

Abstract
During cycles of fasting and feeding, liver function is regulated by both transcriptional and post-translational events. Regulated protein degradation has recently emerged as a key mechanism to control abundance of specific hepatic proteins under different nutritional conditions. As glucagon signaling through cAMP and PKA is central to glucose output during fasting, we hypothesized that this signaling pathway may also regulate ubiquitin ligases in the fasted state. Here we show that fasting stimuli promote expression of the short isoform of the E3 ubiquitin ligase Nedd4l in primary mouse hepatocytes. Nedd4l-short mRNA and NEDD4L (short isoform) protein accumulate in glucagon-treated primary mouse hepatocytes and in liver tissues during fasting. We identified a functional cAMP response element in the alternate Nedd4l-short promoter; mutation of this element blunts cAMP-induced expression of a Nedd4l reporter construct. CREB occupies the endogenous Nedd4l locus near this element. CREB and its co-activator CRTC2, both activated by fasting stimuli, contribute to glucagon-stimulated Nedd4l-short expression in primary hepatocytes. siRNA-mediated Nedd4l depletion in primary hepatocytes did not affect gluconeogenic gene expression, glucose output or glycogen synthesis. Our findings reveal a new mechanism of Nedd4l transcriptional regulation in liver cells.
Introduction
Intermittent food availability requires mammals to store nutrients after feeding and to liberate stored nutrients during fasting. The liver is a major organ responsible for maintenance of normoglycemia and energy balance during cycles of fasting and feeding. In the fasted state, glucagon and catecholamines prevent hypoglycemia by stimulating hepatic glycogen breakdown and gluconeogenesis, in part via the cAMP response element binding protein (CREB) and its co-activator CRTC2 (CREB-regulated transcription coactivator 2) [1]. CREB/ CRTC2 directly and indirectly not only stimulate hepatic gluconeogenesis by transcriptional induction of Pepck (encoding phosphoenolpyruvate carboxykinase), G6pase (encoding glucose 6-phosphatase) and Pgc1α (Ppargc1a, encoding PPAR-gamma coactivator 1-alpha, PGC1α) [2,3], but also inhibit de novo lipogenesis [4] and exert a priming effect on post-prandial hepatic insulin sensitivity by transcriptional induction of Irs2 (encoding Insulin Receptor Substrate 2) [5]. There are more than 4,000 predicted CREB binding sites in the mouse genome [6], so identification of additional CREB/ CRTC2 target genes could shed light on new mechanisms by which these transcriptional activators exert profound effects on hepatic metabolism.
In addition to transcriptional regulation of metabolic enzymes and regulatory factors, counter-regulatory hormone (e.g. catecholamines and glucagon) signaling during fasting also results in selective post-translational regulation of protein stability through the ubiquitin-proteasome pathway. For example, during fasting, cyclin C/ Cdk8 complexes phosphorylate and stimulate ubiquitin-dependent degradation of Srepb1-c [7]. Similarly, fasting stimulates the p38 MAP kinase-COP1 complex to ubiquitylate hepatic fatty acid synthase (FASN), leading to degradation by the proteasome [8]. In C. elegans, fasting induces genes encoding thirty-two different SCF (Skp1-cullen-F box) E3 ligase complex components [9]. Moreover, fasting stimuli induce transcription of the ubiquitin-specific protease Usp2 in mammalian liver, which contributes to hepatic gluconeogenesis [10]. Finally, the CREB co-activator CRTC2 is targeted for ubiquitin-dependent degradation during late fasting in liver by the E3 ubiquitin ligase COP1 [11]. Thus, signal-induced ubiquitin-dependent degradation, either via transcriptional regulation of E3 ligases or phosphorylation-dependent assembly of E3-substrate complexes, is an emerging mechanism for dynamic control of hepatic metabolism.
We hypothesized that additional E3 ubiquitin ligases may contribute to regulation of hepatic metabolism during fasting and feeding in the liver. Because the cAMP-PKA pathway is a major intracellular signaling pathway that regulates hepatocyte responses to fasting, we sought E3 ubiquitin ligases expressed in liver that are known to be regulated by cAMP signaling. The HECT family E3 ubiquitin ligase NEDD4L (also called NEDD4-2) is expressed in liver [12] and is acutely inhibited by direct PKA phosphorylation in epithelial cells treated with vasopressin [13]. In vivo roles of NEDD4L in liver have not been examined, but NEDD4L is best known for inhibition of the epithelial sodium channel (ENaC) in the kidney [14]. Loss of Nedd4l function results in sodium-sensitive hypertension, either due to de-repression of ENaC and other ion channels in the kidney [15,16] or in the brain [17]. Nedd4l is also expressed in lung, where it is required for clearance of fluid [18]. Three recent population-based studies found human NEDD4L variants associated with type 2 diabetes, obesity and diabetic nephropathy [19–21]. In this study, we identified an unexpected role for cAMP-PKA signaling in regulation of a specific isoform of Nedd4l in hepatocytes and liver tissue. We explored transcriptional regulation of this gene by CREB/ CRTC2 and possible roles of NEDD4L in regulation of glucose metabolism in primary mouse hepatocytes.
Results
The Nedd4l-short isoform is induced by fasting stimuli in hepatocytes and liver
To identify PKA-sensitive ubiquitin ligases that may contribute to hepatocyte responses during fasting, we searched the literature for E3 ligases regulated by cAMP signaling that are expressed in liver. NEDD4L (Neural precursor cell expressed, developmentally down-regulated gene 4-like, also known as NEDD4-2) satisfied both of these criteria, but nothing was known about the possible roles of this protein in liver. To begin to characterize regulation of NEDD4L by PKA signaling in liver cells, we tested expression of mouse NEDD4L in primary hepatocytes treated with glucagon, a potent activator of cAMP-PKA signaling in this cell type [1]. There are two isoforms of NEDD4L (a short isoform of 110 kDa and a long isoform of 130 kDa), which are encoded by separate transcripts of the same gene (Ensembl v71 [22], Figure 1A; see also Methods). mRNA and protein of both isoforms are present in unstimulated primary mouse hepatocytes (Figure 1B, C). We observed that both the mRNA and protein of the NEDD4L short isoform were selectively induced by glucagon within 1 and 4 hours, respectively (Figure 1B, C, S1A). The Nedd4l-long isoform (Nedd4l-l) mRNA and protein amounts remained unchanged (Figure 1B, C, S1A). Both Nedd4l-short mRNA and protein returned to baseline levels after longer treatment, similar to the mRNA pattern for known glucagon-responsive genes Pepck and Pgc1α (Figure S1B). NEDD4L short isoform (110 kDa) protein induction in hepatocytes was also sensitive to the dose of glucagon, whereas the NEDD4L long isoform protein (130kDa) remained unchanged from control (Figure S1C).
Figure 1. Nedd4l short isoform is induced by glucagon in primary hepatocytes and during fasting in mouse liver.
