

Abstract
Maleimide is a stable and easy-to-handle moiety that rapidly and covalently conjugates thiol groups of cysteine residues in proteins or peptides. Herein, we use maleimide to modify the surface of liposomes in order to obtain an advanced drug delivery system. Employing a small amount (0.3 mol%) of maleimide-polyethylene glycol (PEG) to modify the surface of the liposomes M-GGLG-liposomes, composed of 1,5-dihexadecyl N,N-diglutamyl-lysyl-L-glutamate (GGLG)/cholesterol/poly(ethylene glycol) 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (PEG5000-DSPE)/maleimide-PEG5000-Glu2C18 at a molar ratio of 5:5:0.03:0.03, drug delivery efficiency was remarkably improved both in vitro and in vivo compared to unmodified liposomes (GGLG-liposomes, composed of GGLG/cholesterol/PEG5000-DSPE/PEG5000-Glu2C18 at a molar ratio of 5:5:0.03:0.03). Moreover, this modification did not elicit any detectable increase in cytotoxicity. The maleimide-modification did not alter the physical characteristics of the liposomes such as size, zeta potential, pH sensitivity, dispersibility and drug encapsulation efficiency. However, M-GGLG-liposomes were more rapidly (≥2-fold) internalized into HeLa, HCC1954, and MDA-MB-468 cells compared to GGLG-liposomes. In vivo, M-GGLG-liposomes encapsulating doxorubicin (M-GGLG-DOX-liposomes) also showed a more potent antitumor effect than GGLG-DOX-liposomes and the widely used 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC)-DOX-liposomes after two subcutaneous injections around breast cancer tissue in mice. The biodistribution of liposomes in this model was observed using an in vivo imaging system, which showed that M-GGLG-liposomes were present for significantly longer at the injection site compared to GGLG-liposomes. The outstanding biological functions of the maleimide-modified liposomes as a novel drug delivery system make them ideally suited to a wide range of applications.
Keywords: maleimide, liposome, thiol-reactive, pH responsive, drug delivery system
Introduction
Liposomal drug delivery systems have been extensively investigated. Due to their nanoscale size range (usually 50 to 300 nm in diameter), liposomes can extravasate from tumor blood vessels and accumulate in tumors, infarcted, or inflamed regions instead of normal tissues via the enhanced permeability and retention (EPR) effect.1 However, nanoparticles such as liposomes can quickly bind to opsonin proteins in the blood serum which leads to rapid clearance from the mononuclear phagocytic system (MPS) after intravenous (iv) injection.2 Therefore, a new generation of so called “long-circulating liposomes” has been developed.3 These “long-circulating liposomes” include protective polymer chains (eg, polyethylene glycol [PEG]) that modify the outer surface of the liposomes to shield them from opsonization, and subsequently slow down their clearance by the MPS and increase the blood circulation time. Unfortunately, this PEGylation effect is limited in clinical applications. Usually only about 5% of administrated nanoparticles remain in the blood circulatory system 12 hours after iv injection; the majority of nanoparticles being eliminated within a few hours by the liver, spleen, and other organs.4 Although there is no doubt that PEGlyation is beneficial in most cases, the mechanism is still not fully understood. However, for most nanoparticles the subcutaneous (sc) environment is much less severe compared with that of the blood circulatory system. Indeed, in the sc region the distribution of large liposomes (>100 nm) is mainly localized, followed by destabilization at regional lymph nodes.5 Thus, the relatively long retention of liposomes at the injection site can reduce side effects of the encapsulated drug caused by exposure to other organs.
Over the past few decades, stimuli-sensitive liposomes have attracted a great deal of attention due to the requirement for controllable drug delivery and release systems.6 For example, liposomes sensitive to pH,7,8 temperature,9 magnetic fields,10 redox potential,11,12 light,13,14 ultrasound,15,16 or shear.17 In our previous study, we developed a pH-responsive liposome, which uses an amino acid-based zwitterionic liposome containing 1,5-dihexadecyl N,N-diglutamyl-lysyl-L-glutamate (GGLG) as the main lipid.7 This GGLG-liposome exhibits enhanced drug release from endosomes compared to conventional 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC)-liposomes. As such, pH responsive GGLG-liposomes display enhanced potency during anticancer therapy. However, reduced efficiency of cellular uptake has hindered the effective application of most nanoparticle-based carrier systems including liposomes. Hence, there is a need for simple and efficient design methods for improved liposome delivery systems.
Recently, cell surface thiols are being considered as a novel target for enhanced cellular uptake of a series of substrates that have thiol-reactive moieties such as peptides, oligonucleotides, nanoparticles, polymers, and some dyes.18,19 One of the key promoters may include protein disulfide isomerase (PDI), a thiol-disulfide oxidoreductase, which plays an important role in maintaining the redox potential of cells, and catalyzes the disulfide and thiol interchange between a substrate and a cell surface.18,20 Although the cellular uptake mechanism of thiol-reactive substrates is still unclear, the importance of cell surface thiols in cellular internalization is undeniable. Indeed, we believe the cell surface thiols could become a powerful target for enhanced cellular uptake of nanoparticles.
Maleimide, a thiol-reactive moiety, was utilized in lipoplex preparations of N-(4-(p-maleimidophenyl) butyryl) dipalmitoylphosphatidylethanolamine (MPB-DPPE)/lipospermine/deoxyribonucleic acid/lipospermine/deoxyribonucleic acid (DNA) for enhanced gene delivery by Kichler et al in 1995.21 However, further development of this system has not been reported. In the original paper, a large amount of maleimide was applied for enhanced cellular uptake, but more detailed information, such as the physical properties, biocompatibility, and in vivo applications of the system were not given. Nonetheless, the results had already revealed that maleimide-modification could target cell surface thiols and the cellular internalization of particles was greatly increased. In order to develop biocompatible liposomes with improved cellular uptake and release of the carrier drug, we designed pH responsive “smart” liposomes modified with a small amount of maleimide at the surface. Here, we have evaluated the biocompatibility, cellular uptake, and drug delivery efficiency of these liposomes in vitro. Biodistribution and tumor growth inhibition by sc injection of doxorubicin (DOX)-encapsulating liposomes were also investigated in vivo. Our fideings reveal that this novel method for enhanced cellular uptake of nanoparticles is highly efficient and therefore has great potential as an advanced drug delivery system.
