

Abstract
Purpose
In another study, it was demonstrated that NADPH oxidase-derived reactive oxygen species (ROS) are important for ischemia–induced increases in vascular endothelial growth factor (VEGF) and retinal neovascularization. Diabetes–induced increases in retinal ROS, VEGF expression, and vascular permeability are accompanied by increases in the NADPH oxidase catalytic subunit NOX2 within the retinal vessels. The goal of this study was to evaluate the potential role of NOX2 and NADPH oxidase activity in the development of retinal vascular inflammation.
Methods
Studies were performed in wild-type mice, mice lacking NOX2, and mice treated with the NADPH oxidase inhibitor apocynin in models of endotoxemia and streptozotocin-induced diabetes. Intracellular adhesion molecule (ICAM)-1 expression was determined by Western blot analysis. Leukocyte adhesion was assessed by labeling adherent leukocytes with concanavalin A. Vascular permeability was assessed by extravasation of FITC-conjugated albumin. ROS production was determined by dichlorofluorescein imaging.
Results
Both endotoxemia- and diabetes-induced increases in ICAM-1 expression and leukostasis were significantly inhibited by deletion of NOX2, indicating that this enzyme is critically involved in both conditions. Moreover, apocynin treatment and deletion of NOX2 were equally effective in preventing diabetes-induced increases in ICAM-1, leukostasis, and breakdown of the blood–retinal barrier, suggesting that NOX2 is primarily responsible for these early signs of diabetic retinopathy.
Conclusions
These data suggest that NOX2 activity has a primary role in retinal vascular inflammation during acute and chronic conditions associated with retinal vascular inflammatory reactions. Targeting this enzyme could be a novel therapeutic strategy for treatment of the retinopathies associated with vascular inflammation.
Vascular inflammation is a common feature of a variety of potentially blinding retinal diseases associated with retinal vascular hyperpermeability and vitreoretinal neovascularization. The early stages of the inflammatory reaction are characterized by leukocyte adhesion to the vessel wall. The adhesion causes retinal capillary plugging and nonperfusion, which is followed by vascular injury and hyperpermeability.1–3 Increased expression of the adhesion molecule intracellular adhesion molecule (ICAM)-1 has been found to play a major role in mediating leukocyte adhesion to vascular endothelial cells, and this has been linked to vascular injury associated with ischemic retinopathy, diabetic retinopathy, and uveitis.4 –7
Various reports have shown that ROS are obligatory mediators of vascular inflammation induced by growth factors and cytokines.8,9 Furthermore, ROS activate the transcription factors NF-κB and AP-1, which play a central and crucial role in inducing the expression of inflammatory cytokines and ICAM-1.10 –13 Previous work has shown that diabetes-induced increases in VEGF expression, leukocyte adhesion, and breakdown of the blood–retinal barrier (BRB) all correlate with increases in oxidative stress.14 –17 The specific role of oxidative stress in triggering retinal vascular inflammatory processes is not yet known. However, studies in models of atherosclerosis and other forms of peripheral vascular disease have implicated NADPH oxidase in the inflammatory reaction.18
In phagocytic cells NADPH oxidase is a multiprotein complex consisting of membrane-bound cytochrome b558, which is composed of the catalytic subunit NOX2 (formerly known as gp91phox), p22phox, the cytoplasmic subunits p47phox and p67phox, and the small Rho GTPase Rac. Vascular endothelial cells express the same subunits as well as two NOX2 homologues, NOX1, and NOX4 (for review, see Ref. 18). Our previous studies in animal and tissue culture models have shown that NOX2 is expressed at low levels in normal retinas and in retinal endothelial cells maintained in control conditions, but is substantially increased in retinal vessels of animals with diabetic or ischemic retinopathy and in retinal endothelial cells exposed to high glucose or hypoxia. Moreover, we have found that inhibiting NADPH oxidase blocks the upregulation of VEGF expression during diabetic and ischemic retinopathy19,20 and prevents vitreoretinal neovascularization during ischemic retinopathy.20 The goal of the present study was to determine the specific role of NADPH oxidase and NOX2 in retinal vascular inflammation related to acute and chronic vascular inflammatory reactions in endotoxemia and diabetic retinopathy, respectively.
