

Abstract
The goal of this study was to compare the effects of two inflammatory modulators, erythropoietin (EPO) and anakinra, on functional recovery and brain gene expression following a cortical contusion impact (CCI) injury. Dosage regimens were designed to provide serum concentrations in the range obtained with clinically approved doses. Functional recovery was assessed using both motor and spatial learning tasks and neuropathological measurements conducted in the cortex and hippocampus. Microarray-based transcriptional profiling was used to determine the effect on gene expression at 24 h, 72 h, and 7 days post-CCI. Ingenuity Pathway Analysis was used to evaluate the effect on relevant functional categories. EPO and anakinra treatment resulted in significant changes in brain gene expression in the CCI model demonstrating acceptable brain penetration. At all three time points, EPO treatment resulted in significantly more differentially expressed genes than anakinra. For anakinra at 24 h and EPO at 24 h, 72 h, and 7 days, the genes in the top 3 functional categories were involved in cellular movement, inflammatory response and cell-to-cell signaling. For EPO, the majority of the genes in the top 10 canonical pathways identified were associated with inflammatory and immune signaling processes. This was true for anakinra only at 24 h post-traumatic brain injury (TBI). The immunomodulation effects of EPO and anakinra did not translate into positive effects on functional behavioral and lesion studies. Treatment with either EPO or anakinra failed to induce significant beneficial effects on recovery of function or produce any significant effects on the prevention of injury induced tissue loss at 30 days post-injury. In conclusion, treatment with EPO or anakinra resulted in significant effects on gene expression in the brain without affecting functional outcome. This suggests that targeting these inflammatory processes alone may not be sufficient for preventing secondary injuries after TBI.
Keywords: traumatic brain injury, recovery of function, cortical contusion impact, gene expression, erythropoietin, anakinra
Introduction
Traumatic brain injury (TBI) is among the leading causes of acute and chronic disability in the United States. Out of the 1.7 million Americans that endure a TBI each year, over 50,000 die (Faul et al., 2010), making recovery of function a major public health issue. The first or primary injury relates to the initial injury caused by direct damage to the brain and only injury prevention will reduce the consequences of the primary injury. The second is indirect and progressive and is referred to as secondary injury. The etiology of the secondary cascade resulting from TBI is likely due to interrelated processes including mitochondrial energy failure, excessive generation of reactive oxygen species, activation of destructive enzymes, membrane disruption, neuronal death, thrombosis due to intravascular coagulation in small vessels, increased synaptic concentrations of excitatory amino acids, and activation of innate inflammatory responses (Wang et al., 2006; Jennings et al., 2008; McConeghy et al., 2012). Preventing the inflammatory response is considered a high potential target for neuroprotection after TBI (Schouten, 2007; Jennings et al., 2008). During secondary injury, early inflammation is triggered by multiple complex signals including, intracellular signaling, reactive oxygen and nitrogen species and loss of cellular homeostasis, all of which is mediated by microglia and astrocytes (Mathew et al., 1994; Juliet et al., 2008; Rock and Kono, 2008). An increase in the brain and cerebral spinal fluid pro-inflammatory cytokines [tumor necrosis factorα (TNFα), interleukin (IL)-1β, IL-6], and anti-inflammatory cytokines [transforming growth factor (TGF-β, IL-4, IL-10)] occurs after injury in experimental TBI models and in patients with TBI (Morganti-Kossmann et al., 2001). Although the precise mechanisms are unknown, the immune response following inflammation directly induces apoptotic death and indirectly increases cell death by signaling cellular death programs (Shojo et al., 2010; Das et al., 2011).
The IL-1 family is a group of cytokines that are important for initiating and regulating immune and inflammatory responses. Animal models suggest that increasing levels of IL-1α and IL-1β exacerbate brain damage, while administering an IL 1 receptor antagonist (IL-1ra) reduces the infarct and improves recovery of function (Feurstein et al., 1997; Touzani et al., 1999). IL-1ra is a protein that in humans is encoded by the IL1RN gene and in rodent by the IL-1rn gene. Preventing IL-1 from binding to the IL-1 type receptor reduces inflammation (Feurstein et al., 1997; Touzani et al., 1999; So et al., 2007). There have been several experiments examining the role of IL-1ra in injury models other than ischemia, including subarachnoid hemorrhage, cryogenic injury, and excitotoxic injury. Following injury, IL-1ra reduced the number of nitric oxide synthase positive cells and improved both anatomical and functional outcomes (Jones et al., 2005). Elevation of IL-1ra after initiation of the inflammatory response is an important part of the auto-regulatory network controlling the inflammatory response (Bartfai et al., 2007). In patients with TBI, high concentrations of IL-1ra and high ratio of IL-1ra/IL-1β were association with better outcome (Hutchinson et al., 2007). IL-1ra reduces susceptibility to excitotoxic brain injury (Hagan et al., 1996), mediates convulsant actions of seizures in transgenic mice (Vezzani et al., 2004) and reduces damage following excitotoxic injury by attenuating neuronal insult and microglial activation in hippocampal slice cultures (Hailer et al., 2005) and in rodent models (Relton and Rothwell, 1992).
Altering this inflammatory pathway, preventing or reducing the inflammatory signal with an IL-1ra following TBI is a promising strategy in limiting the secondary injury. Erythropoietin (epoetin alfa; EPO) and anakinra, a recombinant form of non-glycosylated human IL-1 receptor antagonist are inflammatory modulators proposed as promising neuroprotective agents for the treatment of TBI. EPO, a hematopoietic growth factor that regulates red blood cell production also has significant pleiotropic effects (Boogaerts, 2006; Mammis et al., 2009). EPO has been shown to provide protection in a variety of models of neural damage, including focal ischemic stroke, experimental autoimmune encephalomyelitis, chemically-induced seizures, and blunt force trauma (Brines et al., 2000; Cerami et al., 2002). Subsequent studies have established beneficial effects of EPO administration on behavioral recovery from TBI (Brines et al., 2000; Cerami et al., 2002; Meng et al., 2011). EPO administration has been found to improve motor (Zhang et al., 2009), sensory (Xiong et al., 2008; Zhang et al., 2009), and cognitive function in experimental TBI (Lu et al., 2005; Yatsiv et al., 2005; Mahmood et al., 2007; Xiong et al., 2008; Zhang et al., 2009; Meng et al., 2011) and has also been shown to reduce lesion size (Xiong et al., 2008) and cell death after injury (Yatsiv et al., 2005; Zhang et al., 2009; Meng et al., 2011). Proposed mechanisms for the neuroprotective effect have included the ability of EPO to protect nerve cells from glutamate toxicity, reduce the immune response and inflammatory reaction, enhance nerve recovery, play a role in neurogenesis, prevent neuronal apoptosis, inhibit nitric oxide formation, and prevent oxidative stress (for a review, see Mammis et al., 2009). Research has shown that EPO can cross the blood brain barrier and affect neural tissues, increasing oxygenation to vulnerable tissues (Brines et al., 2000).
There is limited pre-clinical research evaluating treatment with IL-1ra in attenuating neuronal injury. IL-1ra has been shown to be a potential neuroprotective treatment in experimental studies in hippocampal slice cultures after excitotoxic damage (Hailer et al., 2005), in models of focal cerebral ischemia (Relton et al., 1996; Loddick et al., 1997), and in a fluid percussion brain injury model (Toulmond and Rothwell, 1995; Relton et al., 1996; Loddick et al., 1997; Lawrence et al., 1998; Sanderson et al., 1999; Hailer et al., 2005).
The objective of this study was to compare the effects of two inflammatory modulators, EPO and anakinra on functional recovery and gene expression following a unilateral controlled cortical impact (CCI) injury. This study is part of a project that assesses the effectiveness of several drugs on TBI and to identify the mechanism of action using microarray analysis. The ultimate goal is to use these data to design a multi-drug treatment therapy that will also be tested in our TBI model.
Prior to the CCI studies, single- and multiple-dose pharmacokinetic studies were performed in uninjured animals to determine the dose regimens needed to provide average serum concentrations in the range of the concentrations obtained clinically with FDA approved doses.
Experimental procedures
Animals and housing
Male, Sprague-Dawley rats (Harlan, Indianapolis, IN) approximately 3.5 months of age (mean body weight 350 g) were used in this study. All animal and surgical procedures were adhered to as described in the NIH Guide for the Care and Use of Laboratory Animals. The Southern Illinois University Institutional Animal Care and Use Committee (IACUC) and the University of Washington's IACUC reviewed and approved all experimental procedures. Animals were housed in a university-maintained, Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) accredited vivarium, with a 12-h light/dark schedule and a controlled environmental temperature of 22°C in standard housing cages with food and water available ad libitum.
Pharmacokinetic studies
Male Sprague-Dawley rats with surgically implanted jugular vein catheters were obtained from Harlan Laboratories. For the single dose studies (n = 4/study), 100 mg/kg anakinra (Kineret™, Amgen, Thousand Oaks, CA) or 2500 IU/kg EPO (Procrit™, Amgen, Thousand Oaks, CA) were administered by subcutaneous (s.c.) or intraperitoneal injection (i.p.). Blood specimens were collected from the jugular catheter, immediately prior to the dose, and at 1, 2, 4, 6, 12, 24, and 30 h. Samples were collected in microtubes, separated using a centrifuge, and stored at −80°C until assayed. The terminal exponential rate constant (β) was used to determine elimination half-life (T1/2) as 0.693/β. Based on the single dose pharmacokinetics, the multiple doses were calculated to attain clinically relevant steady state concentrations. Targeted average concentrations of erythropoietin and anakinra were 5000–10,000 mIU/ml and 10–25 μg/mL, respectively, based on clinical studies in non-TBI patients receiving FDA approved doses. The initial dose was administered i.p. and subsequent doses administered s.c. every 12 h for 72 h. Blood was collected from a jugular catheter at 0, 1, 2 and then immediately before and 1 h after injections for 72 h. Serum concentrations were determined using commercially available ELISA kits (R&D Systems, Minneapolis, MN).
