

Abstract
Quorum sensing is a mechanism of chemical communication among bacteria that enables collective behaviors. In V. cholerae, the etiological agent of the disease cholera, quorum sensing controls group behaviors including virulence factor production and biofilm formation. The major V. cholerae quorum-sensing system consists of the extracellular signal molecule called CAI-1 and its cognate membrane bound receptor called CqsS. Here, the ligand binding activity of CqsS is probed with structural analogs of the natural signal. Enabled by our discovery of a structurally simplified analog of CAI-1, we prepared and analyzed a focused library. The molecules were designed to probe the effects of conformational and structural changes along the length of the fatty acid tail of CAI-1. Our results, combined with pharmacophore modeling, suggest a molecular basis for signal molecule recognition and receptor fidelity with respect to the fatty acid tail portion of CAI-1. These efforts provide novel probes to enhance discovery of anti-virulence agents for the treatment of V. cholerae.
Keywords: autoinducer, quorum sensing, structure-activity relationship, Vibrio cholerae
1. Introduction
Many bacteria control collective behaviors using a cell-to-cell communication process called quorum sensing (QS).1, 2, They sense cell population density through the production, release, and detection of extracellular signal molecules termed autoinducers (AIs). The concentration of AIs grows in proportion to the cell population density; when a threshold AI concentration is achieved, the bacteria initiate a group-wide response that promotes induction and/or repression of target genes. Community-wide synchronization of gene expression results in behaviors that are effective only when undertaken simultaneously by large groups of cells. QS-controlled functions include virulence factor production, bioluminescence, biofilm formation, sporulation, and antibiotic synthesis.3-5
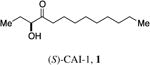
In the human pathogen V. cholerae, QS is mediated by two parallel phospho-relay systems.6 One system, which is the focus of this study, employs the membrane-bound receptor CqsS to detect two related AI molecules, CAI-1 (1, S-3-hydroxytridecan-4-one)7 and Ea-CAI-1 (2, 3-aminotridec-2-en-4-one)8 (Figure 1C). Ea-CAI-1 is produced by the aminotransferase enzyme CqsA and is subsequently converted to CAI-1.9 Under conditions in which signal molecule concentration is below the threshold for detection, such as at low cell density, CqsS functions as a kinase, phosphorylating the response regulator LuxO. The phosphorylation of LuxO results in the expression of a regulon of low cell density genes, including those required for virulence factor production and biofilm formation. At high cell density, when CAI-1 accumulates to an appreciable level, binding of the ligand to CqsS inhibits its kinase activity enabling its phosphatase activity to predominate. As a consequence, phosphate flow in the circuit is reversed, leading to dephosphorylation and inactivation of LuxO; this initiates the high cell density QS gene expression program. Under this condition, production of virulence factors and biofilm formation are repressed. By contrast, activation of the expression of a protease gene occurs, and this protease facilitates the exit of V. cholerae from its host.10
Figure 1.

QS in Vibrio cholerae. a) The CAI-1 AI (green circles)is produced by the CqsA synthase. Under conditions of low CAI-1 concentration the cognate receptor, CqsS acts as a kinase, and transfers phosphate to LuxO which leads to the expression of genes underpinning individual behaviors. b) At high CAI-1 concentrations, CqsS binds CAI-1 and this converts CqsS from a kinase to a phosphatase. LuxO is dephosphorylated which leads to the expression of genes required for group behaviors. c) The native CAI-1 QS molecules in V. cholerae.
CAI-1 and Ea-CAI-1 each contain a 3-carbon unit derived from (S)-adenosyl methionine (SAM) (highlighted in red, Figure 2) attached to an acyl tail.9 Likewise, Legionella pneumophila employs an analog of CAI-1 (LAI-1, 3-hydroxypentadecan-4-one) with a C-12 acyl tail,11 and Vibrio harveyi employs an analog of Ea-CAI-1 (C8-Ea-CAI-1, 3-aminoundec-2-en-4-one) with a C-8 acyl tail.8 Based on these discoveries, we propose that while the conserved polar portion (in red) of this family of signaling molecules certainly plays a role in receptor binding, the variable acyl tail defines receptor specificity and is required to maintain signaling fidelity. This analysis has a strong analogy in the widely studied QS acyl-homoserine lactone AIs (AHLs).2 The AHLs are similarly comprised of a structurally conserved polar fragment, the homoserine lactone, derived from SAM, and a variable fragment, the acyl tail, derived from fatty-acid biosynthesis (Figure 2).
Figure 2.

Simplified analysis of the structural features of the CAI-1 family of QS signal molecules. An equivalent of SAM (3) is incorporated into CAI-1 providing the structurally conserved portion of the final molecules (highlighted in red), along with an equivalent of a fatty acid in the form of a fatty acid thioester that provides the structurally variable portion of the signaling molecules (highlighted in blue). (
 = fatty acid; R′ = CoA or ACP)
= fatty acid; R′ = CoA or ACP)
Bacterial histidine kinases, including CqsS, are ubiquitous and they relay extracellular sensory information into cells. These so-called two-component sensors generally have complicated membrane-spanning domains, which has made structural analysis difficult. In the absence of detailed structural information, chemical genetics approaches can provide useful information about ligand-receptor interactions enabling the design and optimization of small molecule agonists and antagonists that serve as useful probes of signal transduction.
To define the roles of different structural features of V. cholerae CAI-1 in signal recognition and specificity, we previously examined three distinct chemical features of this ligand: (a) the ethyl side chain, (b) the 3-heteroatom substituent and (c) the chain length of the acyl tail.12 The structure-activity relationships can be summarized as follows: (a) the introduction of phenyl substitution in the ethyl side-chain results in molecules that behave as antagonists, (b) the 3-heteroatom position is important for agonist activity with 3-OH or 3-NH2 substitution being optimal, and (c) the receptor is sensitive to changes in the chain-length of the fatty acid tail with a C-9 or C-10 acyl tail being most potent.
We identified distinct amino acid residues in the CqsS trans-membrane region that play corresponding roles in the recognition of each of these three structural features (Fig 3a).13 In summary: (a) F162 recognizes changes in the ethyl side-chain, (b) W104 and S107 dictate the receptor's response to alterations at the 3-heteroatom position, and (c) C170 controls the receptor response to variations in the length of the acyl tail. These studies presumably reveal the location of the ligand binding pocket in the CqsS receptor as well as the constraints on allowable ligand alterations.
Figure 3.

a) SAR targeting three structural features of the CAI-1 signaling molecule. b) Chemical-genetic analysis of CqsS enabling localization of the putative ligand binding pocket and identification of specific amino acid binding determinants.
Here we expand our understanding of the SAR of CAI-1 with a focus on the effect of conformational and structural changes along the length of the fatty acid tail (Library C in Figure 3a). As detailed below, the synthesis of a focused library of analogs was facilitated by the discovery that replacing the keto unit in CAI-1 with an ester group maintains full activity. These efforts provide a foundation for understanding signaling molecule specificity in the V. cholerae CqsS receptor and provide novel structures to serve as probes of this signaling circuit.
2. Results and Discussion
2.1. Assay Methods and Analysis
To quantify the induction of QS activity in V. cholerae, each of the CAI-1 analogs was added in triplicate to V. cholerae MM920 (ΔcqsA, ΔluxQ, pBB1 (a plasmid harboring the V. harveyi luxCDABE operon)) at increasing concentrations (typically using a 4-fold dilution series), and the light output from the heterologous luciferase reporter was monitored using a scintillation counter.14 Two parameters are described: (a) the EC50 values for activation of the QS response, and (b) the maximum level of activation compared to the native signal CAI-1 as 100%. Percent response does not necessarily correlate with EC50 as can be observed in Tables 1 and 3 – 6. Presumably this occurs because changes in the structure of the ligand or in the structure of the receptor binding site can alter the balance between kinase and phosphatase activities.12
Table 1. Simple ester and amide CAI-1 analogs.
| Entry | Compound | EC50 (nM)a | % Responseb |
|---|---|---|---|
| 1 |
|
38±3 | 100±4 |
| 2 |
|
24±2 | 106±15 |
| 3 |
|
670±240 | 94±35 |
| 4 |
|
180±170 | 108±4 |
| 5 |
|
1,500±220 | 81±11 |
| 6 |
|
28±2 | 106±4 |
| 7 |
|
30±2 | 108±3 |
| 8 |
|
33±2 | 107±3 |
Values were determined using a light-based CqsS agonist bioassay, see Supporting Information. All EC50 values are the mean of triplicate analyses with the range defining the 95% confidence interval.
Percent maximal bioluminescence, with respect to CAI-1, which is set at 100%. The range defines the 95% confidence interval.
Table 3. Polar CAI-1 ester analogs.
| Entry | Compound | EC50 (nM)a | % Responseb |
|---|---|---|---|
| 1 |
|
4,400±450 | 96±8 |
| 2 |
|
>50,000 | 31c |
| 3 |
|
2,400±300 | 91±9 |
| 4 |
|
10,000±1,800 | 95±15 |
Values determined using a light-based CqsS agonist bioassay, see Supporting Information. All EC50 values are the mean of triplicate analyses with the range defining the 95% confidence interval.
Percent maximal bioluminescence, with respect to CAI-1, which is set at 100%. The range defines the 95% confidence interval.
The maximal % response at the maximum concentration tested (50,000 nM).
Table 6. Activity of CAI-1 ester analogs in mutant CqsS receptors.
| Compound | wild-type CqsS | CqsS C170A | CqsS C170Y | |||
|---|---|---|---|---|---|---|
| EC50 (nM)a | % Responseb | EC50 (nM)a | % Responseb | EC50 (nM)a | % Responseb | |
| (S)-CAI-1, 1 | 62±3 | 100±4 | 72±21 | 100±44 | >50,000 | 20c |
| C8-CAI-113 | 9,400±1,600 | 83±46 | 2,100±940 | 61±70 | 260±34 | 100±8 |
| C8-ester, 9 | 180±170 | 108±4 | 110±22 | 99±34 | >50,000 | 66c |
| 4-Ph-ester, 37 | 1,100±270 | 59±22 | >50,000 | 16c | >50,000 | 22c |
| 5-Ph-ester, 38 | 231±15 | 93±6 | 180±33 | 83±29 | >50,000 | 19c |
| 3-Cy-ester, 39 | 310±51 | 82±10 | 790±220 | 73±28 | 420±96 | 93±14 |
| 4-Cy-ester, 40 | 84±6 | 97±6 | 360±84 | 77±32 | 960±320 | 51±48 |
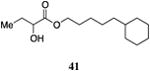
| 5-Cy-ester, 41 | 190±27 | 95±9 | 1,000±180 | 101±53 | 11,000±9,800 | 66±57 |
Values determined using standard light-based agonist bioassay with the designated CqsS mutant strain, see Supporting Information. All EC50 values are the mean of triplicate analyses with the range defining the 95% confidence interval.
Percent maximal bioluminescence, with respect to CAI-1, which is set at 100%. The range defining the 95% confidence interval.
The maximal % response at the maximum concentration tested (50,000 nM).