Materials and Methods
Treatment of Animals
All procedures with animals were performed in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and were approved by the institutional animal care and use committee (Animal Welfare Assurance no. A3307-01). Experiments were performed with C57Bl/6J mice and age-matched NOX2-deficient mice backcrossed on a C57Bl/6 background (Jackson Laboratories, Bar Harbor, ME). Genotyping of NOX2-deficient mice was performed before the experiment.
Mouse Model of Retinal Vascular Inflammation
Mice lacking NOX2 and age-matched wild-type mice (25 g) were injected with lipopolysaccharide from Salmonella typhimurium (LPS, 0.1 mg/kg in PBS, IP; Sigma-Aldrich, St. Louis, MO) as a model of acute retinal vascular inflammation. Age-matched control mice received vehicle alone. Mice were killed 24 hours later and prepared for quantification of leukocyte adhesion and ICAM-1 expression in the retina.
Mouse Model of Diabetic Retinopathy
Additional groups of age-matched wild-type and NOX2−/−mice (25 g) were made diabetic by intraperitoneal injections of 55 mg/kg streptozotocin (STZ; Sigma-Aldrich) dissolved in 0.1 M fresh citrate buffer (pH 4.5). The animals were considered diabetic when the plasma glucose level exceeded 250 mg/dL. Some diabetic mice were treated with apocynin (10 mg/kg/d, dissolved in drinking water) for the duration of the experiments. After 5 weeks of hyperglycemia diabetic and age-matched normoglycemic control mice were processed for quantification of permeability or leukocyte adhesion. Identical groups of animals were prepared, and retinas were processed separately for protein analysis or for frozen sectioning and immunohistochemical analysis.
Analysis of Leukocyte Adhesion
Adhesion of leukocytes to the wall of the retinal vessels was evaluated as described previously, with slight modification.2 After the induction of deep anesthesia, the chest cavity was carefully opened, and a 20-gauge perfusion cannula was introduced into the aorta. Drainage was achieved by opening the right atrium. The animals were then perfused with 10 mL of phosphate-buffered saline (PBS) to wash out nonadherent blood cells. Next, the animals were perfused with 10 mL fluorescent isothiocyanate (FITC)–labeled concanavalin A (Con A) lectin (40 μg/mL in PBS, pH 7.4; Vector Laboratories, Burlingame, CA) to label the adherent leukocytes and vascular endothelial cells. Residual unbound Con A was removed by perfusion with PBS. The eyeballs were removed and fixed with 4% paraformaldehyde. The retinas were then observed by fluorescence microscopy, and the total number of adherent leukocytes per retina was determined. CD45 immunohistochemistry was performed with a specific anti-CD45 antibody (BD-PharMingen, San Diego, CA) to confirm that Con A-stained cells within the vasculature were leukocytes.
Analysis of BRB Permeability
Retinal vascular permeability was measured as described previously.21 Both diabetic and control mice received external jugular vein injections of 10 mg/kg bovine serum albumin (BSA)–Alexa-Fluor 488 conjugate (Invitrogen-Molecular Probes, Eugene, OR). After 30 minutes, the eyes were removed, embedded in optimal cutting temperature (OCT) embedding medium, and snap frozen in liquid nitrogen, and 12-μm-thick sections were prepared. Images (three per section) were collected at 60-μm intervals from 10 sections of each retina. Tracer extravasation was measured by morphometric analysis of fluorescein levels in nonvascular areas in the retinal images. Plasma was assayed for BSA-fluorescein concentration using a spectrofluorometer (model 4000; CytoFluor, Foster City, CA). Fluorescein intensity in the retinal sections was normalized according to the concentration of the tracer in the plasma of the same animal.
Western Blot Analysis
For analysis of ICAM-1 pooled retinas were homogenized in a modified RIPA buffer (20 mM Tris-HCl [pH 7.4], 2.5 mM EDTA, 50 mM NaF, 10 mM Na4P2O7, 1% Triton X-100, 0.1% sodium dodecyl sulfate, 1% sodium deoxycholate, and 1 mM phenyl methyl sulfonyl fluoride), and 50-μg protein samples were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to nitrocellulose membrane, and reacted with anti-ICAM-1 (1:200 rabbit anti-human antibody; Santa Cruz Biotechnology, Santa Cruz, CA) followed by horseradish peroxidase–linked secondary antibody and enhanced chemiluminescence (GE Healthcare, San Francisco, CA). Membranes were stripped and reprobed for β-actin to demonstrate equal loading, and the results were analyzed using densitometry.