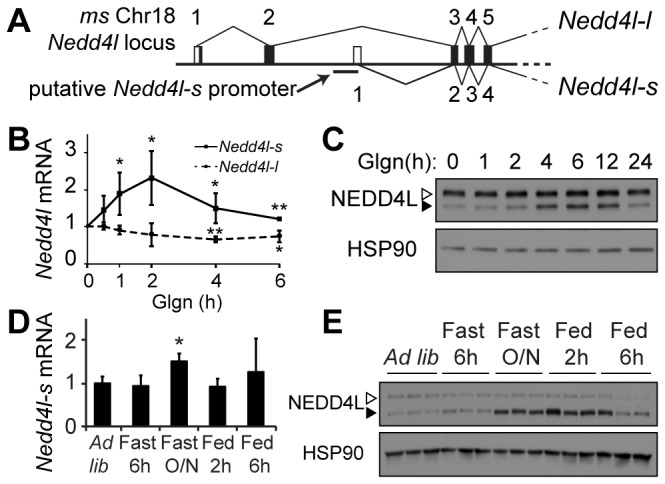
(A) Diagram of the first several exons of the mouse Nedd4l locus on chromosome 18. Exons incorporated into Nedd4-long and Nedd4l-short transcripts are indicated by connected lines and numbered above and below the DNA, respectively. The putative alternate promoter for Nedd4l-short is shown. Only the first several exons are shown; the rest are shared by the two transcripts. (B) Primary mouse hepatocytes were treated with glucagon (100nM) for the indicated time (h) for analysis of Nedd4l isoform mRNA expression. mRNA amounts are normalized to Gapdh, represented as mean fold change from 0 h treatment ±stdev. *p<0.05, **p<0.01 to 0 h. (C) NEDD4L proteins and HSP90 loading control in primary hepatocytes treated as in (B). (D) Nedd4l-short mRNA from liver tissue of ad libitum fed, fasted (6 h or overnight ‘O/N’) or O/N fasted and re-fed (2 and 6 h) male C57Bl6/J mice (n=3 per condition). Mean fold change to ad lib, *p<0.05 to ad lib, 6 h fasted and 2h re-red. All other combinations were not significant. (E) NEDD4L proteins and HSP90 from liver tissue as in (D). Filled arrowheads, NEDD4L-short; open arrowheads, NEDD4L-long. See Figure S1A, S1D for quantification of western blots.
During fasting, glucagon released by the α-cells of the pancreas stimulates cAMP-PKA signaling. To test whether Nedd4l is regulated by fasting in vivo, we compared mRNA and protein levels of Nedd4l short and long isoforms in mouse liver after fasting and re-feeding. Consistent with our findings in primary hepatocytes, Nedd4l-short (Nedd4l-s) mRNA was also induced in liver tissue after an overnight fast, and the expression returned to control levels following 2 to 6 hours of re-feeding (Figure 1D). Similarly, NEDD4L short isoform protein levels were induced fourfold after overnight fasting and persisted for at least 2 hours after re-feeding (Figure 1E, S1D). By 6 hours after re-feeding, NEDD4L-short protein abundance began to decline, but did not yet return to ad libitum levels (Figure 1E, S1D). The effects of fasting on Nedd4l expression in liver were specific to the short isoform; there was no significant change in NEDD4L long isoform protein with fasting and re-feeding in vivo (Figure 1E, S1D). These data show that the short isoform of Nedd4l is selectively regulated by fasting stimuli in hepatocytes.
There is a paucity of information about the functional or regulatory differences between the long and short NEDD4L isoforms. Both isoforms contain the substrate-targeting WW domains, but the short isoform lacks the N-terminal C2 domain, which is required for calcium-stimulated plasma membrane targeting in epithelial cells [23]. More recently, calcium was shown to release an inhibitory C2-HECT domain interaction [24]. Although the NEDD4L-short isoform is predicted to lack the N-terminal C2 domain, it has not been reported to have differential substrate selectivity or subcellular localization from the long isoform. In primary hepatocytes, both isoforms were localized to the cytosolic/ membrane fraction and were excluded from the nucleus, irrespective of glucagon treatment (Figure S1E). The functional difference between these two isoforms remains to be determined.
The short isoform of Nedd4l is a CREB target gene
The rapid and acute regulation of Nedd4l-s mRNA by glucagon signaling is consistent with kinetics of CREB/ CRTC2 activity [1] and known CREB target gene (Pepck, Pgc1α) induction in hepatocytes, so we investigated whether Nedd4l may be a CREB target gene. The two known mouse Nedd4l transcripts (each encoding a distinct isoform, short or long) are transcribed with alternate first exons from the same gene and share most exons (Ensembl v71 [22], Figure 1A; see also Methods). To determine if Nedd4l-short is regulated by CREB, we queried the publicly available CREB target gene database [6] for predicted cAMP response elements (CRE) in or near the Nedd4l locus. We noted that two consensus half CRE sites are present in the putative proximal promoter region of the short Nedd4l transcript (Figure 1A). We therefore hypothesized that Nedd4l-short is directly regulated by CREB in glucagon-treated hepatocytes.
To evaluate the contribution of the predicted CREB binding sites to cAMP-stimulated promoter activity, we tested activity of a luciferase reporter encoding the genomic region surrounding the putative Nedd4l-short promoter (-532 to +321, Figure 2A, 2B top) in HEK-293T cells. This region contains the two consensus CRE sites (CRE1 -412~-407 ‘TGACG’ and CRE2 +196~+201 ‘CGTCA’). Treatment of cells with a cocktail of the adenylyl cyclase agonist forskolin (FSK) and the phosphodiesterase inhibitor IBMX, which induces sustained cAMP production, stimulated Nedd4l-short luciferase activity but not luciferase activity of the empty vector control (Figure 2A, S2A). We mutated each CRE in the putative Nedd4l-short promoter singly and in combination and found that cAMP-stimulated Nedd4l-short luciferase activity was unaffected by mutation of CRE1. Mutation of the second CRE site (CRE2) resulted in reduced Nedd4l-luciferase activity in both vehicle- and FSK/ IBMX-stimulated cells. Mutation of both CRE sites (no CRE) yielded similar luciferase activity to the constructs with mutation of only CRE2 (Figure 2A, S2B). These data show that the first CRE site is likely not functional and the second site accounts for a portion of cAMP-stimulated luciferase activity. To test whether CREB is necessary for Nedd4l-luciferase activity, we co-transfected a dominant negative CREB mutant (ACREB) to block CREB activity. ACREB reduced basal Nedd4l-short luciferase activity and completely abrogated FSK/ IBMX-stimulated Nedd4l-luciferase activity (Figure 2A, S2B). Thus, full cAMP-stimulated induction of the Nedd4l-short promoter requires the second CREB binding site in the DNA and CREB activity. The fact that dominant-negative CREB completely blocks promoter activity suggests that additional, non-canonical CREB responsive elements exist in the promoter region.
Figure 2. Nedd4l-short is a CREB target gene.
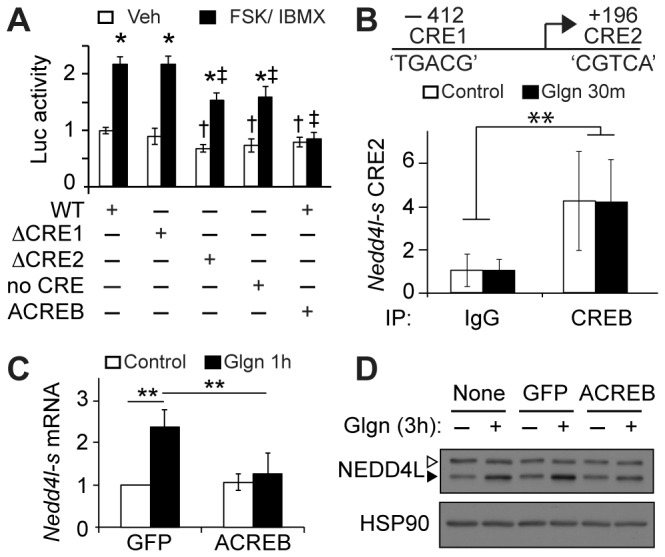
(A) Nedd4l-short luciferase reporter activity in HEK293T cells treated with FSK/ IBMX (6 h). Wild type (WT) Nedd4l-short luciferase reporter compared with individual CRE site mutations (ΔCRE1 or ΔCRE2) or both CREs deleted (no CRE). Effect of co-transfected ACREB is shown. *p<0.01 between veh and FSK/ IBMX treated; ‡p<0.01 to FSK/ IBMX-treated WT; †p<0.01 to veh-treated WT (n=6 replicates shown, representative of three independent experiments). See Figure S2A, B for empty vector control and luciferase data expressed in A.U. (B) Top, predicted CREB binding sites (CRE1: -412 ‘TGACG’; CRE2: +196 ‘CGTCA’) in mouse Nedd4l-short promoter. Bottom, chromatin immunoprecipitation from primary mouse hepatocytes treated without or with glucagon (100nM, 30 min) using non-specific IgG (IgG) or anti-CREB IgG. Recovery of Nedd4l-short genomic DNA containing CRE2 was quantified in chromatin immunoprecipitates, normalized to the input and expressed as mean fold enrichment ± stdev over matched IgG controls among 3 replicates. **, p<0.01 comparing all IgG and all CREB samples. See Figure S2C for Pepck and Gapdh controls. (n=3 independent experiments) (C and D) Nedd4l-short mRNA expression (C, n=4, **p< 0.01) and NEDD4L protein levels (D, n=3) in primary hepatocytes infected with Ad-GFP or Ad-ACREB and treated with glucagon (100nM). Filled arrowheads, NEDD4L-short; open arrowheads, NEDD4L-long. See Figure S2F for quantification of western blots.
