

Abstract
Evidence has accumulated over the past several years demonstrating that lung injury following inhalation of irritants like ozone is due, not only to direct effects of the chemical, but also indirectly to the actions of inflammatory mediators released by infiltrating macrophages. Among the mediators involved in the cytotoxic process, reactive nitrogen species (RNS) are of particular interest because of their well-documented cytotoxic potential. Findings that macrophage suppression blocks RNS production and ozone-induced toxicity provide strong support for a role of these cells and inflammatory mediators in lung injury. Recent investigations have focused on understanding pathways by which macrophages become activated to release RNS. One protein that has attracted considerable attention is caveolin-1, a membrane scaffolding molecule that functions to negatively regulate cell signaling. The fact that expression of caveolin-1 is down-regulated in macrophages after ozone inhalation suggests a mechanism controlling the release of cytotoxic mediators by these inflammatory cells.
Keywords: macrophages, ozone, nitric oxide, caveolin, TNFα
Introduction
Ozone is a ubiquitous urban air pollutant and the main component of photochemical smog. It remains one of the most problematic air pollutants to control because it is formed from intermediates originating from many different sources.1 Inhaled ozone has been shown to irritate and damage the lung in both healthy and susceptible individuals, including children and the elderly.2 Ozone causes inflammation and constriction of the airways, thus reducing pulmonary function leading to respiratory symptoms. Ozone also exacerbates asthma and suppresses nonspecific immunity increasing the susceptibility of exposed individuals to respiratory infections.2 Epidemiologic studies in the United States have demonstrated that for every 10 ppb increase in daily ozone levels, the total death rate for that day and for the two following days increases by 0.87%.3 Potential adverse effects of ozone are even greater in large cities in the developing world where ozone levels can be significantly higher than in the United States. Thus, elucidating the specific pro- and anti-inflammatory mechanisms contributing to ozone-induced tissue injury is highly relevant in terms of designing strategies for reducing morbidity and mortality caused by exposure to air pollutants.
Macrophages and inflammatory mediators
Macrophages are key effectors of innate immunity. In addition to ridding the body of pathogens, dead cells and debris, apoptotic cells, and some tumor cells, they are one of the most active secretory cells in the body, releasing a wide range of mediators that regulate inflammation, adaptive immunity, and homeostasis.4–7 They are also considered professional antigen-presenting cells, triggering specific immune responses of T lymphocytes. It is now well recognized that these diverse actions of macrophages are mediated by distinct subpopulations of cells that are phenotypically polarized by inflammatory mediators in their microenvironment.8 Polarized macrophages are broadly grouped into two major subtypes: classically activated cytotoxic/proinflammatory M1 cells, and alternatively activated anti-inflammatory M2 cells. Whereas M1 cells release mediators that promote inflammation, pathogen destruction, cytotoxicity, and in some instances, tissue injury, M2 cells release anti-inflammatory, and immunosuppressive mediators that facilitate wound repair, tissue remodeling, and angiogenesis. Under pathologic conditions, M2 cells are also key to chronic inflammation and the development of fibrosis.9
Macrophages and ozone toxicity