Experimental injury model
All surgeries were performed under aseptic conditions. The CCI injury model utilized in the present study was based on previous studies (Anderson et al., 2011, 2013; Peterson et al., 2012). Animals were anesthetized using a mixture of isoflurane (2–4%) and oxygen (0.8 L/min). Once the animal was no longer responsive (no ocular or pedal reflexes) the head was shaved and scrubbed with 70% alcohol followed by betadine and placed into a stereotaxic device. A midline incision was made through the skin and the underlying fascia. A circular craniotomy (5.0 mm) was centered 2.4 mm posterior to and 2.4 mm lateral (left) to bregma. The contusion injury was created with a sterile stainless steel impactor tip (4.0 mm in diameter) that was attached to the Benchmark™ stereotaxic impactor (www.myneurolab.com, St. Louis, MO). The injury was induced with an impact speed of 3.0 m/s and depth of 2.5 mm, coming in contact with the dura for 0.5 s before retraction. To maintain normal body temperature (37°C) during surgery and recovery the rats were placed on a warm water recycling bed and pump system (EZ Anesthesia, Palmer, PA). Rats receiving sham surgeries underwent identical surgical preparation as injured animals, with the exception of the impact (received craniotomies), were sutured, and then transferred to recovery. The design was completely counter-balanced to ensure that all animals received the same number of injections, regardless of treatment condition.
Drug administration
Animals were randomly assigned to one of four groups: (a) Sham, (b) CCI-injured erythropoietin (EPO) 2500 IU/kg (c) CCI-injured anakinra at 100 mg/kg or (d) CCI-injured vehicle. Based on the results of the pharmacokinetic studies, in order to obtain peaks concentration within 1–2 h, the initial dose was administered by i.p. injection and subsequent injections were administered by s.c. Doses were administered 2 h, 12 h, 24 h, 36 h, 48 h, 60 h, and 72 h after the CCI injury. Both EPO and anakinra were diluted into 0.9% sterile saline (Sigma Aldrich Co, St. Louis, MO) such that the amount of fluid per kg body weight was the same. Blood samples (750 μ L) were collected in microtubes from the tail vein 1 h after the final injection, separated using a microcentrifuge, and stored at −80°C until assayed. For the functional behavioral studies, a power analysis determined that group sizes of nine results in a power score >0.85 to detect significant differences between the groups.
Gene expression studies
At specified time point's post-CCI (24 h, 72 h, and 7 days) 5 intact sham animals (no craniotomy) and 5 animals in each treatment group were overdosed with a mixture of CO2 (80%) and O2 (20%). The rats were then decapitated, a cardiac blood sample collected and brains were rapidly extracted. To maintain quality control and to assure that all of the brains were injured, each brain was assigned a rating score (1 = no visual sign of trauma; 2 = bruised and swollen cortex; 3 = no remaining cortex or extensive damage) (see Figure 1 in Anderson et al., 2011). Only brains with a score of 2 were used in the subsequent analyses. The brain was then cut into a 4.0 mm coronal slab containing the injury site in a brain matrix (Braintree Scientific, Inc., Braintree, MA) and placed onto an RNAase free cold plate. A 5.0 mm biopsy punch was used to collect the injury site and surrounding cortical tissue (Hoane et al., 2006; Anderson et al., 2011). The tissue punch included all injured cortical tissue and a small strip of pericontusional tissue, with the ventral extent of the punch extending to the corpus callosum. Tissue punches were placed into microcentrifuge tubes, snap frozen and then stored at −80°C. All samples were shipped by overnight carrier to the University of Washington on dry ice.
Processing of the RNA samples, hybridization to the microarray, scanning with the AffymetrixGeneChip® 3000 scanner and univariate analysis of the data using various Bioconductor packages were performed as previously described (Anderson et al., 2011, 2013). Venn diagrams were generated with the Bioconductor limma package. Ingenuity Pathway Analysis (IPA) (Build 131235; Version 11904312; Database Status 02.20.2012; www.ingenuity.com) was used to analyze differentially expressed genes (>1.5-fold up or down-regulated, p < 0.05) using the Core Analysis feature. IPA is a commercial tool that is based on a proprietary database (www.ingenuity.com/) to facilitate the identification of biological themes in microarray gene expression data. IPA maps the rat genes to their human orthologs and uses the human orthologs in its pathway analysis. IPA uses a Right-tailed Fisher's exact test to determine the probability that the number of genes annotated to a given biological function, canonical pathway or transcriptional network in the set of significant genes is due to chance alone. The validation of the data obtained with the microarrays was performed using fluorogenic 5′-nuclease-based assay and quantitative RT-PCR as previously described (Anderson et al., 2011, 2013). The RT-PCR data was normalized to the housekeeping gene, β-actin.
Functional behavioral studies
Sensorimotor/motor assessment
Locomotor placing task.To assess recovery of coordinated, locomotor limb movement, this test was administered on days 2, 4, 6, 8, and 10 post-CCI following methods outlined in previous studies (Hoane et al., 2008b; Goffus et al., 2010; Peterson et al., 2012). Rats were pre-tested to establish baseline levels for 2 days before surgery. On each test day, the rat was placed on an elevated grid floor (56.0 × 54.0 cm) with openings measuring 3.2 × 3.2 cm in size and allowed to freely explore for 120 s. A “foot-fault” occurred when a rat inaccurately placed a limb through one of these openings. Total movement on the grid as measured by the number of lines crossed as well as foot-faults for each limb was recorded. Rats were administered one trial each test day. The total number of foot-faults for the limb contralateral to the injury was the primary dependent variable of interest. The following equation was used to calculate the foot faults as a function of total movement on the grid: (Right sforelimb faults - Left forelimb faults)/lines crossed.
Rotorod. Automated ROTOR-ROD™ (San Diego Instruments, San Diego, CA) testing was performed on post-injury days 8–12 to assess gross motor coordination. Two days of training occurred prior to surgery to establish a pre-injury baseline on the rotorod. Animals were placed on the rotating cylinder (7 cm diameter) against the rotation and the latency to fall (1.3 m) was recorded. On the first day of training, animals received one training trial where they were placed on the cylinder, which was stationary, and replaced when they fell, until they were able to remain on the cylinder for 60 consecutive seconds so that they become acclimated to the device. Following this trial and on all other days of training, the trial was terminated and the latency was recorded when the animal fell off of the cylinder. The initial speed of 5 rpm was maintained for 15 s to allow experimenters to load the rat. Following this 15 s, the speed of the rotation increased from 5 to 50 rpm over the course of 300 s (acceleration rate of 0.15 rpm/s). An infrared beam sensor located below the cylinder recorded the latency for the animal to fall. Each animal received four trials each day with a 15 min inter-trial interval.
Cognitive assessment
Morris water maze (MWM)—reference memory task. All animals were tested in the MWM using a reference memory paradigm which has been widely utilized to assess cognitive performance following TBI (Lindner et al., 1998; Hoane et al., 2003, 2006; Vonder Haar et al., 2011; Peterson et al., 2012). The apparatus consisted of a circular, 180 cm diameter blue plastic pool partially filled with water (22°C) to a depth of approximately 32 cm. A clear plastic platform (10 × 10 cm) was submerged approximately 2 cm below the surface located in the northeast quadrant of the pool for the entirety of the reference memory testing sessions. The animal's progress on the task was evaluated by a video camera affixed above the pool and this data was processed using computer software SMART (San Diego Instruments, San Diego, CA). Animals were assessed on the acquisition of a reference memory task beginning on day 14 post-CCI and were tested for 4 subsequent days. On each testing day, animals received four trials to locate the submerged platform in the pool, starting at one of four release points in random order. The trial was terminated when the rat reached the submerged platform in the northeast quadrant or when 90 s had elapsed. Latencies were recorded, averaged and used as the main dependent variable. If the rat did not find the platform within the 90 s, it was guided to the platform and remained on it for at least 10 s. Each rat remained on the platform for 10 s, after which it was placed in a warm holding cage for at least 15 min before the next trial.
MWM—working memory task. Animals were tested on days 21–23 post-CCI using established methods (Lindner et al., 1998; Hoane et al., 2003, 2006; Vonder Haar et al., 2011; Peterson et al., 2012). The procedure was the same as above, except the platform was submerged at the center of a new randomly chosen quadrant (south-west, north-west, and south-east) each day. Each animal was given four trials per day, starting from one of four randomly chosen release points (inter-trial interval was 15 min). The first trial on each of these days was considered an information trial and was not included in subsequent analyses; the latencies from the last three trials were averaged to form a score for the day. Each trial was terminated when the animal located the platform, or when 90 s had elapsed. Animals not reaching the platform were guided to the platform after the trial ended.
Lesion analysis
At 30 days post-CCI, the rats were euthanized with Euthasol (Virbac Animal Health; 0.3 mL i.p.) and transcardially perfused with 0.9% phosphate-buffered saline (PBS), followed by 10% phosphate buffered formalin (PBF). Brains were post-fixed in PBF for 2 days following removal from the cranium. A 30% sucrose solution was used to cryopreserve the brains 3 days prior to frozen sectioning. Serial, coronal sections (40 μm thickness) were sliced using a sliding microtome on a frozen stage and collected into a cryopreservative solution and stored at −20°C. A series of sections were brush mounted on gelatin-subbed microscope slides, stained with cresyl violet, dehydrated, and cover slipped. The extent of the lesion was analyzed with an Olympus microscope (BX-51) and an Olympus 13.5 megapixel digital camera (DP-70). Images of sections throughout the extent of the injury coordinates were captured using the digital capturing system and area measures of the lesioned tissue were determined using the ImageJ software package (1.43 u, NIH). The Calvalieri method was used to calculate the volumes of the ipsilateral cortex and the contralateral cortex (Coggeshall, 1992; Vonder Haar et al., 2011; Peterson et al., 2012). Four stereotaxic coordinates throughout the lesion, at approximately −0.80, −1.80, −2.80, and −3.80 mm relative to bregma, were selected for lesion analysis. The number of sections and the section thickness (40 μm) were multiplied by the mean area of the remaining cortex. The extent of cortical injury was measured by calculating the percent reduction in the injured ipsilateral cortex compared to the contralateral cortex at each level using the formula: [1 − (ipsi/contra) × 100] and which has been shown to be sensitive enough to detect treatment-induced reductions in injury size (Hoane et al., 2004, 2006, 2008a, 2009).
