

Abstract
Mutation Position Imaging Toolbox (MuPIT) Interactive is a browser-based application for single nucleotide variants (SNVs), which automatically maps the genomic coordinates of SNVs onto the coordinates of available three-dimensional protein structures. The application is designed for interactive browser-based visualization of the putative functional relevance of SNVs by biologists who are not necessarily experts either in bioinformatics or protein structure. Users may submit batches of several thousand SNVs and review all protein structures that cover the SNVs, including available functional annotations such as binding sites, mutagenesis experiments, and common polymorphisms. Multiple SNVs may be mapped onto each structure, enabling 3D visualization of SNV clusters and their relationship to functionally annotated positions. We illustrate the utility of MuPIT Interactive in rationalizing the impact of selected polymorphisms in the PharmGKB database, somatic mutations identified in the Cancer Genome Atlas study of invasive breast carcinomas, and rare variants identified in the Exome Sequencing Project. MuPIT Interactive is freely available for non-profit use at http://mupit.icm.jhu.edu.
Keywords: mutation, protein structure, single nucleotide variant, visualization
Introduction
Modern genomics studies have identified a very large number of human single nucleotide variants (SNVs), many of which reside in protein-coding exons and alter the protein product of the gene via amino acid substitution. Computational prediction of those amino acid substitutions that impact on protein structure and/or function is a growing sub-specialty in bioinformatics (Sunyaev et al. 2001; Ng and Henikoff 2003; Ferrer-Costa et al. 2005; Mi et al. 2005; Bromberg and Rost 2007; Karchin et al. 2007; Carter et al. 2009; Adzhubei et al. 2010; Schwarz et al. 2010). Most prediction methods generate a score for each SNV, which is potentially useful for experimental prioritization. A major challenge in this field is to generate hypotheses to explain why specific SNVs predicted to impact protein structure and/or function are biologically important. One approach is to assess an SNV within the context of the three-dimensional structure of the protein in which it occurs.
Here we present a new web-based application designed to enable users who are neither bioinformatics nor protein structure experts to explore mutation data through dynamic visualization. This application represents a significant improvement on existing web tools (Yue et al. 2006; Singh et al. 2008; Reva et al. 2011) designed to map and visualize SNVs on protein structure in terms of (i) ease of use, (ii) interactive and customizable display, (iii) ability to map multiple mutations onto a single structure in order to visualize clustering patterns, and (iv) visualization of the three-dimensional distances between mutations and position-specific annotations.
Methods and Implementation
Mapping between protein and genomic coordinates
MuPIT is driven by an underlying relational database, which contains mappings from genomic positions to PDB structure coordinates, using methods developed in our LS-SNP/PDB application (Ryan et al. 2009). Briefly, a software pipeline aligns protein sequences from PDB structures to human protein sequences in UniProtKB (UniProt 2012), using BLAT (Kent 2002). UniProtKB protein sequences are aligned to RefSeq mRNA transcript sequences with tBLASTn (Gertz et al. 2006), and mRNA transcript sequences are aligned to human genomic DNA with BLAT. Currently, only the canonical isoform of each UniProtKB protein sequence is supported. PDB sequence information is taken from a local mirror of the PDB database (Berman et al. 2000). The UniProt sequences and feature annotations are taken from a local mirror of the UniProt KnowledgeBase (UniProt 2012). Alignments are calculated on a high-performance computing (HPC) cluster, managed by the Son of Grid Engine (SGE) batch-queuing system. The resulting annotations and alignments are stored in a MySQL database, which is updated on a monthly basis to ensure synchrony with its external sources.
Coverage
MuPIT currently provides at least one viewable PDB structure for 3,641 human proteins (18% of the 20,226 SwissProt-curated proteins in UniProtKB (UniProt 2012) and 924,857 codons in protein-coding exons (8.2% of the 11,291,814 total in SwissProt). Coverage of cancer-related genes in particular is somewhat higher: 45% of 487 genes in the Cancer Gene Census (Futreal et al. 2004)(March 2012 version) and 16% of the codons in these genes are covered in the current version of MuPIT.
User Interface
After input of a list of SNVs, by genomic coordinates, via text box or file upload, MuPIT returns a list of all PDB structures that contain at least one SNV. Queries take from one second to one minute, depending on the number of variants submitted, up to a maximum of 2500. Users can preview a description of each PDB structure and all annotations available, to enable prioritization of interesting hits (Figure 1). After selecting a PDB structure, the user is led to an interactive visualization page (Figure 2). An enhanced Jmol applet displays all mutations mapped onto the structure (Jmol is an open-source Java viewer for three-dimensional chemical structures). A menu of Drawing options enables highlighting of mutations and annotated positions with text, color maps, and space-fill display. The Drawing options menu is designed to make interactive viewing substantially easier than if the user had to work directly with the rather complex native menu system utilized by Jmol (Figure 2). Note that most operating systems will ask users for permission before allowing a Java applet to run. Users can export publication quality images from MuPIT using the native Jmol menu (detailed instructions at http://mupit.icm.jhu.edu/Help.html).