Our laboratories have been investigating the role of macrophages and inflammatory mediators in the pathogenesis of lung injury induced by exposure to pulmonary toxicants such as ozone. We hypothesize that macrophages accumulating in the lung in response to ozone-induced injury are classically activated into M1 cells, and release RNS which contribute to toxicity. To test this hypothesis, we used a whole body exposure system for rodents.10,11 Acute (3 h) inhalation of ozone by both rats (2 ppm) and mice (0.8 ppm) was found to cause structural alterations in the lung including disruption of the alveolar epithelial barrier, followed by Type II cell hypertrophy and hyperplasia. These changes were most notable 24 h post-ozone exposure and were accompanied by alterations in lung functioning including decreases in compliance (Fig. 1). Increases in tissue damping and tissue elastance were also noted in lungs of animals exposed to ozone. These findings indicate that early functional alterations induced by ozone were evident in airways, as well as lung parenchyma. Further analysis of lung sections, as well as bronchoalveolar lavage fluid (BAL) revealed that ozone-induced alterations in lung structure and function were correlated with an accumulation of inflammatory macrophages in the tissue. When these cells were isolated, they were found to exhibit morphologic and functional characteristics of classically activated macrophages. Thus, they were significantly enlarged, relative to macrophages from control animals, were highly vacuolated and possessed ruffled membranes and an increased cytoplasmic:nuclear ratio. In addition, they released increased quantities of proinflammatory/cytotoxic mediators including tumor necrosis factor (TNF)-α, eicosanoids (e.g., PGE2), RNS (e.g., nitric oxide and peroxynitrite), and reactive oxygen species (e.g., superoxide anion and hydrogen peroxide).12,13 They also expressed high levels of mRNA for inducible nitric oxide synthase (iNOS), the enzyme-mediating nitric oxide production by macrophages. In contrast, expression of interleukin (IL)-10 and arginase was suppressed in these cells (Fig. 2, and not shown). These are prototypical characteristics of M1 macrophages.14 To assess whether proinflammatory/cytotoxic M1 macrophages contribute to ozone-induced nitric oxide production and lung injury, we used gadolinium chloride, a rare earth metal reported to block the activity of these cells.15 Pretreatment of animals with gadolinium chloride was found to suppress ozone-induced iNOS expression in the lung, as well as macrophage production of RNS.15 Moreover, animals pretreated with gadolinium chloride were protected from ozone-induced lung injury. These data demonstrate a role for M1 macrophages in the pathogenic response to ozone.
Figure 1.

Effects of ozone on lung function. Female Wistar rats were exposed to air (white bars) or ozone (black bars; 2 ppm, 3 h) in whole body Plexiglas divided chambers. After 24 h, animals were anesthetized and tracheotomy performed. Animals were attached to a flexiVent (SciReq) and measurements of whole lung resistance and compliance, and tissue damping and elastance were made. Each bar is the mean ± SE (n = 3).
Figure 2.

Effects of ozone on iNOS and Arginase1 mRNA expression. Alveolar macrophages collected from animals exposed to ozone were analyzed by real-time PCR for iNOS and Arginase1 (Arg1) gene expression. Data are presented relative to GAPDH and expressed as fold-change over air control. Each bar is the mean ± SE (n = 3).
RNS and lung injury
RNS, including nitric oxide and peroxynitrite, have been implicated in lung injury induced by diverse pulmonary toxicants including silica, particulate matter, and bleomycin.16–18 They have also been shown to be important in the pathogenesis of diseases such as ARDS, COPD, bronchitis, and asthma.19,20 To analyze the role of RNS in ozone toxicity, we used transgenic mice with a targeted disruption of the iNOS gene. Macrophages isolated from iNOS knockout mice were unable to generate nitric oxide or peroxynitrite.12 Ozone-induced expression of TNFα was also suppressed in these mice. Importantly, iNOS knockout mice were protected from ozone-induced lung injury, as measured by levels of BAL protein and inflammatory cells. There was also no evidence of peroxynitrite-mediated lung damage in these animals. Similar results were observed in mice overexpressing superoxide dismutase (SOD), which are unable to generate peroxynitrite.21 Ozone-induced increases in macrophage expression of iNOS, as well as TNFα, and decreases in IL-10 were also suppressed in SOD overexpressing mice.21 These data demonstrate that nitric oxide generated via iNOS and its reactive oxidative product, peroxynitrite, play critical roles in ozone-induced inflammation, production of inflammatory mediators, and lung injury.
