.g., endothelial senescence and atherosclerosis (15). However, the functional outcomes resulting from the concerted effects of the signaling pathways mediated by the distinct S1P receptor subtypes are not fully understood and await elucidation.
The involvement of sphingolipid signaling in the tumor biology of various cancers has been extensively investigated. Previously, it was shown that the activation of sphingosine kinase-1 (SphK1) induced anchorage-independent growth of fibroblasts in vitro and enhanced subcutaneous tumor growth in a xenograft animal experiment (16). Increased cellular levels of sphingosine kinase, a key enzyme for S1P biosynthesis, have been shown to contribute to chemi- and radio-resistance of prostate tumors (17–20). Further, the transactivation between S1P and growth factor receptor signaling pathways has been functionally implied in the invasiveness and metastasis of tumors including breast, glioma, and pancreas (21–24). Recently, an elegant study showed that the S1P1-STAT3 signaling axis may play an important role in the tumorigenesis of several tumor types (25). These observations together suggest that sphingolipid signaling may play an important role in the regulation of tumor initiation, progression, and radio-/chemo-resistance.
In the present study, we observed that S1P3 receptors are markedly increased in a subset of cultured lung adenocarcinoma cells. Knockdown of S1P3 receptors reduced the proliferation and clonogenesis of lung adenocarcinoma cells in vitro. Mechanistically, we report that S1P3 signaling transcriptionally activates the expression of epidermal growth factor receptor (EGFR) via the Rho kinase (ROCK) pathway. Moreover, activation of S1P-S1P3 signaling greatly enhances carcinogenic activities of EGF in stimulating proliferation, clonogenesis, and invasion of lung adenocarcinoma cells. Collectively, these data present a novel S1P-regulated feed-forward signaling pathway, i.e., S1P3 transcriptionally up-regulates EGFR expression, which consequently augments EGFR-stimulated carcinogenic responses. Therefore, our study suggests that the S1P3-EGFR signaling axis may contribute to the tumorigenesis of lung tumors.
Materials and methods
Reagents
Sphingosine-1-phosphate purchased from Biomol was dissolved in 4 mg/ml of fatty acid-free BSA (bovine serum albumin, Sigma). VPC 23019 (Avanti) was solublized in ethanol (20 mg/ml) and aliquoted. Aliquots were vacuumed dried and stored at −20°C. When needed, aliquots were resuspended in 4% fatty acid-free BSA by sonication to make a stock solution of 1 mg/ml. CAY10444 (Cayman Chemical) was dissolved in dimethylformamide. Polyclonal anti-EGFR was purchased from Cell Signaling Technology and anti-β-actin (H-300) was from Santa Cruz Biotechnology. Peroxidase-conjugated sheep anti-rabbit IgG and peroxidase-conjugated goat anti-mouse IgG was from MP Biomedicals and Pierce Biotechnology, respectively. Matrigel was from BD Biosciences. Other reagents, unless specified, were purchased from Sigma.
Cell lines and growth conditions
Lewis Lung carcinoma cells (LLC) and human A549 lung carcinoma cells were obtained from Dr Bodduluri Haribabu (University of Louisville). Both LLC and A549 were cultured and maintained in RPMI-1640 (Hyclone) supplemented with 10% fetal bovine serum (Hyclone), penicillin (50 IU/ml) and streptomycin (50 mg/ml) (Cellgro). Immortalized normal human lung epithelial cells (HBEC2-KT and HBEC3-KT) and human lung adenocarcinoma cells (H23, H1792, H1793) were cultured as we previous described (26). Primary SAEC normal human small airway epithelial cells were purchased from Clonetics and cultured in SAEC culture medium (Clonetics Corp.). All of the cells listed above were cultured in a humidified atmosphere of 37°C and 5% CO2.
RT-PCR and real-time PCR
Total RNA was isolated from cultured cells, reverse-transcribed with an oligo-dT primer (Promega), and PCR amplification of S1P receptor subtypes as we previously described (15). For real-time PCR quantitation, 50 ng of reverse-transcribed cDNAs were amplified with the ABI 7500 system (Applied Biosystems) in the presence of TaqMan DNA polymerase. The sense and anti-sense primers used to detect the gene expression of S1P receptor subtypes and GAPDH were purchased from Applied Biosystems. The qPCR reaction was performed by using a universal PCR Master Mix (Applied Biosystems) according to the manufacturer’s instructions.
Western blot analysis
Cells treated with or without S1P were collected with cell scrapers in ice-cold PBS followed by centrifugation (250 g, 5 min). Cell extracts were prepared in TBST/OG buffer (5), resolved on a 10% SDS-PAGE, and transferred to nitrocellulose membranes. Subsequently, membranes were blocked in 5% non-fat dry milk (Lab Scientific) in TBST (5), washed and incubated with the indicated primary antibodies on a rotary shaker at 4°C overnight. Blots were then incubated with HRP-conjugated second antibody for 1 h at room temperature, incubated with enhanced chemiluminescence reagent (ECL) (Amersham) for 1 min, and visualized by exposure to Kodak X-OMAT film.
siRNA mediated gene silencing
To specifically knockdown the S1P3 receptor, cells were transfected with 1 μg of shRNA vector constructs (Origene) using Lipofectamine™ 2000 (Invitrogen) according to manufacturer’s instructions. The shRNA constructs (5′-GCTTC ATCGT CTTGG AGAAC CTGAT GGTT-3′) used to silence the S1P3 receptor and the negative control shRNA pRS plasmid were purchased from Origene (Rockville, MD). Stable S1P3 knocked-down H1793 cells were isolated with puromycin (1 μg/ml) selection.
