

Background: Class-18A myosins share a unique N-terminal extension comprising a PDZ module and a KE-rich region.
Results: Human myosin-18A binds F-actin via its motor domain in a nucleotide-dependent manner and via the KE-rich region, modulated by direct interaction between the PDZ module and GOLPH3.
Conclusion: Myosin-18A binds F-actin and recruits interaction partners to the cytoskeleton.
Significance: This work establishes a molecular basis for myosin-18A mediated membrane-cytoskeleton interplay.
Keywords: Actin, Cytoskeleton, Myosin, Protein Domains, Protein-Protein Interactions
Abstract
Molecular motors of the myosin superfamily share a generic motor domain region. They commonly bind actin in an ATP-sensitive manner, exhibit actin-activated ATPase activity, and generate force and movement in this interaction. Class-18 myosins form heavy chain dimers and contain protein interaction domains located at their unique N-terminal extension. Here, we characterized human myosin-18A molecular function in the interaction with nucleotides, F-actin, and its putative binding partner, the Golgi-associated phosphoprotein GOLPH3. We show that myosin-18A comprises two actin binding sites. One is located in the KE-rich region at the start of the N-terminal extension and appears to mediate ATP-independent binding to F-actin. The second actin-binding site resides in the generic motor domain and is regulated by nucleotide binding in the absence of intrinsic ATP hydrolysis competence. This core motor domain displays its highest actin affinity in the ADP state. Electron micrographs of myosin-18A motor domain-decorated F-actin filaments show a periodic binding pattern independent of the nucleotide state. We show that the PDZ module mediates direct binding of myosin-18A to GOLPH3, and this interaction in turn modulates the actin binding properties of the N-terminal extension. Thus, myosin-18A can act as an actin cross-linker with multiple regulatory modulators that targets interacting proteins or complexes to the actin-based cytoskeleton.
Introduction
Myosins constitute a large superfamily of molecular motors that use the chemical energy provided by ATP hydrolysis to cyclically interact with filamentous F-actin and generate force and movement (1). All myosins share a generic motor domain that harbors the binding sites for ATP and F-actin. Based on sequence alignments of the motor domain, myosins can be grouped into 35 classes (2). In humans, 40 genes are found that encode for myosins from 13 of these classes. The molecular details of the mechanochemical transduction of energy by myosins from different classes have been unraveled with great accuracy. Nevertheless, most myosins have not been characterized in depth, in particular members of the myosin family, such as class-18 myosins, which show distinct structural features setting them apart. Class-18 myosins are found in various species, from vertebrates to arthropods. They contain protein interaction domains that are located at their N terminus outside the motor domain (3). Like the founding member of this myosin class, MysPDZ (now termed mouse myosin-18A), human myosin-18A comprises a region rich in lysine and glutamate residues (KE) and a PDZ3 module in its N-terminal extension. This domain is followed by a generic motor domain with an adjacent neck domain that can bind essential and regulatory light chains (4). The tail domain contains long stretches of coiled-coils that support heavy chain dimerization (5). The molecular mass of the protein varies between 180 kDa for the shortest isoform, which lacks the N-terminal extension (myosin-18Aβ), and 233 kDa for the longest isoform, termed myosin-18Aα. A recent study identified the gene encoding human myosin-18A to be alternatively spliced in non-small cell lung cancer, leading to in-frame variations in the protein sequence (6). Furthermore, the gene was identified as a partner in the three-way chromosomal translocation of stem cell leukemia-lymphoma syndrome (7) and forms the fusion gene MYO18A-PDGFRB in eosinophilia-associated atypical myeloproliferative neoplasms (8). A three-way translocation of the highly promiscuous oncogene MLL, a histone methyltransferase, and the reciprocal partner gene MYO18A was described in acute myeloid leukemia (9). These studies suggest that functional myosin-18A protein is required for the normal regulation of the cell cycle and the suppression of key processes involved in cancer progression.
Up to now, information on the biochemical properties of class-18 myosins is scarce and in parts controversial. The function of the unique N-terminal extension is only poorly understood (10, 11). In a recent study, Drosophila myosin-18 has been found to be an actin-binding protein that does not bind nucleotide, has no ATPase activity, and cannot actively translocate over actin filaments (12). A current publication of the same group on functional features of mouse myosin-18A reports actin and nucleotide binding properties but no significant ATPase activity and suggests that this myosin is not a traditional motor (4). Moreover, the amino acid sequence of active site elements of the myosin-18 motor domain exhibits changes in highly conserved regions, which can prevent myosin-18 from productively interacting with ATP in the same way as other myosins.
Furthermore, it has been shown that the N-terminal extension of human myosin-18A has an ATP-insensitive actin-binding site outside the PDZ module (5). It was suggested that the motor domain of human myosin-18A does not bind to actin, because YFP-tagged motor domain constructs obtained from cell lysates do not cosediment with actin. This observation is in contrast to the studies on Drosophila myosin-18 and mouse myosin-18A that attribute actin binding properties to the motor domain.
Recently, it has been shown that myosin-18Aα is a novel binding partner of the PAK2·βPIX·GIT1 complex (13). This suggests that myosin-18A may play an important role in regulating epithelial cell migration. The Rac/Cdc42-binding kinase MRCK (myotonic dystrophy kinase-related Cdc42-binding kinase) has been shown to associate with the myosin-18Aα PDZ domain via a linker protein, LRAP35a (14). The resulting phosphorylation of the non-muscle RLC2A (myosin-2A regulatory light chain) suggests an association of myosin-18Aα with RLC2A. This hypothesis is supported by the fact that in vitro mouse myosin-18A binds essential and regulatory light chains via its neck region (4). The tripartite MRCK·LRAP35a·myosin-18Aα complex localizes to lamellar actomyosin bundles, where non-muscle myosin-2A drives the retrograde flow (14, 15). Therefore, myosin-18A can play a role the regulation and organization of the actin cytoskeleton within lamellipodia.
Additionally, Dippold et al. (16) have shown that GOLPH3 binds to myosin-18A and connects the Golgi apparatus to F-actin to provide a tensile force required for efficient tubule and vesicle formation. However, this function would presumably implicate active motor properties for myosin-18A.
Here, we show that human myosin-18A contains two distinct actin binding sites per heavy chain (four per dimer), one of which is regulated by nucleotide binding and is capable of targeting interacting proteins to the actin cytoskeleton, where it can function as an efficient and adjustable actin cross-linker. The PDZ module is shown to mediate direct binding of myosin-18A to GOLPH3, and this interaction modulates the actin binding properties of the unique N-terminal extension of myosin-18A.