2.2. Design and evaluation of hetero substitution in the alkyl tail: CAI-1 ester and amide derivatives
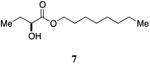
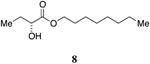
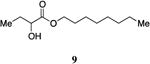
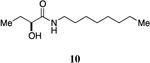
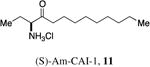
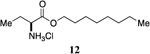
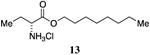
An attractive approach for the preparation of a library differing in the alkyl tail involves the introduction of a heteroatom connector between the hydroxyketone portion and the alkyl tail (7-10, 12, 13, Figure 4). These molecules were prepared by conventional methods of solution-phase synthesis (Scheme 1 (4 -> 6) and Supporting Information) and assayed for their biological activity (Table 1). The amide 10 displayed diminished activity both in terms of its EC50 value (EC50 = 1,500 nM) and maximal activity (81% of CAI-1 maximal response). The lower activity of the amide analog compared to the parent CAI-1 ketone may be due to either the geometrical constraints imposed by the amide functionality or due to unfavorable electronic interactions with the polarized amide structure. However we were pleased to find that ester analog 7 closely maintained the activity profile of native CAI-1 both in terms of EC50(7, EC50 = 24 nM; CAI-1 (1) EC50 = 38 nM) and maximal induction of the QS response (7 = 106% response). The stereochemistry of the analog plays a role in its potency, with the (S)-C8-ester-CAI-1 (7) displaying a 28-fold lower EC50 than the corresponding (R)-C8-ester-CAI-1 analog (8). Both analogs were found to be capable of fully activating the QS response and the racemate (9) showed intermediate activity and full activation, as expected. Similar to previous observations, changing the 3-hydroxy substituent to amino (11-13) resulted in a potent QS agonist (EC50 30-33 nM; 107-108% response). To assess if these esters were serving as prodrugs23, 24 we assayed the activity of the alcohol tail of 9 (1-octanol) and 2-hydroxybutyric acid and found that these molecules show no QS activity up to 50,000 nM. Accordingly, for the systematic evaluation of tail structure, we have employed racemic mixtures of the 3-hydroxyester derivatives (Figure 5).
Scheme 1.
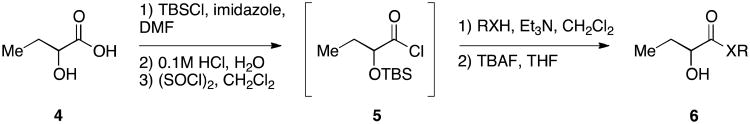
Initial synthesis of structurally simplified CAI-1 analogs.
2.3. Refinement of the synthesis of ester analogs
Building on the discovery that the presence of an oxygen linker in the acyl tail (CAI-1 ester analog 9) provides activity comparable to the native α-hydroxyl ketone signal, we prepared a larger library of CAI-1 ester analogs using an efficient one-step trans-esterification reaction of ethyl 2-hydroxybutyrate with a series of structurally diverse alcohols employing N-heterocyclic carbenes as catalysts.15 We optimized this reaction by varying the carbene catalyst, reaction time, and solvent (see Supporting Information). Catalyst 15 (10 mol%) was generally effective for the selective coupling of a series of primary alcohol tails with unprotected racemic ethyl 2-hydroxybutyrate (14, Scheme 2). Notably, under none of the conditions examined did we detect the formation of the undesired product involving esterification of the secondary alcohol on ethyl 2-hydroxybutyrate.16, 17 Utilizing this methodology, four focused libraries of racemic CAI-1 esters were prepared by introducing: (a) varying tail length, (b) ether substitution along the tail, (c) unsaturation in the tail, and (d) cycloalkyl and arene units in the tail (Tables 2-5).
Scheme 2.
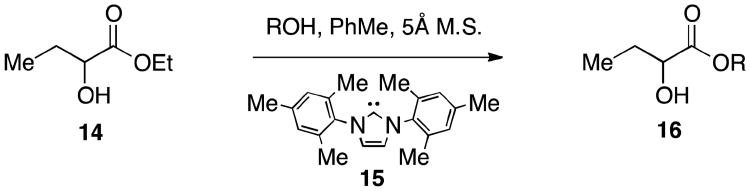
Second-generation synthesis of CAI-1 ester analogs.
Table 2. CAI-1 ester chain-length analogs.
| Entry | Compound | EC50 (nM)a | % Responseb |
|---|---|---|---|
| 1 |
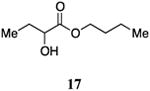
|
>50000 | 23c |
| 2 |
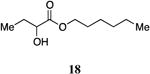
|
1,000±160 | 84±27 |
| 3 |
|
350±59 | 104±9 |
| 4 |
|
73±12 | 92±26 |
| 5 |
|
720±270 | 74±9 |
Values were determined using a light-based CqsS agonist bioassay, see Supporting Information. All EC50 values are the mean of triplicate analyses with the range defining the 95% confidence interval.
Percent maximal bioluminescence, with respect to CAI-1, which is set at 100%. The range defines the 95% confidence interval.
The maximal % response at the maximum concentration tested (50,000 nM).
Table 5. Aryl- and cyclohexyl-substituted CAI-1 ester analogs.
| Entry | Compound | EC50 (nM)a | % Responseb |
|---|---|---|---|
| 1 |
|
1,100±270 | 59±22 |
| 2 |
|
231±15 | 93±6 |
| 2a |
|
151±20 | 92±13 |
| 3 |
|
310±51 | 82±10 |
| 4 |
|
84±6 | 97±6 |
| 5 |
|
190±27 | 95±9 |
| 6 |
|
190±19 | 100±6 |
| 6a |
|
117±12 | 94±11 |
| 7 |
|
3,700±580 | 83±13 |
| 8 |
|
>50,000 | llc |
| 9 |
|
680±84 | 85±8 |
| 10 |
|
1,600±210 | 79+10 |
| 11 |
|
190±17 | 98±6 |
| 12 |
|
9,400±3,600 | 60±53 |
| 13 |
|
>50,000 | 28c |
Values determined using a light-based CqsS agonist bioassay, see Supporting Information. All EC50 values are the mean of triplicate analyses with the range defining the 95% confidence interval.
Percent maximal bioluminescence, with respect to CAI-1, which is set at 100%. The range defining the 95% confidence interval.
The maximal % response at the maximum concentration tested (50,000 nM).
2.4. CAI-1 ester acyl tail length analogs
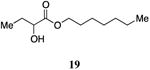
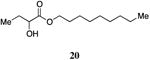
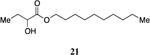
The simple saturated CAI-1 ester analogs display a pattern of biological activity comparable to the previously reported series of CAI-1 tail length analogs (Table 2).12 Specifically, the removal of a single methylene group from the saturated ester tail reduces the activity of analog 19 2-fold compared to the racemic ester 9. In contrast, the addition of a single methylene as in 20 results in a 2-fold enhancement in the EC50 of the analog (9, EC50=180 nM; 20, EC50=73 nM), whereas the addition of a single methylene in the hydroxyketone series of molecules was previously reported to result in a 5-fold decrease in EC50.12 The precise chain lengths in the ester series are not exactly the same as in the alkyl series; the C-O bond length (ca. 1.35 Å) in an ester is less than the typical C-C bond length (1.54 Å); the 10-atom acyl chain in ester 9 is slightly shorter than the 10-carbon acyl chain in 1. This feature may help rationalize why in the ester analog the addition of a methylene unit has a beneficial impact on analog activity (2-fold decrease in EC50) whereas the removal of a methylene group results in lower activity (4-fold increase in EC50). The addition or removal of two methylene units (21 and 18) results in a greater than 4-fold decrease in activity from 9, along with a decrease in the maximal %-response (entries 2 and 5, Table 2). The removal of four methylene units (17) results in the nearly complete loss of QS activity.
As summarized in Figure 6, the SAR presented here closely mirrors that in our previous report involving a series of α-hydroxy ketone acyl tail length analogs. These data support our proposal that the α-hydroxy ester motif is a suitable framework for further exploration of the influence of tail structure on the activation of the CqsS receptor.
Figure 6.

Comparison of EC50 values as a function of acyl tail length for CAI-1 analogs. CAI-1 ketone acyl tail chain-length analogs EC50 values (●)12 and CAI-1 ester chain-length analogs EC50 values (■).
2.4. CAI-1 ester analogs with ether substitution
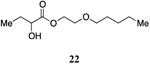
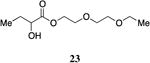
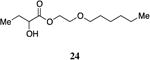
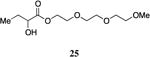
The highly lipophilic nature of the native CAI-1 and existing analogs may limit application as chemical probes in complex systems (e.g., in an animal host). In an effort to uncover effective agonists with polar functionality in the tail, we prepared a series of linear CAI-1 esters containing oxygen atoms at various positions along the ester tail. In general, the incorporation of the ether functionality results in the loss of biological activity (Table 3). A single additional oxygen atom in the CAI-1 ester tail, as in 22 and 24 results in a >13-fold decrease in activity from the C8-ester-CAI-1 (9); however, at sufficient concentration, these analogs did induce the maximal QS response (Table 3, entries 1 and 3). The diethylene glycol derived analog, 23, displays only minimal QS activity at 50,000 nM. Interestingly, however, the triethylene glycol derived analog, 25, stimulated the maximal QS response although at relatively high concentrations (EC50=10,000 nM).
2.2.5. Unsaturated CAI-1 ester analogs
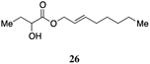
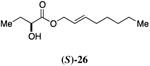
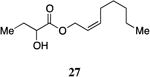
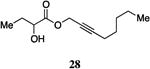
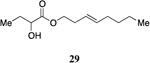
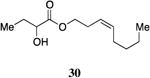
A series of CAI-1 esters was prepared bearing units of unsaturation, including (E)- and (Z)-olefin isomers and alkynes, at specific positions along the alkyl tail portion. We expected these derivatives would clarify whether a linear (E-alkene or alkyne) or bent conformation (Z-alkene) of the tail is preferred. With one exception, full activation of the receptor was observed, but the EC50 values varied as a function of both the configuration and position of the alkene unit (Table 4). The introduction of an (E)-alkene at the 2-position (2-(E)-ester-CAI-1, 26) provided an analog that was 3-fold more active than the fully saturated ester, 9 (Table 4, entry 1). In contrast, the presence of a (Z)-alkene at the 2-position (27; entry 2) led to an analog that is almost 4-fold less active than C8-ester-CAI-1 (9). With an alkene in the 3-position, both the (E)- and (Z)-isomers displayed similarly diminished activity (4-fold lower EC50 than 9) (entries 4 and 5, respectively). Interestingly, in the 4-position, while the (E)-alkene (32, entry 7) was comparable in activity to the saturated C8-ester-CAI-1 with a 1.5-fold higher EC50 value, the (Z)-isomer displayed significantly diminished activity (10-fold less active), similar to the 2-unsaturated alkenes, 23 and 24. The 5-alkene isomers 34 and 35, again showed lower activity for both (E)- and (Z)-olefin isomers, analogous to the 3-unsaturated analogs 29 and 30. The analogs bearing an alkyne at either the 2- or 3-position displayed diminished activity with the 3-isomer activating only 77% of the maximum response (entry 3 and 6). The 2-trans-cyclopropyl ester analog 33 was also diminished in activity, almost 7-times less active than 9 (entry 11). Overall, the (Z)-alkene isomers (27, 30, 33 and 35) all show diminished activity compared to the simple ester analog 9 by a factor of 3 or more. In contrast, the (E)-alkene containing analogs display activities that are highly dependant on the position of the olefin, with the 2-(E) and 4-(E) analogs (26 and 32) exhibiting the greatest activity. Among this series of analogs that possess single points of conformational restriction along the length of the acyl tail portion of CAI-1, the 2-(E) analog 26 uniquely displays enhanced potency compared to the fully saturated analog, suggesting that the presence of a linear conformation of the alkyl tail proximal to the ester is favorable for binding. However, this effect is not simply conformational as the 2-(trans)-cyclopropyl analog 36 displays diminished activity. As was observed in the series of saturated esters (7-9) the (S)-isomer of the most potent compound from this series (S)-26 (entry 1a) displays a lower EC50 value than 26 (entry 1).