The precise biochemical mechanisms underlying the pathogenic actions of RNS in the lung following ozone inhalation have not been elucidated. Nitric oxide is a highly reactive molecule that readily diffuses into membranes and cells where it reacts with molecular targets such as heme- and thiol-containing proteins and amines, and these actions may be important in its cytotoxic activity.22 RNS can also posttranslationally modify proteins resulting in altered functioning. For example, RNS-mediated S-nitrosylation of the pulmonary collectin, surfactant protein-D (SP-D) results in a change in its biological activity from anti-inflammatory to proinflammatory.23 Following ozone inhalation, we detected increased levels of S-nitrosylated SP-D in BAL. This was correlated with increases in the ability of BAL from ozone-treated animals to induce macrophage chemotaxis and production of reactive oxygen species. These findings suggest that one mechanism whereby RNS contributes to pulmonary inflammation and oxidative stress following ozone inhalation involves alterations in protein functioning.
Mechanisms regulating RNS production by lung macrophages
As described earlier, nitric oxide is generated in macrophages by an inducible form of the enzyme nitric oxide synthase. This enzyme is upregulated by inflammatory mediators such as bacterially derived lipopolysaccharide, and Type I cytokines like interferon-γ and TNFα. TNFα is of particular interest because it has direct cytotoxic and proapoptotic activity. It also stimulates the release of other proinflammatory and cytotoxic mediators including IL-1, IL-6, platelet activating factor, colony-simulating factor, and eicosaniods, as well as reactive oxygen and nitrogen species from inflammatory phagocytes which can exacerbate tissue injury.24 TNFα also regulates cellular proliferation and, at later stages of inflammation, is thought to activate antioxidants and initiate wound healing.
In humans, TNFα plays a key role in allergic reactions in the pulmonary airways, and in the pathogenesis of ARDS. It also mediates bronchial hyperresponsiveness in rats following exposure to aerosolized endotoxin and ozone.24 TNFα production by alveolar macrophages is increased in experimental models of tissue injury induced by inhaled particulates, as well as endotoxin and is thought to be the major cytotoxic effector in bleomycin and silica-induced fibrosis.24
Following ozone inhalation, we found that TNFα was rapidly upregulated in the lung, predominantly in alveolar macrophages.10,11,25 To investigate the role of TNFα in ozone-induced iNOS induction and lung injury, we used knockout mice with a targeted deletion of the gene for TNFα. In contrast to wild-type mice, ozone inhalation had no effect on macrophage expression of iNOS induction in TNFα knockout mice. Ozone-induced increases in nitric oxide and peroxynitrite production by macrophages was also suppressed in TNFα knockout mice. These data, together with the observation that TNFα knockout mice were protected from ozone-induced toxicity, demonstrate that TNFα produced early after ozone inhalation contributes to lung injury, in part, by stimulating the production of cytotoxic inflammatory mediators such as RNS.
TNFα exerts its biological activity by binding to a receptor on macrophages; two major receptors have been identified: TNFR1 (p55) and TNFR2 (p75). Evidence suggests that the proinflammatory actions of TNFα are mediated predominantly via activation of TNFR1. This receptor is localized in both lipid raft and nonraft regions of the plasma membrane. In response to TNFα, TNFR1 rapidly associates with lipid rafts where it complexes with TNFα.26 This leads to the recruitment of adaptor proteins and activation of a cascade of signaling molecules including phosphatidylinositol 3 (PI 3)-kinase, p44/42 mitogen-activated protein (MAP) kinase, and consequent activation of NF-κB, a transcription factor important in regulating many inflammatory genes including iNOS.27
Following ozone inhalation, we observed rapid and persistent activation of NF-κB in alveolar macrophages.10,28 This was associated with induction of PI 3-kinase and its down stream target, protein kinase B (PKB). Treatment of alveolar macrophages from ozone exposed mice with the PI 3-kinase inhibitors, wortmanin, and LY294002, blocked excessive nitric oxide production by these cells; NF-κB activation was also suppressed. These data suggest that PI 3-kinase and PKB are important in regulating NF-κB activation and expression of iNOS. To investigate this further, we used transgenic mice with a targeted disruption of the NF-κB p50 gene. Ozone-induced increases in NF-κB nuclear binding activity were significantly attenuated in NF-κB p50 knockout mice.10 In addition, the loss of NF-κB p50 was associated with a marked reduction in the ability of ozone to upregulate iNOS in the lung. We also found that alveolar macrophages from NF-κB p50 knockout mice were unable to generate nitric oxide or peroxynitrite, even after ozone inhalation. Moreover, mice lacking NF-κB p50 were protected against ozone-induced toxicity. These data demonstrate that ozone-induced expression of iNOS and nitric oxide production in the lung are dependent on NF-κB.