Soft agar colony formation
Cells were collected and adjusted to 1×104 cells and resuspended in 1 ml of RPMI-1640 (Hyclone) containing 0.3% Noble agar (BD). Cell suspension was plated in 6-well plates (BD Falcon) above a 1 ml layer of solidified 0.6% Noble agar in RPMI and allowed to set. The standard H1793 culture media (1 ml) (26) containing treatments was added to the solidified cell/agar mixture and changed every 3 days. Cultures were incubated in a humidified 37°C atmosphere with 5% of CO2. Fourteen days later, cultures were fixed with 4% paraformaldehyde overnight. Colonies were then scored through blinded study.
Cell proliferation assay
Cells (8×103) were resuspended in RPMI containing 0.01% FBS, supplemented with or without treatments, and plated into each well of a 96-well plate (Corning). Each treatment was performed in triplicates and cultured at 37°C, in 5% CO2 incubator for 72 h. Subsequently, the medium was removed and replaced with 100 μl of phenol-free RPMI containing 10 μl of MTS/PMS (Promega) and further incubated for 1.5 h at 37°C. The absorbance of the plate was read at 492 nm with a plate reader (Tecan Genios Plus).
Tumor invasion assay in vitro
The invasion ability of cells was determined by using Neuro Probe A-series 96-well chamber and standard framed filters (8 μm pore size) following the manufacturer’s instructions (Neruro Probe, Gaithersburg, MD). The filters were coated with Matrigel™ (250 μg/ml, BD Bioscience) at 37°C for 1 h and then air dried. Cells were washed 3 times with plain medium and cultured in serum-free medium for 12 h. Subsequently, cell suspensions (2×105 cells/ml) were prepared in plain RPMI-1640 medium and plated in the upper chambers. Chemoattractants were added to the lower chambers. Cells were allowed to invade through Matrigel plugs for 8 h at 37°C. Cells on the upper surfaces of filters were removed with a cotton swab. Filters were fixed with 4% paraformaldehyde and stained with 0.5% crystal violet for 30 min. After washes, crystal violet dye was eluted with 10% acetic acid, and absorbance was measured at 595 nm.
Results
Elevated S1P3 expression in a subset of cultured lung adenocarcinoma cells
Our interest in the role of S1P signaling in lung cancer initially prompted us to screen for the expression of S1P receptor subtypes in SAEC (primary culture of normal human small airway epithelial cells), A549 (human lung adenocarcinoma epithelial cells), and LLC (mouse Lewis lung carcinoma cells) using RT-PCR. In both A549 and LLC cell lines, the expression of S1P3 was higher than in SAEC cells (Fig. 1A). Levels of S1P1 were similar in all 3 cell lines. Levels of S1P2 were relatively low, while the expression of S1P4 and S1P5 were almost undetectable (data not shown).
Figure 1.

Increased expression of S1P3 in cultured human lung adenocarcinoma cells. (A) Expression levels of S1P receptors in normal SAEC (primary culture of normal human small airway epithelial cells) and A549 adenocarcinoma cells (left panel), as well as LLC cells (right panel). -ve, PCR were performed without cDNA template. Note that S1P3 receptors are markedly increased in cancerous lung cells, compared to normal lung epithelial cells. (B) Real-time PCR quantitation of S1P3 expression in 3 immortalized normal lung epithelial and 9 lung adenocarcinoma cells lines. Note that S1P3 is elevated, ranging from 2–18 fold increases, in lung adenocarcinoma cells compared to immortalized normal lung epithelial cells. 3-KT, HBEC3-KT; 2-E, HBEC2-E; 2-KT, HBEC2-KT. MCF human breast cancer cells were used as a positive control. (C) RT-PCR analysis for the expression of S1P receptor subtypes in normal lung epithelial and lung carcinoma cells. h3bB4, the constantly expressed acidic ribosomal phosphoprotein P0 was used as the loading control.
To examine whether the increase in S1P3 expression is not only unique to A549 and LLC cells, we performed real-time PCR analysis on a panel of immortalized normal human bronchial epithelial cell lines (HBEC) (26,27) and lung adenocarcinoma cell lines (26). As seen in Fig. 1B, the S1P3 mRNA level remained relatively low amongst the three immortalized normal lung epithelial cell lines, HBEC3-KT, HBEC2-E, and HBEC2-KT. In contrast, S1P3 mRNA levels were elevated within the majority of the lung adenocarcinoma cell lines, especially in H1792 and H1793. A screening for mRNA levels of other S1P receptor subtypes within these cell lines revealed that the expression level of S1P1, 2, 4, 5 receptors remained similar to that of A549 and LLC with an elevation only in S1P3 (Fig. 1C).