Fig 1. Input toy example of the genomic positions of six SNVs on chromosome 21.

MuPIT returns a table of SNV positions that could not be mapped to PDB structures and a table of positions that were successfully mapped to PDB structures. For each PDB structure, a description and list of available annotations is included in the table.
Fig 2. Interactive visualization page for a selected PDB structure.

A “Mutations” table lists all positions mapped to the structure. Clicking on a row of the table produces a text label for that mutation on the protein display. A second table lists all positional and region-based features. A protein viewer window running an enhanced Jmol applet can be manipulated interactively by the user. The protein viewer includes a Drawing Options menu to simplify display configuration.
Hardware/software requirements
MuPIT runs on Windows, Mac OS, and Unix operating systems within a web browser. No specialized hardware or software is required. To our knowledge, all current web browsers are supported. A Java plug-in for the browser is necessary to run Jmol. Installing the latest version of the plug-in is recommended.
Discussion
Rationalizing mutation impact
MuPIT visualization of mutations can be biologically informative. Here we provide several examples of how MuPIT can elucidate the biochemical implications of variants found in the database of pharmacogenomic variants, PharmGKB (McDonagh et al. 2011), Cancer Genome Atlas studies of somatic mutations, and the ESP6500 [Exome Variant Server, NHLBI GO Exome Sequencing Project, Seattle, URL http://evs.gs.washington.edu/EVS/ (15/12/2012 accessed)]. Table 1 lists all SNV positions described in the examples below, in both genomic and protein structure coordinates. The datasets of variants from TCGA and ESP6500 used in these worked examples are included in the Supplementary Materials.
Table 1. Examples of genes and SNVs mapped by MuPIT onto protein structures.
SNVs were taken from PharmGKB,
| Source | Gene Symbol | Genomic Coordinate (NCBI GRCh37) | Protein Structure Residue Coordinate |
|---|---|---|---|
|
| |||
| PharmGKB | GSTP1 | chr11: 67352689 | 3gss: 104 |
|
| |||
| NAT2 | chr8: 18258370 | 2pfr: 286 | |
|
| |||
| Cancer Genome Atlas breast invasive cancer study | RUNX1 | chr21: 36252860 | 1h9d: 141 |
| chr21: 36252946 | 1h9d: 112 | ||
| chr21: 36252880 | 1h9d: 134 | ||
| chr21: 36231791 | 1h9d: 171 | ||
| chr21: 36252958 | 1h9d: 108 | ||
|
| |||
| ERBB2 | chr17: 37880220 | 3pp0: 755 | |
| chr17: 37881000 | 3pp0: 777 | ||
| chr17: 37881332 | 3pp0: 842 | ||
| chr17: 37880261 | 3pp0: 769 | ||
|
| |||
| CHEK2 | chr22: 29092948 | 2cn5: 346 | |
|
| |||
| ESP6500 with MAF<1% | HRH1 | chr3: 11300949 | 3rze:76 |
| chr3: 11300958 | 3rze:79 | ||
| chr3: 11301303 | 3rze:194 | ||
| chr3: 11301309 | 3rze:196 | ||
|
| |||
| SCN5A | chr3: 38592152 | 4dck: 1904 | |
| chr3: 38592162 | 4dck: 1901 | ||
| chr3: 38592170 | 4dck: 1898 | ||
| chr3: 38592171 | 4dck: 1898 | ||
| chr3: 38592174 | 4dck: 1897 | ||
| chr3: 38592339 | 4dck: 1842 | ||
| chr3: 38592356 | 4dck: 1836 | ||
| chr3: 38592369 | 4dck: 1832 | ||
| chr3: 38592386 | 4dck: 1826 | ||
| chr3: 38592387 | 4dck: 1826 | ||
| chr3: 38592428 | 4dck: 1812 | ||
| chr3: 38592474 | 4dck: 1797 | ||
PharmGKB
GSTP1
Several common polymorphisms in this gene, encoding the enzyme glutathione S-transferase Pi (GST), have been associated with differential toxicity subsequent to platinum compound chemotherapy, a standard-of-care treatment for many solid tumors (Booton et al. 2006; Kim et al. 2009; Ruzzo et al. 2007; Stoehlmacher et al. 2004). One mechanism of resistance to platinum drugs involves their conjugation with glutathione by GST and subsequent export from the cell via the ABC transporter MRP2 (Dietel 2007). All GSTP1 variants in PharmGKB were mapped onto 53 unique PDB structures by MuPIT. We focused our attention on PDB 3gss, in which GST is co-complexed with platinum and ethacrynic acid conjugated with glutathione (Oakley et al. 1997). In this structure, the variant I104V (rs1695) is adjacent to the glutathione binding site, which could help to explain why variation at this position could interfere with conjugation of glutathione to platinum, thereby leading to resistance to platinum compound toxicity (Figure 3).