EXPERIMENTAL PROCEDURES
Reagents
Standard chemicals, TRITC-phalloidin, and anti-FLAG antibody were purchased from Sigma-Aldrich; restriction enzymes, polymerases and DNA-modifying enzymes were purchased from MBI-Fermentas and Roche Applied Science.
Plasmid Construction
The full-length cDNA clone IRATp790A0771D (imaGenes GmbH, Berlin; GenBankTM entry BC039612.1) of human myosin-18A was used as a template for the PCR amplification of DNA fragments encoding for the constructs used in this study. Sequences encoding for the N-terminal subdomains KE (amino acids 1–219; upstream primer, GCGGATCCATGGCTAATGCTCCCTCCTGC; downstream primer, GCTCGAGCCGGAGGGTAGGTGGGGGCAG), PDZ (amino acids 220–311; upstream primer, GCGGATCCGAGCTGGAGCTGCAACGACGG; downstream primer, GCTCGAGTGGAATGGGCTGCACCTTGAG), and KEPDZ (amino acids 1–398; upstream primer as for KE; downstream primer, GCTCGAGCTTCTCAACGTCATCCTCATCC) were amplified by PCR, digested with BamHI and XhoI, and subcloned into the plasmid pET23a(+) (Novagen) for the expression of C-terminal His-tagged proteins.
The sequence coding for human GOLPH3 was amplified using the full-length cDNA clone IRAUp969C1265D (imaGenes GmbH, Berlin; GenBankTM entry BC012123.1) as a template for PCR and subcloned into the plasmids pET23a(+) (upstream primer, GCGGATCCATGACCTCGCTGACCCAGCGCAGC; downstream primer, GCTCGAGCTTGGTGAACGCCGCCACCACC) and pGEX-6P2 (upstream primer as for pET23a(+); downstream primer, GCTCGAGTTACTTGGTGAACGCCGCCACCACC), respectively.
For the generation of human myosin-18A motor domain constructs, the DNA sequences encoding for amino acids 399–1185 (M18A-MD; upstream primer, GCGGATCCATGGCTAATGCTCCCTCCTGC; downstream primer, GAAGCTTTTATTTATCATCATCATCTTTATAATCTGTACATAATGCATCCCGCTGCTCCTCTAGACGTGC) or 220–1185 (PDZ-M18A-MD; upstream primer, GCGGATCCGAGCTGGAGCTGCAACGACGGCCC; downstream primer as for M18A-MD) were amplified by PCR, digested with BamHI and HindIII, and subcloned into the plasmid pFastBacDual (Invitrogen). The downstream primers introduce C-terminal FLAG tags (DYKDDDDK) to facilitate purification. All plasmids were confirmed by sequencing.
Protein Production and Purification
Plasmids for the production of the N-terminal subdomains of Homo sapiens myosin-18A and the H. sapiens GOLPH3 protein were transformed into Escherichia coli Rosetta pLys-S cells (Merck). Cells were grown at 30 °C in LB medium (10 g of peptone, 10 g of yeast extract, and 5 g of NaCl per liter, pH 7.0), induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside at A600 = 0.6, grown at 21 or 30 °C for 3–16 h, and harvested by centrifugation (4 °C, 4000 × g).
For the purification of His-tagged proteins, the cells were resuspended in buffer (50 mm HEPES, pH 8.0, 300 mm NaCl, 10 mm imidazole, Complete inhibitor mixture (Roche), 1 mg/ml lysozyme, 1000 units of benzonase, 12 mm MgCl2, 2 mm ATP, 3 mm benzamidine, 1 mm TCEP, 0.02% NaN3). After sonication and the addition of 1% Triton X-100, cell lysis was performed at 4 °C for 30 min. Following centrifugation at 20,000 × g at 4 °C (Avanti J-30I, Beckmann Coulter), the supernatant was applied to a nickel-nitrilotriacetic acid column (Qiagen, Hilden, Germany). The resin was washed with buffer (50 mm HEPES, pH 8.0, 300 mm NaCl, 20 mm imidazole, 3 mm benzamidine, 1 mm TCEP, 0.02% NaN3), and the bound protein was eluted with a linear gradient of this buffer with 500 mm imidazole. The protein-containing fractions were dialyzed against storage buffer (50 mm HEPES, pH 8.0, 300 mm NaCl, 3 mm benzamidine, 1 mm TCEP, 0.02% NaN3), concentrated, and applied to a gel filtration column (HiLoad 16/600 Superdex 200 pg; GE Healthcare). The eluted pure protein was supplemented with 3% sucrose, flash-frozen in liquid nitrogen, and stored at −80 °C.
The GST fusion construct of GOLPH3 was expressed in E. coli Rosetta pLys-S cells. Cells were grown at 30 °C in LB medium, induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside at A600 = 0.6, grown at 30 °C for 3 h, and harvested by centrifugation. Cell lysis was performed at 4 °C for 30 min in buffer (50 mm Tris-HCl, pH 7.5, 300 mm NaCl, 12 mm MgCl2, 1 mm benzamidine, Complete inhibitor mixture, 1 mg/ml lysozyme, 1000 units of benzonase, 2 mm ATP, 1 mm PMSF, 1 mm TCEP) and lysed with Triton X-100 (1% final concentration). After centrifugation at 20,000 × g at 4 °C, the supernatant was applied to a glutathione-Sepharose column and washed with buffer A (50 mm Tris-HCl, pH 7.5, 300 mm NaCl, 2 mm MgCl2, 1 mm benzamidine, 1 mm TCEP). A linear gradient of buffer A and buffer B (buffer A containing 10 mm reduced glutathione) eluted the protein-containing fractions. GST-fused PreScission protease was added to the protein solution that subsequently was dialyzed against storage buffer (50 mm Tris, pH 7.5, 200 mm NaCl, 1 mm EDTA, 1 mm benzamidine, 1 mm TCEP). Cleaved GST and PreScission protease were removed by passing the protein solution over a glutathione-Sepharose column, and the pure GOLPH3 protein was concentrated, snap-frozen in liquid nitrogen, and stored at −80 °C.