Measurement of NADPH Oxidase Activity
Activity of NADPH oxidase was assayed using dichlorofluorescein imaging of retinal frozen sections. Dichlorofluorescein (DCF), the oxidation product of 2,7-dichlorodihydrofluorescein diacetate (DHDCF; Invitrogen) emits a green fluorescent signal and is a marker of cellular oxidation by hydrogen peroxide, peroxynitrite, and hydroxy radicals.22 Retinal sections were incubated with NADPH (100 μM) and DHDCF (10 μM) for 20 minutes at 37°C. The specificity of the reaction was determined by incubating the retinal sections in buffer containing DHDCF with or without PEG-SOD, catalase, or apocynin (Sigma-Al-drich). DCF formation was measured with fluorescence microscopy, to collect the images, and computer-assisted morphometry, to determine fluorescence intensity.
Statistical Analysis
Group differences were evaluated by using ANOVA and post hoc comparisons. Results were considered significant at P <0.05. Data are presented as the mean ± SE.
Results
Abrogation of LPS-Induced Increases in Leukocyte Adhesion and ICAM-1 Expression in the Retinas of NOX2-Deficient Mice
To evaluate whether activity of NOX2-containing NADPH oxi-dase plays a role in retinal vascular inflammation, we analyzed leukocyte adhesion in the retinal vessels of LPS-treated wild-type and NOX2-deficient mice. The number of adherent leukocytes in LPS-injected mice was reduced by ~60% in the mice lacking NOX2 compared with wild-type (mean ± SE ± 53 ± 17 vs. 127 ± 13, respectively; Fig. 1A). Immunoreactivity for CD45 confirmed that the adherent cells were leukocytes (Fig. 1B). Because ICAM-1 is a key mediator of leukocyte adhesion, we tested whether deletion of NOX2 affects its expression in the retina. Our Western blot analyses showed a significant increase in retinal expression of ICAM-1 after the LPS treatment. However, this increase was significantly reduced in mice lacking NOX2 (Fig. 2).
Figure 1.

Evaluation of leukocyte–endothelial interaction in LPS-injected mice. Wild-type mice and mice lacking NOX2 were injected with LPS (0.1 mg/kg, intraperitoneal) as a model of acute retinal vascular inflammation. Flatmounted retinas labeled with Con A lectin (A) show an increased number of adherent leukocytes in retinal vessels (arrows) of LPS-injected wild-type mice (+/+) compared with the mice lacking NOX2 (−/−). *P < 0.05 vs. WT (n = 5). (B) Immunolabeling with Con A and an antibody against CD45 confirms that the adherent cells were leukocytes.
Figure 2.

Western blot analysis of ICAM-1 in LPS-injected mice. ICAM-1 is increased in LPS-injected wild-type mice (+/+) compared with the control mice (C). The effect was significantly inhibited in mice lacking NOX2 (LPS−/−). *P < 0.05 vs. control; #P < 0.05 vs. LPS+/+(n = 3).
Blockade of Diabetes-Induced Increases in Leukocyte Adhesion and ICAM-1 Expression by Deletion of NOX2 or Inhibition of NADPH Oxidase
To test whether NOX2 expression and NADPH oxidase activity have a role in vascular inflammatory processes associated with diabetic retinopathy, we examined leukocyte– endothelial cell attachment and expression of ICAM-1 in the retinas of diabetic mice lacking NOX2 or treated with apocynin. Analysis of leukocyte adhesion showed a significant increase in the number of adherent leukocytes in the diabetic retinas. This increase was substantially reduced by deletion of NOX2 or apocynin treatment (Fig. 3). Furthermore, Western blot analysis showed a significant increase in expression of ICAM-1 in the diabetic retina, which also was completely blocked by apocynin or deletion of NOX2 (Fig. 4). These results indicate that activity of NOX2-containing NADPH oxidase is critically involved in leukocyte– endothelial cell attachment in the diabetic retina.
Figure 3.

Evaluation of leukocyte–endothelial interaction using Con A lectin in diabetic mice. Flatmounted retinas labeled with Con A lectin show increased of adherent leukocytes in retinal vessels of diabetic wild-type mice (DM+/+) compared with the control wild-type mice (C) and decreased leukocytes in diabetic mice lacking NOX2 (DM−/−) or treated with apocynin (DM+apocynin). *P <0.05 vs. C; #P < 0.05 vs. DM+/+(n = 7).
Figure 4.