Fig 3. Glutathione S-transferase in complex with platinum and ethacrynic acid conjugated with glutathione.

A PharmGKB polymorphic variant known to be associated with resistance to platinum compound toxicity (green) can be seen proximal to the binding site of glutathione (pink). Glutathione, ethacrynic acid and platinum are shown as ball and stick.
NAT 2
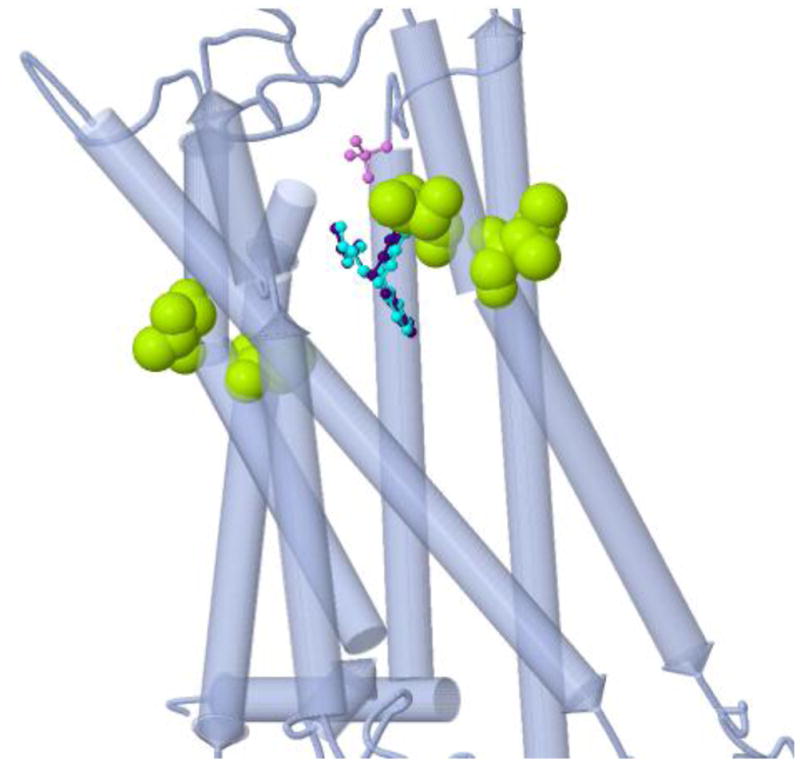
This gene encodes the enzyme N-acetyltransferase 2, which is involved in detoxification of carcinogens and of hydrazine and arylamine drugs (Weber 1987). The detoxification reaction involves removal of an acetyl group from the co-factor acetyl coenzyme A, which is transferred to the toxic substrate. The polymorphic variant G286E (rs1799931) is associated with a slow acetylator phenotype, which confers increased risk of bladder, breast, and esophageal cancers and increased toxicity for aromatic amine and hydrazine drugs (Garcia-Closas et al. 2005; Marcus et al. 2000; van der Hel et al. 2003; Sillanpaa et al. 2005; Morita et al. 1998). MuPIT maps the variant onto the PDB structure of the NAT2-CoA complex (2pfr)(SGX unpublished), revealing its close proximity to the CoA binding site, and hence its potential to interfere with cofactor binding (Suppl. Figure 1).
Cancer Genome Atlas
Mutations from The Cancer Genome Atlas breast invasive carcinoma study (Cancer Genome Atlas 2012) were downloaded from the Firehose website at the Broad Institute (Acknowledgments). The 20,333 mutations identified as “Missense” by Firehose were scored with CHASM software (Carter et al., 2009). The top-ranked 2500 mutations were then submitted to MuPIT Interactive. MuPIT mapped these mutations onto 2929 PDB structures, representing 447 unique proteins. We filtered our results to include only 676 PDB structures (85 unique proteins) for which MuPIT reported position-specific annotations about binding sites and mutagenesis experiments. We used MuPIT’s structure viewer to inspect these 85 proteins and describe three results of interest below.
RUNX1
Five somatic RUNXI mutations were mapped onto PDB structures of RUNX1: 1e50 (dimer of the RUNX1-CBFβ heterodimer complex), 1h9d (RUNX1-CBFβ-DNA ternary complex), 1hjb (RUNX1 andC/EBPbeta bzip homodimer bound to DNA), 1io4(RUNX1, CBFβ, and C/EBPbeta bzip homodimer bound to DNA), and 1ljm (RUNX1 homo-dimer). RUNX1 structural rearrangements have previously been associated with lymphoid carcinomas (Liu et al. 2005; Zhang et al. 2012). The superimposition of mutations and annotations in the MuPIT structure viewer suggests that two of the RUNX1 missense mutations are likely to be functional: one occurs at a DNA-binding site and another at a chloride-binding site (Figure 2). The importance of RUNX1 (and its binding partner CBFβ) in breast cancer was only recently discovered (Cancer Genome Atlas 2012; Ellis et al. 2012).