Knockdown of S1P3 receptors diminishes clonogenesis and proliferation of lung adenocarcinoma cells
To determine whether S1P3 signaling has a functional role in the development of tumors, we knocked-down S1P3 receptors in H1793 human lung adenocarcinoma cells using shRNA-mediated gene silencing technique. As shown in Fig. 2A, transfection of si-S1P3 specifically knocked-down S1P3 receptors, whereas si-S1P3 transfection did not alter the expression of other S1P receptor subtypes. Knockdown of S1P3 receptors significantly reduced cell proliferation by ~30% at day 2 and ~40% at day 3 (p<0.05, t-test) (Fig. 2B). Moreover, ablation of S1P3 receptors also significantly inhibited H1793 colony formation in soft agar (Fig. 2C).
Figure 2.
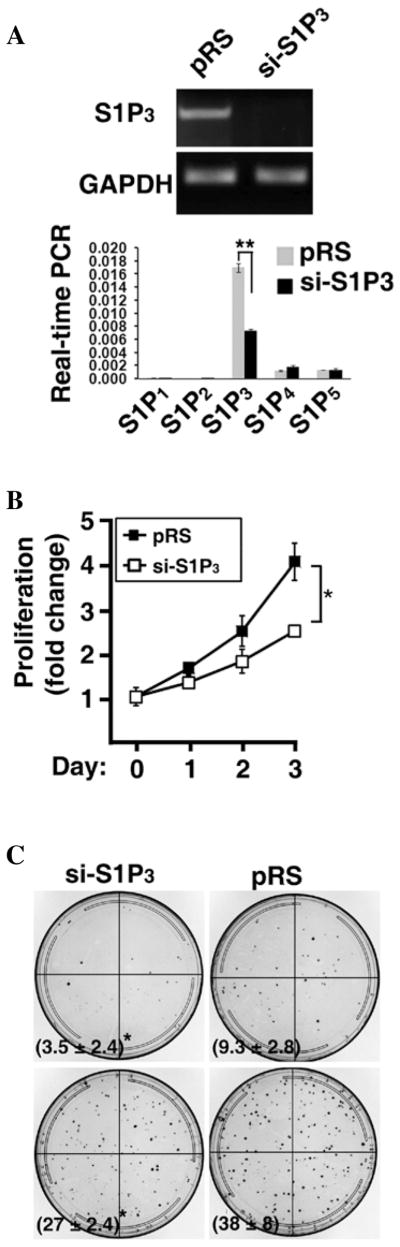
Knockdown of S1P3 diminishes lung adenocarcinoma cell proliferation and colony formation. (A) H1793 human lung adenocarcinoma cells were stably transfected with si-S1P3 or pRS control vector. The expression of S1P receptor subtypes was measured by semi-qunatitative RT-PCR (upper panel) and real-time PCR (lower panel). Note that S1P3 receptors were profoundly knocked-down in si-S1P3 stably transfected cells. The knockdown is specific since si-S1P3 transfection had no effect on the expression of GAPDH (upper panel) or S1P1, 2, 4, 5 receptors (lower panel). Real-time PCR data (mean ± SD of triplicate determinations. **p<0.01, t-test) are shown by 2−ΔCt values (ΔCt = Ct of S1P receptor - Ct of GAPDH) to represent the relative abundance of S1P receptor subtypes in H1793 cells. (B) Cell proliferation in control vector (pRS) and si-S1P3 transfected H1793 cells. Data represent mean ± SD of four determinants. The knockdown of S1P3 receptors markedly reduced cell proliferation. *p<0.05, t-test. (C) Colony formation assay. 100 (upper panels) and 500 (lower panels) cells were seeded for clonogenic assay. Numbers in parenthesis are mean ± SD of colony numbers (n=4; *p<0.05, t-test).
S1P-S1P3 signaling up-regulates EGFR in lung adenocarcinoma cells
To investigate the mechanism of the S1P3-mediated proliferation and clonogenesis in lung adenocarcinoma cells, we compared the expression of genes between HBEC2-KT lung epithelial cells and H1793 lung carcinoma cells, which had the highest expression of S1P3 receptors (Fig. 1B), in the presence or absence of S1P stimulation. The genes examined are known to be involved in inflammation, tumorigenesis, adhesion, proliferation, vascular development, and extracellular matrix modifications; normal cellular functions that are generally dysregulated within cancer. In our initial screening through RT-PCR (data not shown) and qPCR (Fig. 3A), we observed that there was a significant increase of epidermal growth factor receptor (EGFR) expression (~3.5 fold; p<0.01, t-test) in H1793 cells following S1P treatment for 4 h. In contrast, there is no marked alteration of EGFR mRNA in HBEC2-KT cell following S1P treatment (Fig. 3A). Next, we performed nuclear run-off analysis (28) to examine whether the S1P-induced EGFR up-regulation is controlled at the transcriptional level. As shown in Fig. 3B, nuclei isolated from S1P-stimulated H1793 cells were able to de novo synthesize the EGFR mRNA, whereas the newly synthesized EGFR mRNA was undetected in nuclei isolated from control serum-starved H1793 cells. This result suggests that S1P treatment transcriptionally activates EGFR expression.
Figure 3.