The myosin-18A motor domain constructs (M18A-MD and PDZ-M18A-MD) were overproduced in the baculovirus/Sf9 system. The corresponding transfer vector was transformed in DH10Bac E. coli cells to generate recombinant bacmid, which was isolated and transfected in Sf9 insect cells using Cellfectin II (Invitrogen). Recombinant baculovirus was produced as described by the manufacturer. Sf9 cells were infected with recombinant baculovirus, collected 66 h postinfection, and stored at −80 °C. For purification, cells were lysed in buffer (50 mm HEPES (pH 7.3), 300 mm KCl, 3 mm MgCl2, 0.1 mm EGTA, 2 mm ATP, 5 mm β-mercaptoethanol, 5 mm benzamidine, Complete inhibitor mixture (Roche Applied Science)) by sonication and incubation at 4 °C for 30 min. The lysate was ultracentrifuged (138,000 × g, 1 h), and the supernatant was applied to αFLAG-M2 affinity gel (Sigma-Aldrich) and rotated for 2 h at 4 °C to ensure binding. The resin was transferred to a column and washed with ATP buffer (50 mm HEPES (pH 7.5), 300 mm KCl, 0.5 mm ATP, 0.1 mm EGTA, 3 mm MgCl2, 3 mm benzamidine, 0.2% Triton X-100), wash buffer 1 (50 mm HEPES (pH 7.5), 300 mm KCl, 0.1 mm EGTA, 3 mm MgCl2, 3 mm benzamidine), and wash buffer 2 (50 mm HEPES (pH 7.5), 600 mm KCl, 0.1 mm EGTA, 3 mm MgCl2, 3 mm benzamidine). The protein was eluted with wash buffer 1 containing 0.1 mg/ml FLAG peptide and dialyzed against storage buffer (50 mm HEPES (pH 7.5), 300 mm KCl, 0.5 mm EDTA, 0.2 mm EGTA, 1 mm MgCl2, 1 mm benzamidine, 1 mm DTT, 3% trehalose). The pure protein was concentrated, flash-frozen in liquid nitrogen, and stored at −80 °C.
Rabbit skeletal muscle actin was purified as described by Lehrer and Kerwar (17). For selected experiments, F-actin was also stabilized by the addition of equimolar concentrations of phalloidin, because a perturbation of the interaction has been described for some unconventional myosins (18). In all actin interaction assays, no significant differences were observed when using phalloidin-stabilized instead of non-treated actin filaments.
Kinetic Measurements
ATPase activities were measured at 25 °C with the NADH-coupled assay as described previously (19). Transient kinetic experiments were performed at 20 °C with either a Hi-tech Scientific SF-61 DX single mixing stopped-flow system (TgK Scientific Ltd., Bradford on Avon, UK) or an Applied Photophysics PiStar 180 instrument in assay buffer (20 mm MOPS (pH 7.0), 100 mm KCl, 5 mm MgCl2, 1 mm DTT) using procedures and kinetic models described previously (Scheme 1) (20, 21). Error bars represent S.E. Mant-nucleotides (Jena Bioscience) were excited at 365 nm and detected after passing through a KV 389-nm cut-off filter.
SCHEME 1.
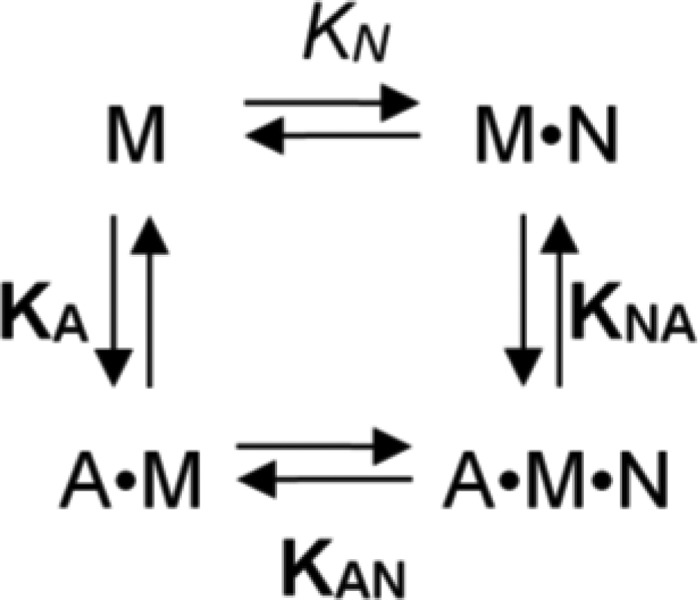
Interaction scheme for actin and nucleotide binding of myosin. A, actin; M, myosin; N, nucleotide (T, ATP; D, ADP). For the equilibrium binding constants, a notation is used that distinguishes between constants in the absence and presence of actin by using italic type (kx, Kx) and boldface type (kx, Kx), respectively.
F-Actin Cosedimentation Assays
The interaction of purified myosin-18A subdomains with F-actin was assayed by cosedimentation. 0.2–2 μm protein was incubated on ice for 15 min in assay buffer (20 mm MOPS (pH 7.0), 100 mm KCl, 5 mm MgCl2, 1 mm TCEP) with varying concentrations of F-actin in the presence or absence of nucleotides. The absence of Mg2+ATP or Mg2+ADP was guaranteed by the addition of 5 mm EDTA and 1 unit/ml apyrase. The samples were subjected to ultracentrifugation for 20 min at 170,000 × g in a Beckman TLA-120.1 rotor (4 °C unless otherwise specified). The pellets were resuspended in assay buffer to the same volume as the supernatants, and both fractions were resolved by SDS-PAGE. Protein content in the supernatants and pellets was quantified by densitometric analysis of Coomassie Blue-stained gels using the program ImageJ (National Institutes of Health, Bethesda, MD). To determine actin affinities, the fraction of bound protein in the pellet relative to the total protein content was plotted as a function of actin concentration, and a hyperbolic function was fitted to the data. In cases where the utilized myosin construct concentration exceeds the affinity, a standard quadratic equation was used to analyze the actin binding data as described previously (22–25). Averaged data of replicate binding experiments using three different preparations are given in the figures. Error bars represent S.E. All data analysis was performed with Origin 7.0 (Originlab).
Differential Static Light Scattering
Protein stability was assayed by following protein aggregation associated with heat denaturation (26). To do so, protein solutions were diluted to a concentration of 0.2 mg/ml in a buffer containing 20 mm MOPS (pH 7.0), 500 mm KCl, 5 mm MgCl2, 1 mm TCEP and heated in steps of 0.6 °C. Static light scattering was measured at each temperature step, and the resulting transition curve was fitted by a sigmoidal function. The transition midpoint gives a measure for the aggregation temperature induced by protein denaturation.
Microscale Thermophoresis
Protein interaction studies using microscale thermophoresis were performed according to Duhr, Braun, and co-workers (27, 28). Protein was labeled according to NanoTemper using the MonolithTM NT.115 protein labeling kit RED-NHS (amine-reactive). Experiments were performed using standard capillaries in the NanoTemper MonolithTM NT instrument for red dye fluorescence in buffer (25 mm HEPES (pH 7.3), 25 mm KCl, 5 mm MgCl2, 1 mm DTT). For high salt conditions, 25 mm KCl was replaced by 300 mm NaCl. Error bars represent S.E.