Western blot analysis of ICAM-1 in diabetic mice. ICAM-1 was increased in diabetic wild-type mice (DM+/+) compared with the control mice (C). This effect was prevented in diabetic mice lacking NOX2 (D−/−) or treated with apocynin (DM+apocynin). *P < 0.05 vs. C; #P < 0.05 vs. DM+/+(n = 7).
Effect of Deletion of NOX2 on ROS Formation in the Diabetic Retina
To assess the potential contribution of NADPH oxidase activity to oxidative stress in the diabetic retina, we studied ROS generation by using real-time DCF imaging of flash-frozen retinas. Retinas of diabetic mice showed increased ROS production. Deletion of NOX2 or treatment of the mice with apocynin prevented the diabetes-induced increases in ROS formation (Fig. 5), indicating the role of NOX2 and NADPH oxidase activity in diabetes-induced increases in oxidative stress in the retina.
Figure 5.
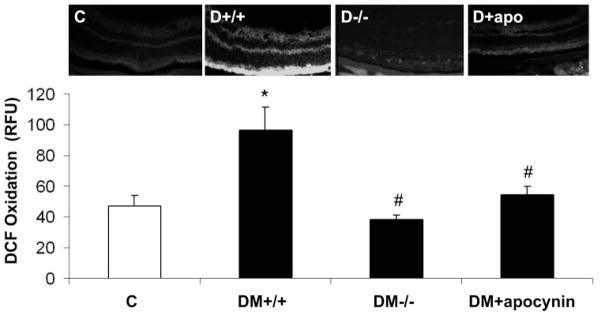
In situ production of ROS as measured by dichlorofluorescein (DCF) imaging. ROS production was increased in retinas of diabetic wild-type mice (D+/+) compared with the control wild-type mice (C). The increase was blocked in NOX2−/−diabetic mice (D−/−) and in diabetic mice treated with apocynin (D+apocynin). *P < 0.05 vs. C; #P < 0.05 vs. D+/+ (n = 7).
Effect of Deletion of NOX2 on the BRB in Diabetic Mice
Because increases in ICAM-1 expression and leukocyte adhesion have been shown to correlate with the breakdown of BRB in diabetes,2,23 we also determined the effects of NOX2 deletion and apocynin treatment on retinal vascular permeability. Evaluation of BRB function by an assay for extravasation of BSA-Alexa-Fluor 488 conjugate showed that NOX2 deletion or apocynin treatment prevented diabetes-induced increases in retinal vascular permeability (Fig. 6), indicating that activity of NOX2-containing NADPH oxidase also plays a role in mediating the vascular permeability increase in diabetes.
Figure 6.
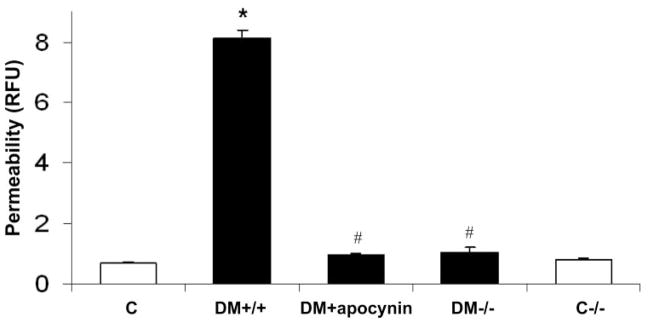
Quantification of BRB breakdown by measuring the extravasation of FITC-conjugated albumin. There was a significant increase in vascular permeability in diabetic mice (DM+/+) compared with the control wild-type (C) or NOX2 knockout (C−/−) mice. The permeability increase was blocked by treatment of the diabetic mice with apocynin (DM-apocynin) or by deletion of NOX2 (DM−/−). *P < 0.05 vs. C; # P < 0.05 vs. DM+/+ (n = 7).
Discussion
These experiments showed that knocking out NOX2 or inhibiting NADPH oxidase activity by apocynin treatment completely blocks diabetes-induced increases in retinal ICAM-1 levels and leukocyte adhesion and preserves the BRB. Furthermore, the NOX2 deletion and apocynin treatment also substantially reduced ROS formation, implying a causal role of NADPH oxidase– derived ROS in the inflammatory reaction. Because generation of ROS has been reported to be a major factor in triggering vascular injury during LPS-induced endotoxemia,24 we also determined the effect of deleting NOX2 on ICAM-1 expression and leukostasis in LPS-injected mice. These studies showed that both ICAM expression and leukostasis are markedly increased after LPS treatment and that this effect is abrogated in mice lacking NOX2. These findings confirm the importance of the activity of NOX2 NADPH oxidase in mediating retinal vascular inflammatory reactions in both acute and chronic disease conditions.