Caveolin-1 and ozone toxicity
Caveolae are small (50–100 nm) vesicular invaginations of the plasma membrane. Because of their unique lipid composition, caveolae are classified as plasma membrane lipid rafts. The chief structural proteins of caveolae are caveolins. To date, three caveolins (Cav-1, Cav-2, and Cav-3) with unique tissue distribution have been characterized. Cav-1 is a 21–24 kDa protein that has been identified as the main structural and functional protein of caveolae, required for their formation and stabilization. Cav-1 functions as a membrane-organizing center, concentrating signaling molecules within a scaffolding domain, and negatively regulating their activation state.29 Functional caveolin-binding motifs have been identified in a number of signaling molecules including TNFR1, in several serine/threonine kinases (e.g., p44/42 MAP kinases, PI 3-kinase) and tyrosine kinases (e.g., src kinase), as well as in endothelial nitric oxide synthase (eNOS), and heme-oxygenase-1 (HO-1).30–33 Downregulation of Cav-1 leads to activation of these signaling pathways.
Alveolar macrophages from control animals constitutively express Cav-1 protein.25 Treatment of mice with ozone caused a marked decrease in Cav-1 expression, which was observed immediately after exposure and persisted for 3 h. Subsequently, levels of Cav-1 returned to control. The suppressive effects of ozone on Cav-1 expression were not observed in TNFR1 knockout mice demonstrating that ozone-induced alterations in Cav-1 are dependent on TNFα signaling via this receptor. This conclusion is supported by our findings that TNFα was effective in suppressing Cav-1 expression in macrophages in culture. To further investigate the role of Cav-1 in ozone toxicity, transgenic mice with a targeted disruption of the Cav-1 gene were used. Ozone-induced toxicity and inflammation were significantly exacerbated in these mice. Thus, it appears that Cav-1 may be key to limiting release of proinflammatory/cytotoxic mediators by macrophages in the lung in response to inhaled ozone.
Model of macrophages and ozone toxicity
Based on our findings we have developed a mechanistic model for the role of macrophages and inflammatory mediators in ozone toxicity (Fig. 3). According to this model, exposure to toxic levels of ozone results in injury to Type I alveolar epithelial cells, classical M1 activation of resident and infiltrating macrophages, and release of TNFα. TNFα binds to TNFR1 and downregulates Cav-1 in macrophages in an autocrine and paracrine manner. This leads to activation of signaling pathways like PI 3 kinase/PKB, subsequent activation of NF-κB, upregulation of iNOS, and excessive production of RNS and tissue injury. Understanding inflammatory mechanisms of ozone-induced tissue injury is critical for the development of clinical approaches for treating or abrogating toxicity induced not only by air pollutants, but also potentially, by chronic and episodic inflammatory lung diseases.
Figure 3.