ERBB2
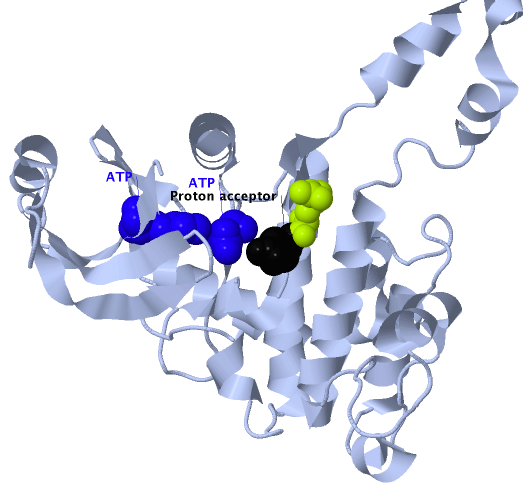
Four somatic ERBB2 mutations were mapped onto the druggable tyrosine kinase domain of ERBB2: PDB structure 3pp0 (Aertgeerts et al. 2011). The viewer shows that one of the mutations is adjacent to the ATP-binding site and another is adjacent to the active (proton acceptor) site (Suppl. Figure 2). All four mutations are located close to somatic mutations previously identified in either glioblastoma multiforme, ovarian cancer, lung adenocarcinoma or gastric adenocarcinoma (UniProt 2012).
CHEK2
A single somatic mutation mapped onto the kinase domain of CHEK2 (found in 24 PDB structures, including 2cn5 and 2cn8) (Oliver et al. 2006). The viewer reveals that the mutated amino acid residue is directly adjacent to the active (proton acceptor) site, and that alanine mutagenesis at this site resulted in loss of kinase activity (UniProt 2012) (Suppl. Figure 3). CHEK2 is a known breast cancer susceptibility gene (Bartek and Lukas 2003).
Exome sequencing project
The ESP6500 is a catalog of variants discovered in next-generation, whole exome sequencing of 6503 individuals with heart, lung and blood disorders. We used MuPIT to map all non-silent ESP6500 variants with MAF < 1% onto PDB structures of all human G-protein coupled receptors (GPCRs) and ion channels listed in the IUPhar database(Harmar et al. 2009) of pharmacologically interesting genes.
HRH1
Nine variants mapped onto the structure of the histamine H1 GPCR, in complex with the antihistamine drug doxepin (PDB ID 3rze)(Shimamura et al. 2011) (Figure 4). Four of the variants (at residue positions 76, 79, 194, 196) are clustered around the doxepin in 3D, but not in one-dimensional space. These variants are interesting candidates for modulators of doxepin response.
Fig 4. Histamine H1 receptor in complex with doxepin (antihistamine drug).
Nine variants from the ESP6500 (all with MAF < 1%) were mapped onto the PDB structure 3rze (Shimamura et al. 2011). Four variants that clustered proximal to the doxepin binding site are shown. Variant positions appear as green balls. Doxepin E-isomer is shown in dark blue. Doxepin Z-isomer is shown in light blue. Phosphate ion is shown in violet.
SCN5A
Sixteen variants mapped onto a structure of this voltage-gated sodium channel, in complex with fibroblast growth factor 13 (FGF13) and a calcium signaling protein, calmodulin (CaM) (PDB ID 4dck)(Wang et al. 2012). Surprisingly, twelve of the variants form a distinct cluster proximal to the three-way binding interface of the sodium channel, the fibroblast growth factor and calmodulin. The positional annotations show that multiple variants associated with both long QT and Brugada syndromes and with sodium channel regulation, are also close to this interface (Figure 5). It is likely that mutations near the interface alter the binding affinities of the three proteins for each other. In particular, two of the variants (at positions 1897 and 1898) are close to position 1895, a amino acid residue known to make a critical contact between the sodium channel and FGF13 (Wang et al. 2012). A possible explanation for this cluster of variants is that many ESP6500 individuals have heart disorders, and would therefore as a group be more likely to harbor inherited genetic variants relevant to an altered sodium channel current than the general population. This example shows how MuPIT can be used to identify putative functionally important variants, within a large collection of variants from a cohort of interest, by their co-location in three dimensions on protein structures.
Fig 5. Voltage-gated sodium channel in complex with fibroblast growth factor 13 and calmodulin.

Twelve variants from ESP6500 (MAF<1%) were mapped onto PDB structure 4dck (Wang et al. 2012). The variants are proximal to the interface of the three proteins and variants previously observed in Brugada and long QT syndromes. ESP6500 variant positions appear as green balls. Brugada and long QT variants appear as magenta balls.