Materials and methods
Materials
The following reagents were purchased for use in this study: poly(ethylene glycol) (N-hydroxysuccinimide 5-pentanoate) ether N′-(3-maleimidopropionyl)aminoethane (maleimide-PEG5000-carboxyl-NHS) from NOF Corporation (Tokyo, Japan); DPPC, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE), and cholesterol from Nippon Fine Chemicals (Osaka, Japan); doxorubicin hydrochloride from Sigma-Aldrich (St Louis, MO, USA); 1,1′-dioctadecyltetramethyl indotricarbocyanine iodide (XenoLight DiR) from Caliper Life Sciences (Hopkinton, MA, USA); and 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) (ammonium salt) (NBD-PE) lipid from Avanti Polar Lipids Inc (Alabaster, AL, USA). 1,5-dioctadecyl L-glutamate (Glu2C18), PEG5000-Glu2C18, PEG5000-DSPE, GGLG and maleimide-PEG5000-Glu2C18 (Figure 1) were synthesized in our laboratory using previously published methods.7,22
Figure 1.
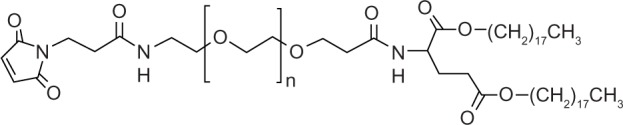
Structure of Mal-PEG5000-Glu2C18.
Abbreviations: Mal, maleimide moiety; PEG, polyethylene glycol; Glu2C18, 1,5-dioctadecyl L-glutamate.
Liposome preparation
Maleimide (M)-GGLG-liposomes were prepared from GGLG, cholesterol, PEG5000-DSPE, and maleimide-PEG5000-Glu2C18 at a molar ratio of 5:5:0.03:0.03, stirring at room temperature (rt) for 4 hours in a citrate solution (pH 2.2) according to the referenced method.7 For preparation of GGLG-liposomes, the lipid PEG5000-Glu2C18 was used instead of maleimide-PEG5000-Glu2C18 with the molar ratio of 5:5:0.03:0.03. After hydration and extrusion, the liposome dispersion was subject to ultracentrifugation at 49,000 rpm for 30 minutes. The supernatant was then removed and the pellet was re-dispersed in Dulbecco’s phosphate buffered saline (DPBS; without magnesium and calcium) for further characterization. DOX, a potent anticancer drug, has a limited clinical application due to its severe side effects, such as myelosuppression, cardiotoxicity, and gastrointestinal toxicity23,24 We utilized the liposomes as a carrier of DOX to protect the drug from rapid metabolism and reduce its side effects by enhancing selectivity for tumor tissues. For the preparation of DOX-encapsulating liposomes, a liposome suspension ([lipid] = 3 mg/mL, 1 mL) and a DOX solution (300 μg/mL, 1 mL) were pre-heated at 60°C for 15 minutes and then mixed at 60°C for a further 15 minutes. The resulting mixed solution was allowed to cool to rt overnight and the DOX-encapsulated liposomes were separated from free DOX by gel filtration chromatography using a Sephadex G-25 column. The lipid concentration of the liposomes was calculated from the concentration of cholesterol using a Cholesterol kit from WAKO Pure Chemical Industries, Ltd (Osaka, Japan) according to the manufacturer’s instructions. The concentration of maleimide moiety was quantified by a reverse-Ellman’s test, which analyzes the concentration of residual thiols of glutathione after interaction with maleimide-liposomes. The concentration of DOX encapsulated in the liposomes was calculated from fluorescent measurements after solubilization of the DOX-liposomes in 0.5% Triton X-100 solution (excitation wavelength, 485 nm; emission wavelength, 590 nm).
Size of the liposomes
The dispersion of liposomes (2 μL) containing 3 mg/mL of total lipids was diluted in DPBS (1 mL). The mean particle diameter was measured in a disposable plastic cuvette using a dynamic light scattering spectrophotometer (N4 PLUS Submicro Particle Size Analyzer; Beckman-Coulter, Fullerton, FL, USA). All measurements were performed in triplicate.
Cell culture
HeLa, HCC1954, and MDA-MB-468 HeLa cells (a human cervical carcinoma cell line), HCC1954 cells and MDA-MB-468 cells (human breast carcinoma cell lines) were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. The cells were grown at 37°C in an atmosphere containing 5% CO2 and passaged by trypsinization with 0.1% trypsin-ethylenediaminetetraacetic acid (EDTA).
Cellular uptake of liposomes
To quantify the concentration of liposomes internalized in the cells, lipids (1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[7-nitro-2-1,3-benzoxadiazol-4-yl] [ammonium salt]; lipids NBD-PE) were homogeneously inserted into liposomes during the preparation of the lipid mixture using a molar ratio of 2%. A standard fluorescence curve for each type of liposome was made to calculate the concentration of liposomes.
Cells were seeded in 24-well cell culture plates (5 × 104 cells/well) and incubated in an atmosphere of 5% CO2 at 37°C for 24 hours. Then the medium in the cell culture dish was exchanged with 500 μL of fresh DMEM (pH 8.3, 7.8 and 6.6, respectively) containing 72 μg/mL NBD-liposomes in the presence of 10% FBS. Upon incubation at 37°C for 2 hours, the cells were washed twice with DPBS and 350 μL of 0.5% Triton X-100 buffer was added. The amount of liposomes in the cells was fluorometrically determined from the lysate using a fluorescence spectrometer (Multi-Detection Microplate Reader, BioTek Instruments Inc, Winooski, VT, USA) with an excitation wavelength of 485 nm and an emission wavelength of 590 nm. A standard curve of fluorescent intensity versus concentration of each type of NBD-liposomes was made by monitoring the fluorescent intensity of standard NBD-liposome solutions at different concentrations (eg, 72, 144, 360, 720, and 1440 μg/L) which were quantified using a Cholesterol Kit (Wako Pure Chemical Industries Ltd Osaka, Japan). The protein concentration of the lysate was determined by a standard protein assay (660 nm Pierce Protein Assay; Pierce Biotechnology, Rockford IL, USA). The cellular uptake efficiency of the liposomes was expressed as lipid-μg per cellular protein-mg.
Cytotoxicity of DOX-liposomes and empty liposomes
HeLa, HCC1954, and MDA-MB-468 cells were seeded in 96-well cell culture plates (1 × 104 cells/well) and incubated in an atmosphere of 5% CO2 at 37°C for 24 hours. Then the medium in the cell culture dish was exchanged with 100 μL fresh DMEM containing the DOX-liposomes ([DOX] = 0.01, 0.05, 0.1, 0.5, 1.0, 5.0, 10.0 μg/mL) or empty liposomes in the presence of 10% FBS. After incubation at 37°C for 24 hours, the cells were washed twice with DPBS and 100 μL of fresh DMEM was then added. The cells were then incubated for a further 24 hours. Cell viability was tested using a WST-8 assay kit (Cell Counting Kit-8; Dojindo Molecular Technologies, Inc, Osaka, Japan), according to the manufacturer’s instructions. After adding 10 μL WST-8 to each well followed by a 2-hour incubation, the absorbance of each sample was measured at a wavelength of 450 nm using a microplate reader (Benchmark Plus, Bio-Rad Laboratories Inc, Hercules, CA, USA). The cell viability was calculated using the equation:
| (1) |
where Abs represents absorbance value and WST represents WST-8 solution.