To extend our findings to endogenous Nedd4l regulation, we first tested whether CREB associates with CRE2 in the Nedd4l-short promoter. We stimulated primary mouse hepatocytes with glucagon for 30 min and performed chromatin immunoprecipitation with unspecific or anti-CREB antiserum (Figure 2B, S2C). The Nedd4l-short genomic DNA containing CRE2 was enriched by fourfold in the CREB immunoprecipitates compared to control antiserum. Similar to other CREB target genes, including Pepck (Figure S2C), glucagon did not appear to affect CREB occupancy at the Neddl-short locus near the CRE2 site. We were unable to detect CREB association with CRE1 (not shown), consistent with the luciferase assay results, and unrelated genomic DNA sequence from the Gapdh locus was not recovered in the immunoprecipitates (Figure S2C). We next asked whether CREB is required for glucagon-stimulated Nedd4l-short expression in primary hepatocytes. We infected the cells with adenovirus encoding GFP control or ACREB to block CREB activity. We confirmed that ACREB was expressed and blocked induction of Sik1 (encoding salt inducible kinase 1, SIK1), a known CREB target gene in hepatocytes [2] (Figure S2D, E). In agreement with the luciferase assay data, ACREB inhibited both Nedd4l-short mRNA (Figure 2C) and protein (Figure 2D, S2F) induction by glucagon. The Nedd4l-long isoform protein was unaffected (Figure 2D, S2F). We observed marginal induction of NEDD4L-short protein in glucagon-treated hepatocytes expressing dominant-negative CREB (Figure 2D, S2F) that failed to reach statistical significance compared to the GFP-expressing controls. It is therefore possible that glucagon stimulates NEDD4L protein accumulation by an additional mechanism, such as a post-translational effect on protein stability. Nonetheless, our data show that endogenous CREB associates with a 150-bp region surrounding the CRE site in the Nedd4l-short locus, and CREB activity is required for full induction of Nedd4l-short mRNA and protein by glucagon within 1-3 hours.
The CREB co-activator CRTC2 regulates Nedd4l-short
In liver, the CREB co-activator CRTC2 contributes to CREB target gene expression in early and late fasting [2,5,25,26]. To test whether CRTC2 is required for Nedd4l-short expression in primary hepatocytes, we infected the cells with adenoviral vectors encoding an unspecific shRNA or CRTC2-specific shRNA (‘CRTC2i’) to knockdown CRTC2 [2]. Similar to our results with ACREB, depletion of CRTC2 blocked Nedd4l-short mRNA induction within 1 hour of glucagon treatment (Figure 3A). Similarly, CRTC2 knockdown reduced both basal and glucagon-stimulated NEDD4L-short protein in primary hepatocytes, but some NEDD4L-short protein was still induced by glucagon (Figure 3B); the low expression of NEDD4L-short in CRTC2i-infected cells precludes accurate densitometry. CRTC2 protein was nearly undetectable in the cells infected with Ad-CRTC2i (Figure 3B). In resting hepatocytes, CRTC2 is known to exist in latent cytoplasmic complexes with 14-3-3 proteins. Upon glucagon stimulation, CRTC2 becomes dephosphorylated and moves to the nucleus, where it associates with CREB and CBP [1,2]. As expected, we observed a downshift of CRTC2 protein in glucagon-treated hepatocytes (Figure 3B). The NEDD4L long isoform protein levels were not altered by CRTC2 knockdown (Figure 3B). These data show that CRTC2 is required for the acute induction of Nedd4l-short mRNA by glucagon in primary hepatocytes. Taken with our other findings, our results support a model in which CREB and CRTC2 regulate Nedd4l-short via CRE or CRE-like elements in the alternate short isoform promoter region.
Figure 3. CRTC2 is required for Nedd4l short isoform regulation.
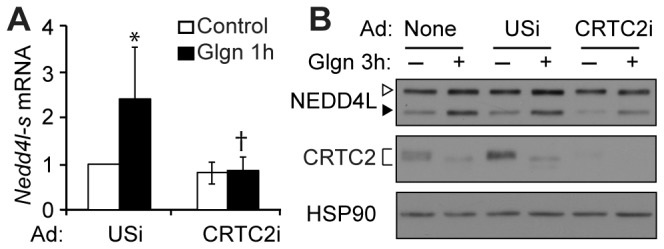
Primary hepatocytes were infected with adenovirus encoding unspecific shRNA (Ad-USi) or CRTC2-specific shRNA (Ad-CRTC2i) and treated with glucagon (100 nM). (A) Nedd4l-short mRNA expression, expressed as mean fold change to GFP control ± stdev (n=4 samples averaged among three independent experiments). *p<0.05 to unstimulated, †p<0.05 to glucagon-treated Ad-USi-infected. (B) Western blots of NEDD4L isoforms, CRTC2 and HSP90 loading control (representative of four experiments). Filled arrowheads, NEDD4L-short; open arrowheads, NEDD4L-long; bracket, phospho- and dephospho-CRTC2.
Nedd4l is not required for gluconeogenesis, glycogen synthesis or lipogenesis in primary hepatocytes
We have shown that Nedd4l-short is a CREB target gene that is induced by glucagon in hepatocytes and by fasting in the liver. In other cell types, NEDD4L is known to target several proteins for ubiquitin-dependent degradation or altered localization, including numerous ion channels (ENaC is the major example) [27] and activated Smad2/3 transcription factors [28]. ENaC is not known to be highly expressed in hepatocytes, and we observed no change in Smad2 or Smad3 abundance or turnover after TGF-β treatment in primary hepatocytes transfected with NEDD4L-selective siRNAs (not shown). Recent proteomics analysis showed that human NEDD4L is capable of ubiquitylating more than 100 proteins, most of which contain consensus PPxY recognition motifs [29]. The role of NEDD4L-dependent regulation of many of these targets in cells or tissues is not yet validated, and in vivo roles of NEDD4L in hepatic metabolism have not yet been evaluated.
Based on the striking induction of NEDD4L short isoform abundance in fasted liver and the known roles of CREB/ CRTC2 to regulate hepatic glucose production [1], we hypothesized that NEDD4L may contribute to regulation of glucose metabolism in hepatocytes. We used siRNAs specific to a common region of both Nedd4l isoforms to deplete NEDD4L in hepatocytes, as the mRNAs of these two isoforms are nearly identical; any observed effects could subsequently be assigned to the short or long isoform by rescue studies or isoform-selective siRNAs. Transfection of two different Nedd4l-specific siRNAs abrogated both basal and glucagon-stimulated NEDD4L protein expression (Figure 4A). We first tested whether NEDD4L affects glucagon-induced expression of mRNAs encoding Pgc1α and Pepck in primary hepatocytes [30]. As expected, glucagon strongly induced both Pgc1α and Pepck. Knockdown of NEDD4L did not affect glucagon-stimulated expression of these genes, but one Nedd4l-selective siRNA slightly reduced basal Pgc1α expression (Figure 4B). Because this reduction was limited to a single siRNA, we do not believe it is a biologically meaningful effect. We further directly tested glucose output from control and Nedd4l-deficient hepatocytes under control or glucagon-stimulated conditions. Consistent with expression of gluconeogenic genes, we found that glucagon-stimulated gluconeogenesis from pyruvate and lactate was similar in untransfected hepatocytes and those expressing either unspecific or Nedd4l-specific siRNAs (Figure 4C). Because NEDD4L can regulate numerous transporters and ion channels, we also tested whether NEDD4L may be required for glucose production from the amino acid alanine. However, glucose synthesis from alanine was also unaffected by knockdown of NEDD4L (Figure 4C), suggesting that alanine import is unimpaired. Thus, NEDD4L isoforms are not required for glucagon-stimulated gluconeogenesis in primary hepatocytes.