Table 4. Unsaturated CAI-1 ester analogs.
| Entry | Compound | EC50 (nM)a | % Responseb |
|---|---|---|---|
| 1 |
|
54±8 | 98±9 |
| 1a |
|
39±5 | 99±13 |
| 2 |
|
710±120 | 90±7 |
| 3 |
|
550±86 | 104±8 |
| 4 |
|
770±110 | 97±6 |
| 5 |
|
780±130 | 100±10 |
| 6 |
|
2,000±670 | 76±34 |
| 7 |
|
270±33 | 103±5 |
| 8 |
|
1,800±380 | 95±13 |
| 9 |
|
830±100 | 101±6 |
| 10 |
|
570±110 | 98±11 |
| 11 |
|
1,200±600 | 98±21 |
Values determined using a light-based CqsS agonist bioassay, see Supporting Information. All EC50 values are the mean of triplicate analyses with the range defining the 95% confidence interval.
Percent maximal bioluminescence, with respect to CAI-1, which is set at 100%. The range defining the 95% confidence interval.
2.2.6. CAI-1 ester analogs with cyclohexane or benzene units incorporated in the tail
We prepared a series of CAI-1 esters containing a cyclohexane or benzene unit at different positions of the CAI-1 ester tail. We anticipated that this series of analogs would provide information about the steric demands of the binding pocket for the tail, as well as evidence regarding whether linear or bent conformations are favored.
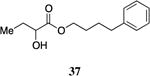
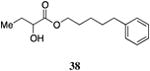
One series of analogs bears substitution at the terminus of the ester tail, linked to the α-hydroxy ester head group by a three, four or five-carbon chain (34 – 38). The activity of these analogs varies as a function of both the linker length and the terminating functionality (Table 5). The analog bearing a five-carbon linker and a terminal phenyl substituent (38, entry 2) displayed the maximal response with an EC50 value of 110 nM. In contrast, the one-carbon shorter analog bearing a terminal phenyl group (37) was diminished in activity (EC50=1,100 nM) and elicited a response of only 59% of the maximum value.
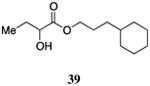
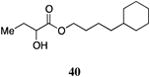
The saturated versions of these analogs, bearing a terminal cyclohexyl group, were universally more potent than the corresponding analogs bearing a terminal phenyl substituent. The analog with a terminal cyclohexyl group and a four-carbon linker (40, entry 4) was the most potent in this series with an EC50 value of 84 nM, 2-fold more active than the simple ester 9. The analogs containing a one-carbon shorter linker (39) or a one-carbon longer linker (41) displayed EC50 values of 310 nM and 190 nM, respectively.
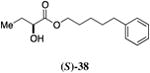
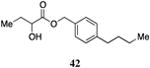
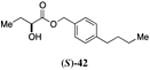
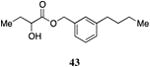
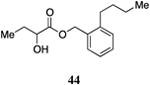
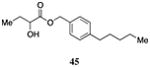
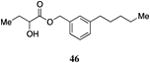
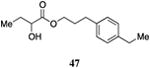
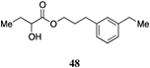
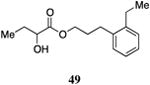
Inserting a benzene ring into the alkyl chain with a linear 1,4-substitution pattern led to a series of active agonists. Two examples with equivalent overall chain length, 42 and 47, displayed equal activity (190 nM) and both achieved the maximal QS response (entries 6 and 11). The related structure with one additional methylene group in the chain (45), was less active (680 nM; entry 9). Introducing a non-linear conformational restriction into the acyl tail of these analogs in the form of the 1,3-substituted aryl units, as in analogs 43, 46, and 48, resulted in a strong diminution in activity, with EC50 values that were >9-fold lower than the parent C8-ester-CAI-1, 9. More dramatically, the 1,2-disubstituted phenyl analogs 44 and 49 showed very weak responses even at high concentrations (50,000 nM). These data suggest a binding pocket for the CAI-1 acyl tail that is somewhat permissive of greater steric demands (incorporation of the phenyl and cyclohexyl rings), but appears to strongly favor an overall linear conformation of the tail. In these analogs, the aromatic ring appears to contribute to activity beyond simply defining the linear substitution pattern as other analogs displaying an overall linear orientation of the alkyl tail were found to be less active (ie. compare 42 with 28 (Table 4)). Consistent with our expectations, the (S)-isomer of two of the most potent analogs in this series, (S)-38 and (S)-42 (entries 2a and 6a, respectively) were found to be more potent than the corresponding racemic compounds.
2.3. Mechanism of Action of the CAI-1 ester analogs
We previously described a competitive antagonist (Ph-CAI-1) of the CAI-1 QS receptor CqsS.13 To confirm that the CAI-1 ester agonists exert their action by binding in the CAI-1 binding pocket in CqsS, we examined the effect of varying concentrations of Ph-CAI-1 on the activity of the potent analogs (S)-C8-ester-CAI-1 (7), 5-Ph-ester-CAI-1 (38), 4-Cy-ester-CAI-1 (40), and 1-(p-n-butyl-Ph)-ester-CAI-1, (42). All of the ester agonists examined displayed diminished potency in the presence of increasing concentrations of the competitive antagonist, Ph-CAI-1 (see Supporting Information).
In an effort to elucidate the specific binding interactions that are maintained by the CAI-1 ester agonists within the CqsS binding pocket, we examined the activity of a series of agonists toward mutant CqsS receptors (Table 6). We had reported that variation in the chain length of the CAI-1 acyl tail plays a significant role in the ligand activity and that the CqsS mutant C170Y displays a preference for ligands bearing shorter acyl tails whereas the mutant C170A has a modest preference for molecules bearing longer acyl tails.13
The wild type CqsS receptor shows a preference for 4-Cy-ester-CAI-1 (40) with both the 5-Cy-ester-CAI-1 analog (41) and the 3-Cy-ester-CAI-1 molecule (39) being less active (EC50 values higher by 2.2 to 3.7-fold, respectively). When these analogs are tested in the CqsS C170Y mutant, the higher activity shifts to the shorter 3-Cy-ester-CAI-1 analog (39); the 4-Cy-ester-CAI-1, 40, and 5-Cy-ester-CAI-1, 41, showed EC50 values 2.3-fold and 27-fold higher, respectively, compared to 39. Additionally, while the 3-Cy-ester-CAI-1, 39 was capable of inducing nearly the maximal QS response (93%), here defined with reference to C8-CAI-1 as 100% (EC50 260 nM), the longer chain analogs 40 and 41 acted as partial agonists of this mutant receptor, inducing 51% and 66% of the maximal QS response, respectively.
While the variation in ligand preference in the C170A mutant receptor is subtle, we find that, consistent with our expectations, the longer chain 5-Cy-ester-CAI-1 analog (41) is, among this series of ligands, uniquely capable of inducing the maximal QS response (101% maximal induction), here given with reference to CAI-1 (EC50=72 nM), while the shorter chain analogs, 39 and 41 act as partial agonists, inducing 73% and 77% of the maximal response, respectively.
In contrast, the CqsS C170A mutant receptor displayed a significantly altered response to 4-Ph-ester-CAI-1 37 and 5-Ph-ester-CAI-1 38 when compared to the wild-type response to these molecules. While both CqsS receptors prefer the longer chain, 5-Ph-ester-CAI-1 analog (wild type CqsS EC50=231 nM, 93% response; C170A CqsS EC50=180 nM, 83% response) the response to the shorter 4-Ph-ester-CAI-1 analog (37) varies significantly. Whereas 37 is a partial agonist of the wild type CqsS receptor, achieving 59% maximal response with an EC50 value of 1100 nM, its activity is severely diminished in the C170A mutant CqsS receptor (EC50 >50000 nM, 16% maximal response at 50000 nM).
Taken together these observations provide further support for the importance of the C170 amino acid of CqsS in ligand recognition. Additionally, the CAI-1 ester analogs display a pattern of varying activity toward the CqsS C170Y and C170A mutant receptors which supports the hypothesis that these analogs bind in the CAI-1 binding pocket and elicit the QS response in a manner similar to the native CAI-1 molecule.
2.4 Pharmacophore Modeling
In support of the development of an anti-virulence agent against V. cholerae that targets the CqsS QS receptor, we generated a pharmacophore model using the activity patterns observed with the CAI-1 ester analogs.18-20
The molecules used to generate the pharmacophore model display EC50 values <250 nM and induced >92% of maximal response, including CAI-1 (1) and the CAI-1 ester analogs 9, 26, 38, 40, 41, 42 and 47 (Figure 9). Each of the molecules in this series has a large number of energetically favorable conformations, making the identification of an optimal binding conformation difficult for any individual compound in this series. When assembled in sum, however, we find that the structures of the active compounds converge to provide a unified model of the CqsS binding pocket. Indeed, among the best-fit pharmacophore models generated, only subtle variation was observed in the overall molecular topology of the ligand binding surface. Together with our chemical genetic analyses of CqsS, this model provides reasonable detail of the ligand-receptor binding interaction within the trans-membrane region of CqsS in the absence of more detailed structural information about this receptor (e.g. X-ray crystallographic analysis).
Figure 9.

Pharmacophore model generated from CAI-1 (1, light blue) and the CAI-1 ester analogs 9 (blue-grey), 26 (yellow), 38 (light green), 40 (purple), 41 (orange), 42 (blue) and 47 (pink).
Consistent with our SAR, this model suggests the presence of a fairly large hydrophobic binding pocket into which the acyl tail of the CAI-1 molecule projects. From the benzene and cyclohexane analogs, it is clear that the binding pocket tolerates significant additional steric bulk along the entire length of the hydrophobic tail. This feature results in a combined hydrophobic surface described by the model that is significantly larger then the van der Waals surface of the saturated acyl tail of CAI-1.
To test the validity of this model, we examined the fit of ligands that displayed diminished activity, predicting that these analogs would not fit well in the optimized pharmacophore model. We selected a series of compounds that varied in their ability to agonize CqsS in order to test the prediction that compounds displaying moderate activity would be subtly perturbed from this model whereas compounds that displayed low activity would possess greater deviations from the ideal molecular topology described by this model. The less potent CAI-1 ester analogs deviate from the model (Figure 10); the extent of deviation appears to generally correlate with the biological activity. For example, 2-(Z)-, 3-(Z)-, 4-(Z)-, and 5-(Z)-C8-ester-CAI-1 (27, 30, 33, and 35) that fully activate the QS response but are >3-fold less active than the saturated analog 9, all deviate clearly from the pharmacophore model (Figure 10a). However, the extent to which these analogs deviate from the model is less pronounced than the deviation of the still less potent 1,3-substituted phenyl analogs 43 and 48 (<83% maximal response, EC50 values >21-fold higher than 9). The 1,2-substituted phenyl analogs 44 and 49 (<28% maximal response, EC50 values >278-fold higher than 9) display even more pronounced deviations from the model (Figure 10b and 10c). While the pharmacophore model correlates with the biological activity of the majority of the analogs, there are two analogs for which the model is not consistent. The 3-(E)-, and 5-(E)-C8-ester-CAI-1 analogs 29 and 34 appear to fully satisfy the model and yet these analogs are only modest agonists, fully activating the QS response but with EC50 values >4-fold higher than the saturated analog 9 (see Supporting Information). Notwithstanding these exceptions, this model approximates the molecular topology of the binding pocket for the fatty acid tail portion of the CAI-1 ligands and effectively correlates with the biological activity for most of the analogs examined; it represents a finding that should be of general interest for further studies of this class of membrane associated receptors.