Data analysis
Behavioral data was analyzed using a mixed model factorial ANOVA and histological data was analyzed using a one-way between-subjects ANOVA (SPSS v. 15 for Windows). The between factor of Treatment (EPO-treated, anakinra-treated, vehicle-injured, and sham) and the within-group factor was day of testing. Both the main effects and the interaction effects were considered. Huynh-Feldt corrections and Tukey's Honestly Significant Different test (Tukey's HSD) were used to control for Type I error in the repeated measures and post-hoc means comparison, respectively. A significant level of p = 0.05 was used for all statistical analyses. A One-Way ANOVA was completed where the between-subject factor of Treatment (EPO-treated, anakinra-treated, vehicle-injured, and sham) was used to analyze the anatomical data. Tukey's HSD was used to control for Type I error and a significant level of p ≤ 0.05 was used for all statistical analyses. Inter-rater reliability measures were evaluated using the bivariate correlation of scores obtained between the two raters. A power analysis indicated that n = 10 for the behavioral studies would give a power rating of 90% and that n =5 for the pharmacokinetic and molecular studies would also allow for detection of significant differences (Anderson et al., 2013). The evaluators were blinded to the treatment assignment.
Results
Anakinra and erythropoietin pharmacokinetics
In the non-injured animals, a single dose i.p. or s.c. injection of EPO 2500 IU/kg, resulted in peak concentrations of 4481 ± 1436 mIU/mL and 2086 ± 201 mIU/mL at 2 and 24 h, respectively. Due to the prolonged absorption from the s.c. dose, the apparent T1/2 (mean ± standard deviation) was 16.9 ± 2.6 h compared to 8.9 ± 0.8 h after i.p. Anakinra 100 mg/kg administered i.p. produced peak concentrations of 62.1 ± 31.7 μ g/mL at 1 h post-dose with a T1/2 of 1.8 ± 0.4 h. When administered s.c., anakinra peak concentrations of 23.5 ± 2.9 μ g/mL occurred 2 h post-dose. Therefore, multiple doses, with the initial dose by i.p. followed by s.c. every 12 h, resulted in clinically relevant EPO concentrations with minimal fluctuations with peak and trough concentrations of 13,375 ± 1411 mIU/mL and 13,395 ± 1436 mIU/mL, respectively (Figure 1A). Multiple doses of anakinra produced an initial peak concentration of 51.9 ± 23.6 μ g/mL and then average plasma concentrations of 20.3 ± 6.9 μ g/mL (Figure 1B). In the functional behavioral and gene expression studies, the EPO and anakinra serum concentrations were 9990 ± 1054 mIU/mL and 9127 ± 944 mIU/mL and 40 ± 7 μg/mL and 28 ± 14 μg/mL when sampled 72 h post-CC, respectively.
Figure 1.
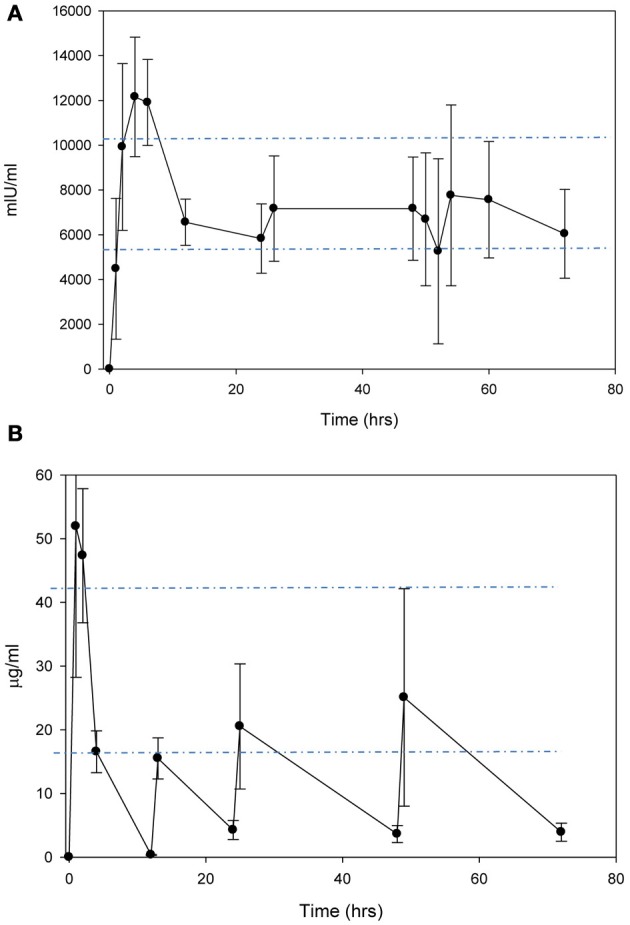
Concentration-time curves after administration of (A) EPO 2500 mg/kg i.p. for the first dose, followed by s.c. every 12 h and (B) anakinra 100 mg/kg i.p. for the first dose, followed by s.c. every 12 h.
Functional behavioral study
In the Locomotor placing task, foot faults score was used as the primary dependent variable (Figure 2A). The within-subjects factor of Day (2, 4 6, 8, and 10) and the between-subjects factor of Treatment (EPO-treated, anakinra-treated, vehicle-injured, and sham) were significant. The interaction of Day × Treatment was significant [F(12, 168) = 2.32, p < 0.05]. Both the main effect of Day [F(4, 168) = 4.39, p < 0.05] and Treatment [F(3, 42) = 106.71, p < 0.001] were significant. The sham group performed significantly better than the EPO-treated group [HSD(19) = 3.37, p < 0.05], the anakinra-treated group [HSD(19) = 4.09, p < 0.05], and the vehicle-injured group [HSD(22) = 4.19, p < 0.05]. There were no significant differences between either of the drug treatment groups and the vehicle-injured group.
Figure 2.

Sensorimotor Assessment. (A) The Locomotor Placing task showing the average fault scores (+SEM) for days 2, 4, 6, 8, and 10 post-CCI. No significant differences between the treated animals and vehicle were found. A trend toward worsening was seen in the anakinra-treated group. (B) The Rotorod test showing the average latency to fall (+SEM) off of the rotating cylinder for day's 8–12 post-CCI. No significant differences between the treated animals and vehicle were found.
On the Rotor-Rod, the latency to fall off the rotating cylinder was averaged over the four trials for each testing day and used as the dependent variable to measure gross motor function with the within-subjects factor of Day (8–12) and the between-subjects factor of Treatment (EPO-treated, anakinra-treated, vehicle-injured, and sham). The interaction of Day × Treatment was not significant [F(11.68, 163.50) = 8.28, p = 0.830]. Both the main effect of Day [F(3.893, 163.5) = 10.75, p < 0.001] and Treatment [F(3, 42) = 3.98, p < 0.05] were significant, see Figure 2B. The sham group performed significantly better than the EPO-treated group [HSD(19) = 41.23, p < 0.05], the anakinra-treated group [HSD(19) = 37.59, p < 0.05], and the vehicle-injured group [HSD(22) = 41.68, p < 0.05]. There were no significant differences between either of the drug treatment groups and the vehicle-injured group.
In the reference memory task, the latency to reach the platform was averaged over the four trials for each testing day during reference memory acquisition (Figure 3A), with the within-subjects factor of Day (14–17) and the between-subjects factor of Treatment (EPO-treated, anakinra-treated, vehicle-injured, and sham). The interaction of Day × Treatment was not significant [F(9, 120) = 0.50, p = 0.87]. Both the main effect of Day [F(3, 120) = 35.14, p < 0.001] and Treatment [F(3, 40) = 5.03, p < 0.05] were significant. The sham group performed significantly better than the EPO-treated group [HSD(19) = 27.08, p < 0.05], the anakinra-treated group [HSD(19) = 20.64, p < 0.05], and the vehicle-injured group, [HSD(22) = 21.11, p < 0.05]. There were no significant differences between either of the drug treatment groups and the vehicle-injured group.
Figure 3.

Cognitive assessment. (A) The MWM reference memory task showing the average latency (+SEM) to reach the platform on day's 14–17 post-CCI. No significant differences between the treated animals and vehicle were found. (B) The MWM working memory task showing the average latency (+SEM) to reach the platform on day's 21–23 post-CCI. No significant differences between the treated animals and vehicle were found.
In the working memory task, the latency to reach the platform was averaged over the last three trials for each testing day during working memory acquisition, with the within-subjects factor of Day (21–23) and the between-subjects factor of Treatment (EPO-treated, anakinra-treated, vehicle-injured, and sham), see Figure 3B. The interaction of Day × Treatment was not significant [F(6, 80) = 1.26, p = 0.291]. The main effect of Day [F(2, 80) = 2.07, p = 0.131] was not significant, but the main effect of Treatment [F(3, 40) = 9.57, p < 0.001] was significant. The sham group performed significantly better than the EPO-treated group [HSD(19) = 22.13, p < 0.001], the anakinra-treated group [HSD(19) = 22.89, p < 0.001], and the vehicle-injured group [HSD(22) = 18.74, p < 0.001]. There were no significant differences between either of the drug treatment groups and the vehicle-injured group.