Comparison to existing tools
Several excellent web tools are already available for mapping and viewing SNVs on 3D protein structures (Yue et al. 2006; Singh et al. 2008; Ryan et al. 2009; Reva et al. 2011). However, MuPIT provides several unique features not found in other tools. It is able both to handle large customized batches of SNVs from genomic positions and to visualize multiple SNVs on the same structure. Thus, users can examine thousands of SNVs from a database or cohort of interest (e.g., ESP6500, COSMIC, TCGA etc), and visualize the relationship between SNVs in 3D (SNVs located close to one another in three-dimensional space may in fact be distant from each other in one-dimensional sequence space). By contrast, SNPs3D (Yue et al. 2006) and mutDB (Singh et al. 2008) provide the user with a pre-determined list of SNVs in a structure of interest that have been reported in public databases, e.g., NCBI’s dbSNP (Sherry et al. 2001) or UniProtKB’s Swiss Variant Pages (UniProt 2012). Similarly, LS-SNP/PDB (Ryan et al. 2009) only supports SNVs reported in dbSNP whilst LS-SNP/PDB, Mutation Assessor, and mutDB do not permit viewing of multiple SNVs on a protein structure. To our knowledge, Mutation Assessor is the only other tool that allows large custom batch inputs of SNVs. Mutation Assessor is a multi-purpose tool that provides multiple sequence alignments, mutation scores, and positional annotations. However, its interactive protein viewer is not integrated with its annotations and is not designed for easy customization, such as that we provide with MuPIT’s Drawing Options toolbar.
Summary and future plans
We have presented MuPIT, a new and publicly available web tool designed to map a large number of SNVs from genomic coordinates to protein structure coordinates. MuPIT enables users to interactively visualize SNVs in conjunction with position-specific annotations, such as binding sites, experimental mutagenesis results, and locations of known polymorphisms and somatic mutations in cancer. Using MuPIT, a researcher should be able to ascertain whether or not a given SNV of interest is likely to be of functional importance.
We plan to continually upgrade the capabilities of MuPIT over time. We propose to add support for alternative splicing isoforms and provide more detailed information about PDB structure quality and evolutionary conservation. We also plan to improve the query interface, so that users will have the option to search with UniProtKB, RefSeq or Ensembl identifiers, to specify alleles at a position of interest, and to download Excel or flat file summaries of results. We plan to integrate MuPIT into our CRAVAT web service (Douville et al. 2013), thereby enabling users to combine visualization with predictive rankings of SNVs as likely cancer drivers (Carter et al. 2009), protein loss of function (Carter et al. 2013) etc.
Supplementary Material
PharmGKB polymorphic variant associated with the slow acetylator phenotype and increased risk for several cancers and drug toxicity, is adjacent to the coenzyme A binding site. Acetylation of carcinogens and other toxic substrates requires transfer of an acetyl group from acetyl coA. The variant potentially interferes with coA binding. Variant positions appear as green balls.
{kind=link}
Four TCGA breast cancer somatic mutations mapped onto the PDB structure 3pp0 (Aertgeerts et al. 2011). One of the mutations lies adjacent to the ATP binding site (pink) whereas another is adjacent to the active site (blue). Variant positions appear as green balls.
{kind=link}
A single TCGA breast cancer somatic mutation mapped onto the PDB structure 2cn5 (Oliver et al. 2006). CHEK2 is a known breast cancer susceptibility gene. The mutation depicted is directly adjacent to the active site. Variant positions appear as green balls.
{kind=link}
Table 2. Comparison of MuPIT interactive with similar available tools.
Only tools with functional websites as of February 2013 are included.
| MuPIT Interactive | LS-SNP/PDB | MutDB | SNPs3D | Mutation Assessor | |
|---|---|---|---|---|---|
| Website URL | mupit.icm.jhu.edu/ | http://ls-snp.icm.jhu.edu/ls-snp-pdb/ | mutdb.org/ | snps3d.org/ | mutationassessor.org |
| Custom input? | Yes. Accepts user genomic coordinates | No. Limited to variants in dbSNP | No. Limited to variants in dbSNP and SwissProt | No. Limited to variants in dbSNP | Yes. Accepts user genomic or protein coordinates |
| 3D-Display | Interactive and customizable via GUI* | Not interactive | Interactive | Interactive and customizable for power users by programming | Interactive |
| Simultaneous display of multiple variants on same structure | Yes | No | No | Yes | No |
| Position-Specific Annotation on protein structure | Yes | No | No | No | No |
| Batch Entry of Variants | Yes (high-throughput) | No | No | No | Yes (high-throughput) |
| Interface | Browser-based | Browser-based | Browser-based | Browser-based | Browser-based |
Graphical User Interface
Acknowledgments
We used the Broad Institute Firehose standardization run from 7 July, 2012, which may be foundhere in the TCGA Data Coordination Center: http://gdac.broadinstitute.org/runs/stddata__2012_07_07/data/BRCA/20120707/gdac.broadinstitute.org_BRCA.Mutation_Packager_Calls.Level_3.2012070700.0.0.tar.gz
Funding: National Institutes of Health CA 152432, 3U24CA143858-2S1, National Science Foundation DBI 0845275
Appendix
Server-side Implementation Details
MuPIT is a Java servlet, accessible to users via a web browser equipped with the Java plugin. The servlet encapsulates a series of Django applications: A Jmol applet for PDB visualization, a database (DB) interfacer, and an overview page constructor (Howard 2013). Javascript functions are used to allow for user interactions between the overview tables and the 3D visualization.