Figure 10.

Images highlighting the deviation of less active CAI-1 ester analogs (shown in grey) from the pharmacophore model. Active analogs used to generate the pharmacophore model are shown in black with the combined VDW surface highlighted with points. a) The CAI-1 ester analogs containing a (Z)-olefin in the 2, 3, 4, or 5 position (27, 30, 33, and 35). b) The ortho- and meta-butyl substituted aryl CAI-1 ester analogs 43 and 44. c) The ortho- and meta-ethyl substituted aryl CAI-1 ester analogs 48 and 49.
3. Conclusion
Synthetic molecules that act as agonists of QS in V. cholerae represent useful tools to enhance our understanding of QS and the mechanisms for controlling virulence in this globally relevant human pathogen. A series of rationally designed CAI-1 analogs was prepared to examine the preference of the receptor to structural changes within the fatty acid-derived tail portion of the CAI-1 signaling molecule. The construction of this compound library was facilitated by the discovery that the CAI-1 α-hydroxy ester analogs display comparable activity and SAR to the parent CAI-1 α-hydroxy ketones. We note that the conformational and electronic differences between the ester and ketone functionality make this an interesting example of an ester serving as a suitable bioisostere for a ketone.21, 22 Further complicating the use of esters as biological probes, esters are subject to the activity of ubiquitous esterases, a feature which has been capitalized on in the design of prodrugs.23, 24 We find that the alcohol tails for several of the most potent analogs, the products of esterase cleavage, show no QS activity up to 50,000 nM. Similarly, the conserved esterase cleavage product, 2-hydroxybutyric acid, displays no activity at concentrations up to 50,000 nM. Accordingly, we believe that the esters described in this study are not undergoing cleavage prior to activation of the QS response. Rather, the esters themselves serve as activators of QS by interacting with the membrane-associated receptor CqsS in a manner similar to the native signal, CAI-1.
The membrane-bound QS receptor in V. cholerae tolerates several structural changes deviating from the fully saturated linear alkyl tail contained in the native signaling molecule, including both conformational restrictions in the acyl tail and the incorporation of branched functionality in the form of phenyl and cyclohexyl rings. Examination of SAR for this series of analogs was used to generate a model for the structural constraints on the ligand allowable by the CqsS QS receptor. This is significant as the receptor is membrane-associated which complicates the acquisition of detailed structural information, for example, through x-ray crystallographic studies. These results, together with chemical genetics, provide crucial information about the molecular topology and localization of the ligand binding pocket of CqsS that will be broadly useful to diverse studies of this important bacterial pathogen.
Experimental Methods
General Experimental
Unless otherwise noted, all reactions were performed in flame-dried glassware under an atmosphere of nitrogen or argon using dried reagents and solvents. All chemicals were purchased from commercial vendors and used without further purification, anhydrous solvents were purchased from commercial vendors.
Flash chromatography was performed using standard grade silica gel 60 230-400 mesh from SORBENT Technologies. Analytical thin-layer chromatography was carried out using Silica G TLC plates, 200 μm with UV254 fluorescent indicator (SORBENT Technologies), and visualization was performed by staining and/or by absorbance of UV light.
NMR spectra were recorded using a Bruker Avance II (500 MHz for 1H; 125 MHz for 13C) spectrometer fitted with either a 1H-optimized TCI (H/C/N) cryoprobe or a 13C-optimized dual C/H cryoprobe. Chemical shifts are reported in parts per million (ppm) and were calibrated according to residual protonated solvent. High-resolution mass spectral analysis was performed using an Agilent 1200-series electrospray ionization – time-of-flight (ESI-TOF) mass spectrometer in the positive ESI mode. Analytical high-performance liquid chromatography was performed by Lotus Separations, LLC using a Rainin HPLC with SD-1 pumps and a Dynamax UV-1 detector. Supercritical fluid chromatography was performed by Lotus Separations, LLC using a Berger Multigram II SFC with Rainin SD-1 pumps and a Knauer K-2501 spectrophotometer. All final compounds were determined to be of >95% purity by analysis of their characterization data.
Typical procedure for the synthesis of CAI-1 analogs 9, 10 and 22-25
Method A
To a solution of the α-hydroxy acid (1.0 eq) in DMF (1.5M) at room temperature was added imidazole (3.0 eq) and tert-butyldimethylsilyl chloride (3.0 eq). The mixture was stirred at room temperature overnight. The reaction was quenched with hydrochloric acid (1N), was extracted with ethyl acetate (EtOAc), washed with H2O and brine, dried over Na2SO4 and concentrated in vacuo. The residue was treated with SOCl2 (6.6 eq) and heated to reflux overnight. Excess SOCl2 was removed in vacuo, and the product was used for the next reaction step without further purification. To a solution of primary alcohol (1.0 eq) and DMAP (1.1 eq) in THF (0.13M) at room temperature was added the acid chloride obtained in the previous step (1.1 eq), dropwise. The mixture was allowed to stir overnight at room temperature and was concentrated in vacuo to form a slurry which was purified by silica gel chromatography. The purified TBS-protected intermediate in THF (1M) was treated with TBAF (3 eq) and the mixture was allowed to stir overnight before direct concentration and purification by silica gel chromatography.
Typical procedure for the synthesis of CAI-1 ester analogs 7, 8, 17-21 and 26-49
Method B
Molecular sieves (5Å powdered, 0.5 g/mmol of ester) and the carbene catalyst (0.1 eq) were added to a screw-top vial equipped with a stir bar. The vial was evacuated and purged to N2 three times before being placed under nitrogen. To the vial was added PhMe (0.5M), ethyl 2-hydroxybutyrate (1 eq) and the primary alcohol (2 eq). The reaction mixture was stirred overnight at room temperature, was directly concentrated and the residue was purified by silica gel chromatography.
Octyl 2-hydroxybutanoate, 9
Prepared according to Method A to provide octyl 2-hydroxybutanoate (20 mg, 24% yield, 4 steps). Also prepared according to Method B using 1-octanol to provide octyl 2-hydroxybutanoate (86 mg, 40% yield). 1H NMR (500 MHz, CDCl3) δ 4.22-4.09 (m, 3H), 2.74 (d, J = 5.5 Hz, 1H), 1.87-1.77 (m, 1H), 1.72-1.59 (m, 3H), 1.37-1.18 (m, 10H), 0.94 (t, J = 7.4 Hz, 3H), 0.86 (t, J = 6.9 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 175.6, 77.5, 77.2, 77.0, 66.0, 32.0, 29.4, 29.3, 28.7, 27.7, 26.0, 22.8; HRMS (ESI-TOF) calculated for C12H25O3 [M+H]+, 217.1804; observed, 217.1794.
(S)-2-Hydroxy-N-octylbutanamide, 10
Prepared according to Method A providing (S)-2-hydroxy-N-octylbutanamide (44 mg, 53% yield, 4 steps). 1H-NMR (500MHz, CDCl3) δ 6.69-6.57 (brs, 1H), 4.02 (dd, J = 3.8, 7.0 Hz, 1H), 3.38-3.13 (m, 3H), 1.89-1.76 (m, 1H), 1.71-1.58 (m, 1H), 1.54-1.43 (m, 2H), 1.35-1.14 (m, 10H), 0.95 (t, J = 7.4 Hz, 3H), 0.83 (t, J = 6.7 Hz, 3H). 13C-NMR (125MHz, CDCl3) δ 173.8, 73.0, 39.1, 31.8, 29.6, 29.3, 29.2, 27.9, 26.9, 22.7, 14.1, 9.1. HRMS (ESI-TOF) calculated for C12H26NO2 [M+H]+, 216.1958; found 216.1974.
2-(Pentyloxy)ethyl 2-hydroxybutanoate, 22
Prepared according to Method A giving 2-(pentyloxy)ethyl 2-hydroxybutanoate (77.8 mg, 22% yield, 4 steps). 1H-NMR (500MHz, CDCl3) δ 4.37-4.26 (m, 2H), 4.18 (dd, J = 5.1, 10.5 Hz, 1H), 3.63 (t, J = 4.7 Hz, 2H), 3.43 (t, J = 6.7 Hz, 2H), 2.73 (d, J = 5.4 Hz, 1H), 1.88-1.79 (m, 1H), 1.73-1.64 (m, 1H), 1.61-1.52 (m, 2H), 1.32-1.26 (m, 4H), 0.95 (t, J = 7.4 Hz, 3H), 0.87 (t, J = 6.8 Hz, 3H). 13C-NMR (125MHz, CDCl3) δ 175.3, 71.5, 71.4, 68.3, 64.6, 29.3, 28.2, 27.5, 22.5, 14.1, 8.9. HRMS (ESI-TOF) calculated for C11H23O4 [M+H]+, 219.1591; found 219.1592.
2-Hydroxy-2-(2-ethoxyethoxy)ethylbutanoate, 23
Prepared according to Method A giving 2-hydroxy-2-(2-ethoxyethoxy)ethylbutanoate (0.349 g, 60% yield, 4 steps). 1H-NMR (500MHz, CDCl3) δ 4.39-4.26 (m, 2H), 4.17 (dd, J = 5.7, 10.9 Hz, 1H), 3.72 (t, J = 4.7 Hz, 2H), 3.65-3.61 (m, 2H), 3.59-3.56 (m, 2H), 3.50 (dd, J = 7.0, 14.0 Hz, 2H), 2.74 (d, J = 5.7 Hz, 1H), 1.88-1.79 (m, 1H), 1.74-1.63 (m, 1H), 1.20 (t, J = 7.0 Hz, 3H), 0.95 (t, J = 7.4 Hz, 3H). 13C-NMR (125MHz, CDCl3) δ 175.4, 71.4, 70.7, 69.8, 68.9, 66.8, 64.5, 27.5, 15.2, 8.9. HRMS (ESI-TOF) calculated for C10H21O5 [M+H]+, 221.1384; found 221.1389.
2-(Hexyloxy)ethyl 2-hydroxybutanoate, 24
Prepared according to Method A giving 2-(hexyloxy)ethyl 2-hydroxybutanoate (0.163g, 19% yield, 4 steps). 1H-NMR (500MHz, CDCl3) δ 4.34-4.21 (m, 2H), 4.13 (dd, 1H, J = 4.2, 8.8 Hz), 3.58 (t, 2H, J = 4.7 Hz), 3.38 (t, 2H, J = 6.7 Hz), 2.67 (d, 1H, J = 5.8 Hz), 1.86-1.74 (m, 1H), 1.69-1.57 (m, 1H), 1.51-1.46 (m, 2H), 1.31-1.16 (m, 6H), 0.92-0.88 (m, 3H), 0.82 (t, 3H, J = 6.9 Hz). 13C-NMR (125MHz, CDCl3) δ 175.4, 71.5, 71.4, 68.3, 64.7, 31.7, 29.6, 27.5, 25.8, 22.7, 14.1, 8.9. HRMS (ESI-TOF) calculated for C12H25O4 [M+H]+, 233.1747; found 233.1752.