Figure 4. NEDD4L does not affect glucose metabolism in primary hepatocytes.
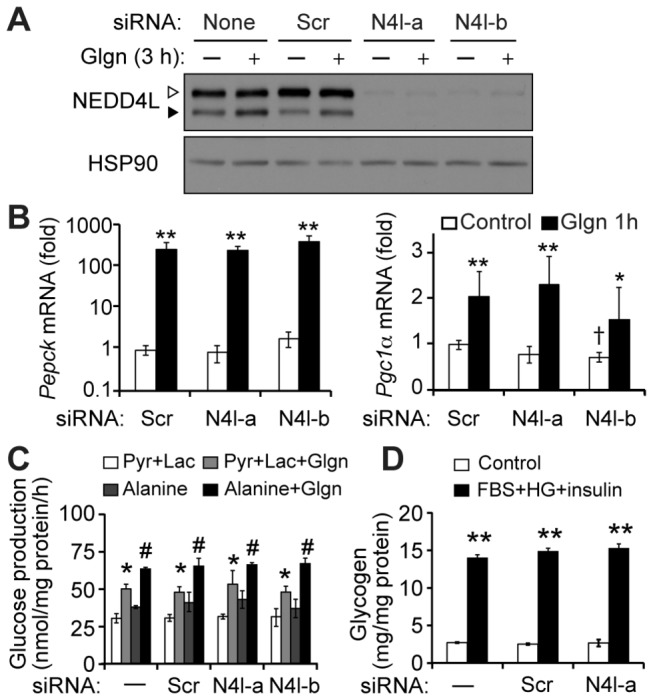
(A) Western blot of NEDD4L isoforms in primary hepatocytes transfected with indicated siRNAs, untreated or treated with glucagon (100nM) for 3 h. Scr: scrambled siRNA control; N4l-a, N4l-b: two different siRNAs specific to Nedd4l. Filled arrowheads, NEDD4L-short; open arrowheads, NEDD4L-long. (B) Basal and glucagon-stimulated gluconeogenic gene expression (Pepck, Pgc1α) in control and NEDD4L-deficient hepatocytes. Data expressed as fold of unstimulated Scr ±stdev; n=4-5 replicates among 2-3 independent experiments; **p<0.01, *p<0.05 compared to unstimulated control for each siRNA, †p<0.05 compared to scrambled siRNA control. (C) Glucose production from pyruvate + lactate (Pyr+Lac) or alanine in control or siRNA-transfected hepatocytes under basal and glucagon-stimulated conditions. Glucagon had a significant stimulatory effect in all groups; within groups of cells receiving the same siRNA, *p<0.05 compared to Pyr+Lac alone, #p<0.05 compared to alanine alone; no significant effect of the Nedd4l siRNAs (mean of 3 biological replicates; representative of two independent experiments). (D) Glycogen synthesis in siRNA-transfected primary hepatocytes treated with FBS, high glucose and insulin (FBS+HG+insulin, 3h). No significant effect of the siRNAs; **p<0.01 to unstimulated control. Mean of 3 biological replicates ±stdev; representative of three independent experiments in triplicate.
In the post-prandial state, insulin stimulates storage of excess glucose as glycogen and lipid in the liver [31]. During fasting, glucagon-stimulated CREB/ CRTC2 complexes sensitize the liver to postprandial insulin signaling by transcriptional induction of Irs2 [5]. We therefore hypothesized that NEDD4L may be induced during fasting for a role in insulin signaling or insulin-induced events during the immediate post-prandial period, when NEDD4L protein levels remain elevated (Figure 1E). We observed no change in acute insulin-stimulated Akt phosphorylation in primary hepatocytes expressing Nedd4l-selective siRNAs (not shown). To test whether NEDD4L impacts dynamics of glycogen storage in hepatocytes, we treated primary hepatocytes with fetal bovine serum (FBS), high glucose and insulin for 3 hours to stimulate glycogen storage [32]. As expected, insulin treatment dramatically stimulated intracellular glycogen accumulation, but NEDD4L knockdown did not affect insulin-stimulated glycogen storage (Figure 4D), glucagon-stimulated glycogen breakdown after this initial loading period (not shown) or insulin-stimulated lipogenesis (Figure S3). These results show that NEDD4L does not regulate glycogen or lipid synthesis in primary hepatocytes cultured ex vivo.
Discussion
We set out to identify PKA-regulated ubiquitin ligases expressed in the liver that may impact hepatic metabolism. Previous studies had shown that PKA directly phosphorylates and regulates the HECT domain E3 ligase NEDD4L [13]. We therefore evaluated Nedd4l expression in primary mouse hepatocytes and liver tissue. We present the first evidence that the short isoform of Nedd4l is regulated by fasting in liver in vivo and by glucagon signaling in primary mouse hepatocytes ex vivo. Nedd4l-short is transcribed from an alternate promoter within the Nedd4l gene. We show that Nedd4l-short is a CREB target gene: the regulatory region contains a consensus cAMP response element (CRE) that is necessary for full cAMP-stimulated transcriptional activity and physically associates with endogenous CREB, and Nedd4l-short transcription is blocked by dominant-negative CREB or knockdown of CRTC2. Our data clearly indicate that the CREB/ CRTC2 complex is required for acute glucagon-stimulated Nedd4l-short mRNA induction in primary hepatocytes and that CREB is required for Nedd4l-short promoter luciferase activity. The finding that mutation of the functional CRE site we identified only partially reduced the promoter activity indicates that additional cAMP- and CREB-sensitive elements exist in the sequence. However, we did not identify additional canonical CRE elements in the promoter region we cloned.
In liver tissue, NEDD4L-short is strongly induced during fasting and declines after re-feeding. Although several studies show that NEDD4L is regulated by post-translational mechanisms in different tissues, there is little known about mechanisms by which the Nedd4l mRNA transcription is regulated. Several of the human NEDD4L transcripts are androgen sensitive in prostate cancer cell lines [33], but other extracellular cues have not been identified. cAMP-dependent regulation of Nedd4l transcription could serve as a mechanism to regulate the abundance or surface expression of specific ion channels or signaling mediators in response to endocrine hormones in liver. Although either expression of dominant-negative CREB or CRTC2-selective shRNA almost entirely blocked acute Nedd4l-short transcriptional induction, NEDD4L-short protein still accumulated to some extent in response to glucagon. It is therefore likely that additional mechanisms contribute to the total amount of NEDD4L-short protein expressed after glucagon treatment in hepatocytes. For example, it is possible that cAMP signaling regulates NEDD4L-short protein stability as well as transcription, as we have recently shown for SIK1 [34]. It is also possible that additional transcriptional regulators contribute to Nedd4l-short induction during fasting in vivo. It is notable in this regard that the gluconeogenic gene G6pase is primarily regulated by CREB/ CRTC2 during early fasting; at later times, these complexes are replaced by FOXO-containing transcriptional complexes [11]. G6pase promoter constructs lacking CREB binding sites are poorly expressed during early fasting, but expressed at normal levels during late fasting [11]. It is not clear whether this model is generalizable to all CREB- and FOXO-regulated genes in liver. For example, Irs2 is regulated by CREB and CRTC2 in liver. Depletion of Crtc2 in liver in vivo completely blocks IRS2 expression after overnight fasting despite the existence of consensus FOXO binding sites in the Irs2 promoter [5]. We did not identify a consensus FOXO binding site in the proximal promoter region of Nedd4l-short. In Crtc2 -/- mice, expression of gluconeogenic mRNAs including G6pase and Pepck is reduced in liver after overnight fasting [25,26]. In vivo studies will reveal the extent to which CREB and CRTC2 are required for Nedd4l-short expression during early and late fasting.