The MuPIT servlet runs on a Dell PowerEdge C1100 server, with four six-core Intel Xeon E5645 2.4GHz cpus (24 cores total), and 96GB of RAM.
Footnotes
Conflict of Interest: none declared.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nature methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aertgeerts K, Skene R, Yano J, Sang BC, Zou H, Snell G, Jennings A, Iwamoto K, Habuka N, Hirokawa A, Ishikawa T, Tanaka T, Miki H, Ohta Y, Sogabe S. Structural analysis of the mechanism of inhibition and allosteric activation of the kinase domain of HER2 protein. The Journal of biological chemistry. 2011;286:18756–18765. doi: 10.1074/jbc.M110.206193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer cell. 2003;3:421–429. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic acids research. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booton R, Ward T, Heighway J, Ashcroft L, Morris J, Thatcher N. Glutathione-S-transferase P1 isoenzyme polymorphisms, platinum-based chemotherapy, and non-small cell lung cancer. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2006;1:679–683. [PubMed] [Google Scholar]
- Bromberg Y, Rost B. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic acids research. 2007;35:3823–3835. doi: 10.1093/nar/gkm238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter H, Chen S, Isik L, Tyekucheva S, Velculescu VE, Kinzler KW, Vogelstein B, Karchin R. Cancer-specific high-throughput annotation of somatic mutations: computational prediction of driver missense mutations. Cancer research. 2009;69:6660–6667. doi: 10.1158/0008-5472.CAN-09-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter H, Douville C, Yeo G, Stenson PD, Cooper DN, Karchin R. Identifying Mendelian disease genes with the Variant Effect Scoring Tool. BMC genomics. 2013 doi: 10.1186/1471-2164-14-S3-S3. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietel M. Recent Results in Cancer Research. Springer; 2007. Targeted Therapies in Cancer; p. 176. [Google Scholar]
- Douville C, Carter H, Kim R, Niknafs N, Diekhans M, Stenson PD, Cooper DN, Ryan M, Karchin R. CRAVAT: cancer-related analysis of variants toolkit. Bioinformatics. 2013;29:647–648. doi: 10.1093/bioinformatics/btt017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, Van Tine BA, Hoog J, Goiffon RJ, Goldstein TC, Ng S, Lin L, Crowder R, Snider J, Ballman K, Weber J, Chen K, Koboldt DC, Kandoth C, Schierding WS, McMichael JF, Miller CA, Lu C, Harris CC, McLellan MD, Wendl MC, DeSchryver K, Allred DC, Esserman L, Unzeitig G, Margenthaler J, Babiera GV, Marcom PK, Guenther JM, Leitch M, Hunt K, Olson J, Tao Y, Maher CA, Fulton LL, Fulton RS, Harrison M, Oberkfell B, Du F, Demeter R, Vickery TL, Elhammali A, Piwnica-Worms H, McDonald S, Watson M, Dooling DJ, Ota D, Chang LW, Bose R, Ley TJ, Piwnica-Worms D, Stuart JM, Wilson RK, Mardis ER. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012;486:353–360. doi: 10.1038/nature11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer-Costa C, Gelpi JL, Zamakola L, Parraga I, de la Cruz X, Orozco M. PMUT: a web-based tool for the annotation of pathological mutations on proteins. Bioinformatics. 2005;21:3176–3178. doi: 10.1093/bioinformatics/bti486. [DOI] [PubMed] [Google Scholar]
- Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR. A census of human cancer genes. Nat Rev Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Closas M, Malats N, Silverman D, Dosemeci M, Kogevinas M, Hein DW, Tardon A, Serra C, Carrato A, Garcia-Closas R, Lloreta J, Castano-Vinyals G, Yeager M, Welch R, Chanock S, Chatterjee N, Wacholder S, Samanic C, Tora M, Fernandez F, Real FX, Rothman N. NAT2 slow acetylation, GSTM1 null genotype, and risk of bladder cancer: results from the Spanish Bladder Cancer Study and meta-analyses. Lancet. 2005;366:649–659. doi: 10.1016/S0140-6736(05)67137-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertz EM, Yu YK, Agarwala R, Schaffer AA, Altschul SF. Composition-based statistics and translated nucleotide searches: improving the TBLASTN module of BLAST. BMC biology. 2006;4:41. doi: 10.1186/1741-7007-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmar AJ, Hills RA, Rosser EM, Jones M, Buneman OP, Dunbar DR, Greenhill SD, Hale VA, Sharman JL, Bonner TI, Catterall WA, Davenport AP, Delagrange P, Dollery CT, Foord SM, Gutman GA, Laudet V, Neubig RR, Ohlstein EH, Olsen RW, Peters J, Pin JP, Ruffolo RR, Searls DB, Wright MW, Spedding M. IUPHAR-DB: the IUPHAR database of G protein-coupled receptors and ion channels. Nucleic Acids Res. 2009;37:D680–685. doi: 10.1093/nar/gkn728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard M. Jmol: an open-source Java viewer for chemical structures in 3D. 2013 http://www.jmol.org/