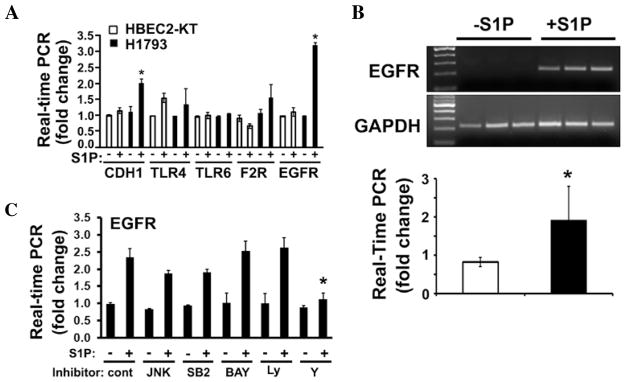
S1P transcriptionally activates EGFR expression via ROCK pathway in lung adenocarcinoma cells. (A) HBEC2-KT and H1793 cells were stimulated with or without S1P (300 nM) for 4 h. The expression of indicated genes was measured by real-time PCR. Note that S1P stimulation significantly induced EGFR in H1793 cells, whereas S1P was unable to induce EGFR expression in HBEC2-KT cells. Data are mean ± SD (n=3). CDH1, E-cadherin; TLR, toll-like receptor; F2R, thrombin receptor. *p<0.01 (t-test). (B) Nuclear run-off analysis indicates that S1P activates EGFR transcription. Nuclei were isolated from H1793 with or without S1P treatment (300 nM) for 4 h. Nuclear run-off assays were performed as we previously described (28). Upper panel, RT-PCR; lower panel, real-time PCR (mean ± SD, n=3). *p<0.05 (t-test). (C) H1793 were pre-treated for 1 h with or without pharmacological inhibitors of various signaling molecules, followed by stimulating with S1P (200 nM) for 4 h. Inhibitors used are: SP600125 for JNK, SB203580 for p38 kinase, Bay 11–7085 for NFκB, LY294002 for PI3-kinase, and Y-27632 for Rho kinase (ROCK). Data are mean ± SD of triplicate determinations. *p<0.01, t-test.
Subsequently, we employed pharmacological inhibitors to investigate the signaling pathways involved in the S1P-mediated EGFR up-regulation. Treatment with inhibitor of JNK, p38 kinase, NFκB, or PI3-kinase did not significantly abrogate the S1P-stimulated EGFR expression (Fig. 3C). In sharp contrast, Rho kinase (ROCK) inhibitor, Y-27632, diminished ~92% of the S1P-induced EGFR expression (p<0.01, t-test) (Fig. 3C), suggesting that the S1P-induced EGFR expression is mediated by the ROCK signaling pathway.
In addition, S1P treatment time- and dose-dependently induced EGFR expression in H1793 human lung adenocarcinoma cells (Fig. 4A and B). In contrast, S1P did not up-regulate EGFR expression in HBEC2-KT immortalized normal lung epithelial cells (Fig. 4A). Similarly, S1P also increased EGFR polypeptides in H1793 cells in a time-dependent manner (Fig. 4C). The S1P-induced increase in EGFR was completely abolished by VPC23019 (Fig. 4D), a competitive antagonist of S1P1 and S1P3 receptors (29,30). S1P1 is barely detected in H1793 cells (Fig. 2A), indicating that the effect of VPC23019 on inhibition of the S1P-induced EGFR expression is mediated by antagonizing S1P3 receptors. Indeed, this notion was further supported by the observation that specific knockdown of S1P3 receptors by shRNA-mediated gene-silencing completely inhibited the S1P-stimulated EGFR up-regulation in H1793 cells (Fig. 4E). Furthermore, the S1P-mediated EGFR up-regulation was observed in four other human lung adenocarcinoma cell lines: A549, H23, H1792, and H1650 (Fig. 4F). In contrast, S1P did not induce EGFR in HBEC3-KT, another immortalized normal bronchial epithelial cell line (Fig. 4F). Together, these data identify a novel signaling cascade in which S1P/S1P3 signaling transcriptionally up-regulates EGFR via ROCK pathway in lung adenocarcinoma cells.
Figure 4.

S1P-enhanced EGFR expression is mediated by S1P3 receptors. (A) H1793 and HBEC2-KT cells were treated with S1P (300 nM) for various times. The expression of EGFR was quantitated by real-time PCR analysis. Note that S1P induces EGFR expression in a time-dependent manner in H1793 cells, whereas S1P was unable to enhance EGFR in HBEC2-KT cells. Data are mean ± SD of triplicate determinations. *p<0.05, vs. control untreated cells, t-test. (B) H1793 cells were treated with indicated concentrations of S1P for 4 h. Note that S1P dose-dependently induced EGFR expression. *p<0.05, vs. control untreated cells, t-test (n=3). (C) H1793 cells were treated with S1P (300 nM) for indicated times. EGFR polypeptides were detected by Western blotting with anti-EGFR. The nitrocellulose membrane was reprobed with β-actin antibody to show the loading control. Lower panel, the intensity of immunoreactive EGFR band was quantified by a densitometer, normalized to β-actin, and represent as fold increase (n=4). *p<0.05, vs. control untreated cells, t-test. (D) Treatment of VPC23019 inhibits S1P-induced EGFR up-regulation. H1793 were stimulated with or without S1P (300 nM), in the presence or absence of VPC23019 (5 μM) for 4 h. *p<0.05 (t-test, n=3). (E) Knockdown of S1P3 abolishes S1P-induced EGFR expression. H1793 cells were stably transfected with si-S1P3 or pRS control vector as described in the Materials and methods section. Following S1P stimulation (300 nM, 4 h), the expression of EGFR (upper panel) and S1P3 (lower panel) was measured by real-time PCR. *p<0.05, vs. untreated cells, t-test (n=3). (F) S1P induced EGFR expression in four other lung carcinoma cell lines; however, S1P was unable to induce EGFR in the normal HBEC3-KT lung epithelial cells. *p<0.01 (t-test, n=3).