Negative Stain Electron Microscopy
Electron micrographs of negatively stained complexes of F-actin decorated with the motor domain of myosin-18A were obtained as follows: H. sapiens myosin-18A-MD was incubated with F-actin in EM-buffer (20 mm HEPES (pH 7.0), 10 mm KCl, 5 mm MgCl2; optional: 5 mm ADP or ATP), adsorbed to freshly glow-discharged carbon-coated grids at a concentration of 0.1 mg/ml (1.1 μm M18A-MD and equimolar F-actin), and negatively stained with 0.75% uranyl formate. The specimen was visualized with a Jeol JEM-1400 transmission electron microscope (Jeol, Tokyo, Japan) at 120 kV.
Bioinformatics
The alignment of the motor domain sequence of Dictyostelium discoideum myosin-2 (amino acids 1–765) with human (amino acids 1–1185) and mouse (amino acids 1–1180) myosin-18A and Drosophila melanogaster myosin-18 (amino acids 1–1319) was performed with the ClustalW algorithm as implemented in MegAlign 5.07 in the DNAStar software package. The homology model of the human myosin-18A motor domain (residues 399–1185 of myosin-18Aα) was prepared using the I-TASSER protein structure and function prediction server (29, 30) with the D. discoideum myosin-2 motor domain as a template (31) (Protein Data Bank code 1G8X). Figures were prepared with PyMOL (version 1.5.0.4 Schrödinger, LLC).
RESULTS
Protein Expression and Purification
To study biochemical properties and protein interaction features of the different human myosin-18A domains, the respective regions were expressed separately as discrete proteins (Fig. 1A). Expression of the human myosin-18A motor domain (M18A-MD) in the baculovirus/Sf9 system was 0.5–1 mg/liter of Sf9 cells at a density of 1.6 × 106 cells/ml. The purity of M18A-MD following anti-FLAG affinity chromatography was >95% based on Coomassie-stained SDS-polyacrylamide gels (cf. Fig. 4A). To ensure proper folding of the motor domain construct, temperature-dependent unfolding of the protein was analyzed by differential static light scattering (data not shown). The transition process is highly cooperative, and the transition temperature (TM = 47.6 ± 0.9 °C) is consistent with values previously reported for myosin (32, 33). The motor domain construct including the preceding PDZ module, PDZ-M18A-MD, was expressed in considerably lower quantities (<0.1 mg/liter of Sf9 cells). The N-terminal subdomain constructs of myosin-18A, KEPDZ, KE, and PDZ and the human GOLPH3 protein were expressed in E. coli at 2–10 mg/liter of bacteria culture. The purity of the respective proteins after nickel-nitrilotriacetic acid affinity and size exclusion chromatography was 90–95%, as estimated from Coomassie-stained SDS-polyacrylamide gels.
FIGURE 1.
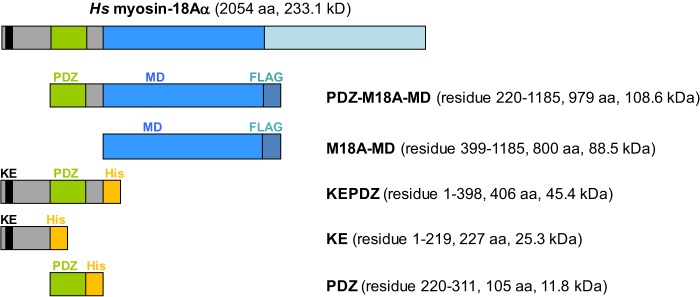
Constructs used in this study. The schematic depicts the modular structure of the myosin-18A constructs investigated in this study. The abbreviated construct names, number of amino acids, and calculated molecular masses are listed. The motor domain constructs PDZ-M18A-MD and M18A-MD are expressed in the baculovirus/Sf9 system and purified via a C-terminal FLAG tag, whereas the N-terminal extension constructs are produced in E. coli and purified via C-terminal His tags. Hs, H. sapiens.
FIGURE 4.
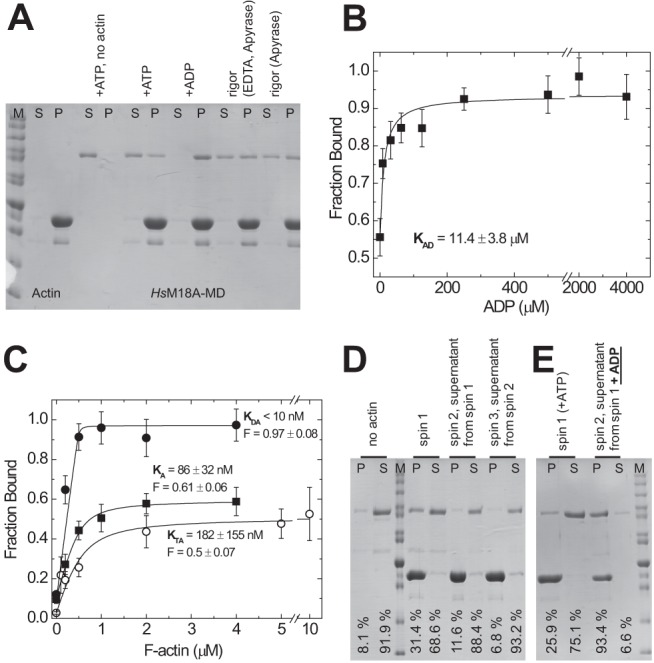
Interaction of H. sapiens myosin-18A-MD with F-actin in different nucleotide states. The interaction of M18A-MD with F-actin was investigated by cosedimentation assays. A, F-actin binding ability of M18A-MD in different nucleotide states. 0.5 μm M18A-MD was incubated with ATP or ADP (5 mm each) or in the presence of apyrase (with and without EDTA to deplete Mg2+) and ultracentrifuged in the absence or presence of F-actin (4 μm). The resulting supernatant (S) and pellet (P) fractions were resolved by SDS-PAGE. M, protein standard. B, affinity of acto-M18A-MD to ADP. Cosedimentation assays of 0.5 μm M18A-MD and 4 μm F-actin at increasing ADP concentrations were conducted as in A, and the resulting M18A-MD amount in supernatant and pellet was determined by densitometry. The fraction of M18A-MD bound to F-actin was calculated, and the data were plotted against ADP concentration. A hyperbolic fit yields an ADP affinity for acto-M18A-MD of KAD = 11.4 ± 3.8 μm. C, determination of actin affinity of M18A-MD in the presence of 5 mm ATP (○) or 5 mm ADP (●) and in the absence of nucleotide (■; rigor conditions). Densitometric analysis of supernatant and pellet after ultracentrifugation was used to determine the fraction of 0.5 μm M18A-MD bound to F-actin as a function of F-actin concentration in the presence or absence of nucleotides. Quadratic fits to the data give actin affinities and allow estimation of the maximum F-actin-bound fraction (F) of M18A-MD. D, sequential actin rebinding experiment. Lanes 1 and 2, pellet (P) and supernatant (S) of 1 μm M18A-MD centrifuged at high speed in the absence of F-actin. spin 1, pellet and supernatant of 1 μm M18A-MD sedimented in the presence of 4 μm F-actin and 1 mm ATP. spin 2, the supernatant from spin 1, containing 1 mm ATP, was mixed with 4 μm F-actin and sedimented again to yield supernatant and pellet fractions. spin 3, the supernatant from spin 2 was repeatedly treated as mentioned before. This experiment was performed at 25 °C, and the setups were incubated for 1 h at 25 °C before centrifugation. E, actin rebinding experiment with nucleotide exchange. Lanes 1 and 2, pellet and supernatant of 1 μm M18A-MD sedimented in the presence of 2 μm F-actin and 100 μm ATP. Lanes 3 and 4, the supernatant from the first sedimentation (lane 2) was mixed with 2 μm F-actin and 1 mm ADP and sedimented again. The relative amount of M18A-MD in the pellet and supernatant fractions is indicated for each lane. All equilibrium constants are summarized in Table 1. Error bars, S.E.