2-Hydroxy-2-(2-(2-methoxyethoxy)ethoxy)ethylbutanoate, 25
Prepared according to Method A giving 2-hydroxy-2-(2-(2-methoxyethoxy)ethoxy)ethylbutanoate (412 mg, 54% yield, 4 steps). 1H-NMR (500MHz, CDCl3) δ 4.37-4.29 (m, 2H), 4.19-4.15 (m, 1H), 3.70 (t, J = 4.7 Hz, 2H), 3.66-3.60 (m, 4H), 3.55-3.51 (m, 2H), 3.36 (s, 2H), 2.90 (d, J = 5.8 Hz, 1H), 1.88-1.78 (m, 1H), 1.73-1.64 (m, 1H), 1.62 (s, 3H), 0.98-0.92 (t, J = 7.4 Hz, 3H). 13C-NMR (125MHz, CDCl3) δ 175.2, 71.9, 71.5, 70.6, 70.6, 70.6, 68.9, 64.5, 59.1, 27.5, 9.0. HRMS (ESI-TOF) calculated for C11H22O6Na [M+Na]+, 273.1309; found 273.1316.
(S)-2-aminooctylbutanoate hydrochloride, 12
To L-aminobutyric acid (500 mg, 4.83 mmol) was added octanol (40.3 mL) and 2M HCl/ether (7.0 mL). The mixture was heated to reflux (150°C) for 24 hours and was concentrated in vacuo. Upon cooling, a solid was formed which was dissolved in ethyl acetate with warming. The solution was left overnight to form crystals which were subsequently filtered and air-dried, giving (S)-2-aminooctylbutanoate hydrochloride (0.611g, 50% yield). 1H-NMR (500MHz, CDCl3) δ 8.97-8.83 (brs, 2H), 4.31-4.16 (m, 2H), 4.06 (t, J = 5.8 Hz, 1H), 2.22-2.06 (m, 2H), 1.76-1.64 (m, 2H), 1.42-1.23 (m, 10H), 1.15 (t, J = 7.5 Hz, 3H), 0.91 (t, J = 6.9 Hz, 3H). 13C-NMR (125MHz, CDCl3) δ 169.2, 66.8, 54.5, 32.0, 29.4, 29.3, 28.6, 25.9, 24.1, 22.8, 14.3, 9.7. HRMS (ESI-TOF) calculated for C12H26NO2 [M+H]+, 216.1958; found 216.1959.
(R)-2-aminooctylbutanoate hydrochloride, 13
Prepared following the procedure for the synthesis of octyl 2-hydroxybutanoate (12) from D-aminobutyric acid. 1H-NMR (500MHz, CDCl3) δ 8.96-8.84 (brs, 2H), 4.30-4.14 (m, 2H), 4.07 (t, J = 5.8, 1H), 2.20-2.08 (m, 2H), 1.75-1.64 (m, 2H), 1.39-1.24 (m, 10H), 1.14 (t, J = 7.5 Hz, 3H), 0.91 (t, J = 6.9 Hz, 3H). 13C-NMR (125MHz, CDCl3) δ 169.1, 66.7, 54.3, 31.8, 29.2, 29.2, 28.4, 25.8, 24.0, 22.7, 14.2, 9.5. HRMS (ESI-TOF) calculated for C12H26NO2 [M+H]+, 216.1958; found 216.1965.
(S)-Octyl 2-hydroxybutanoate, 7
Prepared according to Method B using (S)-methyl 2-hydroxybutanoate25 and octanol to provide (S)-Octyl 2-hydroxybutanoate as a clear colorless oil (55 mg, 40%). 1H NMR (500 MHz, CDCl3) δ 4.17-4.11 (m, 3H), 2.74 (d, J = 5.5 Hz, 1H), 1.88-1.77 (m, 1H), 1.72-1.59 (m, 3H), 1.38-1.19 (m, 10H), 0.94 (t, J = 7.4 Hz, 3H), 0.86 (t, J = 6.9 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 175.6, 77.5, 77.2, 77.0, 66.0, 32.0, 29.4, 29.3, 28.7, 27.7, 26.0, 22.8; HRMS (ESI-TOF) calculated for C12H25O3 [M+H]+, 217.1804; observed, 217.1794.
(R)-Octyl 2-hydroxybutanoate, 8
Prepared according to Method B from (R)-methyl 2-hydroxybutanoate and octanol to provide (R)-Octyl 2-hydroxybutanoate as a clear colorless oil. 1H NMR (500 MHz, CDCl3) δ 4.17-4.11 (m, 3H), 2.73 (d, J = 5.7 Hz, 1H), 1.87-1.77 (m, 1H), 1.72-1.54 (m, 3H), 1.38-1.19 (m, 10H), 0.94 (t, J = 7.4 Hz, 3H), 0.86 (t, J = 7.0 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 175.6, 77.5, 77.2, 77.0, 66.0, 32.0, 29.4, 29.3, 28.7, 27.7, 26.0, 22.8; HRMS (ESI-TOF) calculated for C12H25O3 [M+H]+, 217.1804; observed, 217.1793.
Butyl 2-hydroxybutanoate, 17
Prepared according to Method B using 1-butanol to provide butyl 2-hydroxybutanoate (29 mg, 20% yield). 1H NMR (500 MHz, CDCl3) δ 4.22-4.04 (m, 3H), 2.88 (bs, 1H), 1.86-1.74 (m, 1H), 1.68-1.56 (m, 3H), 1.38-1.30 (m, 2H), 0.92 (t, J = 7.5 Hz, 3H), 0.89 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 175.5, 71.5, 65.6, 30.7, 27.7, 19.2, 13.8, 9.1; HRMS (ESI-TOF) calculated for C8H17O3Na [M+Na]+, 183.0997; observed, 183.0992.
Hexyl 2-hydroxybutanoate, 18
Prepared according to Method B using 1-hexanol (250 μL, 2.0 mmol) to provide hexyl 2-hydroxybutanoate (105 mg, 56% yield). 1H NMR (500 MHz, CDCl3) δ 4.22-4.10 (m, 3H), 2.75 (d, J = 5.7 Hz, 1H), 1.87-1.77 (m, 1H), 1.70-1.59 (m, 3H), 1.38-1.22 (m, 6H), 0.94 (t, J = 7.4 Hz, 3H), 0.87 (t, J = 6.8 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 175.6, 71.5, 66.0, 31.5, 28.7, 27.7, 25.7, 22.7, 14.2, 9.1; HRMS (ESI-TOF) calculated for C10H21O3[M+H]+, 189.1491; observed, 189.1500.
Heptyl 2-hydroxybutanoate, 19
Prepared according to Method B using 1-heptanol to provide heptyl 2-hydroxybutanoate (60 mg, 30% yield). 1H NMR (500 MHz, CDCl3) δ 4.21-4.10 (m, 3H), 2.75 (d, J = 5.7 Hz, 1H), 1.87-1.76 (m, 1H), 1.70-1.59 (m, 3H), 1.37-1.20 (m, 8H), 0.94 (t, J = 7.4 Hz, 3H), 0.86 (t, J = 7.0 Hz, 3H). 13C-NMR (125 MHz, CDCl3) δ 175.6, 71.5, 66.0, 31.9, 29.1, 28.7, 27.7, 26.0, 22.8, 14.3, 9.1. HRMS (ESI-TOF) calculated for C11H23O3 [M+H]+, 203.1647; observed, 203.1650.
Nonyl 2-hydroxybutanoate, 20
Prepared according to Method B using 1-nonanol to provide nonyl 2-hydroxybutanoate (143 mg, 62% yield). 1H NMR (500 MHz, CDCl3) δ 4.23-4.09 (m, 3H), 2.77 (d, J = 5.2 Hz, 1H), 1.87-1.76 (m, 1H), 1.70-1.56 (m, 3H), 1.38-1.16 (m, 12H), 0.94 (t, J = 7.4 Hz, 3H), 0.85 (t, J = 6.9 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 175.6, 71.5, 66.0, 32.0, 29.6, 29.4, 29.4, 28.7, 27.7, 26.0, 22.9, 14.3, 9.1; HRMS (ESI-TOF) calculated for C13H27O3[M+H]+, 231.1960; observed, 231.1961.
Decyl 2-hydroxybutanoate, 21
Prepared according to Method B using 1-decanol to provide decyl 2-hydroxybutanoate (158 mg, 65% yield). 1H NMR (500 MHz, CDCl3) δ 4.22-4.09 (m, 3H), 2.75 (d, J = 5.5 Hz, 1H), 1.87-1.76 (m, 1H), 1.70-1.59 (m, 3H), 1.37-1.17 (m, 14H), 0.94 (t, J = 7.4 Hz, 3H), 0.85 (t, J = 6.9 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 175.4, 71.3, 65.9, 31.9, 29.5, 29.5, 29.3, 29.2, 28.6, 27.5, 25.8, 22.7, 14.2, 8.9; HRMS (ESI-TOF) calculated forC14H29O3 [M+H]+, 245.2117; observed, 245.2122.
(E)-Oct-2-en-1-yl 2-hydroxybutanoate, 26
Prepared according to Method B using (E)-2-octen-1-ol to provide (E)-oct-2-en-1-yl 2-hydroxybutanoate (134 mg, 63% yield). 1H NMR (500 MHz, CDCl3) δ 5.83-5.74 (m, 1H), 5.59-5.49 (m, 1H), 4.65-4.55 (m, 2H), 4.14 (dd, J = 6.2, 4.7 Hz, 1H), 2.73 (bs, 1H), 2.03 (q, J = 7.1 Hz, 2H), 1.87-1.77 (m, 1H), 1.71-1.61 (m, 1H), 1.40-1.31 (m, 2H), 1.31-1.21 (m, 4H), 0.94 (t, J = 7.4 Hz, 3H), 0.86 (t, J = 6.9 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 175.3, 138.0, 123.2, 71.6, 66.6, 32.4, 31.5, 28.7, 27.7, 22.7, 14.3, 9.1; HRMS (ESI-TOF) calculated for C12H22O3Na [M+Na]+, 237.1466; observed, 237.1470.
(Z)-Oct-2-en-1-yl 2-hydroxybutanoate, 27
Prepared according to Method B using (Z)-2-octen-1-ol to provide (Z)-oct-2-en-1-yl 2-hydroxybutanoate (180 mg, 84% yield). 1H NMR (500 MHz, CDCl3) δ 5.71-5.62 (m, 1H), 5.55-5.46 (m, 1H), 4.77-4.65 (m, 2H), 4.14 (s, 1H), 2.74 (s, 1H), 2.08 (q, J = 7.3 Hz, 2H), 1.87-1.76 (m, 1H), 1.73-1.60 (m, 1H), 1.40-1.21 (m, 6H), 0.93 (t, J = 7.4 Hz, 3H), 0.86 (t, J = 6.9 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 175.4, 136.7, 122.6, 71.6, 61.6, 31.6, 29.2, 27.7, 27.7, 22.7, 14.3, 9.1; HRMS (ESI-TOF) calculated for C12H22O3Na[M+Na]+, 237.1466; observed, 237.1471.