- Karchin R, Monteiro AN, Tavtigian SV, Carvalho MA, Sali A. Functional impact of missense variants in BRCA1 predicted by supervised learning. PLoS computational biology. 2007;3:e26. doi: 10.1371/journal.pcbi.0030026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ. BLAT--the BLAST-like alignment tool. Genome research. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Kim MK, Chung HH, Kim JW, Park NH, Song YS, Kang SB. Genetic polymorphisms affecting clinical outcomes in epithelial ovarian cancer patients treated with taxanes and platinum compounds: a Korean population-based study. Gynecologic oncology. 2009;113:264–269. doi: 10.1016/j.ygyno.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Liu S, Shen T, Huynh L, Klisovic MI, Rush LJ, Ford JL, Yu J, Becknell B, Li Y, Liu C, Vukosavljevic T, Whitman SP, Chang KS, Byrd JC, Perrotti D, Plass C, Marcucci G. Interplay of RUNX1/MTG8 and DNA methyltransferase 1 in acute myeloid leukemia. Cancer research. 2005;65:1277–1284. doi: 10.1158/0008-5472.CAN-04-4532. [DOI] [PubMed] [Google Scholar]
- Marcus PM, Vineis P, Rothman N. NAT2 slow acetylation and bladder cancer risk: a meta-analysis of 22 case-control studies conducted in the general population. Pharmacogenetics. 2000;10:115–122. doi: 10.1097/00008571-200003000-00003. [DOI] [PubMed] [Google Scholar]
- McDonagh EM, Whirl-Carrillo M, Garten Y, Altman RB, Klein TE. From pharmacogenomic knowledge acquisition to clinical applications: the PharmGKB as a clinical pharmacogenomic biomarker resource. Biomarkers in medicine. 2011;5:795–806. doi: 10.2217/bmm.11.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Lazareva-Ulitsky B, Loo R, Kejariwal A, Vandergriff J, Rabkin S, Guo N, Muruganujan A, Doremieux O, Campbell MJ, Kitano H, Thomas PD. The PANTHER database of protein families, subfamilies, functions and pathways. Nucleic acids research. 2005;33:D284–288. doi: 10.1093/nar/gki078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita S, Yano M, Tsujinaka T, Ogawa A, Taniguchi M, Kaneko K, Shiozaki H, Doki Y, Inoue M, Monden M. Association between genetic polymorphisms of glutathione S-transferase P1 and N-acetyltransferase 2 and susceptibility to squamous-cell carcinoma of the esophagus. International journal of cancer Journal international du cancer. 1998;79:517–520. doi: 10.1002/(sici)1097-0215(19981023)79:5<517::aid-ijc12>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic acids research. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley AJ, Rossjohn J, Lo Bello M, Caccuri AM, Federici G, Parker MW. The three-dimensional structure of the human Pi class glutathione transferase P1-1 in complex with the inhibitor ethacrynic acid and its glutathione conjugate. Biochemistry. 1997;36:576–585. doi: 10.1021/bi962316i. [DOI] [PubMed] [Google Scholar]
- Oliver AW, Paul A, Boxall KJ, Barrie SE, Aherne GW, Garrett MD, Mittnacht S, Pearl LH. Trans-activation of the DNA-damage signalling protein kinase Chk2 by T-loop exchange. The EMBO journal. 2006;25:3179–3190. doi: 10.1038/sj.emboj.7601209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic acids research. 2011;39:e118. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzzo A, Graziano F, Loupakis F, Rulli E, Canestrari E, Santini D, Catalano V, Ficarelli R, Maltese P, Bisonni R, Masi G, Schiavon G, Giordani P, Giustini L, Falcone A, Tonini G, Silva R, Mattioli R, Floriani I, Magnani M. Pharmacogenetic profiling in patients with advanced colorectal cancer treated with first-line FOLFOX-4 chemotherapy. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2007;25:1247–1254. doi: 10.1200/JCO.2006.08.1844. [DOI] [PubMed] [Google Scholar]