Steady-state ATPase Activity of the Myosin-18A Motor Domain
To explore the ability of M18A-MD to hydrolyze ATP, we performed NADH-coupled ATPase assays in the absence and presence of F-actin (Fig. 2). We did not detect any significant basal ATPase activity for the motor domain construct, M18A-MD, or the motor domain including the preceding PDZ domain, PDZ-M18A-MD. Furthermore, ATPase activity could not be detected in the presence of up to 60 μm F-actin for both motor domain constructs. These data imply that the myosin-18A motor domain has no intrinsic ATPase activity. Nevertheless, a specific allosteric trigger different from actin could be required to switch on its enzymatic activity. Therefore, we investigated if the addition of purified KE or KEPDZ domain constructs (1 μm) or purified GOLPH3 protein (up to 10 μm) to M18A-MD or PDZ-M18A-MD stimulates ATPase activity. Neither of these possible intra- and intermolecular binding partners activated myosin-18A motor domain ATPase; nor did variations in pH and ionic strength over a broad range (data not shown).
FIGURE 2.
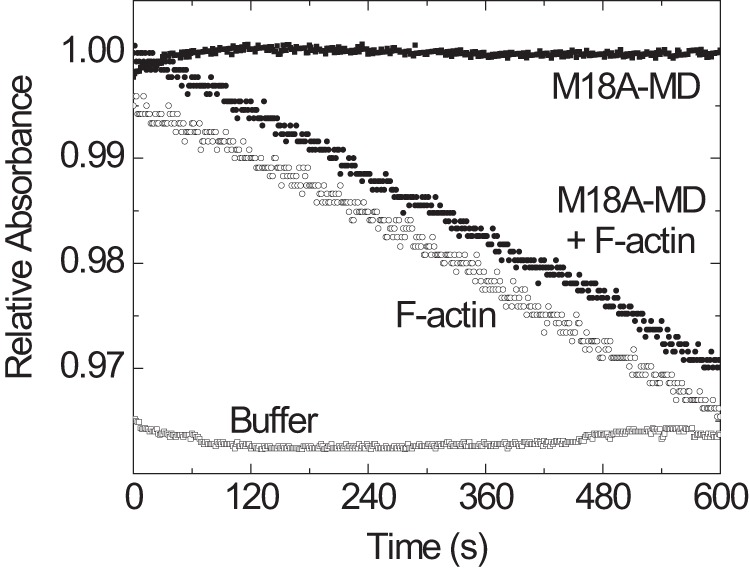
ATPase activity of the myosin-18A motor domain. The ability of M18A-MD to hydrolyze ATP was assayed using an NADH-coupled detection system with 1 mm ATP. No significant time-dependent decrease in absorbance in the presence of 2.6 μm M18A-MD (■) was detected in comparison with buffer alone (□). The linear decline in absorbance that is generated by the ATPase activity of 10 μm F-actin (○) is not further increased by the presence of 2.6 μm M18A-MD (●), implying that there is no actin-mediated initiation of M18A-MD ATPase.
ATP and ADP Binding to the Motor Domain of Myosin-18A
We sought to elucidate if the myosin-18A motor domain binds nucleotide. The absence of a reliable intrinsic signal that reports nucleotide binding prompted us to test the extrinsic fluorescence signal of mant-coupled nucleotides to sense binding in stopped-flow transient kinetic measurements.
Rapid mixing of 0.5 μm M18A-MD with increasing concentrations of mant-ATP in the range of 1–10 μm (all postmix concentrations) resulted in a ∼1% increase in mant fluorescence (Fig. 3A, inset). The observed transients were best fit by two exponentials (relative amplitudes of 45 and 55%) with linearly increasing rate constants for both fast and slow phase (Fig. 3A). Linear fits yield apparent second order rate binding constants k+T of 1.8 ± 0.3 and 0.05 ± 0.01 μm−1 s−1 for the fast and slow phase, respectively. The y axis intercepts was used to estimate dissociation rate constants k−T of 5.2 ± 1.9 and 0.12 ± 0.08 s−1. From the ratio of k−T/k+T, affinity constants KT of 2.9 and 2.4 μm were calculated (Table 1). In a similar experimental setup, the binding of mant-ADP to M18A-MD was assayed. Here, a comparable 0.6–1% mant fluorescence increase was detected upon mixing that was best described by a double exponential function with relative amplitudes of 63 and 27% for the fast and slow rates, respectively (Fig. 3B, inset). Linear fits to the data from the fast and slow phase were used to calculate an apparent second order rate binding constant for ADP k+D of 1.5 ± 0.1 and 0.29 ± 0.02 μm−1 s−1, respectively (Fig. 3B). The y axis intercepts gave dissociation rate constants k−D of 4.2 ± 0.5 and 0.14 ± 0.1 s−1. The calculated values for ADP affinity KD correspond to 2.8 and 0.5 μm (Table 1). To test if bound ATP can be exchanged by ADP and vice versa, we premixed M18A-MD with mant-labeled nucleotide and performed a chasing experiment with an excess of the other nucleotide (Fig. 3C). The observed transients can be described by single exponential functions in both cases and define dissociation rate constants k−T of 0.21 ± 0.07 s−1 and k−D of 0.18 ± 0.04 s−1. Although we were not able to resolve a fast phase in the chasing experiments, the obtained off-rates for the direct displacement are in good agreement with the values obtained from the y intercepts of the slow phase linear fits in Fig. 3, A and B. These data demonstrate the ability of the myosin-18A motor domain to bind and interchange ATP and ADP nucleotides.
FIGURE 3.