Oct-2-yn-1-yl 2-hydroxybutanoate, 28
Prepared according to Method B using 2-octyn-1-ol to provide oct-2-yn-1-yl 2-hydroxybutanoate (102 mg, 48% yield). 1H NMR (500 MHz, CDCl3) δ 4.80-4.70 (m, 2H), 4.19 (dd, J = 6.3, 4.4 Hz, 1H), 2.67 (s, 1H), 2.22-2.15 (m, 2H), 1.89-1.79 (m, 1H), 1.76-1.65 (m, 1H), 1.53-1.44 (m, 2H), 1.37-1.24 (m, 4H), 0.95 (t, J = 7.4 Hz, 3H), 0.87 (t, J = 7.0 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 174.9, 88.7, 73.4, 71.5, 54.1, 31.2, 28.2, 27.6, 22.4, 18.9, 14.2, 9.0; HRMS (ESI-TOF) calculated for C12H20O3Na [M+Na]+, 235.1310; observed, 235.1310.
(E)-Oct-3-en-1-yl 2-hydroxybutanoate, 29
Prepared according to Method B using (E)-3-octen-1-ol to provide (E)-oct-3-en-1-yl 2-hydroxybutanoate (88 mg, 98% yield). 1H NMR (500 MHz, CDCl3) δ 5.54-5.44 (m, 1H), 5.36-5.26 (m, 1H), 4.23-4.07 (m, 3H), 2.79 (s, 1H), 2.32 (q, J = 6.7 Hz, 2H), 2.00-1.90 (m, 2H), 1.84-1.74 (m, 1H), 1.70-1.58 (m, 1H), 1.34-1.19 (m, 4H), 0.93 (t, J = 7.4 Hz, 3H), 0.85 (t, J = 7.0 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 175.5, 134.2, 124.7, 71.5, 65.4, 32.5, 32.1, 31.7, 27.7, 22.3, 14.1, 9.1; HRMS (ESI-TOF) calculated for C12H22O3Na [M+Na]+, 237.1466; observed, 237.1475.
(Z)-Oct-3-en-1-yl 2-hydroxybutanoate, 30
Prepared according to Method B using (Z)-3-octen-1-ol to provide (Z)-oct-3-en-1-yl 2-hydroxybutanoate (60 mg, 30% yield). 1H NMR (500 MHz, CDCl3) δ 5.55-5.43 (m, 1H), 5.34-5.23 (m, 1H), 4.21-4.08 (m, 3H), 2.79 (d, J = 3.4 Hz, 1H), 2.38 (q, J = 7.0 Hz, 2H), 2.08-1.95 (m, 1H), 1.84-1.74 (m, 1H), 1.69-1.59 (m, 1H), 1.34-1.24 (m, 4H), 0.93 (t, J = 7.5 Hz, 3H), 0.87 (t, J = 7.0 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 175.5, 133.5, 123.9, 71.6, 65.2, 31.9, 27.7, 27.2, 27.0, 22.5, 14.1, 9.1; HRMS (ESI-TOF) calculated for C12H22O3Na [M+Na]+, 237.1466; observed, 237.1475.
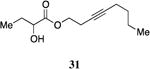
Oct-3-yn-1-yl 2-hydroxybutanoate, 31
Prepared according to Method B using 3-octyn-1-ol to provide oct-3-yn-1-yl 2-hydroxybutanoate (110 mg, 52% yield). 1H NMR (500 MHz, CDCl3) δ 4.29-4.13 (m, 3H), 2.70 (s, 1H), 2.54-2.46 (m, 2H) 2.14-2.07 (m, 2H), 1.88-1.77 (m, 1H), 1.74-1.62 (m, 1H), 1.47-1.31 (m, 4H), 0.95 (t, J = 7.4 Hz, 3H), 0.88 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 175.3, 82.5, 75.2, 71.5, 64.1, 31.1, 27.7, 22.1, 19.5, 18.5, 13.8, 9.1; HRMS (ESI-TOF) calculated for C12H20O3Na [M+Na]+, 235.1310; observed, 235.1313.
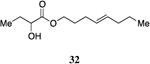
(E)-Oct-4-enyl 2-hydroxybutanoate, 32
Prepared according to Method B using (E)-4-octyn-1-ol to provide (E)-oct-4-enyl 2-hydroxybutanoate (69.9 mg, 75% yield). 1H-NMR (500MHz, CDCl3) δ 5.46-5.29 (m, 2H), 4.22-4.09 (m, 3H), 2.75 (s, 1H), 2.04 (q, J = 7.0 Hz, 2H), 1.93 (q, J = 6.9 Hz, 2H), 1.87-1.77 (m, 1H), 1.74-1.62 (m, 3H), 1.39-1.29 (m, 2H), 0.94 (t, J = 7.4 Hz, 3H), 0.86 (t, J = 7.4 Hz, 3H). 13C-NMR (125MHz, CDCl3) δ 175.6, 131.9, 128.7, 71.5, 65.3, 34.8, 28.9, 28.6, 27.7, 22.8, 13.9, 9.1. HRMS (ESI-TOF) calculated for C12H23O3 [M+H]+, 215.1647; observed, 215.1644.
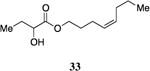
(Z)-Oct-4-enyl 2-hydroxybutanoate, 33
Prepared according to Method B using (Z)-4-octyn-1-ol to provide (Z)-oct-4-enyl 2-hydroxybutanoate (42.7 mg, 55% yield). 1H-NMR (500MHz, CDCl3) δ 5.45-5.27 (m, 2H), 4.23-4.09 (m, 3H), 2.76 (d, J = 5.7 Hz, 1H), 2.09 (q, J = 7.3 Hz, 2H), 1.97 (q, J = 7.3 Hz, 2H), 1.88-1.77 (m, 1H), 1.76-1.62 (m, 3H), 1.39-1.28 (m, 2H), 0.95 (t, J = 7.4 Hz, 3H), 0.87 (t, J = 7.2 Hz, 3H). 13C-NMR (125MHz, CDCl3) δ 175.5, 131.4, 128.2, 71.5, 65.4, 29.5, 28.7, 27.7, 23.6, 23.0, 14.0, 9.1. HRMS (ESI-TOF) calculated for C12H23O3 [M+H]+, 215.1647; observed, 215.1646.
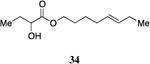
(E)-Oct-5-enyl 2-hydroxybutanoate, 34
Prepared according to Method B using (E)-5-octyn-1-ol to provide (E)-oct-5-enyl 2-hydroxybutanoate (41.3 mg, 65% yield). 1H-NMR (500MHz, CDCl3) δ 5.49-5.29 (m, 2H), 4.23-4.09 (m, 3H), 2.74 (d, J = 5.7 Hz, 1H), 2.04-1.93 (m, 4H), 1.86-1.78 (m, 1H), 1.71-1.59 (m, 3H), 1.44-1.35 (m, 2H), 0.94 (t, J = 7.4 Hz, 6H). 13C-NMR (125MHz, CDCl3) δ 175.6, 133.0, 128.5, 71.5, 65.8, 32.2, 28.2, 27.7, 25.9, 25.8, 14.2, 9.1. HRMS (ESI-TOF) calculated for C12H23O3 [M+H]+, 215.1647; observed, 215.1642.
(Z)-Oct-5-en-1-yl 2-hydroxybutanoate, 35
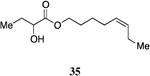
Prepared according to Method B using (Z)-5-octen-1-ol to provide (Z)-oct-5-en-1-yl 2-hydroxybutanoate (116 mg, 78% yield). 1H NMR (500 MHz, CDCl3) δ 5.42-5.34 (m, 1H), 5.32-5.23 (m, 1H), 4.23-4.09 (m, 3H), 2.09-1.96 (m, 4H), 1.86-1.77 (m, 1H), 1.70-1.61 (m, 3H), 1.45-1.35 (m, 2H), 0.99-0.88 (m, 6H). 13C NMR (126 MHz, CDCl3) δ 175.6, 132.6, 128.4, 71.5, 65.8, 28.3, 27.7, 26.7, 26.1, 20.7, 14.6, 9.1; HRMS (ESI-TOF) calculated for C12H22O3Na [M+Na]+, 237.1466; observed, 237.1472.
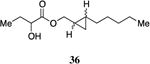
(2-Pentylcyclopropyl)methyl 2-hydroxybutanoate, 36
Prepared according to Method B using (2-pentylcyclopropyl)methanol to provide (2-pentylcyclopropyl)methyl 2-hydroxybutanoate as a mixture of stereoisomers (23.7 mg, 60% yield). 1H NMR (500 MHz, CDCl3) δ 4.17-4.11 (m, 1H), 4.10-3.90 (m, 2H), 2.76 (d, J = 5.6 Hz, 1H), 1.89-1.78 (m, 1H), 1.73-1.62 (m, 1H), 1.39-1.20 (m, 7H), 1.18-1.08 (m, 1H), 0.95 (t, J = 7.4 Hz, 3H), 0.86 (t, J = 6.9 Hz, 3H), 0.70-0.61 (m, 1H), 0.45-0.38 (m, 1H), 0.38-0.31 (m, 1H). 13C NMR (126 MHz, CDCl3) δ 175.63, 71.6, 71.5, 70.4, 70.3, 33.6, 31.8, 29.3, 29.3, 27.7, 22.9, 18.1, 18.0, 17.3, 17.3, 14.3, 10.7, 10.7, 9.1, 9.1. HRMS (ESI-TOF) calculated for C13H25O3 [M+H]+, 229.1804; observed, 229.1810.
4-Phenylbutyl 2-hydroxybutyrate, 37
Prepared according to Method B using 4-phenyl-1-butanol to provide 4-phenylbutyl 2-hydroxybutyrate (200 mg, 85% yield). 1H NMR (500 MHz, CDCl3) δ 7.30-7.22 (m, 2H), 7.21-7.13 (m, 3H), 4.25-4.14 (m, 2H), 4.13 (dd, J = 6.6, 4.4 Hz, 1H), 2.70 (s, 1H), 2.67-2.57 (m, 2H), 1.87-1.76 (m, 1H), 1.73-1.61 (m, 5H), 0.94 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 175.4, 141.8, 128.4, 128.4 125.9, 71.4, 65.5, 35.4, 28.2, 27.6, 27.5, 8.9. HRMS (ESI-TOF) calculated for C14H21O3 [M+H]+, 237.1491; observed, 237.1494.
5-Phenylpentyl 2-hydroxybutyrate, 38
Prepared according to Method B using 5-phenyl pentan-1-ol to provide 5-phenylpentyl 2-hydroxybutyrate (220 mg, 88% yield). 1H NMR (500 MHz, CDCl3) δ 7.29-7.11 (m, 2H), 7.19-7.12 (m, 3H), 4.22-4.07 (m, 3H), 2.71 (d, J = 5.7 Hz, 1H), 2.60 (t, J = 7.7 Hz, 2H), 1.84-1.76 (m, 1H), 1.71-1.58 (m, 5H), 1.43-1.33 (m, 2H), 0.93 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 175.6, 142.4, 128.6, 128.5, 126.0, 71.5, 65.8, 35.9, 31.2, 28.6, 27.7, 25.6, 9.1. HRMS (ESI-TOF) calculated for C15H23O3 [M+H]+, 251.1647; observed, 251.1655.