Confocal laser scanning microscopic observation of cellular internalization of DOX-liposomes
HeLa cells (1 × 105 cells/well) were seeded in a 35-mm glass bottom cell culture dish and incubated in an atmosphere of 5% CO2 at 37°C for 24 hours. After removing the medium, fresh DMEM containing DOX-liposomes ([DOX] = 150 μg/mL) was added to the dish and incubated for 5 minutes. The cells were then washed three times with DPBS and observed under a confocal laser scanning microscope (FV1000; Olympus, Japan).
In vivo evaluation of tumor growth inhibition by DOX-liposomes
The antitumor effects of DOX-liposomes were evaluated using tumor-bearing mice. HCC1954 cells (5 × 105) in 50 μL DPBS were mixed with 50 μL basement membrane matrix (BD Biosciences, Matrigel™; Franklin Lakes, NJ, USA) on ice, and then subcutaneously injected into the flank of female BALB/c nu/nu mice (5 weeks old 17–19 g) purchased from Sankyo Labo Service Corp (Tokyo, Japan). The mice were fed and housed under standard conditions with free access to water and food. In order to distinguish the subtle differences of antitumor effects among various DOX-liposomes, the injection amount, times, and frequency were limited. After confirming that the tumor size had increased to 250 mm3, 2 mg DOX/kg was subcutaneously administered twice around the tumor tissue with 2-week intervals between treatments. DPPC-DOX-liposome was applied as a positive control and saline was used as a negative control. The tumor volume (V) was measured and calculated using the following equation:
| (2) |
where L and W indicated the long and short diameters of the tumor tissue, respectively. All the animal experiments were supervised and approved by the local ethical committee of Waseda University.
Live imaging of the biodistribution of liposomes
To visualize the biodistribution and measure the rate of removal of liposomes at the injection site, fluorescent liposomes were prepared by adding XenoLight DiR to the mixed lipids at a molar ratio of 1% of the lipid mixture. The fluorescent liposomes of GGLG or M-GGLG (16 μg) in a volume of 200 μL were injected subcutaneously across the longer diameter of the tumor. The biodistribution of liposomes was observed using an in vivo imaging system (IVIS, Caliper Life Sciences, Hopkinton, MA, USA). The proportion of unmetabolized liposomes at the injection site was expressed as the rate of residual fluorescence efficiency, calculated using the following equation:
| (3) |
where t represents the time post injection and t=0 represents the initial time point.
Statistical analysis
Two-way analysis of variance (ANOVA) was performed for the statistical analysis of the results of time dependence of cellular uptake of liposomes, cytotoxicity of DOX- and empty liposomes, and biodegradation of liposomes at injection site. The Student’s t-test was applied in the analysis of pH dependence of cellular uptake of liposomes and the tumor growth inhibition at the 60th day by sc injection of DOX-liposomes. A P-value of less than 0.05 was considered statistically significant.
Results
Preparation and characterization of liposomes
The size of liposomes is an important factor for their physical and biological properties, such as stability, cellular uptake pathway, and metabolism in vivo. Nanoparticles, with a mean diameter around 200 nm, are thought to have a similar mechanism of cellular uptake.25 As shown in Table 1, similar size distributions of M-GGLG- and GGLG-liposomes were observed, and the mean diameters were around 160–190 nm before and after encapsulation of DOX. Therefore, it was suggested that the particle size would not result in significant change in physical or biological functions of maleimide-modified liposomes. Moreover, M-GGLG-liposomes were as stable as the GGLG-liposomes during storage (ie, 4°C in refrigerator). Specifically, after 1-month of storage at 4°C, no aggregation of the liposomes was detected by measuring the size distribution (Supplementary Table S1). Furthermore, the drug encapsulation efficiency of M-GGLG-DOX-liposomes was as high as 95%, and the leakage ratio of DOX at 37°C 1-month after drug encapsulation was around 10%, which was slightly lower than that of GGLG-DOX-liposomes (Supplementary Figure S1). Thus, we judged the M-GGLG-DOX-liposomes to be sufficiently stable to conduct further in vitro and in vivo measurements.
Table 1.
Size and characteristics of empty liposomes and DOX-encapsulating liposomes
| Samples | Size (nm) | DOX/lipid (μg/mg) | Encapsulation efficiency (%) |
|---|---|---|---|
| GGLG-liposome | 167.5 ± 56.03 | – | – |
| GGLG-DOX-liposome | 175.9 ± 53.40 | 94.2 | 93.3 |
| M-GGLG-liposome | 178.6 ± 68.79 | – | – |
| M-GGLG-DOX-liposome | 188.6 ± 66.41 | 99.2 | 95.0 |
Notes: The composition of GGLG-liposome was GGLG/cholesterol/PEG5000-DSPE/PEG5000-Glu2C18 whereas that of M-GGLG-liposome was GGLG/cholesterol/PEG5000-DSPE/Mal-PEG5000-Glu2C18 at a molar ratio of 5:5:0.03:0.03 in DPBS at room temperature (n = 3). M-GGLG-liposomes showed similar size distribution, ratio of DOX-lipid and drug-encapsulation efficiency to GGLG-liposomes. The size of liposomes expresses the mean and distribution of diameter; DOX/lipid expresses the ratio of drug to total lipids in the composition of DOX-encapsulating liposomes; encapsulation efficiency evaluates the ability of drug loading in liposomes, which is expressed as the percentage of the loaded amount to the total amount of DOX.
Abbreviations: DOX, anticancer drug doxorubicin; DPBS, Dulbecco’s phosphate buffered saline; DSPE, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine; GGLG, pH sensitive lipid 1,5-dihexadecyl N,N-diglutamyl-lysyl-L-glutamate; Mal/M, maleimide moiety; PEG, polyethylene glycol.
As we reported previously, the GGLG-liposomes are pH sensitive owing to the presence of zwitterionic lipids on the liposomal surface.7 The amount of maleimide in the M-GGLG-liposomes was so low (0.3 mol%) that the zeta potential of M-GGLG-liposomes showed similar values to those of GGLG-liposomes at various pHs, and became zero at about pH 5.5 (Supplementary Figure S2). Thus, the M-GGLG-liposomes have the same pH sensitivity as GGLG-liposomes.
Cellular uptake efficiency of liposomes
The surface charge of nanoparticles is an important factor in terms of their cellular internalization. Positively charged nanoparticles have a tendency to attach themselves to the negatively charged cell membrane. For HCC1954 and MDA-MB-468 cells, GGLG- and M-GGLG-liposomes showed the highest cellular uptake efficiencies at pH 6.6 rather than pH 7.8 or 8.3 (Figure 2A). The surfaces of the M-GGLG and GGLG-liposomes were less negatively charged at pH 6.6 than at the higher pH values (Supplementary Figure S2). Thus, the liposomes were more likely to attach to the cell surface and be internalized by cells at the lower pH (eg, pH 6.6). It should be noted that the M-GGLG-liposomes showed significantly increased cellular uptake efficiencies over the GGLG-lipo-somes in all three cell lines and pH conditions tested.