Lesion analysis
The ratio of lesion volume in the cortices, hippocampus, and hemisphere for each group were each compared with a one-way ANOVA [Group (EPO-treated, anakinra-treated, vehicle-injured, and sham)], see Figure 4A. There was a significant difference between the groups [F(3, 35) = 42.13, p < 0.001] for lesion volume in the cortices. Post-hoc analysis showed that the sham group had significantly more cortical volume compared to the EPO-treated group [HSD(19) = 29.56, p < 0.001], the anakinra-treated group [HSD(19) = 28.87, p < 0.001], and the vehicle group [HSD(22) = 29.65, p < 0.001]. There were no significant differences between either of the drug treatment groups and the vehicle-injured group.
Figure 4.

Lesion Analysis. (A) The average (+SEM) percent reduction in hemispherical, cortical, and hippocampal volumes between the ipsilateral and contralateral sides of the injury. No significant differences between the treated animals and vehicle were found. (B) Representative images of cresyl violet stained tissue throughout the injury coordinates; −0.08 mm, −1.8 mm, −2.8 mm, and −3.8 mm, relative to bregma. Scale bar = 2.0 mm.
There was a significant difference between the groups [F(3, 35) = 11.19, p < 0.001] for lesion volume in the hippocampus. Post-hoc analysis showed that the sham group had significantly more hippocampal volume compared to the EPO-treated group [HSD(19) = 32.22, p < 0.05], the anakinra-treated group [HSD(19) = 41.59, p < 0.001], and the vehicle group [HSD(22) = 45.82, p < 0.001]. There were no significant differences between either of the drug treatment groups and the vehicle-injured group.
There was a significant difference between the groups [F(3, 35) = 11.9, p < 0.001] for lesion volume in the hemispheres. Post-hoc analysis showed that the sham group had significantly more hemispherical volume compared to the EPO-treated group [HSD(19) = 16.55, p < 0.001], the anakinra-treated group [HSD(19) = 18.35, p < 0.001], and the vehicle group [HSD(22) = 19.75, p < 0.001], see Figure 4. There were no significant differences between either of the drug treatment groups and the vehicle-injured group. See Figure 4B for representative lesion images for the experimental groups.
Gene expression
The microarray data passed all the standard and advanced quality control metrics. The number of differentially expressed genes (>1.5-fold change, p < 0.05) at 24 h, 72 h and 7 days are presented in Table 1. The vehicle to sham comparison reflects the effect of the TBI without treatment relative to sham controls. The EPO or anakinra (CCI animals that received treatment) to vehicle (CCI animals that received vehicle) comparison evaluates the effect of treatment on gene expression in the context of TBI. Both EPO and anakinra treatments resulted in significant changes in brain gene expression in the CCI model demonstrating acceptable brain penetration. At all three time points, EPO treatment resulted in significantly more differentially expressed genes than anakinra. At 7 days post-injury (4 days after the last dose of EPO or anakinra), 337 genes were differentially expressed by EPO with 96% (322/337) showing increased expression. Similarly, at 7 day post injury, 209 gene were differentially expressed by anakinra with 85% of genes (178/209) showing increased expression. The Venn diagrams in Figure 5 show the number of differentially expressed genes (1.5-fold, p < 0.05) that are unique for each of the EPO/vehicle and ankinra/vehicle contrasts, as well as the number of genes that are shared between these contrasts for each of the three time points. At 24 h and 7 days post-TBI, approximately a quarter of the genes differentially expressed with EPO treatment also were affected by anakinra treatment. At 72 h post-TBI, only 8 (3%) of the 58 genes differentially expressed by anakinra treatment compared to vehicle were also differentially expressed with EPO treatment.
Table 1.
The number of differentially expressed genes probes (>1.5-fold up or down, p < 0.05).
| 24 h | 72 h | 7 days | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Down | Up | Total | Down | Up | Total | Down | Up | Total | |
| Vehicle/Sham | 1014 | 1457 | 2471 | 1330 | 1956 | 3286 | 318 | 1104 | 1422 |
| EPO/Vehicle | 137 | 58 | 195 | 178 | 47 | 245 | 15 | 322 | 337 |
| Anakinra/Vehicle | 103 | 17 | 120 | 33 | 25 | 58 | 31 | 178 | 209 |
Figure 5.
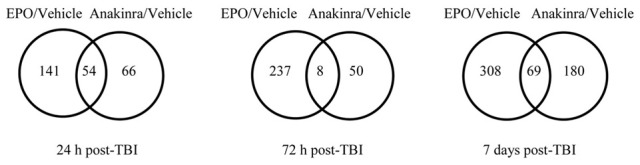
The Venn diagrams show the number of genes whose expression was up or down regulated more than 1.5-fold (p < 0.05) in the EPO/Vehicle and Anakinra/Vehicle contrasts at the 24 h, 72 h, and 7 day time points. Venn diagrams were generated with the Bioconductorlimma package.
Ingenuity Pathway Analysis (IPA) was used to facilitate the identification of biological themes in the microarray data. IPA shifts the emphasis from the evaluation of single genes to an evaluation of molecular pathways, networks and biological functions. Functional categories are identified by molecular and cellular functions. For anakinra at 24 h and EPO at all three times points, the regulated genes in the top 3 functional categories were involved in cellular movement (primarily inflammatory cells), inflammatory response and cell-to-cell signaling. For anakinra, at 7 d post-TBI, the top functional categories were molecular transport, organismal injury and abnormalities and carbohydrate metabolism. Differentially expressed genes of interest were selected and their specific fold changes in expression for the contrast EPO/vehicle, anakinra/vehicle and vehicle/shame are presented in Table 2. Of note, IL 1b, IL 1 receptor, type II, IL 1 receptor antagonist (IL-1rn), several chemokines and chemokine receptors and nitric oxide synthase were significantly increased by TBI and decreased by both EPO and anakinra. Specifically, administration of anakinra decreased gene expression of endogenous IL-1rn only at the 24 h with no effect at 72 h or 7 days and EPO decreased the gene expression of IL-1rn at both 24 and 72 h post-injury.
Table 2.
The effect of Erythropoietin, Anakinra and TBI (vehicle/sham) on genes of interest (1.5 fold change, p < 0.05).
| Affymetrix ID | Gene symbol | Genes | EPO vehicle | Anakinra vehicle | Vehicle sham |
|---|---|---|---|---|---|
| 24 H | |||||
| 10775968 | Alb | Albumin | 1.91 | n.s. | 0.50 |
| 10911380 | Aldh1a2 | Aldehyde dehydrogenase 1 family, member A2 | 1.97 | n.s. | n.s. |
| 10744425 | Alox15 | Arachidonate 15-lipoxygenase | 3.76 | 1.95 | n.s. |
| 10821016 | Ccnb1 | Cyclin B1 | 1.50 | n.s. | n.s. |
| 10717195 | Ccnb2 | Cyclin B2 | 1.53 | n.s. | n.s. |
| 10921163 | Ccr1 | Chemokine (C-C motif) receptor 1 | 0.49 | 0.59 | 9.78 |
| 10829761 | Cdk1 | Cyclin-dependent kinase 1 | 1.50 | n.s. | 2.36 |
| 10775896 | Cxcl3 | Chemokine (C-X-C motif) ligand 2 | 0.48 | n.s. | 11.92 |
| 10924245 | Cxcr2 | Chemokines (C-X-C motif) receptor 2 | 0.49 | 0.47 | 18.82 |
| 10825153 | Fcgr1a | Fc fragment of IgG, high affinity Ia, receptor | 0.66 | n.s. | 7.36 |
| 10755148 | Fetub | Fetuin B | n.s. | 0.64 | 2.48 |
| 10926967 | Gsta2 | Glutathione-S-transferase alpha 2 | 1.56 | n.s. | 0.42 |
| 10849841 | IL1b | Interleukin 1 beta | 0.48 | 0.59 | 6.82 |
| 10922816 | IL1r2 | Interleukin 1 receptor, type II | 0.53 | 0.64 | 7.10 |
| 10834109 | IL1rn | Interleukin 1 receptor antagonist | 0.46 | 0.59 | 17.23 |
| 10922882 | IL18rap | Interleukin 18 receptor accessory protein | 0.64 | 0.66 | 2.38 |