3-Cyclohexylpropyl 2-hydroxybutyrate, 39
Prepared according to Method B using 3-cyclohexyl-1-propanol to provide 3-cyclohexylpropyl 2-hydroxybutyrate (218 mg, 96% yield). 1H NMR (500 MHz, CDCl3) δ 4.21-4.08 (m, 3H), 2.72 (s, 1H), 1.87-1.77 (m, 1H), 1.73-1.58 (m, 8H), 1.25-1.05 (m, 6H), 0.94 (t, J = 7.4 Hz, 3H), 0.90-0.78 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 175.6, 71.6, 66.3, 37.5, 33.6, 33.5, 27.7, 26.8, 26.5, 26.2, 9.1. HRMS (ESI-TOF) calculated for C13H25O3 [M+H]+, 229.1804; observed, 229.1809.
4-Cyclohexylbutyl 2-hydroxybutyrate, 40
Prepared according to Method B using 4-cyclohexyl-1-butanol to provide 4-cyclohexylbutyl 2-hydroxybutyrate (225 mg, 93% yield). 1H NMR (500 MHz, CDCl3) δ 4.22-4.09 (m, 3H), 2.78 (s, 1H), 1.87-1.76 (m, 1H), 1.70-1.56 (m, 8H), 1.37-1.28 (m, 2H), 1.24-1.03 (m, 6H), 0.94 (t, J = 7.4 Hz, 3H), 0.89-0.76 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 175.6, 71.5, 66.0, 37.7, 37.2, 33.5, 29.0, 27.7, 26.9, 26.6, 23.2, 9.1. HRMS (ESI-TOF) calculated for C14H27O3 [M+H]+, 243.1960; observed, 243.1962.
5-Cyclohexylpentyl 2-hydroxybutyrate, 41
Prepared according to Method B using 5-cyclohexyl-1-butanol to provide 5-cyclohexylpentyl 2-hydroxybutyrate (151 mg, 59% yield). 1H NMR (500 MHz, CDCl3) δ 4.21-4.10 (m, 3H), 2.73 (d, J = 5.7 Hz, 1H), 1.87-1.76 (m, 1H), 1.70-1.57 (m, 8H), 1.35-1.25 (m, 4H), 1.23-1.06 (m, 6H), 0.94 (t, J = 7.4 Hz, 3H), 0.88-0.76 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 175.6, 71.5, 66.0, 37.8, 37.5, 33.6, 28.8, 27.7, 26.9, 26.6, 26.6, 26.3, 9.1. HRMS (ESI-TOF) calculated for C15H29O3 [M+H]+, 257.2117; observed, 257.2120.
1-(p-Butyl-phenyl)methyl 2-hydroxybutyrate, 42
Prepared according to Method B using (4-butylphenyl)methanol to provide (1-(p-butyl-phenyl)methyl 2-hydroxybutyrate) (193 mg, 74% yield). 1H NMR (500 MHz, CDCl3) δ 7.25 (d, J = 7.1 Hz, 2H), 7.17 (d, J = 8.0 Hz, 2H), 5.16 (s, 2H), 4.17 (dd, J = 6.7, 4.3 Hz, 1H), 2.59 (dd, J = 7.8, 7.8 Hz, 2H), 1.88-1.78 (m, 1H), 1.72-1.62 (m, 1H), 1.61-1.52 (m, 2H), 1.38-1.28 (m, 2H), 0.91 (t, J = 7.4 Hz, 3H), 0.90 (t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 175.4, 143.7, 132.5, 128.9, 128.7, 71.6, 67.6, 35.6, 33.8, 27.7, 22.5, 14.2, 9.1; HRMS (ESI-TOF) calculated for C15H22O3Na [M+Na]+, 237.1466; observed, 273.14715.
1-(m-Butyl-phenyl)methyl 2-hydroxybutyrate, 43
Prepared according to Method B using (3-butylphenyl)methanol to provide 1-(m-butyl-phenyl)methyl 2-hydroxybutyrate) (29 mg, 47% yield). 1H NMR (500 MHz, CDCl3) δ 7.29-7.22 (m, 1H), 7.18-7.12 (m, 3H), 5.19 (A of AB, J = 12.1 Hz, 1H), 5.15 (B of AB, J = 12.1 Hz, 1H), 4.19 (dd, J = 10.8, 5.9 Hz, 1H), 2.72 (d, J = 5.8 Hz, 1H), 2.60 (dd, J = 7.7, 7.7 Hz, 2H), 1.89-1.78 (m, 1H), 1.73-1.62 (m, 1H), 1.61-1.51 (m, 2H), 1.39-1.28 (m, 2H), 0.95-0.87 (m, 6H). 13C NMR (126 MHz, CDCl3) δ 175.4, 143.7, 135.2, 128.9, 128.8, 128.7, 125.8, 71.6, 67.7, 35.7, 33.8, 27.7, 22.6, 14.2, 9.1. HRMS (ESI-TOF) calculated for C15H22O3Na [M+Na]+, 273.1467; observed, 273.1478.
1-(o-Butyl-phenyl)methyl 2-hydroxybutyrate, 44
Prepared according to Method B using (2-butylphenyl)methanol to provide 1-(o-butyl-phenyl)methyl 2-hydroxybutyrate) (55 mg, 40% yield). 1H NMR (500 MHz, CDCl3) δ 7.33-7.26 (m, 2H), 7.22-7.16 (m, 2H), 5.23 (s, 2H), 4.17 (dd, J = 10.6, 6.1 Hz, 1H), 2.72 (d, J = 5.7 Hz, 1H), 2.63 (dd, J = 7.8, 7.8 Hz, 2H), 1.88-1.78 (m, 1H), 1.72-1.62 (m, 1H), 1.59-1.50 (m, 2H), 1.42-1.33 (m, 2H), 0.92 (t, J = 7.4 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 175.4, 142.1, 132.7, 130.1, 129.8, 129.2, 126.3, 71.6, 65.7, 33.7, 32.5, 27.7, 23.0, 14.2, 9.1. HRMS (ESI-TOF) calculated for C15H22O3Na [M+Na]+, 273.1467;observed, 273.1465.
1-(p-Pentyl-phenyl)methyl 2-hydroxybutyrate, 45
Prepared according to Method B using (4-pentylphenyl)methanol to provide (1-(p-pentyl-phenyl)methyl 2-hydroxybutyrate) (124 mg, 73% yield). 1H NMR (500 MHz, CDCl3) δ 7.25 (d, J = 7.0 Hz, 2H), 7.16 (d, J = 8.0 Hz, 2H), 5.16 (s, 2H), 4.17 (dd, J = 10.8, 6.0 Hz, 1H), 2.72 (d, J = 5.7 Hz, 1H), 2.58 (dd, J = 7.7 Hz, 2H), 1.87-1.77 (m, 1H), 1.72-1.62 (m, 1H), 1.62-1.54 (m, 2H), 1.35-1.25 (m, 4H), 0.91 (t, J = 7.4 Hz, 3H), 0.86 (t, J = 6.9 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 175.4, 143.8, 132.5, 128.9, 128.7, 71.6, 67.6, 35.9, 31.7, 31.3, 27.7, 22.7, 14.3, 9.1. HRMS (ESI-TOF) calculated for C16H24O3Na[M+Na]+, 287.1623; observed, 287.1631.
1-(m-Pentyl-phenyl)methyl 2-hydroxybutyrate, 46
Prepared according to Method B using (3-pentylphenyl)methanol to provide (1-(m-pentyl-phenyl)methyl 2-hydroxybutyrate) (170 mg, 81% yield). 1H NMR (500 MHz, CDCl3) δ 7.31-7.22 (m, 1H), 7.19-7.10 (m, 3H), 5.19 (A of AB, J = 12.1 Hz, 1H), 5.16 (B of AB, J = 12.1 Hz, 1H), 4.19 (dd, J = 10.6, 6.1 Hz, 1H), 2.72 (d, J = 5.7 Hz, 1H), 2.59 (dd, J = 7.7, 7.7 Hz, 2H), 1.89-1.78 (m, 1H), 1.74-1.64 (m, 1H), 1.64-1.53 (m, 2H), 1.37-1.24 (m, 4H), 0.92 (t, J = 7.4 Hz, 3H), 0.87 (t, J = 6.8 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 175.4, 143.7, 135.2, 128.9, 128.8, 128.6, 125.9, 71.6, 67.7, 36.0, 31.7, 31.4, 27.7, 22.7, 14.3, 9.1. HRMS (ESI-TOF) calculated for C16H24O3Na [M+Na]+, 287.1623; observed,287.1631.
3-(p-Ethyl-phenyl)propyl 2-hydroxybutyrate, 47
Prepared according to Method B using 3-(4-ethylphenyl)propan-1-ol to provide 3-(p-ethyl-phenyl)propyl 2-hydroxybutyrate (39 mg, 86% yield). 1H NMR (500 MHz, CDCl3) δ 7.11 (d, J = 7.8 Hz, 2H), 7.07 (d, J = 7.8 Hz, 2H), 4.24-4.10 (m, 3H), 2.70 (d, J = 5.6 Hz, 1H), 2.68-2.55 (m, 4H), 2.01-1.91 (m, 2H), 1.87-1.77 (m, 1H), 1.73-1.61 (m, 1H), 1.20 (t, J = 7.6 Hz, 3H), 0.95 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 175.5, 148.3, 138.2, 128.5, 128.2, 71.5, 65.2, 31.8, 30.4, 28.6, 27.7, 15.9, 9.2. HRMS (ESI-TOF) calculated for C15H23O3 [M+H]+, 251.1647; observed, 251.1660.
3-(m-Ethyl-phenyl)propyl 2-hydroxybutyrate, 48
Prepared according to Method B using (3-(3-ethylphenyl)propan-1-ol to provide 3-(p-ethyl-phenyl)propyl 2-hydroxybutyrate (231 mg, 92% yield). 1H NMR (500 MHz, CDCl3) δ 7.19 (t, J = 7.5 Hz, 1H), 7.03 (d, J = 7.5 Hz, 1H), 7.01-6.95 (m, 2H), 4.23-4.11 (m, 3H), 2.70 (d, J = 5.7 Hz, 1H), 2.68-2.57 (m, 4H), 2.03-1.93 (m, 2H), 1.89-1.78 (m, 1H), 1.72-1.62 (m, 1H), 1.21 (t, J = 7.6 Hz, 3H), 0.96 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 175.5, 144.7, 141.0, 128.7, 128.2, 125.9, 125.8, 71.6, 65.3, 32.3, 30.4, 29.0, 27.7, 15.8, 9.2. HRMS (ESI-TOF) calculated for C15H23O3 [M+H]+, 251.1647;observed, 251.1653.
3-(o-Ethyl-phenyl)propyl 2-hydroxybutyrate, 49
Prepared according to Method B using 3-(2-ethylphenyl)propan-1-ol to provide 3-(o-ethyl-phenyl)propyl 2-hydroxybutyrate (37 mg, 74% yield). 1H NMR (500 MHz, CDCl3) δ 7.19-7.09 (m, 4H), 4.30-4.07 (m, 4H), 2.76-2.58 (m, 4H), 1.99-1.90 (m, 2H), 1.89-1.78 (m, 1H), 1.73-1.63 (m, 1H), 1.20 (t, J = 7.6 Hz, 3H), 0.97 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 175.5, 142.1, 138.6, 129.3, 128.8, 126.7, 126.2, 71.6, 65.5, 30.1, 29.0, 27.7, 25.7, 15.6, 9.2. HRMS (ESI-TOF) calculated for C15H23O3 [M+H]+, 251.1647; observed, 251.1652.