Transient kinetic analysis of the interaction of nucleotides with H. sapiens myosin-18A-MD. A, the addition of excess mant-ATP to M18A-MD causes a ∼1% increase in mant fluorescence that can be fitted with a double exponential function (inset; shown is the fluorescence transient after mixing 1/0.5 μm M18A-MD with 10/5 μm mant-ATP, pre-/postmix concentrations). The observed rate constants kobs increase with increasing mant-ATP concentrations in the range from 1 to 10 μm, and the data can be fitted with a linear function. B, binding of mant-ADP to M18A-MD causes a fluorescence increase that could be fitted by two exponentials (inset; shown is the fluorescence transient resulting from mixing of 1/0.5 μm M18A-MD with 8/4 μm mant-ADP). The rate constants of the slow and fast phase are linearly dependent on mant-ADP concentration. C, fluorescence transients observed after chasing bound mant-ATP from M18A-MD with excess ADP (top trace, black line) and mant-ADP from M18A-MD with excess ATP (bottom trace, gray transient). Single exponential fits to the data define nucleotide release rate constants. All resulting kinetic parameters for nucleotide binding are summarized in Table 1. Error bars, S.E.
TABLE 1.
Summary of the rate and equilibrium constants of the interactions of the myosin-18A motor domain and N-terminal extension constructs with actin and nucleotides
Experimental conditions were as follows: 25 mm MOPS, 100 mm KCl, 5 mm MgCl2, 1 mm DTT, pH 7.0.
| Constant | H. sapiens myosin-18A motor domain | KEPDZ | KE | Mus musculus myosin-18Aβ S1a | D. melanogaster myosin-18 motor domainb | |
|---|---|---|---|---|---|---|
| Nucleotide binding to myosin (fast/slow phase) | ||||||
| Mant-ATP | k+T (μm−1s−1) | 1.8 ± 0.3/0.05 ± 0.01 | 0.12 ± 0.01 | |||
| k−T (s−1)c | 5.2 ± 1.9/0.12 ± 0.08 | 10.16 ± 0.09 | ||||
| k−T (s−1)d | 0.21 ± 0.07 | NDe | ||||
| KT (μm) | 2.9/2.4 | 84.7 | ||||
| Mant-ADP | k+D (μm−1 s−1) | 1.5 ± 0.1/0.29 ± 0.02 | 0.10 ± 0.02 | |||
| k-D (s−1)f | 4.2 ± 0.5/0.14 ± 0.1 | 10.87 ± 0.16 | ||||
| k−D (s−1)g | 0.18 ± 0.04 | 10.3 ± 0.3 | ||||
| KD (μm) | 2.8/0.5 | 108.7 | ||||
| Nucleotide binding to acto-myosin | ||||||
| ADP coupling | KAD (μm) | 11.4 ± 3.8 | ||||
| KAD/KD | 4.1/22.8 | |||||
| Actin binding to myosin | ||||||
| Rigor | KA (μm) | 0.086 ± 0.032 | 0.99 ± 0.29 | 6.53 ± 4.1 | 47.9 ± 9.2 | 1.0 ± 0.2 |
| Fraction bound | 0.61 ± 0.06 | 1.0 ± 0.09 | 1.0 ± 0.27 | 0.248 ± 0.078 | 0.83 ± 0.05 | |
| ADP | KDA (μm) | <0.01 | ND | ND | ND | ND |
| Fraction bound | 0.97 ± 0.08 | |||||
| ATP | KTA (μm) | 0.182 ± 0.155 | 1.02 ± 0.24 | 3.92 ± 6.75 | 54.0 ± 29 | 1.0 ± 0.1 |
| Fraction bound | 0.5 ± 0.07 | 0.99 ± 0.1 | 0.87 ± 0.22 | 0.245 ± 0.071 | 0.81 ± 0.09 | |
Actin Binding Properties of the Myosin-18A Motor Domain
We made use of F-actin cosedimentation assays to test the actin binding ability of M18A-MD in different nucleotide states (Fig. 4A). In the absence of actin, the isolated motor domain (0.5 μm) was completely soluble under our assay conditions. When incubated with 4 μm F-actin at high ATP concentrations (5 mm) a fraction of 35% of M18A-MD was bound to actin (as determined by densitometry, described under “Experimental Procedures”). Under nucleotide-depleted rigor conditions, 44% of total M18A-MD remained unbound, whereas 56% was in the actin-bound state. In the ADP state, M18A-MD displayed the highest actin affinity and was almost completely bound to actin (97%; Fig. 4A). Essentially the same results were obtained using the motor domain construct including the N-terminal PDZ module, PDZ-M18A-MD. These data provide evidence that the protein binds nucleotide and couples the conformational information to the actin binding site. Because ADP binding seems to strengthen actin binding of M18A-MD, we analyzed its effect on F-actin cosedimentation in more detail. We found that the addition of increasing ADP concentrations to the cosedimentation assay (0.5 μm myosin, 4 μm F-actin) shifted the extent of bound M18A-MD from 56% in the absence of nucleotide to ∼95% at saturating ADP (Fig. 4B). Fitting of a hyperbola to the data defines the ADP affinity of acto-M18A-MD as KAD = 11.4 ± 3.8 μm (see Scheme 1 and legend for equilibrium binding constant notation). These data suggest a switching of myosin-18A motor domain conformation from a state that is compromised in actin binding to a fully actin binding-competent state upon ADP binding. The F-actin cosedimentation assay results (Fig. 4A) imply a partitioning of myosin-18A between an actin-bound and dissociated state depending on bound nucleotide. To analyze the actin affinity of M18A-MD in different nucleotide states in more detail, we mixed 0.5 μm M18A-MD with increasing concentrations of F-actin in the cosedimentation assay, and the fraction of bound myosin was plotted against actin concentration (Fig. 4C). In all cases, a standard quadratic equation was fitted to the data to determine dissociation equilibrium constants (cf. Scheme 1); in the absence of nucleotide, M18A-MD has an actin affinity of KA = 86 ± 32 nm that is only marginally changed in the presence of ATP with KTA = 182 ± 155 nm. In the ADP state, the highest actin affinity could be observed with KDA < 10 nm. To test the possibility of a slow equilibrium between an actin binding-competent state of M18A-MD and a state that is unable to bind actin, we performed sequential actin rebinding experiments in the presence of 1 mm ATP (Fig. 4D). In contrast to the mouse isoform, the human myosin-18A motor domain does not re-equilibrate to give the same distribution between incompetent and competent actin binding states. In contrast, the M18A-MD fraction that is incompetent to bind F-actin preserves this state even after 1 h of incubation at 25 °C. Essentially the same result was observed in the absence of nucleotide (data not shown). Nevertheless, the fraction of bound M18A-MD is influenced by the nucleotide state. As shown in Fig. 4A, only about 50–60% of the myosin binds to actin in the presence of ATP or in the absence of nucleotide, whereas ADP binding enables complete fractional binding of myosin-18A to actin. Therefore, we assayed the ability of the actin binding-incompetent fraction of M18A-MD to gain actin binding competence upon the addition of ADP (Fig. 4E). The supernatant of an F-actin cosedimentation assay in the presence of 100 μm ATP (75.1%) was mixed with F-actin and excess (1 mm) ADP and subjected to a second sedimentation. The fraction of bound M18A-MD redistributed to 93.4% in the ADP-bound state. All data are summarized in Table 1.