| 10907689 | Itga5 | Integrin, alpha 5 | 0.66 | n.s. | 4.63 |
| 10711299 | Itgax | Integrin, alpha X | 0.61 | 0.44 | 4.35 |
| 10810736 | Lcat | Lecithin cholesterol acyltransferase | 1.55 | n.s. | 0.44 |
| 10736312 | Nos2 | Nitric oxide synthase 2, inducible | 0.42 | 0.51 | 7.71 |
| 10873341 | Pla2g2a | Phospholipase A2, group IIA | n.s. | 1.77 | 2.23 |
| 10708214 | Prc1 | Protein regulator of cytokinesis 1 | 1.52 | 1.64 | 1.54 |
| 10835817 | Ptgs1 | Prostaglandin-endoperoxide synthase 1 | n.s. | 1.52 | n.s. |
| 10764551 | Ptgs2 | Prostaglandin-endoperoxide synthase 2 | 0.56 | n.s. | 2.67 |
| 10742194 | Pttg1 | Pituitary tumor-transforming 1 | 1.57 | n.s. | n.s. |
| 10738130 | Rara | Retinoic acid receptor, alpha | 0.66 | n.s. | 1.83 |
| 10786108 | Rarb | Retinoic acid receptor, beta | 1.94 | n.s. | 0.25 |
| 10827686 | RT1-M6-1 | RT1 class I, locus M6, gene 1 | 0.46 | n.s. | 2.13 |
| 10827691 | RT1-M6-2 | RT1 class I, locus M6, gene 2 | 0.48 | n.s. | 2.21 |
| 10817071 | S100a8 | S100 calcium binding protein A8 | n.s. | 0.66 | 3.28 |
| 10934608 | Tir13 | Toll-like receptor 13 | 0.64 | 0.17 | 6.31 |
| 10770577 | Tgfb2 | Transforming growth factor, beta 2 | 0.63 | n.s. | n.s. |
| 10746976 | Top2a | Topoisomerase (DNA) II alpha | 1.50 | n.s. | 3.26 |
| 72 H | |||||
| 10885393 | Akap5 | A kinase (PRKA) anchor protein 5 | 1.54 | n.s. | 0.57 |
| 10932912 | Alas2 | Aminolevulinate, delta-, synthase 2 | 2.96 | 1.87 | n.s. |
| 10744425 | Alox15 | Arachidonate 15-lipoxygenase | 14.84 | 1.75 | n.s. |
| 10736697 | Ccl2 | Chemokines (C-C motif) ligand 2 | 0.66 | n.s. | 45.91 |
| 10736702 | Ccl7 | Chemokines (C-C motif) ligand 7 | 0.52 | n.s. | 9.29 |
| 10858566 | Clec4a2 | C-type lectin domain family 4, member A2 | 0.61 | n.s. | 7.66 |
| 10866019 | Clec7a | C-type lectin domain family 7, member A | 0.59 | n.s. | 30.82 |
| 10914614 | Ccr2 | Chemokine (C-C motif) receptor 2 | 0.51 | n.s. | 11.00 |
| 10865585 | Cd4 | Cd4 molecule | 0.53 | n.s. | 2.86 |
| 10856274 | Cd8a | CD8a molecule | 0.57 | n.s. | 5.90 |
| 10856265 | Cd8b | CD8b molecule | 0.56 | n.s. | 5.85 |
| 10802013 | Cd72 | Cd74 molecule, major histocompatibility complex, class II invariant chain | 0.64 | n.s. | 3.93 |
| 10897428 | Csf2rb | Colony stimulating factor 2 receptor, beta, low-affinity (granulocyte-macrophage) | 0.60 | n.s. | 10.88 |
| 10767373 | Cxcr4 | Chemokine (C-X-C motif) receptor 4 | 0.63 | n.s. | 2.65 |
| 10811571 | Cyba | Cytochrome b-245, alpha polypeptide | 0.66 | n.s. | 3.95 |
| 10936899 | Cybb | Cytochrome b-245, beta polypeptide | 0.65 | n.s. | 17.68 |
| 10732652 | Dusp1 | Dual specificity phosphatase 1 | n.s. | 1.51 | 0.40 |
| 10828344 | HLA-DRAa | Major histocompatibility complex, class II, DR alpha | 0.55 | 0.65 | 7.18 |
| 10831657 | HLA-DRB1a | Major histocompatibility complex, class II, DR beta 1 | 0.63 | 0.69 | 2.50 |
| 10828351 | HLA-DQA1a | Major histocompatibility complex, class II, DQ alpha 1 | 0.60 | 0.71 | 3.92 |
| 10767518 | Ikbke | Inhibitor of kappa light polypeptide gene enhancer in B-cells, kinase epsilon | 0.66 | n.s. | 2.30 |
| 10834109 | ILr1n | Interleukin 1 receptor antagonist | 0.55 | n.s. | 9.09 |
| 10938654 | IL2rg | Interleukin 2 receptor, gamma | 0.65 | n.s. | 4.34 |
| 10832306 | Itgb2 | Integrin, beta 2 | 0.65 | n.s. | 7.52 |
| 10907689 | Itgb5 | Integrin, alpha 5 (fibronectin receptor, alpha polypeptide) | 0.60 | n.s. | 4.13 |
| 10907881 | Mmp3 | Matrix metallopeptidase 3 | 0.43 | 0.51 | 3.22 |
| 10780205 | Mmp14 | Matrix metallopeptidase 14 (membrane-inserted) | 0.61 | n.s. | 2.66 |
| 10796476 | Mrc1 | Mannose receptor, C type 1 | 0.63 | n.s. | 4.64 |
| 10757898 | Ncf1 | Neutrophil cytosolic factor 1 | 0.60 | n.s. | 6.81 |
| 10764722 | Ncf2 | Neutrophil cytosolic factor 2 | 0.65 | n.s. | 4.32 |
| 10873336 | Pla2g2a | Phospholipase A2, group IID | 0.54 | n.s. | 4.76 |
| 10828351 | RT1-Ba | RT1 class II, locus Ba | 0.60 | n.s. | 3.92 |
| 10831557 | RT1-Db1 | RT1 class II, locus Db1 | 0.63 | n.s. | 2.50 |
| 10797509 | Syk | Spleen tyrosine kinase | 0.66 | n.s. | 4.14 |
| 7 DAYS | |||||
| 10819379 | Adh1ca | Alcohol dehydrogenase (class 1) | 1.64 | n.s | 1.61 |
| 10854406 | Akr1b10a | Aldo-ketoreductase family 1, member B10 | 1.90 | n.s. | n.s. |
| 10911380 | Aldh1a2 | Aldehyde dehydrogenase 1 family, member A2 | 1.81 | n.s. | 1.61 |
| 10722992 | Anpep | Alanyl (membrane) aminopeptidase | 2.44 | n.s. | 2.74 |
| 10744425 | Alox15 | Arachidonate 15-lipoxygenase | 18.75 | n.s. | n.s. |
| 10719524 | Apolipoprotein C-I (Apoc1) | 1.69 | n.s. | n.s. | |
| 10848030 | Bbox1 | Butyrobetaine (gamma), 2-oxoglutarate dioxygenase 1 | n.s. | 1.61 | n.s. |
| 10914614 | Ccr2 | Chemokine (C-C motif) receptor 2 | n.s. | 0.63 | n.s. |
| 10803991 | Cd14 | CD14 molecule | 1.59 | n.s. | 2.06 |
| 10940473 | Cd36 | CD36 molecule (thrombospondin receptor) | 3.97 | n.s. | 2.62 |
| 10935177 | Cldn2 | Claudin 2 | 3.27 | 3.85 | 0.19 |
| 10767373 | Cxcr4 | Chemokine (C-X-C motif) receptor 4 | 1.51 | n.s. | n.s. |
| 10936899 | Cybb | Cytochrome b-245, beta polypeptide | 1.65 | n.s. | 10.08 |
| 10765212 | F5 | Coagulation factor V (proaccelerin, labile factor) | 2.22 | 3.03 | 0.46 |
| 10794734 | F13a1 | Coagulation factor XIII, A1 polypeptide | 2.00 | n.s. | n.s. |
| 10810778 | Dpep2 | Dipeptidase 2 | 1.67 | n.s. | 1.84 |
| 10928761 | Fn1 | Fibronectin 1 | 2.37 | n.s. | 2.23 |
| 10939901 | Gpr101 | G protein-coupled receptor 101 (Gpr101) | n.s. | 1.83 | 0.54 |
| 10804221 | Gpr151 | G protein-coupled receptor 151 | n.s. | 1.82 | n.s. |
| 10825915 | Gstm2 | Glutathione S-transferase mu 2 | 1.78 | n.s. | n.s. |
| 10806122 | Hmox1 | Hemeoxygenase (decycling) 1 | 1.98 | n.s. | 5.90 |
| 10937302 | Htr2c | 5-hydroxytryptamine (serotonin) receptor 2C | 1.90 | 0.46 | |
| 10841693 | Lbp | Lipopolysaccharide binding protein | 2.19 | n.s. | 1.94 |
| 10791250 | Lpl | Lipoprotein lipase | 1.55 | 1.71 | 0.56 |
| 10874918 | Ly96 | Lymphocyte antigen 96 | 1.51 | n.s. | 2.63 |
| 10859392 | Mgst1 | Microsomal glutathione S-transferase 1 | 1.67 | n.s. | 1.72 |
| 10842239 | Mmp9 | Matrix metallopeptidase 9 | 1.85 | n.s. | n.s. |
| 10907869 | Mmp12 | Matrix metallopeptidase 12 | 1.91 | n.s. | 5.62 |
| 10780205 | Mmp14 | Matrix metallopeptidase 14 (membrane-inserted) | 1.60 | n.s. | 1.79 |
| 10893231 | Mmp19 | Matrix metallopeptidase 19 | 1.51 | n.s. | 2.91 |
| 10788427 | Msr1a | Macrophage scavenger receptor 1 | 2.53 | n.s. | 3.36 |
| 10821698 | Osmr | Oncostatin M receptor | 1.51 | n.s. | 3.69 |
| 10860867 | Pon1 | Paraoxonase 1 | 3.55 | 3.53 | n.s. |
| 10860878 | Pon3 | Paraoxonase 3 | 1.79 | n.s. | n.s. |
| 10873341 | Pla2g2a | Phospholipase A2, group IIA | 1.61 | n.s. | n.s. |
| 10873336 | Pla2g2d | Phospholipase A2, group IID | 2.17 | n.s. | n.s. |
| 10751295 | Pla1a | Phospholipase A1 member A | 1.70 | n.s. | 1.68 |
| 10782271 | Plau | Plasminogen activator, urokinase | 1.70 | n.s. | 4.36 |
| 10823819 | Rxfp1 | Relaxin/insulin-like family peptide receptor 1 | n.s. | 0.62 | n.s. |
| 10912439 | Rbp1 | Retinol binding protein 1, cellular | 1.81 | n.s. | 1.54 |
| 10761047 | Serpine1 | Serpin peptidase inhibitor, clade E, member 1 | 2.71 | n.s. | n.s. |
| 10819139 | Tacr3 | Tachykinin receptor 3 | 0.56 | 0.62 | n.s. |
A rat gene symbol was not provided by the Affymetrix annotation file for this transcript, however, IPA identified a human ortholog. Therefore, the gene symbol for the human ortholog is listed here.