A potential link between NADPH oxidase activity and vascular inflammatory reactions has been suggested by previous studies showing that diabetes-induced increases in oxidative stress, expression of VEGF and ICAM-1, leukostasis, and breakdown of the BRB are all blocked by simvastatin, which is known to inhibit NADPH oxidase.19,23 Studies showing that diabetes-induced increases in systemic oxidative stress are blocked by apocynin also support the role of NADPH oxidase in diabetic tissue damage.25 Studies showing that retinal leukostasis induced by diabetes or intravitreous injection of angiotensin II is prevented by treatment with apocynin also support a role for NADPH oxidase activity in retinopathy.16 Further support for the role of NADPH oxidase in retinal vascular inflammatory reactions comes from studies showing that blockade of angiotensin II type 1 receptor signaling, a known stimulus for NADPH oxidase activation, blocks leukocyte adhesion to the retinal vessels and ICAM-1 expression in models of uveitis or diabetes.26,27 Studies of coronary arteries from diabetic pigs have shown that diabetes-induced increases in NADPH oxidase activity are accompanied by upregulation of inflammatory cytokines (IL-6 and TNF-α), chemokines (MCP-1), and vascular cell adhesive molecules (VCAM-1), providing further support for the role of NADPH oxidase in vascular inflammation.28 Overall, these reports suggest that NADPH oxidase-derived ROS are critically involved in triggering leukocyte–endothelial cell adhesion and vascular injury during diabetes or other inflammatory conditions.
To the best of our knowledge, the present study is the first to show that NOX2 is critically involved in retinal vascular inflammation and diabetes-induced breakdown of the BRB. However, consistent with our finding that diabetes-induced ROS formation is reduced in the NOX2 knockout mouse retina, others have reported that deletion of NOX2 inhibits ROS production in various tissues, including aorta in DOCA (deoxycorticosterone acetate)-salt–induced hypertension,29 brain with intracerebral hemorrhage,30 intrapulmonary arteries with chronic hypoxia, and ischemic hindlimb.31 Furthermore, knocking out NOX2 has been shown to reduce leukocyte adhesion in a mouse model of hypercholesterolemia or ischemia–reperfusion injury in brain.32
Various sources of oxidative stress have been identified in experimental models of diabetes. Studies in diabetic animals and retinal cells exposed to high-glucose conditions in vitro have shown that activation of the mitochondrial electron transport chain is a primary source of oxidative stress.33 Mitochondria-derived ROS trigger the activation of multiple pathways of hyperglycemic damage, including NADPH oxidase via protein kinase C.34,35 Moreover, studies in other models indicate that although mitochondrial-derived ROS are important for the initiation of oxidative stress responses, activity of NADPH oxidase is needed to sustain sufficient levels of ROS formation for the transduction of specific cellular responses.36 –38 Further study is needed to determine the potential role of mitochondrial ROS in activating NADPH oxidase in models of diabetes and endotoxemia. However, our finding that increases in retinal expression of ICAM-1 and leukocyte adhesion to endothelial cells were substantially inhibited in the LPS model and completely blocked in mice with diabetes that lacked NOX2 or in diabetic mice treated with apocynin provides strong evidence of the critical role played by NOX2 and NADPH oxidase activity in mediating retinal vascular inflammatory reactions in both models.
Further work is also needed to identify the specific cellular source(s) of the NADPH oxidase activity responsible for the retinal vascular inflammatory reactions observed in our experiments. NOX2 is expressed by both phagocytic and vascular endothelial cells. During conditions of inflammation or host defense reactions, ROS are produced at high levels by phagocyte NADPH oxidase. In normal endothelial cells ROS are produced mainly by activity of the NOX4 enzyme.39,40 However, previous studies have shown that diabetes and high-glucose treatment promote significant increases in NOX2 levels in retinal endothelial cells.19 Thus, it seems likely that both phagocyte- and endothelial cell-derived ROS are involved in the pathologic response.
In summary, our present results suggest that reactive oxygen species produced by the NOX2 NADPH oxidase mediate retinal vascular inflammatory reactions associated with both acute and chronic models of retinal vascular disease. Inhibition of this enzyme may offer a novel therapeutic strategy for the amelioration of vascular injury during retinopathy.