Figure 2.

Cellular internalization of liposomes. (A) Cellular uptake efficiency of M-GGLG- and GGLG-liposomes (72 μg/mL) for 2 hour incubation in HeLa, HCC1954, and MDa-MB-468 cells at various pH levels. (B) Time dependence of cellular uptake efficiency of M-GGLG- and GGLG-liposomes (72 μg/mL) in HeLa cells at pH 7.8. (n = 4). Error bars represent standard error of the mean. *P < 0.05, **P < 0.01. (C) Confocal microscopic observation of rapid cellular uptake of M-GGLG-DOX- and GGLG-DOX-liposomes ([DOX] = 150 μg/mL) after 5 minutes incubation with HeLa cells. No morphological change of cells was confirmed after short-time incubation with M-GGLG-DOX- and GGLG-DOX-liposomes.
Notes: Both empty and DOX-encapsulating M-GGLG-liposomes showed significantly increased cellular uptake efficiency by comparison with that of GGLG-liposomes.
Abbreviations: DOX, anticancer drug doxorubicin; GGLG, pH sensitive lipid 1,5-dihexadecyl N,N-diglutamyl-lysyl-L-glutamate; M, maleimide moiety; min, minutes.
The internalization of liposomes occurred as soon as the liposomes were attached to the cell surface. For example, M-GGLG-liposomes exhibited a rapid cellular internalization within a short time of attaching to the cell surface (Figure 2C), which then continued for at least 8 hours (Figure 2B). By contrast, the cellular internalization of GGLG-liposomes was much lower, reaching a maximum after 2 hours.
Cytotoxicity of DOX-liposomes and empty liposomes
Doxorubicin is a low molecular weight compound that enters the cell nucleus, intercalates into the DNA and thereby triggers apoptosis. Thus, the drug delivery efficiency of DOX-liposomes could be evaluated in terms of cytotoxicity caused by the release of DOX from the liposomes into the cytoplasm and ultimately the nucleus. As shown in Figure 3A, the cytotoxicity of M-GGLG-DOX-liposomes was significantly higher than that of GGLG-DOX-liposomes in all the cell lines tested. The IC50 (the concentration required for 50% inhibition of cell proliferation) of GGLG-DOX-liposomes was estimated to be >18.4 μM (10 μg/mL), while that of M-GGLG-DOX-liposomes was approximately 9.2 μM (5 μg/mL) for HeLa and HCC1954 cells, and 1.84 μM (1 μg/mL) for MDA-MB-468 cells.
Figure 3.

Cytotoxicity of (A) DOX-liposomes after 24 hour incubation and (B) empty liposomes after 48 hour incubation with HeLa, HCC1954, and MDA-MB-468 cells at 37°C. Error bars represent standard deviation; (n = 4); **P < 0.01.
Notes: Empty liposomes of both M-GGLG- and GGLG-liposomes were biocompatible at all experimental concentrations used for drug delivery (at [DOX] of 10 μg/mL, the lipid concentration of DOX-liposomes was around 100 mg/L). The significantly increased cytotoxicity of DOX-encapsulating M-GGLG-liposomes revealed advanced drug delivery by maleimide-modification.
Abbreviations: DOX, anticancer drug doxorubicin; GGLG, pH sensitive lipid 1,5-dihexadecyl N,N-diglutamyl-lysyl-L-glutamate; M, maleimide moiety; min, minutes.
The cytotoxicity of empty liposomes was observed after a relatively long (eg, 48 hours) incubation period with the cells (Figure 3B). The IC10 (the concentration required for 10% inhibition of cell proliferation) of empty liposomes was estimated to be 720, 200, and 730 mg/L for GGLG-liposomes, and 1000, 500, and 600 mg/L for M-GGLG-liposomes in HeLa, HCC1954, and MDA-MB-468 cells, respectively. The maximum concentration of the lipids used in the cytotoxicity experiments of DOX-liposomes was around 100 mg/L (calculated by [DOX] and DOX-lipid rate), which was much lower than the IC10 values of empty liposomes (shown above). Therefore, in the cytotoxicity experiments of DOX-liposomes, liposomes alone did not lead to cell death in vitro after a 24-hour incubation, suggesting the observed cytotoxicity was caused by the encapsulated DOX.
In vivo evaluation of tumor growth inhibition by DOX-encapsulating liposomes
The tumor growth inhibition by DPPC-DOX-, GGLG-DOX- and M-GGLG-DOX-liposomes was evaluated against breast cancer using nude mice. As shown in Figure 4, the M-GGLG-DOX-liposomes exhibited an enhanced antitumor effect over GGLG-DOX-liposomes throughout the observation period. In the early stage of tumor growth, the DPPC-DOX-liposomes exerted a relatively strong antitumor activity within a short period after sc injections. However, this inhibition of tumor growth was not maintained and the tumor regrew after the second administration of drug. By comparison, the M-GGLG-DOX-liposomes elicited a slightly weaker inhibition of tumor growth than the DPPC-DOX-liposomes in the early stage of treatment. However, the anticancer effect of M-GGLG-DOX-liposomes was more sustained and became more potent than DPPC-DOX-liposomes after the final administration. Moreover, the inhibition of tumor growth continued for a further 20 days without any additional injection of M-GGLG-DOX-liposomes, and no increase in tumor volume was observed (Table 2). Our results suggest that the inhibition of tumor growth by M-GGLG-DOX-liposomes was delayed but more prolonged than that brought about by an equivalent treatment with DPPC-DOX-liposomes.
Figure 4.
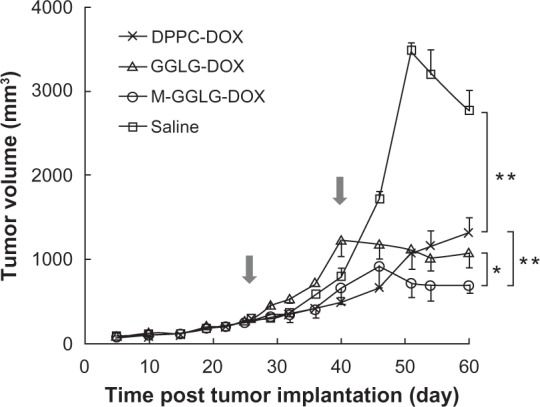
Tumor growth inhibition by sc injection of DOX-liposomes (2 mg DOX/kg) around breast cancer tissues. Arrows indicated the injection dates. Error bars represent standard error of the mean; n = 4; *P < 0.05, **P < 0.01.