AT 7 d post-TBI, anakinra increased the expression of several receptors involved in signaling pathways including 5-hydroxytryptamine (serotonin) receptor 2c, cholinergic receptor, nicotinic, alpha 3, fibroblast growth factor, G-protein-coupled receptor 101 and 105 and prolactin receptor (Table 2). EPO treatment caused an over expression of genes involved in the coagulation system (coagulation factor V (F5), coagulation factor XIII, A1 polypeptide, plasminogen activator, urokinraase and serpin peptidase inhibitor, clade E) and expression of genes involved in acute phase response signaling (hemeoxygenase 1, fibronectin 1, oncostatin M receptor, lipopolysaccharide binding protein, serpin peptidase inhibitor member 1 and retinal binding protein 1) (Table 2).
We used TaqMan-based quantitative-PCR (qPCR) analysis to validate gene expression changes of 14 genes (Alox15, Casp12, CD68, CYP1B1, GAL, HMOX1, IHF2, IL1r2, IL1rn, MDK, MMP9, NIACR1, S100A9, XDH) selected from specific pathways of interest in TBI. The data generated via microarray and quantitative-PCR (qPCR) was normalized to β-actin. Figure 6 shows that the qPCR findings were highly correlated with the microarray data (Slope = 1.352, Pearson's R = 0.859).
Figure 6.
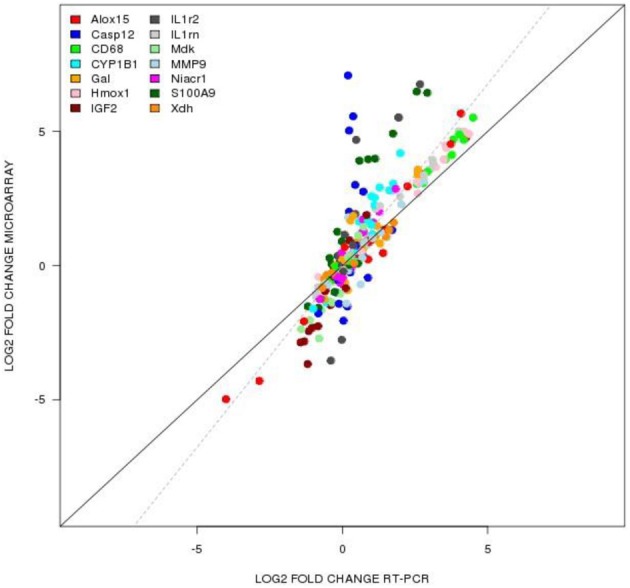
TaqMan based RT-PCR validation of the microarray data for the selected genes: Alox15 (arachidonate 15-lipoxygenase), Casp12 (caspase 12), CD68 (CD 68 molecule), Cyp1b1 (cytochrome P450 1b1), Gal (galanin), Hmox1 (hemeoxygenase 1), IGF2 (insulin like growth factor 2), IL1rn (interleukin 1 receptor antagonist), Mdk (midkine), Mmp9 (matrix metallopeptidase 9), Niacr1 (niacin receptor 1), S100a9 (S100 calcium binding protein A9), Xdh (xanthine dehydrogenase). The RT-PCR data was normalized to the housekeeping gene β-actin. Pearson's coefficient, r = 0.859.
Discussion
The dosage regiment used in our studies of anakinra and EPO were designed to produce concentrations in the range reported in patients receiving FDA approved doses and to reflect initiating therapy within 4 h of a head injury in patient. Clinically, administering an intravenous (i.v.) loading dose of either drug results in rapid peak concentrations, compared to the delay in peak found with our experimental model (i.p) initiated 2 h post-TBI. The FDA approved dosage recommendations for EPO is 150 U/kg administered s.c. three times weekly or 40,000 U weekly. EPO at a dose of 40,000 (~500 IU/kg) proposed for use in critically ill patients (Corwin et al., 2002) and TBI patients (Nirula et al., 2010) when administered i.v. would result in an peak concentration of approximately 10,000 mIU/ml with an average elimination half-life of 17 h and reaching non-detectable concentrations by 3 days post-dose (McCluskey et al., 2009). The recommended dose of anakinra in rheumatoid arthritis is 100 mg/day administered s.c. A pilot study of anakinra in acute stroke patients, receiving 100 mg i.v. bolus followed by a 2 mg/kg·h infusion resulted in median concentrations ranging from 20 to 40 μg/mL (Emsley et al., 2005).
The immunomodulation effects of EPO and anakinra did not translate into effects on functional behavioral and lesion studies. The severities of these deficits were similar to what we have observed using this model of injury previously (Kuypers and Hoane, 2010; Swan et al., 2011; Vonder Haar et al., 2011; Peterson et al., 2012). Treatment with either EPO or anakinra failed to induce significant beneficial effects on recovery of function in all four behavioral tests examined in this study. Additionally, both drugs failed to produce any significant effects on the prevention of injury induced tissue loss in the cortex or hippocampus when examined 30 days post-injury.
The use of microarray analysis to delineate gene expression patterns and profile changes is a powerful tool that can be used to evaluate the potential effect of treatment, in addition to increasing the understanding of the mechanism of the treatment effect. Both EPO and anakinra treatments penetrated the brain and altered the inflammatory and immune pathways.
The lack of effect of EPO and anakinra on functional recovery of cognition and motor behavior is in contrast to the significant effect of other drugs such as progesterone and nicotinamide when administered for 72 h in the same CCI model (Peterson et al., 2012). Both nicotinamide and progesterone treatments also resulted in significant effects on the expression of genes involved in the inflammatory/immune pathways (Anderson et al., 2011, 2013). However, in contrast to EPO and anakinra, both nicotinamide and progesterone also demonstrate significant effects on gene expression of other biological pathways (Anderson et al., 2011). Nicotinamide treatment resulted in the differential expression of genes involved in the inflammatory/immune pathways only at 24 h and not at 72 h and 7 days with IPA analysis also identified over-expression of genes involved in 1L-10 signaling, LXR-RXR activation, TREM1 signaling, communication between innate and adaptive immune cells and PPAR signaling. However, the canonical pathways identified at 72 h after injury for nicotinamide included a significant effect on cell signaling pathways involving neurotransmitters, neuropeptides, growth factors and ion channels (Anderson et al., 2013). In addition to positive and negative effects on inflammatory pathways, progesterone also affected genes involved in cell proliferation, DNA damage response, regulation of apoptosis and blood vessel remodeling (Anderson et al., 2011).
EPO significantly increased the gene expression of arachidonate 15-lipoxygenase (Alox15) at 24 h, 72 h and 7 days post-TBI and anakinra increased the expression of Alox15 at 24 and 72 h post-TBI. Alox15 is considered to have a pro-inflammatory effect and may play a key role in the acute inflammatory response and reportedly leads to the generation of unstable lipid products from arachidonate (Kuhn and O'Donnell, 2006). Previous studies investigating the effect of two different doses of progesterone (Anderson et al., 2011) and nicotinamide (Anderson et al., 2013) on gene expression in the CCI model found that neither nicotinamide infusion nor low dose progesterone increased Alox15. In contrast, the higher dose of progesterone did significantly increase gene expression of Alox15. When low and high dose progesterone and nicotinamide were compared on the functional recovery of cognitive behavior in the same model, the low dose progesterone and nicotinamide produced significant effects on functional recovery that were not found with the higher dose. Nicotinamide and low dose progesterone also reduced tissue loss compared to vehicle (Peterson et al., 2012). As high concentrations of IL-1ra are associated with better outcome in patients (Hutchinson et al., 2007), the decreased gene expression of IL-1rn by EPO at 24 and 72 h and by anakinra at 24 h post-TBI may also be a significant negative effect of the treatment. Both doses of progesterone increased IL-1rn at 72 h (Anderson et al., 2011). Nicotinamide did decrease gene expression of IL-1rn at 24 h with no effect at 72 h or 7 days post-injury (Anderson et al., 2013).
The majority of the pre-clinical research in animal models evaluating the effectiveness of EPO in brain injury utilized a dose of 5000 U/kg, which was tested at various time-points ranging from a pretreatment up to 9 h post-injury, with earlier administration resulting in greater benefits (Brines et al., 2000; Cerami et al., 2002). However, beneficial effects were found with administration as late as 24 h post-injury (Zhang et al., 2009; Meng et al., 2011) and with multiple dose studies administered up to 14 days (Lu et al., 2005; Liao et al., 2008; Xiong et al., 2008). Meng et al. found dose dependent effects of EPO when administered i.p. at 24, 48, and 72 h (Meng et al., 2011). All doses (1000–7000 U/kg) provided some neuroprotection in the form of less hippocampal cell loss and improved sensorimotor and spatial learning performance in comparison to the sham group (Meng et al., 2011). However, the optimal dose was 5000 U/kg for a significant improvement in both histological and functional outcomes. This dose would result in EPO concentration considerably above those associated with the current FDA recommendations (150 U/kg) (Rizzo et al., 2010).