We show that NEDD4L is readily detectable in liver and highly regulated by fasting stimuli, but its physiological role in this tissue remains unknown. Our findings of strong and selective cAMP-CREB dependent regulation of the Nedd4l-short mRNA and protein during fasting suggest that the NEDD4L short isoform functions during fasting or in the postprandial state, probably regulating processes other than glucagon-induced glucose output, insulin- and glucagon-regulated glycogen synthesis and degradation, or lipogenesis. Many major metabolic and functional changes occur in the fasted liver, including enhanced ketogenesis, lipid oxidation, and amino acid catabolism, most of which are best studied in vivo where analysis can be performed in the context of the other metabolic tissues and full complement of endocrine regulatory hormones. Perinatal lethality precludes analysis of metabolic phenotypes in adult Nedd4l knockout mice [18], but a conditional Nedd4l knockout model was recently reported [16]. It will be interesting to investigate other possible metabolic roles of NEDD4L isoforms in liver in vivo in Nedd4l knockout mice to help determine why mutations in human NEDD4L loci are associated with risk for type 2 diabetes [19], obesity [20] and diabetic nephropathy [21].
Methods
Ethics statement
All animal experiments in this study were approved by Animal Welfare Committee of the University of Texas Health Science Center at Houston (HSC-AWC-11-095) following all current NIH guidelines for animal care and welfare.
Primary hepatocytes
Primary mouse hepatocytes were isolated from C57Bl6/J mice (Jackson Laboratories) by the modified collagenase method as described [35]. Briefly, mice were anesthetized using isoflurane and the liver was perfused via the descending vena cava with Hank’s balanced salt solution (HBSS, 5.33 mM KCl, 0.44 mM KH2PO4, 138 mM NaCl, 4.2 mM NaHCO3, 0.34 mM Na2HPO4, 5.6 mM glucose) containing 50 mM HEPES, 5 mM EGTA, pH 7.4, and then with HBSS containing 50 mM HEPES, 5 mM CaCl2 and 100 U/mL collagenase type IV (Sigma, C5138) at a rate of 2 ml/min. Livers were removed, and cells were dissociated and washed with HBSS. This method typically yields ~70% viable hepatocytes, which were plated at 1x105 viable cells/cm2 in plating medium (M199, 10% FBS (v/v), 2 mM glutamine, 100 nM dexamethasone, 100 U/mL penicillin, and 100 µg/mL streptomycin). After cell attachment (3 h), the medium was replaced by fresh maintenance medium (M199, 2 mM glutamine, 100 U/mL penicillin, 100 µg/mL streptomycin). The day after plating, primary hepatocytes were transfected with 5 nM scrambled unspecific (si-Scr) or Nedd4l-specific siRNA duplexes (Sigma, sequences in Table S1) with Lipofectamine 2000 (Life Technologies) or were infected with adenovirus (~20 PFU/cell) encoding GFP (Ad-GFP), ACREB (Ad-ACREB) [3], unspecific shRNA (Ad-USi) or CRTC2-selective shRNA (Ad-CRTC2i) [2], yielding 100% infection based on co-expressed GFP in all viruses. Experiments were performed 24 h after transfection or infection.
Animal tissue
Wild-type 8-12 week-old male C57Bl6/J mice maintained on normal chow diet were fed ad libitum, fasted for 6 or 16 h (overnight, ‘O/N’), or fasted for 16 h and re-fed with normal chow for 2 or 6 h prior to euthanization by CO2 inhalation with immediate removal of the liver tissue. Liver tissue was rinsed quickly in cold 1X PBS, snap frozen in LN2, and stored at -80°C until use. For extraction of protein and mRNA, liver tissue was pulverized with a mortar and pestle under LN2 and homogenized in the appropriate lysis buffer on ice using a rotor-stator prior to further purification.
Plasmids
The Nedd4l-short promoter region (-538 to +321) was amplified from mouse genomic DNA by PCR and cloned into the HindIII-XhoI restriction sites of pXP2 to create Nedd4l- luciferase. CRE sites were mutated by quick-change mutagenesis using PfuTurbo (Agilent): CRE1 (-412 to -407 TGACG to CTAGA) and/ or CRE2 (+196 to +201 CGTCA to CATGG). Oligonucleotide primer sequences are listed in Table S1. CMV-driven Flag-ACREB [36] and RSV-β-galactosidase plasmids were gifts of Dr. Marc Montminy.
Promoter analysis
Nedd4l promoter sequences and transcript data were taken from the Ensembl database (v71) using the Mus musculus GRCm38 assembly [22]. Transcript Nedd4l-201 (referred to in this study as Nedd4l-short because it is predicted to encode the short protein isoform) initiates from an alternate exon downstream of exon 1 of Nedd4l-202 (Figure 1A). Nedd4l-201 comprises 30 exons, contains 8,212 nucleotides and is predicted to encode an 855 amino acid protein (NEDD4L-short). Nedd4l-202 (referred to in this study as Nedd4l-long) comprises 31 exons, contains 8,163 nucleotides and is predicted to encode a 976 amino acid protein (NEDD4L-long). Promoter sequences were further analyzed using the DNAStar Lasergene suite. Nedd4l-201 (Nedd4l-short) promoter numbering is based on the transcription start site (+1) annotated in the Ensembl v71 assembly.
Luciferase assays
HEK293T cells (ATCC) were transfected with promoter luciferase expression constructs, Rous sarcoma virus LTR-driven (RSV) β-galactosidase plasmid, and expression constructs (pZeo-ACREB) or empty vector controls with Lipofectamine 2000 for 24 hours. Cells were stimulated with DMSO vehicle or a mixture of forskolin (FSK, 10 µM, EMD) and 1-isobutyl-3-methylxanthine (IBMX, 18 µM, Sigma) for 6 h. Luciferase and β-galactosidase activities were determined as described [37]. Luciferase activity was normalized to β-galactosidase activity, represented as fold change to vehicle-treated, wild type Nedd4l-short luciferase control or as arbitrary units (A.U.).
Analysis of proteins
Whole cell extracts were prepared from cells and tissues in ice-cold modified RIPA-T buffer, sonicated, clarified by centrifugation and protein concentration determined [34]. For cell fractionation, hepatocytes were washed with cold PBS and lysed in hypotonic lysis buffer (50 mM HEPES pH 7.4, 10 mM NaF, 1 mM EDTA, 0.5% NP-40, 0.25M sucrose, 0.5 mM DTT with protease inhibitors) followed by 30 dounces in a glass homogenizer with a B-type pestle. Nuclei were pelleted (1,200xg, 5 min 4°C), washed several times in hypotonic lysis buffer, and lysed in nuclear extraction buffer (50 mM HEPES pH 7.4, 420 mM NaCl, 10 mM NaF, 1 mM EDTA, 0.5 mM DTT with protease inhibitors) followed by homogenization with a plastic pestle. Final fractions were clarified at 14,000xg, 30 min 4°C. Extracts were boiled, resolved on SDS-PAGE gels, and then transferred to PVDF membrane for western blot and detection by ECL. Antibodies: anti-NEDD4L (Cell Signaling, 4013), anti-CRTC2 (Epitomics, #3565-1), HSP90 (Santa Cruz, sc-7947). Western blots were quantified by densitometry (ImageJ) on unsaturated films; signals normalized to loading control in arbitrary units, expressed as fold change of control.