Electron Microscopic Investigation of Actin Filament Decoration by the Myosin-18A Motor Domain
The results described above suggest an actin binding mechanism of M18A-MD where a moderate actin affinity in the ATP or nucleotide-free state is associated with a partitioning between a binding and a non-binding population. The presence of ADP in the nucleotide binding pocket increases actin affinity and supports full fractional actin binding. We investigated the decoration of F-actin filaments with M18A-MD in different nucleotide states by electron microscopy. We observed a well ordered decoration of most actin filaments in the absence of nucleotide, which is the strong actin binding state for most myosins characterized so far and is generally referred to as the rigor state (Fig. 5A). Also, in the presence of ADP, the actin filaments were completely decorated with M18A-MD (Fig. 5B). Remarkably, M18A-MD also bound to actin in the presence of excess ATP (Fig. 5C). This is in strong contrast to most myosins characterized so far with the exception of myosin-9 (23). The electron micrographs clearly show filaments with the typical arrowhead appearance and confirm the actin binding ability of the myosin-18A motor domain in all nucleotide states.
FIGURE 5.

Representative electron micrographs of negatively stained complexes of F-actin decorated with myosin-18A motor domains. A–C, M18A-MD in its nucleotide-free (A), ADP (B), and ATP state (C). The electron micrographs clearly show actin filaments with bound M18A-MD indicated by the typical arrowhead appearance and prove that the myosin-18A motor domain can bind to actin independent of its nucleotide state. Scale bars, 70 nm.
Actin Binding Properties of the N-terminal KEPDZ Domain
The unique N-terminal extension of human myosin-18A contains a region rich in lysine and glutamate (KE) and a PDZ domain and has previously been shown to interact with F-actin (5). We wanted to investigate the actin binding that is mediated by the N terminus of myosin-18A more precisely and performed cosedimentation assays to quantify actin affinities (Fig. 6). A construct encompassing the complete N-terminal extension (KEPDZ) binds to actin with an affinity of KA = 0.99 ± 0.29 μm (Table 1). We also analyzed the effect of ATP on this interaction and found that it is not influenced by the presence of nucleotide (KTA = 1.02 ± 0.24 μm; data not shown). Next, we explored the actin interaction of a smaller subdomain containing the KE-rich region and a “middle” region but lacking the PDZ module (KE; cf. Fig. 1). This construct displays a weaker actin affinity of KA = 6.53 ± 4.1 μm that is also not influenced by ATP (KTA = 3.92 ± 6.75 μm; data not shown). The fraction of actin-bound protein plateaus at 100% for both constructs, indicating complete actin binding. Because the actin affinity of the N terminus of myosin-18A is increased in the presence of the PDZ module, we tested if this domain itself has actin binding properties. Fig. 6 shows that the isolated PDZ module does not bind to F-actin.
FIGURE 6.
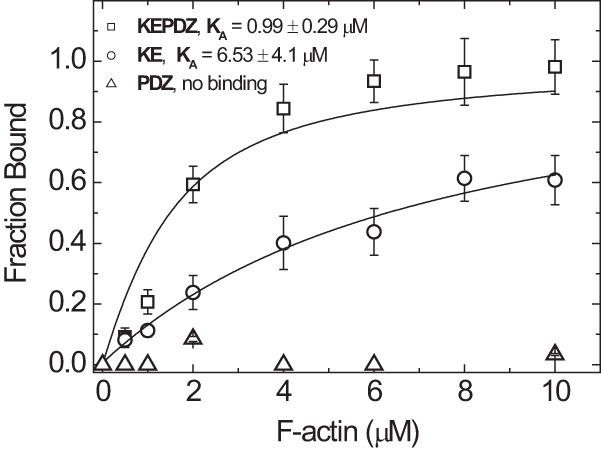
Binding of N-terminal extension constructs of H. sapiens myosin-18A to F-actin. Cosedimentation of F-actin with N-terminal extension constructs of myosin-18A (1 μm) was performed as described in the legend to Fig. 4 and analyzed by fitting a quadratic equation to the data. Direct F-actin binding of the construct KEPDZ was observed with intermediate affinity of KA,KEPDZ = 0.99 ± 0.29 μm. The isolated KE-rich domain has a lower actin affinity of KA,KE = 6.53 ± 4.1 μm and approaches complete binding only at higher actin concentrations. These affinities are not influenced by the presence of ATP (KTA,KEPDZ = 1.02 ± 0.24 μm and KTA,KE = 3.92 ± 6.75 μm; data not shown). The isolated PDZ module does not cosediment with F-actin and thus has no actin binding properties. Error bars, S.E.
Interaction of the PDZ Module with H. sapiens GOLPH3
PDZ modules are known to mediate protein-protein interactions in signaling complexes (34, 35). An interaction of the Golgi phosphoprotein GOLPH3 with myosin-18A has been suggested based on cellular studies (16). We utilized the construct encoding for the N-terminal extension of myosin-18A (KEPDZ) as well as the full-length human GOLPH3 protein to characterize the binding domain and affinity in more detail. Initial cosedimentation assays of F-actin and KEPDZ in the presence of GOLPH3 showed that KEPDZ binds to both actin and GOLPH3 simultaneously as it directs the tripartite complex to the pellet after high speed centrifugation (Fig. 7A). We further tested the influence of GOLPH3 binding to KEPDZ on the actin affinity of the N-terminal extension of human myosin-18A. Quantified cosedimentation assays (as in Fig. 6) showed a modulation of the KEPDZ-actin affinity by GOLPH3 binding. The actin affinity is 5-fold increased from 1.18 ± 0.2 to 0.22 ± 0.08 μm in the presence of GOLPH3 (Fig. 7B). In control experiments, GOLPH3 was completely soluble under the assay conditions and did not bind to F-actin. To analyze and quantify the direct interaction of KEPDZ and GOLPH3, we took advantage of microscale thermophoresis (27, 28). This newly developed method allows the determination of the binding affinity between a fluorescently labeled and a non-labeled protein. We titrated Alexa-647-labeled KEPDZ protein with increasing concentrations of GOLPH3. The observed GOLPH3 concentration-dependent decrease in the thermophoresis fluorescence amplitude (Fig. 7C) was fitted with a hyperbola to give an apparent equilibrium binding constant Kapp of 0.62 ± 0.12 μm at low salt conditions. In the presence of high salt concentrations (300 mm NaCl), the binding affinity was decreased to 22.5 ± 2.9 μm. This result confirms a direct binding event between GOLPH3 and the N-terminal extension of myosin-18A. To test if the isolated PDZ module is sufficient to mediate binding of myosin-18A to GOLPH3, we repeated the thermophoresis experiment with purified fluorescently labeled PDZ protein (Fig. 7D). The results show that the PDZ module binds to GOLPH3 but with lower affinity (4.96 ± 0.78 μm at low salt conditions) than the complete N terminus (KEPDZ construct).