This study is limited by evaluating only one dosage regiment initiated at one time point. Experimental studies have demonstrated both a neurotoxic as well as neuroprotective function of inflammatory response (Lenzlinger et al., 2001). The response is pro-inflammatory during the acute phase and anti-inflammatory during the chronic phase, which theoretically assists in repair and recovery processes(Correale and Villa, 2004). Therefore, the timing of the inflammatory modulator treatment may be critical. The limited pre-clinical data evaluating IL-1ra in TBI, suggests that initiating therapy later for anakinra would not increase efficacy, however, increasing the treatment duration could improve outcome. Treatment of IL-1ra, 100 mg/kg s.c. reduced injury volume by 44% when treated within 15 min, but only 28% when administrated at 4 h in the fluid percussion injury (FPI) model (Toulmond and Rothwell, 1995). In the FPI model, when IL-1ra 100 mg/kg was administered 15 min after injury and animals were treated for 7 days, IL-1ra significantly reduced neuronal loss in the hippocampus and the cortex. A lower dose (10 mg/kg) had no significant effects (Sanderson et al., 1999). As starting therapy within 15 min of a head injury is not feasible, if early initiation is necessary for effect, anakinra would not provide a practical treatment. For EPO, initiating therapy at 24 h post-TBI and treating for a long duration may have improved the effects.
Anakinra and EPO initiated at dosage regiments designed to produce concentrations in the range reported in patients receiving FDA approved doses did result in significant effects on gene expression in the brain reflecting adequate penetration and altered genes involved in inflammatory processes; however, the dose regiments were not sufficient to produce neurorestorative effects. This data suggests that targeting inflammatory processes alone may not be sufficient for preventing secondary injuries after TBI. Ultimately, a polytherapy approach that addresses the multitude of immediate and prolonged symptoms of TBI may be the key to an effective overall treatment.
Author contributions
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The research was supported by a grant from the National Institutes of Health/National Institute of Child, Health and Development (R01 HD061944-01) and by the NIEHS Center for Ecogenetics & Environmental Health (P30ES007033)
References
- Anderson G. D., Farin F. M., Bammler T. K., Beyer R. P., Swan A. A., Wilkerson H. W., et al. (2011). The effect of progesterone dose on gene expression after traumatic brain injury. J. Neurotrauma 28, 1827–1843 10.1089/neu.2011.1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson G. D., Peterson T. C., Farin F. M., Bammler T. K., Beyer R. P., Kantor E. D., et al. (2013). The effect of nicotinamide on gene expression in a traumatic brain injury model. Front. Neurosci. 7:21 10.3389/fnins.2013.00021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartfai T., Sanchez-Alavez M., Andell-Jonsson S., Schultzberg M., Vezzani A., Danielsson E., et al. (2007). Interleukin-1 system in CNS stress: seizures, fever, and neurotrauma. Ann. N.Y. Acad. Sci. 1113, 173–177 10.1196/annals.1391.022 [DOI] [PubMed] [Google Scholar]
- Boogaerts M. (2006). Pleiotropic effects of erythropoietin in neuronal and vascular systems. Curr. Med. Res. Opin. 22Suppl. 4, S15–S22 10.1185/030079906X15958816914072 [DOI] [Google Scholar]
- Brines M., Ghezzi P., Keenan S., Agnello D., de Lanerolle N. C., Cerami C., et al. (2000). Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. Proc. Natl. Acad. Sci. U.S.A. 97, 10526–10531 10.1073/pnas.97.19.10526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami A., Brines M., Ghezzi P., Cerami C., Itri L. M. (2002). Neuroprotective properties of epoetin alfa. Nephrol. Dial. Transplant. 17, 8–12 10.1093/ndt/17.suppl_1.8 [DOI] [PubMed] [Google Scholar]
- Coggeshall R. E. (1992). A consideration of neural counting methods. Trends Neurosci. 15, 9–13 10.1016/0166-2236(92)90339-A [DOI] [PubMed] [Google Scholar]
- Correale J., Villa A. (2004). The neuroprotective role of inflammation in nervous system injuries. J. Neurol. 251, 1304–1316 10.1007/s00415-004-0649-z [DOI] [PubMed] [Google Scholar]
- Corwin H. L., Gettinger A., Pearl R. G., Fink M. P., Levy M. M., Shapiro M. J., et al. (2002). Efficacy of recombinant human erythropoietin in critically ill patients: a randomized controlled trial. JAMA 288, 2827–2835 10.1001/jama.288.22.2827 [DOI] [PubMed] [Google Scholar]
- Das M., Leonardo C., Rangooni S., Pennypacker K., Mohapatra S., Mohapatra S. (2011). Lateral fluid percussion injury of the brain induces CCL20 inflammatory chemokine expression in rats. J. Neuroinflammation 8, 148 10.1186/1742-2094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley H. C., Smith C. J., Georgiou R. F., Vail A., Hopkins S. J., Rothwell N. J., et al. (2005). A randomised phase II study of interleukin-1 receptor antagonist in acute stroke patients. J. Neurol. Neurosurg. Psychiatry 76, 1366–1372 10.1136/jnnp.2004.054882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faul M., Xu L., Wald M., Coronado V. (2010). Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations, and Deaths. Atlanta, GA: Centers for Disease Control and Prevention, National Center for Injury Prevention and Control [Google Scholar]
- Feurstein G. Z., Wang X., Barone F. C. (1997). Inflammatory gene expression in cerebral ischemia and trauma. Potential new therapeutic targets. Ann. N.Y. Acad. Sci. 15, 179–183 10.1111/j.1749-6632.1997.tb48428.x [DOI] [PubMed] [Google Scholar]
- Goffus A. M., Anderson G. D., Hoane M. (2010). Sustained delivery of nicotinamide limits cortical injury and improves functional recovery following traumatic brain injury. Oxid. Med. Cell. Longev. 3, 145–152 10.4161/oxim.3.2.11315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagan P., Barks J. D. E., Yabut M., Davidson B. L., Roessler B., Silverstein F. S. (1996). Adenovirus-mediated over-expression of Interleukin-1 receptor antagonist reduces susceptibility to excitotoxic brain injury in perinatal rats. Neuroscience 75, 1033–1045 10.1016/0306-4522(96)00225-4 [DOI] [PubMed] [Google Scholar]
- Hailer N. P., Vogt C., Korf H. W., Dehghani F. (2005). Interleukin-1beta exacerbates and interleukin-1 receptor antagonist attenuates neuronal injury and microglial activation after excitotoxic damage in organotypic hippocampal slice cultures. Eur. J. Neurosci. 21, 2347–2360 10.1111/j.1460-9568.2005.04067.x [DOI] [PubMed] [Google Scholar]
- Hoane M. R., Akstulewicz S. L., Toppen J. (2003). Treatment with vitamin B3 improves functional recovery and reduces GFAP expression following traumatic brain injury in rats. J. Neurotrauma 20, 1189–1199 10.1089/089771503770802871 [DOI] [PubMed] [Google Scholar]
- Hoane M. R., Becerra G. D., Shank J. E., Tatko L., Pak E. S., Smith M., et al. (2004). Transplantation of neuronal and glial precursors dramatically improves sensorimotor function but not cognitive function in the traumatically injured brain. J. Neurotrauma 21, 163–174 10.1089/089771504322778622 [DOI] [PubMed] [Google Scholar]
- Hoane M. R., Gilbert D. R., Holland M. A., Pierce J. L. (2006). Nicotinamide reduces acute cortical neuronal death and edema in the traumatically injured brain. Neurosci. Lett. 408, 35–39 10.1016/j.neulet.2006.07.011 [DOI] [PubMed] [Google Scholar]
- Hoane M. R., Kaufman N., Vitek M. P., McKenna S. E. (2009). COG1410 improves cognitive performance and reduces cortical neuronal loss in the traumatically injured brain. J. Neurotrauma 26, 121–129 10.1089/neu.2008.0565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoane M. R., Pierce J. L., Holland M. A., Anderson G. D. (2008a) Nicotinamide treatment induces behavioral recovery when administered up to 4 hours following cortical contusion injury in the rat. Neuroscience 154, 861–868 10.1016/j.neuroscience.2008.04.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoane M. R., Pierce J. L., Kauffman N. A., Beare J. (2008b) Variation in chronic nicotinamide treatment after traumatic brain injury can alter components of functional recovery independent of histological damage. Oxid. Med. Cell. Longev. 1, 46–53 10.4161/oxim.1.1.6694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson P. J., O'Connell M. T., Rothwell N. J., Hopkins S. J., Nortje J., Carpenter K. L., et al. (2007). Inflammation in human brain injury: intracerebral concentrations of IL-1alpha, IL-1beta, and their endogenous inhibitor IL-1ra. J. Neurotrauma 24, 1545–1557 10.1089/neu.2007.0295 [DOI] [PubMed] [Google Scholar]
- Jennings J. S., Gerber A. M., Vallano M. L. (2008). Pharmacological strategies for neuroprotection in traumatic brain injury. Mini Rev. Med. Chem. 8, 689–701 10.2174/138955708784567377 [DOI] [PubMed] [Google Scholar]
- Jones N. C., Prior M. J. W., Burden-Teh E., Marsden C. A., Morris P. G., Murphy S. (2005). Antagonism of the interleukin-1 receptor following traumatic brain injury in the mouse reduces the number of nitric oxide synthase-2-positive cells and improves anatomical and functional outcomes. Eur. J. Neurosci. 22, 72–78 10.1111/j.1460-9568.2005.04221.x [DOI] [PubMed] [Google Scholar]