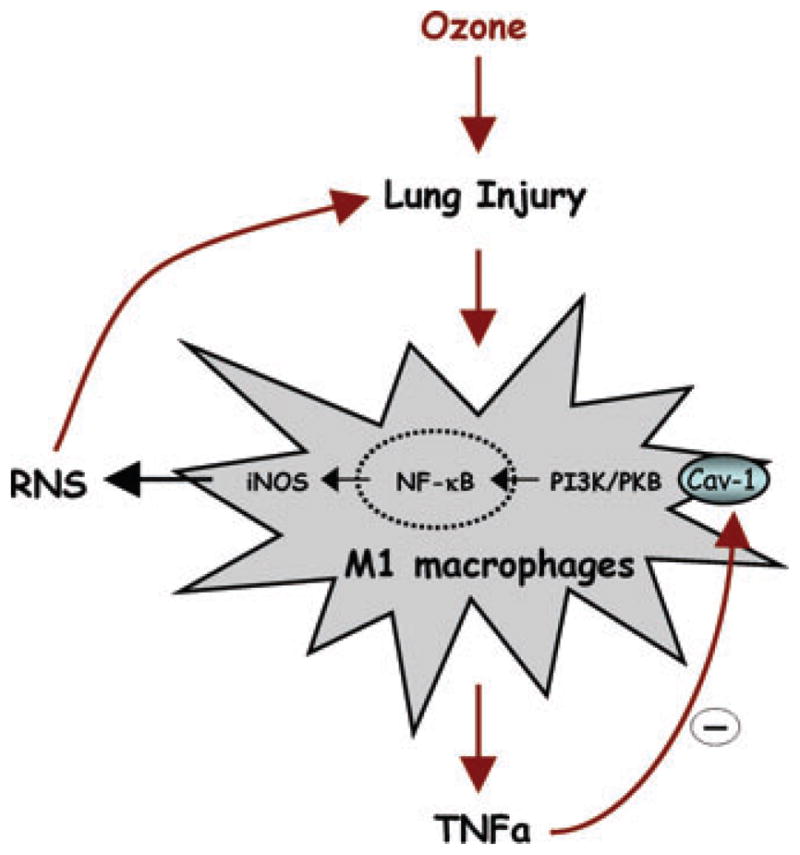
Model for the role of macrophages and RNS in lung injury. Macrophages responding to ozone-induced lung injury become phenotypically polarized into M1 cells; TNFα released from M1 macrophages acts in an autocrine and paracrine manner to downregulate Cav-1, leading to activation of PI3 K/PKB signaling, NF-κB, and induction of iNOS. RNS generated via iNOS contributes to ozone-induced lung injury.
Acknowledgments
This work was supported by National Institutes of Health Grants ES004738, GM034310, CA132624, AR055073, HL086621, and ES005022.
Footnotes
Conflicts of interest
The authors declare no conflict of interest.
References
- 1.Nadadur SS, Costa DL, Slade R, et al. Acute ozone-induced differential gene expression profiles in rat lung. Environ Health Perspect. 2005;113:1717–1722. doi: 10.1289/ehp.7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.U.S. Environmental Protection Agency. Air quality criteria for ozone and related oxidants 2005 [Google Scholar]
- 3.Levy JI, Chemerynski SM, Sarnat JA. Ozone exposure and mortality: an empiric Bayes metaregression analysis. Epidemiology. 2005;16:458–468. doi: 10.1097/01.ede.0000165820.08301.b3. [DOI] [PubMed] [Google Scholar]
- 4.Dale DC, Boxer L, Liles WC. The phagocytes: neutrophils and monocytes. Blood. 2008;112:935–945. doi: 10.1182/blood-2007-12-077917. [DOI] [PubMed] [Google Scholar]
- 5.Laskin DL, Laskin JD. Role of macrophages and inflammatory mediators in chemically induced toxicity. Toxicology. 2001;160:111–118. doi: 10.1016/s0300-483x(00)00437-6. [DOI] [PubMed] [Google Scholar]
- 6.Porcheray F, Viaud S, Rimaniol AC, et al. Macrophage activation switching: an asset for the resolution of inflammation. Clin Exp Immunol. 2005;142:481–489. doi: 10.1111/j.1365-2249.2005.02934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang X, Mosser DM. Macrophage activation by endogenous danger signals. J Pathol. 2008;214:161–178. doi: 10.1002/path.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453–461. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 9.Laskin DL. Macrophages and inflammatory mediators in chemical toxicity: a battle of forces. Chem Res Toxicol. 2009;22:1376–1385. doi: 10.1021/tx900086v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fakhrzadeh L, Laskin JD, Laskin DL. Ozone-induced production of nitric oxide and TNF-alpha and tissue injury are dependent on NF-kappaB p50. Am J Physiol Lung Cell Mol Physiol. 2004;287:L279–L285. doi: 10.1152/ajplung.00348.2003. [DOI] [PubMed] [Google Scholar]
- 11.Pendino KJ, Shuler RL, Laskin JD, Laskin DL. Enhanced production of interleukin-1, tumor necrosis factor-alpha, and fibronectin by rat lung phagocytes following inhalation of a pulmonary irritant. Am J Respir Cell Mol Biol. 1994;11:279–286. doi: 10.1165/ajrcmb.11.3.8086166. [DOI] [PubMed] [Google Scholar]