- Ryan M, Diekhans M, Lien S, Liu Y, Karchin R. LS-SNP/PDB: annotated non-synonymous SNPs mapped to Protein Data Bank structures. Bioinformatics. 2009;25:1431–1432. doi: 10.1093/bioinformatics/btp242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nature methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic acids research. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura T, Shiroishi M, Weyand S, Tsujimoto H, Winter G, Katritch V, Abagyan R, Cherezov V, Liu W, Han GW, Kobayashi T, Stevens RC, Iwata S. Structure of the human histamine H1 receptor complex with doxepin. Nature. 2011;475:65–70. doi: 10.1038/nature10236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillanpaa P, Hirvonen A, Kataja V, Eskelinen M, Kosma VM, Uusitupa M, Vainio H, Mitrunen K. NAT2 slow acetylator genotype as an important modifier of breast cancer risk. International journal of cancer Journal international du cancer. 2005;114:579–584. doi: 10.1002/ijc.20677. [DOI] [PubMed] [Google Scholar]
- Singh A, Olowoyeye A, Baenziger PH, Dantzer J, Kann MG, Radivojac P, Heiland R, Mooney SD. MutDB: update on development of tools for the biochemical analysis of genetic variation. Nucleic acids research. 2008;36:D815–819. doi: 10.1093/nar/gkm659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoehlmacher J, Park DJ, Zhang W, Yang D, Groshen S, Zahedy S, Lenz HJ. A multivariate analysis of genomic polymorphisms: prediction of clinical outcome to 5-FU/oxaliplatin combination chemotherapy in refractory colorectal cancer. British journal of cancer. 2004;91:344–354. doi: 10.1038/sj.bjc.6601975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunyaev S, Ramensky V, Koch I, Lathe W, 3rd, Kondrashov AS, Bork P. Prediction of deleterious human alleles. Human molecular genetics. 2001;10:591–597. doi: 10.1093/hmg/10.6.591. [DOI] [PubMed] [Google Scholar]
- UniProt C. Reorganizing the protein space at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2012;40:D71–75. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Hel OL, Peeters PH, Hein DW, Doll MA, Grobbee DE, Kromhout D, Bueno de Mesquita HB. NAT2 slow acetylation and GSTM1 null genotypes may increase postmenopausal breast cancer risk in long-term smoking women. Pharmacogenetics. 2003;13:399–407. doi: 10.1097/00008571-200307000-00005. [DOI] [PubMed] [Google Scholar]
- Wang C, Chung BC, Yan H, Lee SY, Pitt GS. Crystal structure of the ternary complex of a NaV C-terminal domain, a fibroblast growth factor homologous factor, and calmodulin. Structure. 2012;20:1167–1176. doi: 10.1016/j.str.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber WW. The acetylator genes and drug response. Oxford University Press; New York: 1987. [Google Scholar]
- Yue P, Melamud E, Moult J. SNPs3D: candidate gene and SNP selection for association studies. BMC bioinformatics. 2006;7:166. doi: 10.1186/1471-2105-7-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, Lu C, Chen SC, Wei L, Collins-Underwood JR, Ma J, Roberts KG, Pounds SB, Ulyanov A, Becksfort J, Gupta P, Huether R, Kriwacki RW, Parker M, McGoldrick DJ, Zhao D, Alford D, Espy S, Bobba KC, Song G, Pei D, Cheng C, Roberts S, Barbato MI, Campana D, Coustan-Smith E, Shurtleff SA, Raimondi SC, Kleppe M, Cools J, Shimano KA, Hermiston ML, Doulatov S, Eppert K, Laurenti E, Notta F, Dick JE, Basso G, Hunger SP, Loh ML, Devidas M, Wood B, Winter S, Dunsmore KP, Fulton RS, Fulton LL, Hong X, Harris CC, Dooling DJ, Ochoa K, Johnson KJ, Obenauer JC, Evans WE, Pui CH, Naeve CW, Ley TJ, Mardis ER, Wilson RK, Downing JR, Mullighan CG. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–163. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PharmGKB polymorphic variant associated with the slow acetylator phenotype and increased risk for several cancers and drug toxicity, is adjacent to the coenzyme A binding site. Acetylation of carcinogens and other toxic substrates requires transfer of an acetyl group from acetyl coA. The variant potentially interferes with coA binding. Variant positions appear as green balls.
Four TCGA breast cancer somatic mutations mapped onto the PDB structure 3pp0 (Aertgeerts et al. 2011). One of the mutations lies adjacent to the ATP binding site (pink) whereas another is adjacent to the active site (blue). Variant positions appear as green balls.
A single TCGA breast cancer somatic mutation mapped onto the PDB structure 2cn5 (Oliver et al. 2006). CHEK2 is a known breast cancer susceptibility gene. The mutation depicted is directly adjacent to the active site. Variant positions appear as green balls.