- Juliet P. A., Mao X., Del Bigio M. R. (2008). Proinflammatory cytokine production by cultured neonatal rat microglia after exposure to blood products. Neurosci. Lett. 289, 5–8 [DOI] [PubMed] [Google Scholar]
- Kuhn H., O'Donnell V. B. (2006). Inflammation and immune regulation by 12/15-lipoxygenases. Prog. Lipid Res. 45, 334–356 10.1016/j.plipres.2006.02.003 [DOI] [PubMed] [Google Scholar]
- Kuypers N. J., Hoane M. R. (2010). Pyridoxine administration improves behavioral and anatomical outcome after unilateral contusion injury in the rat. J. Neurotrauma 27, 1275–1282 10.1089/neu.2010.1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence C. B., Allan S. M., Rothwell N. J. (1998). Interleukin-1beta and the interleukin-1 receptor antagonist act in the striatum to modify excitotoxic brain damage in the rat. Eur. J. Neurosci. 10, 1188–1195 10.1046/j.1460-9568.1998.00136.x [DOI] [PubMed] [Google Scholar]
- Lenzlinger P. M., Morganti-Kossmann M. C., Laurer H. L., McIntosh T. K. (2001). The duality of the inflammatory response to traumatic brain injury. Mol. Neurobiol. 24, 169–181 10.1385/MN:24:1-3:169 [DOI] [PubMed] [Google Scholar]
- Liao Z. B., Zhi X. G., Shi Q. H., He Z. H. (2008). Recombinant human erythropoietin administration protects cortical neurons from traumatic brain injury in rats. Eur. J. Neurol. 15, 140–149 10.1111/j.1468-1331.2007.02013.x [DOI] [PubMed] [Google Scholar]
- Lindner M. D., Plone M. A., Cain C. K., Frydel B., Francis J. M., Emerich D. F., et al. (1998). Dissociable long-term cognitive deficits after frontal versus sensorimotor cortical contusions. J. Neurotrauma 15, 199–216 10.1089/neu.1998.15.199 [DOI] [PubMed] [Google Scholar]
- Loddick S. A., Wong M. L., Bongiorno P. B., Gold P. W., Licinio J., Rothwell N. J. (1997). Endogenous interleukin-1 receptor antagonist is neuroprotective. Biochem. Biophys. Res. Commun. 234, 211–215 10.1006/bbrc.1997.6436 [DOI] [PubMed] [Google Scholar]
- Lu D., Mahmood A., Qu C., Goussev A., Schallert T., Chopp M. (2005). Erythropoietin enhances neurogenesis and restores spatial memory in rats after traumatic brain injury. J. Neurotrauma 22, 1011–1017 10.1089/neu.2005.22.1011 [DOI] [PubMed] [Google Scholar]
- Mahmood A., Lu D., Qu C., Goussev A., Zhang Z. G., Lu C., et al. (2007). Treatment of traumatic brain injury in rats with erythropoietin and carbamylated erythropoietin. J. Neurosurg. 107, 392–397 10.3171/JNS-07/08/0392 [DOI] [PubMed] [Google Scholar]
- Mammis A., McIntosh T. K., Maniker A. H. (2009). Erythropoietin as a neuroprotective agent in traumatic brain injury: review. Surg. Neurol. 71, 527–531 10.1016/j.surneu.2008.02.040 [DOI] [PubMed] [Google Scholar]
- Mathew P., Graham D. I., Bullock R., Maxwell W., McCulloch J., Teasdale G. (1994). Focal brain injury: histological evidence of delayed inflammatory response in a new rodent model of focal cortical injury. Acta Neurochir. Suppl. 60, 428–430 [DOI] [PubMed] [Google Scholar]
- McCluskey S., Cheung W., Katznelson R., Poonawal H., Fedorko L., Djaiani G., et al. (2009). The pharmacokinetic profile of recombinant human erythropoietin is unchanged in patients undergoing cardiac surgery. Eur. J. Clin. Pharmacol. 65, 279–279 10.1007/s00228-008-0575-6 [DOI] [PubMed] [Google Scholar]
- McConeghy K. W., Hatton J., Hughes L., Cook A. M. (2012). A review of neuroprotection pharmacology and therapies in patients with acute traumatic brain injury. CNS Drugs 26, 613–636 10.2165/11634020-000000000-00000 [DOI] [PubMed] [Google Scholar]
- Meng Y., Xiong Y., Mahmood A., Zhang Y., Qu C., Chopp M. (2011). Dose-dependent neurorestorative effects of delayed treatment of traumatic brain injury with recombinant human erythropoietin in rats. J. Neurosurg. 115, 550–560 10.3171/2011.3.JNS101721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morganti-Kossmann M. C., Rancan M., Otto V. I., Stahel P. F., Kossmann T. (2001). Role of cerebral inflammation after traumatic brain injury: a revisited concept. Shock 16, 165–177 10.1097/00024382-200116030-00001 [DOI] [PubMed] [Google Scholar]
- Nirula R., Diaz-Arrastia R., Brasel K., Weigelt J. A., Waxman K. (2010). Safety and efficacy of erythropoietin in traumatic brain injury patients: a pilot randomized trial. Crit. Care Res. Pract. 2010:209848 10.1155/2010/209848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson T., Ward J., Logue M., Anderson G. D., Hoane M. R. (2012). A comparison of the effects of nicotinamide and progesterone on functional recovery of cognitive behavior following cortical contusion injury in the rat. J. Neurotrauma 29, 2823–2830 10.1089/neu.2012.2471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relton J. K., Martin D., Thompson R. C., Russell D. A. (1996). Peripheral administration of Interleukin-1 Receptor antagonist inhibits brain damage after focal cerebral ischemia in the rat. Exp. Neurol. 138, 206–213 10.1006/exnr.1996.0059 [DOI] [PubMed] [Google Scholar]
- Relton J. K., Rothwell N. J. (1992). Interleukin-1 receptor antagonist inhibits ischaemic and excitotoxic neuronal damage in the rat. Brain Res. Bull. 29, 243–246 10.1016/0361-9230(92)90033-T [DOI] [PubMed] [Google Scholar]
- Rizzo J. D., Brouwers M., Hurley P., Seidenfeld J., Arcasoy M. O., Spivak J. L., et al. (2010). American society of hematology/American society of clinical oncology clinical practice guideline update on the use of epoetin and darbepoetin in adult patients with cancer. Blood 116, 4045–4059 10.1182/blood-2010-08-300541 [DOI] [PubMed] [Google Scholar]
- Rock K. L., Kono H. (2008). The inflammatory response to cell death. Annu. Rev. Pathol. 3, 99–126 10.1146/annurev.pathmechdis.3.121806.151456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson K. L., Raghupathi R., Saatman K. E., Martin D., Miller G., McIntosh T. K. (1999). Interleukin-1 receptor antagonist attenuates regional neuronal cell death and cognitive dysfunction after experimental brain injury. J. Cereb. Blood Flow Metab. 19, 1118–1125 10.1097/00004647-199910000-00008 [DOI] [PubMed] [Google Scholar]
- Schouten J. W. (2007). Neuroprotection in traumatic brain injury: a complex struggle against the biology of nature. Curr. Opin. Crit. Care 13, 134–142 10.1097/MCC.0b013e3280895d5c [DOI] [PubMed] [Google Scholar]
- Shojo H., Kaneko Y., Mabuchi T., Kibayashi K., Adachi N., Borlongan C. V. (2010). Genetic and histological evidence implicates role of inflammation in traumatic brain injury-induced apoptosis in the rat cerebral cortex following moderate fluid percussion injury. Neuroscience 171, 1273–1282 10.1016/j.neuroscience.2010.10.018 [DOI] [PubMed] [Google Scholar]
- So A., De Smedt T., Revaz S., Tschopp J. (2007). A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res. Ther. 9, 29–34 10.1186/ar2143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swan A. A., Chandrashekar R., Beare J., Hoane M. R. (2011). Preclinical efficacy testing in middle-aged rats: nicotinamide, a novel neuroprotectant, demonstrates diminished preclinical efficacy after controlled cortical impact. J. Neurotrauma 28, 431–440 10.1089/neu.2010.1519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toulmond S., Rothwell N. J. (1995). Interleukin-1 receptor antagonist inhibits neuronal damage caused by fluid percussion injury in the rat. Brain Res. 671, 261–266 10.1016/0006-8993(94)01343-G [DOI] [PubMed] [Google Scholar]
- Touzani O., Boutin H., Chuquet J., Rothwell N. J. (1999). Potential mechanisms of interleukin-1 involvement in cerebra ischemia. J. Neuroimmunol. 100, 203–215 10.1016/S0165-5728(99)00202-7 [DOI] [PubMed] [Google Scholar]
- Vezzani A., Moneta D., Richichi C., Perego C., Simoni M. G. (2004). Functional role of proinflammatory and anti-inflammatory cytokines in seizures. Adv. Exp. Med. Biol. 42, 123–133 10.1007/978-1-4757-6376-8_10 [DOI] [PubMed] [Google Scholar]
- Vonder Haar C., Anderson G. D., Hoane M. R. (2011). Continuous nicotinamide administration improves behavioral recovery and reduces lesion size following bilateral frontal controlled cortical impact injury. Behav. Brain Res. 224, 311–317 10.1016/j.bbr.2011.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K. K., Larner S. F., Robinson G., Hayes R. L. (2006). Neuroprotection targets after traumatic brain injury. Curr. Opin. Neurol. 19, 514–519 10.1097/WCO.0b013e3280102b10 [DOI] [PubMed] [Google Scholar]
- Xiong Y., Mahmood A., Lu D., Qu C., Kazmi H., Goussev A., et al. (2008). Histological and functional outcomes after traumatic brain injury in mice null for the erythropoietin receptor in the central nervous system. Brain Res. 1230, 247–257 10.1016/j.brainres.2008.06.127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsiv I., Grigoriadis N., Simeonidou C., Stahel P. F., Schmidt O. I., Alexandrovich A. G., et al. (2005). Erythropoietin is neuroprotective, improves functional recovery, and reduces neuronal apoptosis and inflammation in a rodent model of experimental closed head injury. FASEB J. 19, 1701–1703 10.1096/fj.05-3907fje [DOI] [PubMed] [Google Scholar]
- Zhang Y., Xiong Y., Mahmood A., Meng Y., Qu C., Schallert T., et al. (2009). Therapeutic effects of erythropoietin on histological and functional outcomes following traumatic brain injury in rats are independent of hematocrit. Brain Res. 1294, 153–164 10.1016/j.brainres.2009.07.077 [DOI] [PMC free article] [PubMed] [Google Scholar]