- 12.Fakhrzadeh L, Laskin JD, Laskin DL. Deficiency in inducible nitric oxide synthase protects mice from ozone-induced lung inflammation and tissue injury. Am J Respir Cell Mol Biol. 2002;26:413–419. doi: 10.1165/ajrcmb.26.4.4516. [DOI] [PubMed] [Google Scholar]
- 13.Pendino KJ, Gardner CR, Shuler RL, et al. Inhibition of ozone-induced nitric oxide synthase expression in the lung by endotoxin. Am J Respir Cell Mol Biol. 1996;14:516–525. doi: 10.1165/ajrcmb.14.6.8652180. [DOI] [PubMed] [Google Scholar]
- 14.Ho VW, Sly LM. Derivation and characterization of murine alternatively activated (M2) macrophages. Methods Mol Biol. 2009;531:173–185. doi: 10.1007/978-1-59745-396-7_12. [DOI] [PubMed] [Google Scholar]
- 15.Pendino KJ, Meidhof TM, Heck DE, et al. Inhibition of macrophages with gadolinium chloride abrogates ozone-induced pulmonary injury and inflammatory mediator production. Am J Respir Cell Mol Biol. 1995;13:125–132. doi: 10.1165/ajrcmb.13.2.7542894. [DOI] [PubMed] [Google Scholar]
- 16.Inghilleri S, Morbini P, Oggionni T, et al. In situ assessment of oxidant and nitrogenic stress in bleomycin pulmonary fibrosis. Histochem Cell Biol. 2006;125:661–669. doi: 10.1007/s00418-005-0116-7. [DOI] [PubMed] [Google Scholar]
- 17.Rimal B, Greenberg AK, Rom WN. Basic pathogenetic mechanisms in silicosis: current understanding. Curr Opin Pulm Med. 2005;11:169–173. doi: 10.1097/01.mcp.0000152998.11335.24. [DOI] [PubMed] [Google Scholar]
- 18.Roberts JR, Young SH, Castranova V, Antonini JM. Soluble metals in residual oil fly ash alter innate and adaptive pulmonary immune responses to bacterial infection in rats. Toxicol Appl Pharmacol. 2007;221:306–319. doi: 10.1016/j.taap.2007.03.022. [DOI] [PubMed] [Google Scholar]
- 19.Ricciardolo FL, Caramori G, Ito K, et al. Nitrosative stress in the bronchial mucosa of severe chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2005;116:1028–1035. doi: 10.1016/j.jaci.2005.06.034. [DOI] [PubMed] [Google Scholar]
- 20.Ricciardolo FL, Di Stefano A, Sabatini F, Folkerts G. Reactive nitrogen species in the respiratory tract. Eur J Pharmacol. 2006;533:240–252. doi: 10.1016/j.ejphar.2005.12.057. [DOI] [PubMed] [Google Scholar]
- 21.Fakhrzadeh L, Laskin JD, Gardner CR, Laskin DL. Superoxide dismutase-overexpressing mice are resistant to ozone-induced tissue injury and increases in nitric oxide and tumor necrosis factor-alpha. Am J Respir Cell Mol Biol. 2004;30:280–287. doi: 10.1165/rcmb.2003-0044OC. [DOI] [PubMed] [Google Scholar]
- 22.Laskin JD, Heck DE, Laskin DL. Nitric Oxide Pathways in Toxic Responses. In: Ballantyne B, Marrs T, Syversen T, editors. General and Applied Toxicology. Chapter 17. Wiley-Blackwell; UK: 2010. [Google Scholar]