Gene expression
Total RNA was extracted from hepatocytes or liver tissue with on-column DNAse digestion (5 PRIME) and cDNA prepared by MMLV reverse transcriptase (Invitrogen) using a Nedd4l gene-specific RT primer and oligo(dT)20 primer in the same reaction. To quantify the mRNAs encoding the two Nedd4l isoforms, a gene-specific RT primer common to both Nedd4l isoforms was used for cDNA production followed by real-time PCR for long or short Nedd4l isoforms using exon-specific primers. For Nedd4l-short, both forward and reverse primers recognize the first exon, which is absent in the long form. For Nedd4l-long, qPCR primers amplify a region encoded by exons 1 and 2 that is absent in the short isoform. Relative mRNA abundance was determined by real-time PCR with SYBR green detection as described [38], normalized to Gapdh internal control, expressed as fold change of control averaged over multiple experiments or experimental replicates, as indicated. See Table S1 for primer sequences.
Chromatin immunoprecipitation
Primary mouse hepatocytes were stimulated for 30 min with vehicle or glucagon (100 nM), crosslinked and neutralized as described [39]. Chromatin in cell lysates was sonicated to an average size of 500 bp using a Covaris S220 UltraSonicator. 1 mg of crosslinked extract was incubated with 2 µg/mL normal rabbit IgG (Cell Signaling #2729) or rabbit anti-CREB IgG (Cell Signaling #9197) at 4°C O/N prior to capture on Protein G Dynabeads (Life Technologies). Beads were washed extensively, crosslinks reversed and protein digested with Proteinase K, and chromatin purified on columns (Zymo Research). Recovery of specific genomic regions (Nedd4l-short CRE2 +126 to +271, positive control Pepck CRE site -96 to +14, and negative control Gapdh +453 to +620) was determined by quantitative real-time PCR with relative quantification compared with input fractions or by visualization of PCR products on agarose gels.
Glucose output assay
Glucose output was determined as described [40]. Primary mouse hepatocytes were washed twice with warm PBS and once with glucose-free DMEM. Cells were incubated with phenol red-free, glucose-free DMEM, supplemented with 20 mM sodium lactate + 2 mM sodium pyruvate or 20 mM alanine for 3 h with or without 100 nM glucagon. Glucose concentration in the medium was measured enzymatically [41] by incubation in 150 mM HEPES, 15 mM MgCl2, 3 mM EDTA, 2.5 mM NADP, 2.5 mM ATP, 2.5 U/ml glucose-6-phosphate dehydrogenase, 5 U/ml hexokinase for 10 min, room temperature. Glucose concentration was calculated based on A340 relative to a standard curve and normalized to protein content (BCA assay).
Glycogen synthesis assay
Glycogen synthesis was determined as described [32,42]. siRNA-transfected primary hepatocytes were stimulated with FBS (10% v/v), high glucose (20 mM) and insulin (50 nM) 3 h and harvested in modified-RIPA buffer; KOH was added to a final concentration of 5M and samples were boiled at 95°C for 2 h. 100% ethanol was added to a final concentration of 80% (v/v) to precipitate glycogen overnight at -20°C. Glycogen was pelleted by centrifugation (15,000xg, 15 min 4°C), pellets dried and suspended in 50 mM sodium acetate, 50 mM acetic acid, pH 4.7. Glycogen was digested to glucose by amyloglucosidase (100 U/mL, Sigma #10115), and glucose was quantified as described above. Glycogen concentration was calculated based on a standard curve, normalized to protein content in the original extract.
Lipogenesis assay
Lipogenesis was stimulated in control or siRNA transfected primary hepatocytes cultured in maintenance medium (low glucose control) or supplemented with 20 mM glucose, 50 nM insulin, 10% FBS (v/v) [43] for 3 days, then fixed in 10% formalin (v/v) for 5 min, washed with PBS and 60% isopropyl alcohol (v/v), and stained with fresh Oil Red O solution for 30 min. Images were captured on a Nikon brightfield microscope with Nikon NIS Elements software.
Statistical analyses
Results are reported as mean ±stdev. Differences between groups were considered significant at p<0.05 by two-tailed Student’s t-test. Data shown are average values of biological replicates within one experiment or averages of independent experiments. Animal data represent biological replicates (independent animals) tested in the same experiment.
Supporting Information
NEDD4L regulation in primary hepatocytes. (A) Quantification of NEDD4L isoform protein abundance from western blots shown in Figures 1C and 2 additional experiments, represented as mean fold induction ±stdev at each time point, *p<0.05 to 0 h control. (B) Time course of Pepck and Pgc1α mRNA expression in primary hepatocytes treated with glucagon (100 nM); representative of three independent experiments. (C) NEDD4L isoform protein levels in primary hepatocytes treated with indicated doses of glucagon for 3 h (n=3). (D) NEDD4L protein abundance from Figure 1E, normalized to HSP90, expressed mean fold change from ad lib, ±stdev, *p<0.05 to ad lib. (E) NEDD4L in cytoplasm/ membrane and nuclear fractions in primary hepatocytes treated with glucagon (100 nM, 3h) (n=3). HSP90 and LAMIN A show relative purity of cytoplasmic and nuclear fractions and equivalent loading. Filled arrowheads, NEDD4L-short; open arrowheads, NEDD4L-long.
(EPS)
Inhibition of CREB activity in primary hepatocytes. (A) pXP2-luciferase and pXP2-Nedd4l-short luciferase activity in HEK293T cells treated with vehicle (veh, DMSO) or FSK/ IBMX for 6 h, expressed in arbitrary units (A.U., luminescence/ β-galactosidase activity) ±stdev (n=2 experiments in triplicate), **p<0.01 compared to veh. (B) Luciferase assay data shown in Figure 2A expressed in arbitrary units (A.U.) ±stdev (n=3 experiments in triplicate), *p<0.01 between veh and FSK/ IBMX treated; ‡p<0.01 to FSK/ IBMX-treated WT; †p<0.01 to veh-treated WT. (C) Genomic DNA recovered in chromatin immunoprecipitates (input, IgG IP or CREB IP) from primary mouse hepatocytes as in Figure 2B. The promoter proximal CRE sites of Nedd4l-short (CRE2) and Pepck and an intragenic region of Gapdh were amplified by PCR, analyzed on an agarose gel. (D-F) Primary mouse hepatocytes were infected with adenovirus (Ad) encoding GFP or ACREB, treated with glucagon for the indicated time. (D) Western blots of Flag-ACREB and GAPDH loading control. (E) Sik1 mRNA normalized to Gapdh (mean fold change ±stdev over GFP-infected control). n>3 samples among three independent experiments. (F) Quantification of NEDD4L isoform protein abundance from western blots shown in Figure 2D and two additional experiments, normalized to HSP90, represented as mean fold change of uninfected control, ± stdev. *p< 0.05.
(EPS)
Lipogenesis in NEDD4L-deficient hepatocytes. Oil Red O (neutral lipid) staining in (A) untransfected primary mouse hepatocytes cultured in control medium [low glucose (5 mM)] or lipogenic medium [high glucose (25 mM), FBS (10% v/v), insulin (50 nM)] and (B) siRNA-transfected hepatocytes cultured in lipogenic medium. No visible effect of siRNAs (n=2). Bar, 100 µm.
(EPS)
Sequences of oligonucleotide primers and siRNAs.
(DOCX)
Acknowledgments
We thank Christopher Robb and Courtney Leiter for technical assistance, Dr. Marc Montminy (Salk Institute) for generous gifts of plasmids and adenovirus, and Drs. Marc Dionne and Jeffrey Frost for helpful discussions.