S1P potentiates EGF effects on proliferation, clonogenesis, and invasion of lung adenocarcinoma cells
We next determined the functional consequences of the S1P3-mediated EGFR up-regulation in lung adenocarcinoma cells. Treatment of S1P (0.1–1 μM) or EGF (0.05–0.5 ng/ml) alone had no effect on H1793 cell proliferation (Fig. 5A). However, co-treatment of H1793 cells with S1P (0.1 μM) and EGF (0.05 ng/ml) significantly stimulated the proliferation (1.9±0.3-fold increase, p<0.01, t-test). The fact that co-treatment of S1P and EGF significantly stimulates proliferation was also observed in A549 (Fig. 5B) and H1650 lung adenocarcinoma cells (data not shown). Similarly, there was no marked increase in colony numbers when H1793 cells were treated with EGF alone (0–10 ng/ml) for 14 days in soft agar (Fig. 5C). However, S1P dose-dependently enhanced the clonogenic capability of H1793 cells. Importantly, there are significantly more colonies in H1793 cells co-treated with S1P and EGF (10 ng/ml), compared to cells treated with S1P alone (Fig. 5C).
Figure 5.

S1P treatment potentiates EGF on proliferation, anchorage-independent growth, and invasion of lung adenocarcinoma cells. H1793 (A) and A549 (B) (8,000 cells in 200 μl) were resuspended in plain RPMI medium containing indicated concentrations of S1P and/or EGF for three days. Cell proliferation was measured as described in the Materials and methods section. Note that neither S1P nor EGF stimulated cell proliferation in this concentration range, whereas S1P plus EGF significantly stimulated cell proliferation (*p<0.01, t-test). Data are mean ± SD (n=3) of a representative experiment, which were repeated three times with similar results. (C) Clonogenic assays were performed as described in Materials and methods section. Note that treatment of S1P and EGF together significantly induced colony formation compared to treatment of S1P alone (n=3; *p<0.05, t-test). Cell invasion was measured in H1793 (D) and A549 (E) cells treated with indicated concentrations of S1P and/or EGF. Note that EGF-induced cell invasion was greatly enhanced in the presence of S1P (n=6; *p<0.05, t-test). (F) Invasive capability of H1793 cells was measured in the indicated combinations of treatments. Note that Cay10444 (5 μM) completely inhibited, whereas gefitinib (0.5 μM) reduced approximately 40% of the S1P (100 nM) and EGF (0.05 ng/ml)-induced H1793 invasion. Data are mean ± SD (n=3) of a representative experiment, which was repeated two times with similar results.
In contrast to the lack of effect of EGF alone on H1793 cell proliferation and clonogenesis, EGF dose-dependently increased the invasiveness of H1793 cells (Fig. 5D). However, S1P significantly enhanced EGF-stimulated cell invasion (Fig. 5D). For example, although 0.05 ng/ml of EGF did not significantly enhance the invasiveness of H1793 (1.3±0.1 vs. 1.0±0.1, EGF vs. control, p=0.18, t-test, n=6), co-treatment of H793 cells with S1P (100 nM) and EGF (0.05 ng/ml) significantly enhanced invasiveness (2.6±0.1 vs. 2.1±0.1, S1P+EGF vs. S1P; p<0.05, t-test, n=6). Also, co-treatment of S1P (100 nM) and EGF (0.05 ng/ml) significantly induced cell invasion in A549 (Fig. 5E) and H1650 lung carcinoma cells (data not shown), whereas S1P (100 nM) and EGF (0.05 ng/ml) had no effect (Fig. 5E). Furthermore, treatment of S1P significantly enhanced EGF (0.05–1 ng/ml) effects on A549 invasion (Fig. 5E). These data support the notion that S1P3 signaling transcriptionally up-regulates EGFR expression, and thus enhances the effects of EGF on cell proliferation, clonogenesis, and invasiveness.
Next, we utilized pharmacological antagonist/inhibitors to further examine the role of the S1P3-EGFR axis in H1793 invasiveness. As shown in Fig. 5F, the stimulation of H1793 cell invasion by co-treatment with S1P (100 nM) and EGF (0.05 ng/ml) was completely inhibited by Cay10444, a S1P3 specific antagonist (31–33), or by Cay10444 plus gefitinib, an EGFR inhibitor. However, there was only an approximate 40% reduction of S1P and EGF stimulated invasion when H1793 cells were treated with getfitnib alone (Fig. 5F). This result suggests that S1P3 signaling activates both EGFR-dependent and EGFR-independent pathways, and both of these pathways are required for H1793 invasiveness. This observation further supports our notion that EGFR activation is downstream of the S1P3 signaling axis. Collectively, these data indicate that S1P3-EGFR signaling plays an important role in the proliferation, anchorage-independent growth, and invasion of lung adenocarcinoma cells.