Acknowledgments
Supported by the Juvenile Diabetes Research Foundation (JDRF), Fight For Sight, and the American Heart Association Grant AHA00104 (MA); National Eye Institute Grants R01 EY04618 and R01 EY11766, and the Veterans Administration Career Development Award (CDA) and Merit Review Award (RBC).
Footnotes
Disclosure: M. Al-Shabrawey, None; M. Rojas, None; T. Sanders, None; A. Behzadian, None; A. El-Remessy, None; M. Bartoli, None; A.K. Parpia, None; G. Liou, None; R.B. Caldwell, None
References
- 1.Adamis AP. Is diabetic retinopathy an inflammatory disease? Br J Ophthalmol. 2002;86:363–365. doi: 10.1136/bjo.86.4.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Joussen AM, Poulaki V, Le ML, et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 2004;18:1450–1452. doi: 10.1096/fj.03-1476fje. [DOI] [PubMed] [Google Scholar]
- 3.Miyamoto K, Hiroshiba N, Tsujikawa A, Ogura Y. In vivo demonstration of increased leukocyte entrapment in retinal microcirculation of diabetic rats. Invest Ophthalmol Vis Sci. 1998;39:2190–2194. [PubMed] [Google Scholar]
- 4.Ishida S, Yamashiro K, Usui T, et al. Leukocytes mediate retinal vascular remodeling during development and vasoobliteration in disease. Nat Med. 2003;9:781–788. doi: 10.1038/nm877. [DOI] [PubMed] [Google Scholar]
- 5.Joussen AM, Murata T, Tsujikawa A, Kirchhof B, Bursell SE, Adamis AP. Leukocytemediated endothelial cell injury and death in the diabetic retina. Am J Pathol. 2001;158:147–152. doi: 10.1016/S0002-9440(10)63952-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koizumi K, Poulaki V, Doehmen S, et al. Contribution of TNF-alpha to leukocyte adhesion, vascular leakage, and apoptotic cell death in endotoxin-induced uveitis in vivo. Invest Ophthalmol Vis Sci. 2003;44:2184–2191. doi: 10.1167/iovs.02-0589. [DOI] [PubMed] [Google Scholar]
- 7.Kern TS. Contributions of inflammatory processes to the development of the early stages of diabetic retinopathy. Exp Diabetes Res. 2007;2007:95103. doi: 10.1155/2007/95103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nagata M. Inflammatory cells and oxygen radicals. Curr Drug Targets Inflamm Allergy. 2005;4:503–504. doi: 10.2174/1568010054526322. [DOI] [PubMed] [Google Scholar]
- 9.Hordijk PL. Regulation of NADPH oxidases: the role of Rac proteins. Circ Res. 2006;98:453– 462. doi: 10.1161/01.RES.0000204727.46710.5e. [DOI] [PubMed] [Google Scholar]
- 10.Wang T, Zhang X, Li JJ. The role of NF-kappaB in the regulation of cell stress responses. Int Immunopharmacol. 2002;2:1509–1520. doi: 10.1016/s1567-5769(02)00058-9. [DOI] [PubMed] [Google Scholar]
- 11.Kitamei H, Iwabuchi K, Namba K, et al. Amelioration of experimental autoimmune uveoretinitis (EAU) with an inhibitor of nuclear factor-kappaB (NF-kappaB), pyrrolidine dithiocarbamate. J Leukoc Biol. 2006;79:1193–1201. doi: 10.1189/jlb.0805453. [DOI] [PubMed] [Google Scholar]
- 12.Fang IM, Yang CH, Lin CP, Yang CM, Chen MS. Effects of pyrrolidine dithiocarbamate, an NF-kappaB inhibitor, on cytokine expression and ocular inflammation in experimental autoimmune anterior uveitis. J Ocul Pharmacol Ther. 2005;21:95–106. doi: 10.1089/jop.2005.21.95. [DOI] [PubMed] [Google Scholar]
- 13.Chen CC, Chow MP, Huang WC, Lin YC, Chang YJ. Flavonoids inhibit tumor necrosis factor-alpha-induced up-regulation of inter-cellular adhesion molecule-1 (ICAM-1) in respiratory epithelial cells through activator protein-1 and nuclear factor-kappaB: structure-activity relationships. Mol Pharmacol. 2004;66:683– 693. doi: 10.1124/mol.66.3.. [DOI] [PubMed] [Google Scholar]