Funding Statement
Research reported in this publication was supported by the National Institutes of Diabetes and Digestive and Kidney Diseases (R01DK092590, P30DK056338) and Arthritis and Musculoskeletal and Skin Diseases (R01AR059847). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Altarejos JY, Montminy M (2011) CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat Rev Mol Cell Biol 12: 141-151. doi: 10.1038/nrm3072. PubMed: 21346730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Koo SH, Flechner L, Qi L, Zhang X, Screaton RA et al. (2005) The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 437: 1109-1111. doi: 10.1038/nature03967. PubMed: 16148943. [DOI] [PubMed] [Google Scholar]
- 3. Herzig S, Long F, Jhala US, Hedrick S, Quinn R et al. (2001) CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 413: 179-183. doi: 10.1038/35093131. PubMed: 11557984. [DOI] [PubMed] [Google Scholar]
- 4. Herzig S, Hedrick S, Morantte I, Koo SH, Galimi F et al. (2003) CREB controls hepatic lipid metabolism through nuclear hormone receptor PPAR-gamma. Nature 426: 190-193. doi: 10.1038/nature02110. PubMed: 14614508. [DOI] [PubMed] [Google Scholar]
- 5. Canettieri G, Koo SH, Berdeaux R, Heredia J, Hedrick S et al. (2005) Dual role of the coactivator TORC2 in modulating hepatic glucose output and insulin signaling. Cell Metab 2: 331-338. doi: 10.1016/j.cmet.2005.09.008. PubMed: 16271533. [DOI] [PubMed] [Google Scholar]
- 6. Zhang X, Odom DT, Koo SH, Conkright MD, Canettieri G et al. (2005) Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci U S A 102: 4459-4464. doi: 10.1073/pnas.0501076102. PubMed: 15753290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhao X, Feng D, Wang Q, Abdulla A, Xie XJ et al. (2012) Regulation of lipogenesis by cyclin-dependent kinase 8-mediated control of SREBP-1. J Clin Invest 122: 2417-2427. doi: 10.1172/JCI61462. PubMed: 22684109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yu J, Deng R, Zhu HH, Zhang SS, Zhu C et al. (2013) Modulation of fatty acid synthase degradation by concerted action of p38 MAP kinase, E3 ligase COP1, and SH2-tyrosine phosphatase Shp2. J Biol Chem 288: 3823-3830. doi: 10.1074/jbc.M112.397885. PubMed: 23269672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Uno M, Honjoh S, Matsuda M, Hoshikawa H, Kishimoto S et al. (2013) A fasting-responsive signaling pathway that extends life span in C. elegans. Cell Rep 3: 79-91. doi: 10.1016/j.celrep.2012.12.018. PubMed: 23352664. [DOI] [PubMed] [Google Scholar]
- 10. Molusky MM, Li S, Ma D, Yu L, Lin JD (2012) Ubiquitin-specific protease 2 regulates hepatic gluconeogenesis and diurnal glucose metabolism through 11beta-hydroxysteroid dehydrogenase 1. Diabetes 61: 1025-1035. doi: 10.2337/db11-0970. PubMed: 22447855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu Y, Dentin; Liu Y, Dentin R, Chen D, Hedrick S, Ravnskjaer K, Schenk S, Milne J, Meyers DJ, Cole P, Yates J, Olefsky J, Guarente L, Montminy MR, Chen D, Hedrick S, Ravnskjaer K, et al (2008) A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange. Nature 456: 269-273. doi: 10.1038/nature07349. PubMed: 18849969. 10.1038/nature07349 PubMed: 18849969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kamynina E, Debonneville C, Bens M, Vandewalle A, Staub O (2001) A novel mouse Nedd4 protein suppresses the activity of the epithelial Na+ channel. FASEB J 15: 204-214. doi: 10.1096/fj.00-0191com. PubMed: 11149908. [DOI] [PubMed] [Google Scholar]
- 13. Snyder PM, Olson DR, Kabra R, Zhou R, Steines JC (2004) cAMP and serum and glucocorticoid-inducible kinase (SGK) regulate the epithelial Na(+) channel through convergent phosphorylation of Nedd4-2. J Biol Chem 279: 45753-45758. doi: 10.1074/jbc.M407858200. PubMed: 15328345. [DOI] [PubMed] [Google Scholar]
- 14. Yang B, Kumar S (2010) Nedd4 and Nedd4-2: closely related ubiquitin-protein ligases with distinct physiological functions. Cell Death Differ 17: 68-77. doi: 10.1038/cdd.2009.84. PubMed: 19557014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shi PP, Cao XR, Sweezer EM, Kinney TS, Williams NR et al. (2008) Salt-sensitive hypertension and cardiac hypertrophy in mice deficient in the ubiquitin ligase Nedd4-2. Am J Physiol Renal Physiol 295: F462-F470. doi: 10.1152/ajprenal.90300.2008. PubMed: 18524855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ronzaud C, Loffing-Cueni D, Hausel P, Debonneville A, Malsure SR et al. (2013) Renal tubular NEDD4-2 deficiency causes NCC-mediated salt-dependent hypertension. J Clin Invest 123: 657-665. PubMed: 23348737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Van Huysse JW, Amin MS, Yang B, Leenen FH (2012) Salt-induced hypertension in a mouse model of Liddle syndrome is mediated by epithelial sodium channels in the brain. Hypertension 60: 691-696. doi: 10.1161/HYPERTENSIONAHA.112.193045. PubMed: 22802227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Boase NA, Rychkov GY, Townley SL, Dinudom A, Candi E et al. (2011) Respiratory distress and perinatal lethality in Nedd4-2-deficient mice. Nat Commun 2: 287. doi: 10.1038/ncomms1284. PubMed: 21505443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hasstedt SJ, Highland HM, Elbein SC, Hanis CL, Das SK (2013) Five linkage regions each harbor multiple type 2 diabetes genes in the African American subset of the GENNID Study. J Hum Genet 58: 378-383. doi: 10.1038/jhg.2013.21. PubMed: 23552671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang YL, Liang HY, Gao YH, Wu XJ, Chen X et al. (2013) A Functional Variant of NEDD4L Is Associated with Obesity and Related Phenotypes in a Han Population of Southern China. Int J Mol Sci 14: 7433-7444. doi: 10.3390/ijms14047433. PubMed: 23549273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McDonough CW, Bostrom MA, Lu L, Hicks PJ, Langefeld CD et al. (2009) Genetic analysis of diabetic nephropathy on chromosome 18 in African Americans: linkage analysis and dense SNP mapping. Hum Genet 126: 805-817. doi: 10.1007/s00439-009-0732-8. PubMed: 19690890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Flicek P, Ahmed I, Amode MR, Barrell D, Beal K et al. (2013) Ensembl 2013. Nucleic Acids Res 41: D48-D55. doi: 10.1093/nar/gks1236. PubMed: 23203987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Itani OA, Campbell JR, Herrero J, Snyder PM, Thomas CP (2003) Alternate promoters and variable splicing lead to hNedd4-2 isoforms with a C2 domain and varying number of WW domains. Am J Physiol Renal Physiol 285: F916-F929. PubMed: 12876068. [DOI] [PubMed] [Google Scholar]