Discussion
In this study, we found that S1P3 receptors are markedly increased in a panel of human lung adenocarcinoma cells and a mouse lung tumor cell line (LLC) comparing to the normal (SAEC, Fig. 1A) or immortalized (Fig. 1B) human lung epithelial cell lines. A recent study suggests that there is no general expression pattern of S1P receptors among colon, breast, melanoma, and lung tumors, based on screening representative cultured tumor cell lines (34). In contrast, our observation implies that increased S1P3 expression may be pathophysiologically and functionally relevant in association with lung tumors. Importantly, specific inhibition of S1P3 axis by shRNA-mediated gene knock-down technique reduced tumor cell proliferation and clonogenesis of human lung adenocarcinoma cells. These data suggest that the S1P3-mediated signaling may play an important role in the tumorigenesis of lung carcinomas.
Studies of sphingolipid singaling in tumor progression and prevention have mainly focused on the ceramide-sphingosine-sphingosine-1-phosphate rheostat and sphingosine kinases (35). Ceramide and sphingosine induce cell apopotosis whereas S1P promotes cell proliferation and survival. Increased sphingosine kinase 1 activity with elevated S1P production has been observed in several types of cancer cells (20–23). However, mounting evidence suggests that the function of sphingosine kinase-S1P in cancer is complex and is influenced by cell type, receptor expression, subcellular localization of sphingosine kinase-S1P and environmental context. The observations made in this study further advance our knowledge of the contribution of receptor levels to cancer cell biology and indicate that S1P3 receptor-mediated signaling may contribute to lung carcinogenesis.
Furthermore, we found that S1P significantly induced EGFR expression in lung adenocarcinoma cells, but not in normal lung epithelial cells. The S1P-mediated EGFR up-regulation was observed both at the mRNA and protein levels and was demonstrated to be specifically mediated by S1P3 receptors. Amplification or mutation of EGFR contributes to lung tumorigenesis (36,37). The observed increase in EGFR mediated by S1P3 receptors thus provides a mechanistic insight of the role of S1P3 signaling in lung tumors. Evidence for cross-transactivation between S1P receptor subtypes and EGFR has been reported. For example, S1P, via S1P2 receptors, induces the secretion of EGF which subsequently activated EGFR in gastric cancer cells (38). Also, S1P, through S1P3, transactivates EGFR in MCF-7 breast cancer cells (39). However, these studies demonstrated an autocrine/paracrine mechanism, in which S1P transactivates EGFR via increasing EGF secretion. In contrast, our study showed that S1P3 signaling directly transcriptionally up-regulates EGFR expression via ROCK pathway. Although a previous report showed that S1P induced EGFR expression in smooth muscle cells (40), the involved receptors and detailed mechanisms were not characterized.
In summary, we demonstrated that S1P3 receptors are significantly up-regulated in a subset of lung adenocarcinoma cells. Several lines of evidence were presented to support a functional role of the S1P-mediated EGFR up-regulation in the carcinogenesis of lung tumor. Individually, S1P (ranging from 0.1–1 μM) and EGF (ranging from 0.05–0.5 ng/ml) had no effect on the proliferation of H1793 lung carcinoma cells (Fig. 5A). However, co-treatment with EGF plus S1P significantly stimulated H1793 cell proliferation. Similar results were observed in A549 and H1650 human lung carcinoma cells. These observations strongly argue that the observed EGFR increase in the presence of S1P treatment is functional, thus enabling EGF to stimulate cell proliferation. This notion is further supported by the fact that EGF stimulated clonogenesis and the invasive ability of lung cancer cells was significantly enhanced by S1P. Lastly, the S1P3-selective antagonist completely inhibited, whereas the EGFR inhibitor only partially inhibited, the S1P and EGF stimulated cancer cell invasion (Fig. 5F). These results suggest that S1P3 receptors function as an upstream activator of EGFR activation, i.e., by increasing EGFR expression. Collectively, these data indicate that the S1P3-EGFR signaling axis is important in the proliferation, colony formation, and invasion of lung cancer cells. Therefore, targeting the S1P3 receptor might provide a novel therapeutic utility in the treatment of cancer.
Acknowledgments
This study is supported by NIH grant R01HL071071 to M.L., and supported in part by grants from Joan’s Legacy Lung Cancer Foundation/Lungevity and the Kentucky Lung Cancer Research Program to C.M.K.