Vibrio cholerae Agonism Bioassay
Reporter strain MM920 (V. cholerae ΔcqsA ΔluxQ carrying pBB1 cosmid, which contains the V. harveyi luxCDABE luciferase operon) was used to assay agonist activity of each synthetic compound. This strain was grown in LB medium containing 10 μg/mL tetracycline at 30 °C for >16 hours and diluted 20-fold with the same medium. Two μL of each synthetic compound dissolved in DMSO in various concentrations was added to 200 μL of the diluted reporter strain in triplicate in a 96-well plate. Bioluminescence and OD600 were measured in a PerkinElmer EnVision Multilabel Reader following 4-hour incubation at 30 °C with shaking. DMSO was used as the negative control.
The CqsS C170Y and CqsS C170A mutants
The cqsSC170Y and cqsSC170A alter the Vibrio cholerae CqsS receptor specificity to CAI-1 type ligands.8, 13 To introduce these mutations to the genome of Vibrio cholerae, these two cqsS alleles were first cloned into the suicide vector pKAS3226 resulting in plasmids WN1957 (C170Y) and WN1961 (C170A), respectively. These two cqsS mutations were subsequently introduced into the genome of the V. cholerae strain WN1170 (ΔcqsA ΔluxQ) as described previously, resulting in WN1977 (ΔcqsA ΔluxQ cqsSC170Y) and WN1982 (ΔcqsA ΔluxQ cqsSC170A). The presence of the desired cqsS mutation was confirmed by sequencing. The luxCDABE operon from Vibrio harveyi carried on cosmid pBB1 was introduced into WN1977 and WN1982 by conjugation, yielding strain WN1989 andWN1994, respectively. Quorum-sensing dependent response from these V. cholerae strains was monitored by measuring bioluminescence in the presence of different ligands as described above.
Pharmacophore modeling
Pharmacophore model generation was carried out using the software Ligand Scout. Ligand Scout version 3.03b (inteligand.com) was used.18-20 The “Training Set” consisting of eight CAI-1 ester analogs that we had identified during our SAR studies. The compounds selected all displayed EC50 values of less then 0.2 μM and were capable of activating greater than 92% of the maximal quorum sensing response. A ligand-based pharmacophore was generated for the selected data. For pharmacophore generation, the sdf files of the data set, all as the (S)-stereoisomer, were provided as an input. Sdf files of the data set were obtained from ChemDraw Ultra using iBabel for file conversion. The sdf files were imported into Ligand Scout and an unbiased pharmacophore model was generated using the default “BEST” settings, generating 500 unique conformers of each of the ligands and a aligning each pharmacophore conformer to provide 10 best fit models which served as the pharmacophore model. Subsequent modeling of inactive analogs (“Test Set” Compounds) was achieved using the pharmacophore model generated above and including a series of analogs displaying lower biological activity as “Test Set” analogs.
Supplementary Material
Acknowledgments
The authors acknowledge Lotus Separations LLC (Christina Kraml) for essential contributions to compound purification and enantiomer resolution. This work was supported by the Howard Hughes Medical Institute, the National Institutes of Health grant 5R01AI054442, the National Science Foundation grant MCB-0343821 to BLB, and an NIH postdoctoral fellowship GM082061 to W.L.N.
Abbreviations Used
- CAI-1
(S)-3-hydroxytridecan-4-one
- Ea-CAI-1
3-aminotridec-2-en-4-one
- Ph-CAI-1
1-hydroxy-1-phenylundecan-2-one
- C8-CAI-1
(S)-3-hydroxyundecan-4-one
Footnotes
Notes: The authors declare no competing financial interest.
Supporting Information: Details of compound synthesis and spectral data (1H-NMR and 13C-NMR) for all compounds; primary bioassay data; pharmacophore modeling. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Miller MB, Bassler BL. Quorum sensing in bacteria. Annual Review of Microbiology. 2001;55:165–199. doi: 10.1146/annurev.micro.55.1.165. [DOI] [PubMed] [Google Scholar]
- 2.Galloway WRJD, Hodgkinson JT, Bowden SD, Welch M, Spring DR. Quorum Sensing in Gram-Negative Bacteria: Small-Molecule Modulation of AHL and Al-2 Quorum Sensing Pathways. Chemical Reviews. 2011;111:28–67. doi: 10.1021/cr100109t. [DOI] [PubMed] [Google Scholar]
- 3.Rasko DA, Sperandio V. Anti-virulence strategies to combat bacteria-mediated disease. Nature Reviews Drug Discovery. 2010;9:117–128. doi: 10.1038/nrd3013. [DOI] [PubMed] [Google Scholar]
- 4.Hentzer M, Givskov M. Pharmacological inhibition of quorum sensing for the treatment of chronic bacterial infections. Journal of Clinical Investigation. 2003;112:1300–1307. doi: 10.1172/JCI20074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cegelski L, Marshall GR, Eldridge GR, Hultgren SJ. The biology and future prospects of antivirulence therapies. Nature Reviews Microbiology. 2008;6:17–27. doi: 10.1038/nrmicro1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ng WL, Bassler BL. Bacterial Quorum-Sensing Network Architectures. Annual Review of Genetics. 2009;43:197–222. doi: 10.1146/annurev-genet-102108-134304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Higgins DA, Pomianek ME, Kraml CM, Taylor RK, Semmelhack MF, Bassler BL. The major Vibrio cholerae autoinducer and its role in virulence factor production. Nature. 2007;450:883–886. doi: 10.1038/nature06284. [DOI] [PubMed] [Google Scholar]
- 8.Ng WL, Perez LJ, Wei Y, Kraml C, Semmelhack MF, Bassler BL. Signal production and detection specificity in Vibrio CqsA/CqsS quorum-sensing systems. Molecular Microbiology. 2011;79:1407–1417. doi: 10.1111/j.1365-2958.2011.07548.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wei Y, Perez LJ, Ng WL, Semmelhack MF, Bassler BL. Mechanism of Vibrio cholerae Autoinducer-1 Biosynthesis. Acs Chemical Biology. 2011;6:356–365. doi: 10.1021/cb1003652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu J, Mekalanos JJ. Quorum sensing-dependent biofilms enhance colonization in Vibrio cholerae. Developmental Cell. 2003;5:647–656. doi: 10.1016/s1534-5807(03)00295-8. [DOI] [PubMed] [Google Scholar]
- 11.Spirig T, Tiaden A, Kiefer P, Buchrieser C, Vorholt JA, Hilbi H. The Legionella autoinducer synthase LqsA produces an alpha-hydroxyketone signaling molecule. Journal of Biological Chemistry. 2008;283:18113–18123. doi: 10.1074/jbc.M801929200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bolitho ME, Perez LJ, Koch MJ, Ng WL, Bassler BL, Semmelhack MF. Small molecule probes of the receptor binding site in the Vibrio cholerae CAI-1 quorum sensing circuit. Bioorganic & Medicinal Chemistry. 2011;19:6906–6918. doi: 10.1016/j.bmc.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ng WL, Wei Y, Perez LJ, Cong J, Long T, Koch M, Semmelhack MF, Wingreen NS, Bassler BL. Probing bacterial transmembrane histidine kinase receptor-ligand interactions with natural and synthetic molecules. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:5575–5580. doi: 10.1073/pnas.1001392107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller MB, Skorupski K, Lenz DH, Taylor RK, Bassler BL. Parallel quorum sensing systems converge to regulate virulence in Vibrio cholerae. Cell. 2002;110:303–314. doi: 10.1016/s0092-8674(02)00829-2. [DOI] [PubMed] [Google Scholar]
- 15.Grasa GA, Kissling RM, Nolan SP. N-heterocyclic carbenes as versatile nucleophilic catalysts for transesterification/acylation reactions. Organic Letters. 2002;4:3583–3586. doi: 10.1021/ol0264760. [DOI] [PubMed] [Google Scholar]
- 16.Grasa GA, Guveli T, Singh R, Nolan SP. Efficient transesterification/acylation reactions mediated by N-heterocyclic carbene catalysts. Journal of Organic Chemistry. 2003;68:2812–2819. doi: 10.1021/jo0267551. [DOI] [PubMed] [Google Scholar]
- 17.Singh R, Kissling RM, Letellier MA, Nolan SP. Transesterification/acylation of secondary alcohols mediated by N-heterocyclic carbene catalysts. Journal of Organic Chemistry. 2004;69:209–212. doi: 10.1021/jo035431p. [DOI] [PubMed] [Google Scholar]
- 18.Wolber G, Langer T. LigandScout: 3-d pharmacophores derived from protein-bound Ligands and their use as virtual screening filters. Journal of Chemical Information and Modeling. 2005;45:160–169. doi: 10.1021/ci049885e. [DOI] [PubMed] [Google Scholar]
- 19.Wolber G, Dornhofer AA, Langer T. Efficient overlay of small organic molecules using 3D pharmacophores. Journal of Computer-Aided Molecular Design. 2006;20:773–788. doi: 10.1007/s10822-006-9078-7. [DOI] [PubMed] [Google Scholar]
- 20.Yang SY. Pharmacophore modeling and applications in drug discovery: challenges and recent advances. Drug Discovery Today. 2010;15:444–450. doi: 10.1016/j.drudis.2010.03.013. [DOI] [PubMed] [Google Scholar]
- 21.Meanwell NA. Synopsis of Some Recent Tactical Application of Bioisosteres in Drug Design. Journal of Medicinal Chemistry. 2011;54:2529–2591. doi: 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- 22.Choi H, Mascuch SJ, Villa FA, Byrum T, Teasdale ME, Smith JE, Preskitt LB, Rowley DC, Gerwick L, Gerwick WH. Honaucins A-C, Potent Inhibitors of Inflammation and Bacterial Quorum Sensing: Synthetic Derivatives and Structure-Activity Relationships. Chemistry & Biology. 2012;19:589–598. doi: 10.1016/j.chembiol.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frezza M, Soulere L, Balestrino D, Gohar M, Deshayes C, Queneau Y, Forestier C, Doutheau A. Ac-2-DPD, the bis-(O)-acetylated derivative of 4,5-dihydroxy-2,3-pentanedione (DPD) is a convenient stable precursor of bacterial quorum sensing autoinducer AI-2. Bioorganic & Medicinal Chemistry Letters. 2007;17:1428–1431. doi: 10.1016/j.bmcl.2006.11.076. [DOI] [PubMed] [Google Scholar]
- 24.Guo M, Gamby S, Nakayama S, Smith J, Sintim HO. A Pro-Drug Approach for Selective Modulation of AI-2-Mediated Bacterial Cell-to-Cell Communication. Sensors. 2012;12:3762–3772. doi: 10.3390/s120303762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moore CG, Murphy PJ, Williams HL, McGown AT, Smith NK. Synthetic studies towards ptilomycalin A: total synthesis of crambescidin 359. Tetrahedron. 2007;63:11771–11780. [Google Scholar]
- 26.Skorupski K, Taylor RK. Positive selection vectors for allelic exchange. Gene. 1996;169:47–52. doi: 10.1016/0378-1119(95)00793-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.