Abbreviations: DOX, anticancer drug doxorubicin; DPPC, 1,2-dipalmitoyl-sn-glycero-3-phosphocholine; M, maleimide moiety; GGLG, pH sensitive lipid 1,5-dihexadecyl N,N-diglutamyl-lysyl-L-glutamate; sc, subcutaneous.
Table 2.
Tumor volume of each experimental group at the 60th day post tumor implantation
| Groups | Tumor volume (mm3) |
|---|---|
| Saline | 2778 ± 236 |
| DPPC-DOX-liposomes | 1323 ± 182 |
| GGLG-DOX-liposomes | 1086 ± 176 |
| M-GGLG-DOX-liposomes | 703 ± 97 |
Notes: The values express the mean and standard error of mean of tumor volumes, which are calculated by measuring the long and short diameters of breast tumor tissue.
Abbreviations: DOX, anticancer drug doxorubicin; DPPC, 1,2-dipalmitoyl-sn-glycero-3-phosphocholine; GGLG, pH sensitive lipid 1,5-dihexadecyl N,N-diglutamyl-lysyl-L-glutamate; M, maleimide moiety.
Biodistribution of liposomes after sc injection at the tumor site
After sc injection, the liposomes with a mean diameter of more than 100 nm were mostly accumulated at the injection site (shown in Figure 5A), following the recognition and metabolism by the local lympH nodes.5 Within 24 hours after injection, the concentration of DPPC- and GGLG-liposomes sharply decreased with more than half of the liposomes eliminated at the local site. Subsequently, the decomposition of DPPC- and GGLG-liposomes slowed down, and finally, almost 90% of the liposomes were eliminated on the fifth day post-injection (Figure 5B). By contrast, M-GGLG-liposomes exhibited a significantly slower clearance rate over the entire period; ie, >30% of liposomes were retained 5 days after sc injection. Our results suggest that M-GGLG-liposomes are retained for much longer at the injection site compared to DPPC- and GGLG-liposomes.
Figure 5.

Biodistribution of liposomes in vivo. (A) IVIS observation of residual DiR-liposomes after sc injection around breast cancer tissue. (B) The rate of residual liposomes at injection site post-injection. The fluorescent intensity was measured from (A) by using Living Image software. Error bars represent standard error of the mean; n = 2; **P < 0.01.
Note: M-GGLG-liposomes showed a significantly enhanced retention after sc injection for 5 days.
Abbreviations: DiR, 1,1′-dioctadecyltetramethyl indotricarbocyanine iodide; DPPC, 1,2-dipalmitoyl-sn-glycero-3-phosphocholine; GGLG, pH sensitive lipid 1,5-dihexadecyl N,N-diglutamyl-lysyl-L-glutamate; IVIS, in vivo imaging system; M, maleimide moiety; max, maximum; min, minimum; sc, subcutaneous.
Discussion
The surface structure of the liposome is an important factor that influences its physical and biological properties, such as stimuli-sensitivity, stability, cellular uptake efficiency, drug release rate, and rate of biodegradation. Maleimide is a thiol-reactive moiety that rapidly, covalently, and specifically conjugates with the thiol group of cysteine residues. Thus, surface modification with maleimide-PEG provides the liposomes with reactive sites to the cell membrane thiols. We reasoned such a modification might strengthen the association of liposomes with the cell surface. This concept was introduced into the design of liposomes with the expectation of enhanced cellular uptake.19
Maleimide-modification at a level of 0.3 mol% of total lipids in the liposomes did not affect their physical characteristics such as size, drug encapsulation efficiency (Table 1), stability (Supplementary Figure S1), and pH sensitivity (Supplementary Figure S2). Therefore, M-GGLG-liposomes exhibit a similar pH response characteristic upon cellular uptake (Figure 2A) and drug release in vitro (Supplementary Figure S4) to that of pH responsive GGLG-liposomes. If we consider that the liposomes are composed of 68.2% GGLG lipid and 4.2% PEG of total lipid weight, 0.3 mol% of maleimide moiety represents only 0.05% of total lipid weight. Thus, it is not surprising that our maleimide modification did not influence the physical properties of the liposomes to any significant extent.
Nonetheless, the maleimide moiety furnished the liposomes with new biological properties and/or functions. The maleimide moiety on the outer surface of the M-GGLG-liposomes could recognize and conjugate with thiol groups on the cell surface. The more rapid and increased duration of cellular uptake observed for the M-GGLG-liposomes (Figure 2) implied that the conjugation of maleimide moieties to the cell surface thiols facilitated their cellular internalization. Given that liposomes with a mean size of 100 to 200 nm enter cells mainly via clathrin- and/or caveolae-mediated endocytosis,25 it is possible the thiol-reactive maleimide moiety strengthens the recognition and/or interaction between liposomes and coat proteins on the plasma membrane. It is also hypothesized that the thiol-mediated cellular uptake is independent from conventional endocytosis. Some other factors such as PDI might also be involved in this movement.
To estimate the safety of the maleimide moiety applied on M-GGLG-liposomes, the cytotoxicity of empty M-GGLG-liposomes was investigated. Cytotoxicity was only observed after incubation for 48 hours at extremely high concentrations of total lipid; ie, more than 1000, 500, and 600 mg/L for HeLa, HCC1954 and MDA-MB-468 cells, respectively (Figure 3B). These fideings were similar to those obtained for GGLG-liposomes. The concentration of total lipid used for the experiments of cellular uptake efficiency and cytotoxicity of DOX-liposomes was 72 mg/L and 100 mg/L for no more than a 24-hour incubation in vitro. Hence, we considered that the biocompatibilities of GGLG- and M-GGLG-liposomes were sufficiently high in all cell lines tested to evaluate their biological functions in vitro. Furthermore, the concentration of the maleimide moiety on M-GGLG-liposomes used in the studies of tumor growth inhibition and biodistribution was 1.25 mg/L and 41.7 μg/L, respectively. Both these concentrations were within the safe concentration limit of N-ethylma-leimide for cells (Supplementary Figure S3 and Table S2). We therefore concluded that the modification of liposomes using 0.3 mol% of maleimide moiety on M-GGLG-liposomes was also biocompatible in vivo. Moreover, the increased cytotoxicity of DOX-liposomes in vitro (Figure 3A) and antitumor effect in vivo (Figure 4) were a result of enhanced cellular uptake (Figure 2) and efficient drug (DOX) release of the M-GGLG-DOX-liposomes (Supplementary Figure S4). Our experiments show that the maleimide moiety did not contribute to the observed cytotoxicity of the modified liposomes.