Abbreviations
- S1P
sphingosine-1-phosphate
- S1P1–5
sphingosine-1-phosphate receptor subtype 1–5
- GPCR
G protein coupled receptor
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- LLC
Lewis lung carcinoma cells
- ROCK
Rho kinase
References
- 1.Hla T, Lee MJ, Ancellin N, et al. Sphingosine-1-phosphate: extracellular mediator or intracellular second messenger? Biochem Pharmacol. 1999;58:201–207. doi: 10.1016/s0006-2952(99)00086-6. [DOI] [PubMed] [Google Scholar]
- 2.Igarashi Y, Yatomi Y. Sphingosine 1-phosphate is a blood constituent released from activated platelets, possibly playing a variety of physiological and pathophysiological roles. Acta Biochim Pol. 1998;45:299–309. [PubMed] [Google Scholar]
- 3.Moolenaar WH. Bioactive lysophospholipids and their G protein-coupled receptors. Exp Cell Res. 1999;253:230–238. doi: 10.1006/excr.1999.4702. [DOI] [PubMed] [Google Scholar]
- 4.Spiegel S. Sphingosine 1-phosphate: a prototype of a new class of second messengers. J Leukoc Biol. 1999;65:341–344. doi: 10.1002/jlb.65.3.341. [DOI] [PubMed] [Google Scholar]
- 5.Lee MJ, Van Brocklyn JR, Thangada S, et al. Sphingosine-1-phosphate as a ligand for the G protein-coupled receptor EDG-1. Science. 1998;279:1552–1555. doi: 10.1126/science.279.5356.1552. [DOI] [PubMed] [Google Scholar]
- 6.An S, Goetzl EJ, Lee H. Signaling mechanisms and molecular characteristics of G protein-coupled receptors for lysophosphatidic acid and sphingosine 1-phosphate. J Cell Biochem. 1998;(Suppl 30–31):S147–S157. [PubMed] [Google Scholar]
- 7.Zondag GC, Postma FR, Etten IV, Verlaan I, Moolenaar WH. Sphingosine 1-phosphate signalling through the G-protein-coupled receptor Edg-1. Biochem J. 1998;330:605–609. doi: 10.1042/bj3300605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chun J, Goetzl EJ, Hla T, et al. International Union of Pharmacology. XXXIV. Lysophospholipid receptor nomenclature. Pharmacol Rev. 2002;54:265–269. doi: 10.1124/pr.54.2.265. [DOI] [PubMed] [Google Scholar]
- 9.Ancellin N, Hla T. Differential pharmacological properties and signal transduction of the sphingosine 1-phosphate receptors EDG-1, EDG-3, and EDG-5. J Biol Chem. 1999;274:18997–19002. doi: 10.1074/jbc.274.27.18997. [DOI] [PubMed] [Google Scholar]
- 10.Im DS, Heise CE, Ancellin N, et al. Characterization of a novel sphingosine 1-phosphate receptor, Edg-8. J Biol Chem. 2000;275:14281–14286. doi: 10.1074/jbc.275.19.14281. [DOI] [PubMed] [Google Scholar]
- 11.Windh RT, Lee MJ, Hla T, An S, Barr AJ, Manning DR. Differential coupling of the sphingosine 1-phosphate receptors Edg-1, Edg-3, and H218/Edg-5 to the G(i), G(q), and G(12) families of heterotrimeric G proteins. J Biol Chem. 1999;274:27351–27358. doi: 10.1074/jbc.274.39.27351. [DOI] [PubMed] [Google Scholar]
- 12.Lee MJ, Thangada S, Claffey KP, et al. Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell. 1999;99:301–312. doi: 10.1016/s0092-8674(00)81661-x. [DOI] [PubMed] [Google Scholar]
- 13.Lee MJ, Thangada S, Paik JH, et al. Akt-mediated phosphorylation of the G protein-coupled receptor EDG-1 is required for endothelial cell chemotaxis. Mol Cell. 2001;8:693–704. doi: 10.1016/s1097-2765(01)00324-0. [DOI] [PubMed] [Google Scholar]
- 14.Lee JF, Gordon S, Estrada R, et al. Balance of S1P1 and S1P2 signaling regulates peripheral microvascular permeability in rat cremaster muscle vasculature. Am J Physiol Heart Circ Physiol. 2009;296:H33–H42. doi: 10.1152/ajpheart.00097.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Estrada R, Zeng Q, Lu H, et al. Up-regulating sphingosine 1-phosphate receptor-2 signaling impairs chemotactic, wound-healing, and morphogenetic responses in senescent endothelial cells. J Biol Chem. 2008;283:30363–30375. doi: 10.1074/jbc.M804392200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pitson SM, Xia P, Leclercq TM, et al. Phosphorylation-dependent translocation of sphingosine kinase to the plasma membrane drives its oncogenic signalling. J Exp Med. 2005;201:49–54. doi: 10.1084/jem.20040559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pchejetski D, Bohler T, Brizuela L, et al. FTY720 (fingolimod) sensitizes prostate cancer cells to radiotherapy by inhibition of sphingosine kinase-1. Cancer Res. 2010;70:8651–8661. doi: 10.1158/0008-5472.CAN-10-1388. [DOI] [PubMed] [Google Scholar]
- 18.Illuzzi G, Bernacchioni C, Aureli M, et al. Sphingosine kinase mediates resistance to the synthetic retinoid N-(4-hydroxyphenyl) retinamide in human ovarian cancer cells. J Biol Chem. 2010;285:18594–18602. doi: 10.1074/jbc.M109.072801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pitman MR, Pitson SM. Inhibitors of the sphingosine kinase pathway as potential therapeutics. Curr Cancer Drug Targets. 2010;10:354–367. doi: 10.2174/156800910791208599. [DOI] [PubMed] [Google Scholar]