- 23.Guo CJ, Atochina-Vasserman EN, Abramova E, et al. S-nitrosylation of surfactant protein-D controls inflammatory function. PLoS Biol. 2008;6:e266. doi: 10.1371/journal.pbio.0060266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laskin DL, Sunil VR, Laumbach RJ, Kipen HM. Inflammatory Cytokines and Lung Toxicity. In: House RV, Descote J, editors. Cytokines in Human Health: Immunotoxicology, Pathology and Therapeutic Applications. Humana Press Inc; Totowa, NJ: 2007. pp. 83–112. [Google Scholar]
- 25.Fakhrzadeh L, Laskin JD, Laskin DL. Regulation of caveolin-1 expression, nitric oxide production and tissue injury by tumor necrosis factor-alpha following ozone inhalation. Toxicol Appl Pharmacol. 2008;227:380–389. doi: 10.1016/j.taap.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doan JE, Windmiller DA, Riches DW. Differential regulation of TNF-R1 signaling: lipid raft dependency of p42MAPK/ERK2 activation, but not NF-kappaB activation. J Immunol. 2004;172:7654–7660. doi: 10.4049/jimmunol.172.12.7654. [DOI] [PubMed] [Google Scholar]
- 27.Aggarwal BB, Shishodia S, Ashikawa K, Bharti AC. The role of TNF and its family members in inflammation and cancer: lessons from gene deletion. Curr Drug Targets Inflamm Allergy. 2002;1:327–341. doi: 10.2174/1568010023344571. [DOI] [PubMed] [Google Scholar]
- 28.Laskin DL, Fakhrzadeh L, Heck DE, et al. Upregulation of phosphoinositide 3-kinase and protein kinase B in alveolar macrophages following ozone inhalation. Role of NF-kappaB and STAT-1 in ozone-induced nitric oxide production and toxicity. Mol Cell Biochem. 2002;234–235:91–98. [PubMed] [Google Scholar]
- 29.Hnasko R, Lisanti MP. The biology of caveolae: lessons from caveolin knockout mice and implications for human disease. Mol Interv. 2003;3:445–464. doi: 10.1124/mi.3.8.445. [DOI] [PubMed] [Google Scholar]
- 30.Engelman JA, Zhang X, Galbiati F, et al. Molecular genetics of the caveolin gene family: implications for human cancers, diabetes, Alzheimer disease, and muscular dystrophy. Am J Hum Genet. 1998;63:1578–1587. doi: 10.1086/302172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hulit J, Bash T, Fu M, et al. The cyclin D1 gene is transcriptionally repressed by caveolin-1. J Biol Chem. 2000;275:21203–21209. doi: 10.1074/jbc.M000321200. [DOI] [PubMed] [Google Scholar]
- 32.Kim HP, Wang X, Galbiati F, et al. Caveolae compartmentalization of heme oxygenase-1 in endothelial cells. FASEB J. 2004;18:1080–1089. doi: 10.1096/fj.03-1391com. [DOI] [PubMed] [Google Scholar]
- 33.Okamoto T, Schlegel A, Scherer PE, Lisanti MP. Caveolins, a family of scaffolding proteins for organizing “preassembled signaling complexes” at the plasma membrane. J Biol Chem. 1998;273:5419–5422. doi: 10.1074/jbc.273.10.5419. [DOI] [PubMed] [Google Scholar]