By contrast to iv injection (Supplementary Picture S1), sc injection can prevent the liposomes with a mean diameter of above 100 nm from entering the blood circulation directly (Figure 5A) and thereby reduce the side effect on normal organs and avoid the rapid clearance of liposomes by MPS in the liver and spleen. Therefore, we reasoned sc injection to be an appropriate means of administering M-GGLG-liposomes in order to study their in vivo effect. Upon sc injection around the tumor site, liposomes directly diffuse into a limited interstitial area and are either taken up by the tumor tissues or pass through a system of lymphatic vessels to arrive at one or more lymph nodes, and then are taken up and degraded by macrophages via phagocytosis.26 Shortly after sc injection of liposomes, the concentration of liposomes in the sc tissues was high, leading to a high rate of degradation by lymph nodes. Gradually, the liposomes were assimilated and the concentration decreased, resulting in a decrease in the degradation rate. Although the clearance of DPPC- and GGLG-liposomes occurred via logarithmic elimination (Figure 5B), the M-GGLG-liposomes exhibited a more complicated and slower mode of degradation. Because the lipid composition of M-GGLG- and GGLG-liposomes was the same and the size distribution and other physical properties were similar (Table 1), the presence of the maleimide-moiety was considered as a key factor that resulted in the different biological properties of M-GGLG-liposomes. It is hypothesized that due to the high reactivity of maleimide with thiols of cysteine residues, M-GGLG-liposomes are likely to conjugate with thiols of the tumor and interstitial tissue (mainly adipose tissue) surface, and/or proteins in the interstitial fluid such as albumin, transferrin, and globulin. In the first instance, maleimides immobilize the liposomes to the tissues at the injection site and consequently decrease the absorption of liposomes into the lymphatic drainage system. Maleimide-conjugation with proteins in the interstitial fluid leads to an increase in particle size, which hinders their passage through the interstitium and into the lymphatic capillaries.27 Therefore, the conjugation of maleimide moiety on the liposome surface might prevent liposomes from lymphatic absorption and destabilization, which results in an extended subcutaneous retention of the M-GGLG-liposomes at the injection site. Hence, the extended inhibition of tumor growth elicited by M-GGLG-DOX-liposomes (Figure 4) appears to be due to the high retention rate of liposomes around the tumor tissue as well as the subsequent enhanced cellular uptake and efficient drug release.
Conclusion
We have designed a maleimide-modified M-GGLG-liposome that takes advantage of thiol-conjugation to enhance cellular uptake. The introduction of a small amount (0.3 mol%) of maleimide did not influence the biocompatibility and physical properties of the liposomes. However, the cellular uptake efficiency and drug delivery ability were remarkably increased both in vitro and in vivo. The improved biological properties are thought to be due to the contribution of the thiol-reactive maleimide-modified surface, which might trigger more potent cellular internalization. Although the detailed mechanism of thiol-mediated cellular uptake still remains unclear, the high biocompatibility and drug delivery efficiency of M-GGLG-liposomes indicated a possible application of maleimide for advanced drug delivery systems.
Supplementary data
Characterization of liposomes
Stability of DOX-liposomes
The stability of DOX-liposomes was evaluated by the DOX leakage from liposomes in saline at 37°C. After column purification and ultracentrifugation, GGLG-DOX- and M-GGLG-DOX-liposomes (final concentration [DOX] = 15 μg/mL) were dispersed in saline (5 mL) and incubated at 37°C for 1, 4, 24, 48, 168 and 720 hours. At each time point, 0.5 mL of a solution of DOX-liposomes was drawn and subject to ultrafiltration at 14,000 g for 20 min using an Amicon Ultra-100k (Millipore Ireland Ltd Tullagreen, Carrigtwohill, CO Cork, Ireland) tube according to the manufacturer’s instruction. The leakage rate of DOX was calculated using equation (1):
| (1) |
Zeta potential of the liposomes
Zeta potentials of the liposomes at various pH values were calculated with a Zetasizer (Zetasizer4; Malvern, UK). The liposome dispersion in acetic acid-sodium acetate buffers (pH 3.0, 3.5, 4.0, 4.5, 5.0, 5.5, 6.0) or disodium hydrogen phosphate-sodium dihydrogen phosphate buffer (pH 6.5, 7.0, 7.5, 8.0, 8.5, 9.0) was loaded in a capillary cell mounted on the apparatus and measured in triplicate at 37°C.
Zeta potentials of GGLG- and M-GGLG-liposomes increased from negative to positive by decreasing pH value from 9.0 to 3.0 (Figure 2). It was suggested that the modification of liposomes with a small amount of maleimide did not change the zeta potential to a large extent, and M-GGLG-liposomes were still sensitive to pH change.
The zeta potentials of both GGLG- and M-GGLG-liposomes became 0 at around pH 5.5. At this pH there was an increase in the rate of aggregation/self-fusion of liposomes, most likely due to the absence of electrostatic repulsion on the liposome surfaces under these conditions. The increasing ratios of the mean diameter of GGLG-liposomes at pH 5.5 were 1.58, 2.09 and 2.35 by comparison to those at physiological pH after storage at 4°C for 0.5 hour, 6 hours and 12 hours, respectively. Likewise, the increasing ratios of the mean diameter of M-GGLG-liposomes under the same conditions described above were 1.34, 1.64 and 1.88, respectively.
Stability of GGLG-DOX- and M-GGLG-DOX-liposomes in saline at 37°C. Errors mean SEM. (n = 3).
Abbreviations: DOX, doxorubicin; GGLG, pH sensitive lipid 1,5-dihexadecyl N,N-diglutamyl-lysyl-L-glutamate; M, maleimide moiety; SEM, standard error of mean.
Zeta potentials of GGLG- and M-GGLG-liposomes under various pH values at 37°C. Errors mean SEM. (n = 5).
Abbreviations: GGLG, pH sensitive lipid 1,5-dihexadecyl N,N-diglutamyl-lysyl-L-glutamate; M, maleimide moiety; SEM, standard error of mean.
Cytotoxicity of N-ethylmaleimide
Cell viability of (square) HeLa, (circle) HCC1954, and (triangle) MDA-MB-468 cells after incubation with N-ethylmaleimide (NEM) for 24 hr at 37°C. Errors mean SD (n = 4).
Abbreviations: NEM, N-ethylmaleimide; SD, standard deviation.
Intracellular drug release efficiency
Rate of [DOX] in nucleus after incubation of DOX-liposomes ([DOX] = 100 μ g/mL) with HeLa cells at 37°C for 8 hr. The drug release efficiency was calculated by measuring the total fluorescent intensity of DOX internalized in the whole cell and DOX released into the nucleus using confocal Microscopy. The ratio of [DOX] released into the nucleus was expressed as the percentage of [DOX]nucleus/[DOX]cell. Errors mean SEM. n = 70. **P < 0.01.
Abbreviations: DOX, doxorubicin; GGLG, pH sensitive lipid 1,5-dihexadecyl N,N-diglutamyl-lysyl-L-glutamate; M, maleimide moiety; SEM, standard error of mean.