- 20.Watson C, Long JS, Orange C, et al. High expression of sphingosine 1-phosphate receptors, S1P1 and S1P3, sphingosine kinase 1, and extracellular signal-regulated kinase-1/2 is associated with development of tamoxifen resistance in estrogen receptor-positive breast cancer patients. Am J Pathol. 2010;177:2205–2215. doi: 10.2353/ajpath.2010.100220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu G, Zheng H, Zhang Z, et al. Overexpression of sphingosine kinase 1 is associated with salivary gland carcinoma progression and might be a novel predictive marker for adjuvant therapy. BMC Cancer. 2010;10:495. doi: 10.1186/1471-2407-10-495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shirai K, Kaneshiro T, Wada M, et al. A role of sphingosine kinase 1 in head & neck carcinogenesis. Cancer Prev Res (Phila) 2011;4:454–462. doi: 10.1158/1940-6207.CAPR-10-0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pyne NJ, Pyne S. Sphingosine 1-phosphate and cancer. Nat Rev Cancer. 2010;10:489–503. doi: 10.1038/nrc2875. [DOI] [PubMed] [Google Scholar]
- 24.Feng H, Stachura DL, White RM, et al. T-Lymphoblastic lymphoma cells express high levels of BCL2, S1P1, and ICAM1, leading to a blockade of tumor cell intravasation. Cancer Cell. 2010;18:353–366. doi: 10.1016/j.ccr.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee H, Deng J, Kujawski M, et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat Med. 2010;16:1421–1428. doi: 10.1038/nm.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ivanova MM, Mazhawidza W, Dougherty SM, Minna JD, Klinge CM. Activity and intracellular location of estrogen receptors [alpha] and [beta] in human bronchial epithelial cells. Mol Cell Endocrinol. 2009;305:12–21. doi: 10.1016/j.mce.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramirez RD, Sheridan S, Girard L, et al. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer Res. 2004;64:9027–9034. doi: 10.1158/0008-5472.CAN-04-3703. [DOI] [PubMed] [Google Scholar]
- 28.Estrada R, Wang L, Jala VR, et al. Ligand-induced nuclear translocation of S1P(1) receptors mediates Cyr61 and CTGF transcription in endothelial cells. Histochem Cell Biol. 2009;131:239–249. doi: 10.1007/s00418-008-0521-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davis MD, Clemens JJ, Macdonald TL, Lynch KR. Sphingosine 1-phosphate analogs as receptor antagonists. J Biol Chem. 2005;280:9833–9841. doi: 10.1074/jbc.M412356200. [DOI] [PubMed] [Google Scholar]
- 30.Foss FW, Jr, Snyder AH, Davis MD, et al. Synthesis and biological evaluation of [gamma]-aminophosphonates as potent, subtype-selective sphingosine 1-phosphate receptor agonists and antagonists. Bioorg Med Chem. 2007;15:663–677. doi: 10.1016/j.bmc.2006.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murakami M, Ito H, Hagiwara K, et al. Sphingosine kinase 1/S1P pathway involvement in the GDNF-induced GAP43 transcription. J Cell Biochem. 2011;112:3449–3458. doi: 10.1002/jcb.23275. [DOI] [PubMed] [Google Scholar]
- 32.Tao R, Hoover HE, Honbo N, et al. High-density lipoprotein determines adult mouse cardiomyocyte fate after hypoxia-reoxy-genation through lipoprotein-associated sphingosine 1-phosphate. Am J Physiol Heart Circ Physiol. 2010;298:H1022–H1028. doi: 10.1152/ajpheart.00902.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tao R, Hoover HE, Zhang J, Honbo N, Alano CC, Karliner JS. Cardiomyocyte S1P1 receptor-mediated extracellular signal-related kinase signaling and desensitization. J Cardiovasc Pharmacol. 2009;53:486–494. doi: 10.1097/FJC.0b013e3181a7b58a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muller R, Berliner C, Leptin J, et al. Expression of sphingosine-1-phosphate receptors and lysophosphatidic acid receptors on cultured and xenografted human colon, breast, melanoma, and lung tumor cells. Tumour Biol. 2010;31:341–349. doi: 10.1007/s13277-010-0043-7. [DOI] [PubMed] [Google Scholar]
- 35.Cuvillier O, Pirianov G, Kleuser B, et al. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature. 1996;381:800–803. doi: 10.1038/381800a0. [DOI] [PubMed] [Google Scholar]
- 36.Yamamoto H, Toyooka S, Mitsudomi T. Impact of EGFR mutation analysis in non-small cell lung cancer. Lung Cancer. 2009;63:315–321. doi: 10.1016/j.lungcan.2008.06.021. [DOI] [PubMed] [Google Scholar]
- 37.Gazdar AF. Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene. 2009;28 (Suppl 1):S24–S31. doi: 10.1038/onc.2009.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shida D, Kitayama J, Yamaguchi H, et al. Sphingosine 1-phosphate transactivates c-Met as well as epidermal growth factor receptor (EGFR) in human gastric cancer cells. FEBS Lett. 2004;577:333–338. doi: 10.1016/j.febslet.2004.10.024. [DOI] [PubMed] [Google Scholar]
- 39.Martin JL, Lin MZ, McGowan EM, Baxter RC. Potentiation of growth factor signaling by insulin-like growth factor-binding protein-3 in breast epithelial cells requires sphingosine kinase activity. J Biol Chem. 2009;284:25542–25552. doi: 10.1074/jbc.M109.007120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hsieh HL, Sun CC, Wu CB, et al. Sphingosine 1-phosphate induces EGFR expression via Akt/NF-kappaB and ERK/AP-1 pathways in rat vascular smooth muscle cells. J Cell Biochem. 2008;103:1732–1746. doi: 10.1002/jcb.21563. [DOI] [PubMed] [Google Scholar]