- 14.El-Remessy AB, Behzadian MA, Abou-Mohamed G, Franklin T, Caldwell RW, Caldwell RB. Experimental diabetes causes breakdown of the blood–retina barrier by a mechanism involving tyrosine nitration and increases in expression of vascular endothelial growth factor and urokinase plasminogen activator receptor. Am J Pathol. 2003;162:1995–2004. doi: 10.1016/S0002-9440(10)64332-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abiko T, Abiko A, Clermont AC, et al. Characterization of retinal leukostasis and hemodynamics in insulin resistance and diabetes: role of oxidants and protein kinase-C activation. Diabetes. 2003;52:829– 837. doi: 10.2337/diabetes.52.3.829. [DOI] [PubMed] [Google Scholar]
- 16.Chen P, Guo AM, Edwards PA, Trick G, Scicli AG. Role of NADPH oxidase in angiotensin II in diabetes-induced retinal leukostasis. Am J Physiol Regul Integr Comp Physiol. 2007;293(4):R1619–R1629. doi: 10.1152/ajpregu.00290.2007. [DOI] [PubMed] [Google Scholar]
- 17.Kern TS, Miller CM, Du Y, et al. Topical administration of nepafenac inhibits diabetes-induced retinal microvascular disease and underlying abnormalities of retinal metabolism and physiology. Diabetes. 2007;56:373–379. doi: 10.2337/db05-1621. [DOI] [PubMed] [Google Scholar]
- 18.Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 19.Al-Shabrawey M, Bartoli M, El-Remessy A, et al. Role of NADPH Oxidase and Stat3 in statin-mediated protection against diabetic retinopathy. Invest Ophthalmol Vis Sci. 2008;49:3231–3238. doi: 10.1167/iovs.08-1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Al-Shabrawey M, Bartoli M, El-Remessy AB, et al. Inhibition of NAD(P)H oxidase activity blocks vascular endothelial growth factor overexpression and neovascularization during ischemic retinopathy. Am J Pathol. 2005;167:599– 607. doi: 10.1016/S0002-9440(10)63001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Antonetti DA, Barber AJ, Khin S, Lieth E, Tarbell JM, Gardner TW. Vascular permeability in experimental diabetes is associated with reduced endothelial occludin content: vascular endothelial growth factor decreases occludin in retinal endothelial cells. Penn State Retina Research Group. Diabetes. 1998;47:1953–1959. doi: 10.2337/diabetes.47.12.1953. [DOI] [PubMed] [Google Scholar]
- 22.Munzel T, Afanas’ev IB, Kleschyov AL, Harrison DG. Detection of superoxide in vascular tissue. Arterioscler Thromb Vasc Biol. 2002;22:1761–1768. doi: 10.1161/01.atv.0000034022.11764.ec. [DOI] [PubMed] [Google Scholar]
- 23.Miyahara S, Kiryu J, Yamashiro K, et al. Simvastatin inhibits leukocyte accumulation and vascular permeability in the retinas of rats with streptozotocin-induced diabetes. Am J Pathol. 2004;164:1697–1706. doi: 10.1016/S0002-9440(10)63728-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brandes RP, Koddenberg G, Gwinner W, et al. Role of increased production of superoxide anions by NAD(P)H oxidase and xanthine oxidase in prolonged endotoxemia. Hypertension. 1999;33:1243–1249. doi: 10.1161/01.hyp.33.5.1243. [DOI] [PubMed] [Google Scholar]
- 25.Sonta T, Inoguchi T, Tsubouchi H, et al. Evidence for contribution of vascular NAD(P)H oxidase to increased oxidative stress in animal models of diabetes and obesity. Free Radic Biol Med. 2004;37:115–123. doi: 10.1016/j.freeradbiomed.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 26.Nagai N, Izumi-Nagai K, Oike Y, et al. Suppression of diabetes-induced retinal inflammation by blocking the angiotensin II type 1 receptor or its downstream nuclear factor-kappaB pathway. Invest Ophthalmol Vis Sci. 2007;48:4342– 4350. doi: 10.1167/iovs.06-1473. [DOI] [PubMed] [Google Scholar]