Biodistribution of M-GGLG-liposomes after iv injection
Biodistribution of M-GGLG-liposomes after intravenous injection for 2 hr.
Abbreviations: GGLG, pH sensitive lipid 1,5-dihexadecyl N,N-diglutamyl-lysyl-L-glutamate; M, maleimide moiety; iv, intravenous.
Table S1.
Size distribution of DPPC-, GGLG- and M-GGLG-liposomes containing lipid DPPC or GGLG/cholesterol/PEG5000-DSPE/PEG5000-Glu2C18 (DPPC- or GGLG-liposome) or maleimide-PEG5000-Glu2C18 (M-GGLG-liposome) at molar ratio of 5:5:0.03:0.03 after preparation for 30 days stocking at 4°C. (n = 3)
| Time (d) | DPPC-liposome | GGLG-liposome | M-GGLG-liposome |
|---|---|---|---|
| 0 | 162.2 ± 67.41 | 167.5 ± 56.03 | 178.6 ± 68.79 |
| 30 | 167.3 ± 76.73 | 167.9 ± 72.3 | 173.7 ± 81.33 |
Notes: The values express the mean and standard deviation of the diameters of liposomes, which are measured instantly after preparation or post 30 days stocking.
Abbreviations: DPPC, 1,2-dipalmitoyl-sn-glycero-3-phosphocholine; GGLG, pH sensitive lipid 1,5-dihexadecyl N,N-diglutamyl-lysyl-L-glutamate; DSPE, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine; Glu2C18, 1,5-dioctadecyl L-glutamate; PEG, polyethylene glycol; M, maleimide moiety; d, day.
Table S2.
IC50 of NEM in HeLa, HCC1954 and MDA-MB-468 cell lines for 24 hr incubation
| Cell lines | HeLa | HCC1954 | MDA-MB-468 |
|---|---|---|---|
| IC50 (mg/L; μM) | 6.8; 54.4 | 3.7; 29.6 | 3.5; 28.0 |
Notes: The cytotoxicity was tested by Cell Counting Kit-8 according to the manufacturer’s instruction. No significant cytotoxicity of N-ethylmaleimide (NEM) was confirmed within the concentration of 1.25 mg/L (10 μM) in HeLa, HCC1954 and MDA-MB-468 cells.
Abbreviations: NEM, N-ethylmaleimide; IC50, the concentration required for 50% inhibition of cell proliferation.
Acknowledgments
Tianshu Li would like to thank China Scholarship Council for supporting her PhD study. This work was partially supported by the GCOE “Practical Chemical Wisdom” and “Grants for Excellent Graduate Schools” project for Waseda University from MEXT, Japan.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv Enzyme Regul. 2001;41:189–207. doi: 10.1016/s0065-2571(00)00013-3. [DOI] [PubMed] [Google Scholar]
- 2.Owens DE, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307(1):93–102. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 3.Allen TM. Long-circulating (sterically stabilized) liposomes for targeted drug delivery. Trends Pharmacol Sci. 1994;15(7):215–220. doi: 10.1016/0165-6147(94)90314-x. [DOI] [PubMed] [Google Scholar]
- 4.Bae YH, Park K. Targeted drug delivery to tumors: myths, reality and possibility. J Control Release. 2011;153(3):198–205. doi: 10.1016/j.jconrel.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oussoren C, Zuidema J, Crommelin DJ, Storm G. Lymphatic uptake and biodistribution of liposomes after subcutaneous injection. II. Influence of liposomal size, lipid compostion and lipid dose. Biochim Biophys Acta. 1997;1328(2):261–272. doi: 10.1016/s0005-2736(97)00122-3. [DOI] [PubMed] [Google Scholar]
- 6.Sawant RR, Torchilin VP. Liposomes as ‘smart’ pharmaceutical nanocarriers. Soft Matter. 2010;6(17):4026–4044. [Google Scholar]
- 7.Obata Y, Tajima S, Takeoka S. Evaluation of pH-responsive liposomes containing amino acid-based zwitterionic lipids for improving intracellular drug delivery in vitro and in vivo. J Control Release. 2010;142(2):267–276. doi: 10.1016/j.jconrel.2009.10.023. [DOI] [PubMed] [Google Scholar]
- 8.Connor J, Yatvin MB, Huang L. pH-sensitive liposomes: acid-induced liposome fusion. Proc Natl Acad Sci U S A. 1984;81(6):1715–1718. doi: 10.1073/pnas.81.6.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Needham D, Anyarambhatla G, Kong G, Dewhirst MW. A new temperature-sensitive liposome for use with mild hyperthermia: characterization and testing in a human tumor xenograft model. Cancer Res. 2000;60(5):1197–1201. [PubMed] [Google Scholar]
- 10.Kikumori T, Kobayashi T, Sawaki M, Imai T. Anti-cancer effect of hyperthermia on breast cancer by magnetite nanoparticle-loaded anti-HER2 immunoliposomes. Breast Cancer Res Treat. 2009;113(3):435–441. doi: 10.1007/s10549-008-9948-x. [DOI] [PubMed] [Google Scholar]
- 11.Ong W, Yang Y, Cruciano AC, McCarley RL. Redox-triggered contents release from liposomes. J Am Chem Soc. 2008;130(44):14739–14744. doi: 10.1021/ja8050469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meng F, Hennink WE, Zhong Z. Reduction-sensitive polymers and bioconjugates for biomedical applications. Biomaterials. 2009;30(12):2180–2198. doi: 10.1016/j.biomaterials.2009.01.026. [DOI] [PubMed] [Google Scholar]
- 13.Liang X, Yue X, Dai Z, Kikuchi J. Photoresponsive liposomal nanohybrid cerasomes. Chem Commun (Camb) 2011;47(16):4751–4753. doi: 10.1039/c1cc00063b. [DOI] [PubMed] [Google Scholar]
- 14.Ohya Y, Okuyama Y, Fukunaga A, Ouchi T. Photo-sensitive lipid membrane perturbation by a single chain lipid having terminal spiropyran group. Supramol Sci. 1998;5(1–2):21–29. [Google Scholar]
- 15.Huang SL. Liposomes in ultrasonic drug and gene delivery. Adv Drug Deliv Rev. 2008;60(10):1167–1176. doi: 10.1016/j.addr.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 16.Schroeder A, Honen R, Turjeman K, Gabizon A, Kost J, Barenholz Y. Ultrasound triggered release of cisplatin from liposomes in murine tumors. J Control Release. 2009;137(1):63–68. doi: 10.1016/j.jconrel.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 17.Holme MN, Fedotenko IA, Abegg D, et al. Shear-stress sensitive lenticular vesicles for targeted drug delivery. Nat Nanotechnol. 2012;7(8):536–543. doi: 10.1038/nnano.2012.84. [DOI] [PubMed] [Google Scholar]