- 24. Wang J, Peng Q, Lin Q, Childress C, Carey D et al. (2010) Calcium activates Nedd4 E3 ubiquitin ligases by releasing the C2 domain-mediated auto-inhibition. J Biol Chem 285: 12279-12288. doi: 10.1074/jbc.M109.086405. PubMed: 20172859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Le Lay J, Tuteja G, White P, Dhir R, Ahima R et al. (2009) CRTC2 (TORC2) contributes to the transcriptional response to fasting in the liver but is not required for the maintenance of glucose homeostasis. Cell Metab 10: 55-62. doi: 10.1016/j.cmet.2009.06.006. PubMed: 19583954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang Y, Inoue H, Ravnskjaer K, Viste K, Miller N et al. (2010) Targeted disruption of the CREB coactivator Crtc2 increases insulin sensitivity. Proc Natl Acad Sci U S A 107: 3087-3092. doi: 10.1073/pnas.0914897107. PubMed: 20133702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Debonneville C, Flores SY, Kamynina E, Plant PJ, Tauxe C et al. (2001) Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na(+) channel cell surface expression. EMBO J 20: 7052-7059. doi: 10.1093/emboj/20.24.7052. PubMed: 11742982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gao S, Alarcón C, Sapkota G, Rahman S, Chen PY et al. (2009) Ubiquitin ligase Nedd4L targets activated Smad2/3 to limit TGF-beta signaling. Mol Cell 36: 457-468. doi: 10.1016/j.molcel.2009.09.043. PubMed: 19917253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Persaud A, Alberts P, Amsen EM, Xiong X, Wasmuth J et al. (2009) Comparison of substrate specificity of the ubiquitin ligases Nedd4 and Nedd4-2 using proteome arrays. Mol Syst Biol 5: 333 PubMed: 19953087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mayr B, Montminy M (2001) Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol 2: 599-609. doi: 10.1038/35085068. PubMed: 11483993. [DOI] [PubMed] [Google Scholar]
- 31. Salway JG (2004) Metabolism at a Glance. Malden, MA: Blackwell Publishing, Ltd. p. 125. [Google Scholar]
- 32. Fleig WE, Enderle D, Steudter S, Nöther-Fleig G, Ditschuneit H (1987) Regulation of basal and insulin-stimulated glycogen synthesis in cultured hepatocytes. Inverse relationship to glycogen content. J Biol Chem 262: 1155-1160. PubMed: 3100527. [PubMed] [Google Scholar]
- 33. Qi H, Grenier J, Fournier A, Labrie C (2003) Androgens differentially regulate the expression of NEDD4L transcripts in LNCaP human prostate cancer cells. Mol Cell Endocrinol 210: 51-62. doi: 10.1016/j.mce.2003.08.009. PubMed: 14615060. [DOI] [PubMed] [Google Scholar]
- 34. Stewart R, Akhmedov D, Robb C, Leiter C, Berdeaux R (2013) Regulation of SIK1 abundance and stability is critical for myogenesis. Proc Natl Acad Sci U S A 110: 117-122. doi: 10.1073/pnas.1212676110. PubMed: 23256157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dentin; Dentin R, Pégorier JP, Benhamed F, Foufelle F, Ferré P, Fauveau V, Magnuson MA, Girard J, Postic CR, Pegorier JP, Benhamed F, Foufelle F, Ferre P, et al (2004) Hepatic glucokinase is required for the synergistic action of ChREBP and SREBP-1c on glycolytic and lipogenic gene expression. J Biol Chem 279: 20314-20326. doi: 10.1074/jbc.M312475200. PubMed: 14985368. 10.1074/jbc.M312475200 PubMed: 14985368 [DOI] [PubMed] [Google Scholar]
- 36. Ahn S, Olive M, Aggarwal S, Krylov D, Ginty DD et al. (1998) A dominant-negative inhibitor of CREB reveals that it is a general mediator of stimulus-dependent transcription of c-fos. Mol Cell Biol 18: 967-977. PubMed: 9447994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Conkright MD, Canettieri G, Screaton R, Guzman E, Miraglia L et al. (2003) TORCs: transducers of regulated CREB activity. Mol Cell 12: 413-423. doi: 10.1016/j.molcel.2003.08.013. PubMed: 14536081. [DOI] [PubMed] [Google Scholar]
- 38. Stewart R, Flechner L, Montminy M, Berdeaux R (2011) CREB is activated by muscle injury and promotes muscle regeneration. PLOS ONE 6: e24714. doi: 10.1371/journal.pone.0024714. PubMed: 21931825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Berdeaux R, Goebel N, Banaszynski L, Takemori H, Wandless T et al. (2007) SIK1 is a class II HDAC kinase that promotes survival of skeletal myocytes. Nat Med 13: 597-603. doi: 10.1038/nm1573. PubMed: 17468767. [DOI] [PubMed] [Google Scholar]
- 40. Miller RA, Chu Q, Le Lay J, Scherer PE, Ahima RS et al. (2011) Adiponectin suppresses gluconeogenic gene expression in mouse hepatocytes independent of LKB1-AMPK signaling. J Clin Invest 121: 2518-2528. doi: 10.1172/JCI45942. PubMed: 21606593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Passonneau JV, Lowry OH (1993) Enzymatic Analysis: A Practical Guide. New Jersey, USA: The Humana Press Inc. [Google Scholar]
- 42. Sun C, Zhang F, Ge X, Yan T, Chen X et al. (2007) SIRT1 improves insulin sensitivity under insulin-resistant conditions by repressing PTP1B. Cell Metab 6: 307-319. doi: 10.1016/j.cmet.2007.08.014. PubMed: 17908559. [DOI] [PubMed] [Google Scholar]
- 43. Osawa Y, Seki E, Kodama Y, Suetsugu A, Miura K et al. (2011) Acid sphingomyelinase regulates glucose and lipid metabolism in hepatocytes through AKT activation and AMP-activated protein kinase suppression. FASEB J 25: 1133-1144. doi: 10.1096/fj.10-168351. PubMed: 21163859. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
NEDD4L regulation in primary hepatocytes. (A) Quantification of NEDD4L isoform protein abundance from western blots shown in Figures 1C and 2 additional experiments, represented as mean fold induction ±stdev at each time point, *p<0.05 to 0 h control. (B) Time course of Pepck and Pgc1α mRNA expression in primary hepatocytes treated with glucagon (100 nM); representative of three independent experiments. (C) NEDD4L isoform protein levels in primary hepatocytes treated with indicated doses of glucagon for 3 h (n=3). (D) NEDD4L protein abundance from Figure 1E, normalized to HSP90, expressed mean fold change from ad lib, ±stdev, *p<0.05 to ad lib. (E) NEDD4L in cytoplasm/ membrane and nuclear fractions in primary hepatocytes treated with glucagon (100 nM, 3h) (n=3). HSP90 and LAMIN A show relative purity of cytoplasmic and nuclear fractions and equivalent loading. Filled arrowheads, NEDD4L-short; open arrowheads, NEDD4L-long.
(EPS)
Inhibition of CREB activity in primary hepatocytes. (A) pXP2-luciferase and pXP2-Nedd4l-short luciferase activity in HEK293T cells treated with vehicle (veh, DMSO) or FSK/ IBMX for 6 h, expressed in arbitrary units (A.U., luminescence/ β-galactosidase activity) ±stdev (n=2 experiments in triplicate), **p<0.01 compared to veh. (B) Luciferase assay data shown in Figure 2A expressed in arbitrary units (A.U.) ±stdev (n=3 experiments in triplicate), *p<0.01 between veh and FSK/ IBMX treated; ‡p<0.01 to FSK/ IBMX-treated WT; †p<0.01 to veh-treated WT. (C) Genomic DNA recovered in chromatin immunoprecipitates (input, IgG IP or CREB IP) from primary mouse hepatocytes as in Figure 2B. The promoter proximal CRE sites of Nedd4l-short (CRE2) and Pepck and an intragenic region of Gapdh were amplified by PCR, analyzed on an agarose gel. (D-F) Primary mouse hepatocytes were infected with adenovirus (Ad) encoding GFP or ACREB, treated with glucagon for the indicated time. (D) Western blots of Flag-ACREB and GAPDH loading control. (E) Sik1 mRNA normalized to Gapdh (mean fold change ±stdev over GFP-infected control). n>3 samples among three independent experiments. (F) Quantification of NEDD4L isoform protein abundance from western blots shown in Figure 2D and two additional experiments, normalized to HSP90, represented as mean fold change of uninfected control, ± stdev. *p< 0.05.
(EPS)
Lipogenesis in NEDD4L-deficient hepatocytes. Oil Red O (neutral lipid) staining in (A) untransfected primary mouse hepatocytes cultured in control medium [low glucose (5 mM)] or lipogenic medium [high glucose (25 mM), FBS (10% v/v), insulin (50 nM)] and (B) siRNA-transfected hepatocytes cultured in lipogenic medium. No visible effect of siRNAs (n=2). Bar, 100 µm.
(EPS)
Sequences of oligonucleotide primers and siRNAs.
(DOCX)