- 27.Nagai N, Oike Y, Noda K, et al. Suppression of ocular inflammation in endotoxin-induced uveitis by blocking the angiotensin II type 1 receptor. Invest Ophthalmol Vis Sci. 2005;46:2925–2931. doi: 10.1167/iovs.04-1476. [DOI] [PubMed] [Google Scholar]
- 28.Zhang L, Zalewski A, Liu Y, et al. Diabetes-induced oxidative stress and low-grade inflammation in porcine coronary arteries. Circulation. 2003;108:472– 478. doi: 10.1161/01.CIR.0000080378.96063.23. [DOI] [PubMed] [Google Scholar]
- 29.Fujii A, Nakano D, Katsuragi M, et al. Role of gp91phox-containing NADPH oxidase in the deoxycorticosterone acetate-salt-induced hypertension. Eur J Pharmacol. 2006;552:131–134. doi: 10.1016/j.ejphar.2006.09.039. [DOI] [PubMed] [Google Scholar]
- 30.Tang J, Liu J, Zhou C, et al. Role of NADPH oxidase in the brain injury of intracerebral hemorrhage. J Neurochem. 2005;94:1342–1350. doi: 10.1111/j.1471-4159.2005.03292.x. [DOI] [PubMed] [Google Scholar]
- 31.Tojo T, Ushio-Fukai M, Yamaoka-Tojo M, Ikeda S, Patrushev N, Alexander RW. Role of gp91phox (Nox2)-containing NAD(P)H oxidase in angiogenesis in response to hindlimb ischemia. Circulation. 2005;111:2347–2355. doi: 10.1161/01.CIR.0000164261.62586.14. [DOI] [PubMed] [Google Scholar]
- 32.Ishikawa M, Stokes KY, Zhang JH, Nanda A, Granger DN. Cerebral microvascular responses to hypercholesterolemia: roles of NADPH oxidase and P-selectin. Circ Res. 2004;94:239–244. doi: 10.1161/01.RES.0000111524.05779.60. [DOI] [PubMed] [Google Scholar]
- 33.Du Y, Miller CM, Kern TS. Hyperglycemia increases mitochondrial superoxide in retina and retinal cells. Free Radic Biol Med. 2003;35:1491–1499. doi: 10.1016/j.freeradbiomed.2003.08.018. [DOI] [PubMed] [Google Scholar]
- 34.Inoguchi T, Sonta T, Tsubouchi H, et al. Protein kinase C-dependent increase in reactive oxygen species (ROS) production in vascular tissues of diabetes: role of vascular NAD(P)H oxidase. J Am Soc Nephrol. 2003;14:S227–S232. doi: 10.1097/01.asn.0000077407.90309.65. [DOI] [PubMed] [Google Scholar]
- 35.Inoguchi T, Tsubouchi H, Etoh T, et al. A possible target of antioxidative therapy for diabetic vascular complications-vascular NAD(P)H oxidase. Curr Med Chem. 2003;10:1759–1764. doi: 10.2174/0929867033457133. [DOI] [PubMed] [Google Scholar]
- 36.Lee SB, Bae IH, Bae YS, Um HD. Link between mitochondria and NADPH oxidase 1 isozyme for the sustained production of reactive oxygen species and cell death. J Biol Chem. 2006;281:36228–36235. doi: 10.1074/jbc.M606702200. [DOI] [PubMed] [Google Scholar]
- 37.Kimura S, Zhang GX, Nishiyama A, et al. Role of NAD(P)H oxidase-and mitochondria-derived reactive oxygen species in cardioprotection of ischemic reperfusion injury by angiotensin II. Hypertension. 2005;45:860– 866. doi: 10.1161/01.HYP.0000163462.98381.7f. [DOI] [PubMed] [Google Scholar]
- 38.Schafer M, Schafer C, Ewald N, Piper HM, Noll T. Role of redox signaling in the autonomous proliferative response of endothelial cells to hypoxia. Circ Res. 2003;92:1010–1015. doi: 10.1161/01.RES.0000070882.81508.FC. [DOI] [PubMed] [Google Scholar]
- 39.Sorescu D, Weiss D, Lassegue B, et al. Superoxide production and expression of Nox family proteins in human atherosclerosis. Circulation. 2002;105:1429–1435. doi: 10.1161/01.cir.0000012917.74432.66. [DOI] [PubMed] [Google Scholar]
- 40.Ago T, Kitazono T, Ooboshi H, et al. Nox4 as the major catalytic component of an endothelial NAD(P)H oxidase. Circulation. 2004;109:227–233. doi: 10.1161/01.CIR.0000105680.92873.70. [DOI] [PubMed] [Google Scholar]