- 18.Torres AG, Gait MJ. Exploiting cell surface thiols to enhance cellular uptake. Trends Biotechnol. 2012;30(4):185–190. doi: 10.1016/j.tibtech.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 19.Aubry S, Burlina F, Dupont E, et al. Cell-surface thiols affect cell entry of disulfide-conjugated peptides. FASEB J. 2009;23(9):2956–2967. doi: 10.1096/fj.08-127563. [DOI] [PubMed] [Google Scholar]
- 20.Saito G, Swanson JA, Lee KD. Drug delivery strategy utilizing conjugation via reversible disulfide linkages: role and site of cellular reducing activities. Adv Drug Deliv Rev. 2003;55(2):199–215. doi: 10.1016/s0169-409x(02)00179-5. [DOI] [PubMed] [Google Scholar]
- 21.Kichler A, Remy JS, Boussif O, et al. Efficient gene delivery with neutral complexes of lipospermine and thiol-reactive phospholipids. Biochem Biophys Res Commun. 1995;209(2):444–450. doi: 10.1006/bbrc.1995.1522. [DOI] [PubMed] [Google Scholar]
- 22.Okamura Y, Maekawa I, Teramura Y, et al. Hemostatic effects of phospholipid vesicles carrying fibrinogen gamma chain dodecapeptide in vitro and in vivo. Bioconjug Chem. 2005;16(6):1589–1596. doi: 10.1021/bc050178g. [DOI] [PubMed] [Google Scholar]
- 23.Voûte PA, Souhami RL, Nooij M, et al. A phase II study of cisplatin, ifosfamide and doxorubicin in operable primary, axial skeletal and metastatic osteosarcoma. European Osteosarcoma Intergroup (EOI) Ann Oncol. 1999;10(10):1211–1218. doi: 10.1023/a:1008361612767. [DOI] [PubMed] [Google Scholar]
- 24.Bruynzeel AM, Niessen HW, Bronzwaer JG, et al. The effect of monohydroxyethylrutoside on doxorubicin-induced cardiotoxicity in patients treated for metastatic cancer in a phase II study. Br J Cancer. 2007;97(8):1084–1089. doi: 10.1038/sj.bjc.6603994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem J. 2004;377(Pt 1):159–169. doi: 10.1042/BJ20031253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oussoren C, Velinova M, Scherphof G, van der Want JJ, van Rooijen N, Storm G. Lymphatic uptake and biodistribution of liposomes after subcutaneous injection. IV. Fate of liposomes in regional lymph nodes. Biochim Biophys Acta. 1998;1370(2):259–272. doi: 10.1016/s0005-2736(97)00275-7. [DOI] [PubMed] [Google Scholar]
- 27.Oussoren C, Storm G. Liposomes to target the lymphatics by subcutaneous administration. Adv Drug Deliv Rev. 2001;50(1–2):143–156. doi: 10.1016/s0169-409x(01)00154-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Stability of GGLG-DOX- and M-GGLG-DOX-liposomes in saline at 37°C. Errors mean SEM. (n = 3).
Abbreviations: DOX, doxorubicin; GGLG, pH sensitive lipid 1,5-dihexadecyl N,N-diglutamyl-lysyl-L-glutamate; M, maleimide moiety; SEM, standard error of mean.
Zeta potentials of GGLG- and M-GGLG-liposomes under various pH values at 37°C. Errors mean SEM. (n = 5).
Abbreviations: GGLG, pH sensitive lipid 1,5-dihexadecyl N,N-diglutamyl-lysyl-L-glutamate; M, maleimide moiety; SEM, standard error of mean.
Cell viability of (square) HeLa, (circle) HCC1954, and (triangle) MDA-MB-468 cells after incubation with N-ethylmaleimide (NEM) for 24 hr at 37°C. Errors mean SD (n = 4).
Abbreviations: NEM, N-ethylmaleimide; SD, standard deviation.
Rate of [DOX] in nucleus after incubation of DOX-liposomes ([DOX] = 100 μ g/mL) with HeLa cells at 37°C for 8 hr. The drug release efficiency was calculated by measuring the total fluorescent intensity of DOX internalized in the whole cell and DOX released into the nucleus using confocal Microscopy. The ratio of [DOX] released into the nucleus was expressed as the percentage of [DOX]nucleus/[DOX]cell. Errors mean SEM. n = 70. **P < 0.01.
Abbreviations: DOX, doxorubicin; GGLG, pH sensitive lipid 1,5-dihexadecyl N,N-diglutamyl-lysyl-L-glutamate; M, maleimide moiety; SEM, standard error of mean.
Biodistribution of M-GGLG-liposomes after intravenous injection for 2 hr.
Abbreviations: GGLG, pH sensitive lipid 1,5-dihexadecyl N,N-diglutamyl-lysyl-L-glutamate; M, maleimide moiety; iv, intravenous.
Table S1.
Size distribution of DPPC-, GGLG- and M-GGLG-liposomes containing lipid DPPC or GGLG/cholesterol/PEG5000-DSPE/PEG5000-Glu2C18 (DPPC- or GGLG-liposome) or maleimide-PEG5000-Glu2C18 (M-GGLG-liposome) at molar ratio of 5:5:0.03:0.03 after preparation for 30 days stocking at 4°C. (n = 3)
| Time (d) | DPPC-liposome | GGLG-liposome | M-GGLG-liposome |
|---|---|---|---|
| 0 | 162.2 ± 67.41 | 167.5 ± 56.03 | 178.6 ± 68.79 |
| 30 | 167.3 ± 76.73 | 167.9 ± 72.3 | 173.7 ± 81.33 |
Notes: The values express the mean and standard deviation of the diameters of liposomes, which are measured instantly after preparation or post 30 days stocking.
Abbreviations: DPPC, 1,2-dipalmitoyl-sn-glycero-3-phosphocholine; GGLG, pH sensitive lipid 1,5-dihexadecyl N,N-diglutamyl-lysyl-L-glutamate; DSPE, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine; Glu2C18, 1,5-dioctadecyl L-glutamate; PEG, polyethylene glycol; M, maleimide moiety; d, day.
Table S2.
IC50 of NEM in HeLa, HCC1954 and MDA-MB-468 cell lines for 24 hr incubation
| Cell lines | HeLa | HCC1954 | MDA-MB-468 |
|---|---|---|---|
| IC50 (mg/L; μM) | 6.8; 54.4 | 3.7; 29.6 | 3.5; 28.0 |
Notes: The cytotoxicity was tested by Cell Counting Kit-8 according to the manufacturer’s instruction. No significant cytotoxicity of N-ethylmaleimide (NEM) was confirmed within the concentration of 1.25 mg/L (10 μM) in HeLa, HCC1954 and MDA-MB-468 cells.
Abbreviations: NEM, N-ethylmaleimide; IC50, the concentration required for 50% inhibition of cell proliferation.