FIGURE 7.

Interaction of the N-terminal extension construct KEPDZ of H. sapiens myosin-18A with H. sapiens GOLPH3. A, cosedimentation of F-actin with KEPDZ (1 μm) in the presence of a fixed molar excess of 4 μm GOLPH3 and subsequent analysis of the resulting supernatant (S) and pellet (P) fractions by SDS-PAGE. At low actin concentrations (0.25 μm; lanes 2 and 3), both KEPDZ and GOLPH3 are soluble and are mainly found in the supernatant. At high actin concentrations (4.5 μm; lanes 4 and 5), KEPDZ is exclusively found in the pellet, and a significant fraction of GOLPH3 cosediments with KEPDZ. B, to investigate the influence of GOLPH3 on the actin affinity of KEPDZ, cosedimentation assays like those in Fig. 6 were repeated in the presence (4 μm fixed concentration) and absence of GOLPH3 and quantified accordingly by densitometry. The data were analyzed by quadratic approximation as in Fig. 6. The actin affinity of KEPDZ (KA,KEPDZ = 1.18 ± 0.2 μm) is 5-fold increased to KA,KEPDZ+GOLPH3 = 0.22 ± 0.08 μm in the presence of 4 μm GOLPH3. Densitometric analysis of the respective SDS gel suggests a 1:1 binding stoichiometry for the GOLPH3·KEPDZ complex. GOLPH3 does not bind to F-actin in the absence of KEPDZ. C, microscale thermophoresis measurements were used to quantify the interaction of fluorescently labeled KEPDZ with human GOLPH3 protein. A fixed concentration of 0.156 μm Alexa-647-labeled KEPDZ was titrated with 100 μm to 0.6 nm H. sapiens GOLPH3 at a salt concentration of 25 mm (■), and the respective relative thermophoresis fluorescence amplitude was plotted against H. sapiens GOLPH3 concentration. By fitting a hyperbola to the data, an equilibrium binding constant of 0.62 ± 0.12 μm could be determined. In the presence of 300 mm salt (●), the binding affinity was decreased to 22.5 ± 2.9 μm. D, microscale thermophoresis measurements of fluorescently labeled PDZ with human GOLPH3 as in C at low salt conditions. A hyperbolic fit defines an equilibrium binding constant of 4.96 ± 0.78 μm. Error bars, S.E.
DISCUSSION
In the present study, we aimed to elucidate the molecular properties of three fundamental human myosin-18A functional domains: the KE-rich region and the PDZ module, which together constitute the N-terminal extension, as well as the generic myosin motor domain. We established the soluble expression and purification of different constructs, using two expression systems; a minimal construct encompassing the core motor domain (M18A-MD) and a motor domain construct including the N-terminally located PDZ module (PDZ-M18A-MD) were successfully produced in the Sf9/insect cell system. The N-terminal extension construct (KEPDZ) as well as the separate KE-rich region (KE) and the PDZ module (PDZ) were produced in E. coli and purified to homogeneity. In addition, we expressed the phosphoprotein GOLPH3 as a GST fusion protein in E. coli and utilized protease cleavage to obtain a “tag-free” version for interaction studies. Using these constructs, we studied the biochemical properties of the individual myosin-18A domains to assemble an overall picture of the protein's molecular mechanism.
We show that the core myosin-18A motor domain binds mant-labeled ATP and ADP nucleotides but does not exhibit intrinsic basal or actin-activated ATPase activity. Nucleotide binding modulates the actin affinity of the motor domain and regulates its partitioning between an actin binding-competent and -incompetent state. The ADP-bound state has the highest actin affinity, and complete fractional binding of the myosin-18A motor domain to the actin filament is only observed in this state. These results are supported by the fact that in electron micrographs, we observe complete decoration of F-actin filaments with the typical arrowhead appearance of the myosin motor domain in all nucleotide states. Like generic myosin motors, the myosin-18A motor domain appears to dock with similar orientation and employing conserved surface contacts to the actin filament. Accordingly, the myosin-18A motor domain has preserved the myosin-inherent ability to bind nucleotide and to couple the conformational information from the nucleotide binding pocket to the actin binding site.
The core motor domain of myosin-18A, omitting the N-terminal extension, comprises 786 amino acids. It is considerably extended compared with human skeletal muscle myosin-2 (701 amino acids without the N-terminal SH3-like subdomain) or D. discoideum myosin-2 (685 amino acids without SH3). The underlying insertions are distributed over the nucleotide binding pocket elements (switch-2 region), the actin binding regions (cardiomyopathy loop (CM-loop) and activation loop), and prominent surface loops (near the SH2 domain) of the motor domain. We prepared a structural model of the motor domain using the I-TASSER protein structure and function prediction server (29, 30). The model shows the location of these inserts and their likely effect on conserved structural elements in the myosin motor domain (Fig. 8, A and B; alignment depicted in Fig. 8C).
FIGURE 8.

Structural model of the H. sapiens myosin-18A motor domain. The D. discoideum myosin-2 motor domain structure (Protein Data Bank code 1G8X) was used as a template to generate a homology model of the human myosin-18A motor domain (residues 399–1185 of the full-length sequence). A, the overall fold of the motor domain of human myosin-18A displays high similarity with generic myosin motor domains. Nevertheless, H. sapiens myosin-18A contains four major insertions in the motor domain sequence, which are located near switch-2 (pink; 6 residues), at the CM-loop (blue; 14 residues), at the activation loop (orange; 13 residues), and preceding the SH2 helix (yellow; 29 residues). ADP is shown in a stick representation with black carbon atoms; the orange sphere designates the location of the Mg2+ ion. B, close-up view of the nucleotide binding pocket. The molecule was subjected to a left-handed rotation of about 45º around a vertical axis through the Mg2+ ion. Important features of the binding pocket are colored as follows: cyan, P-loop (GSSGSGKT); red, switch-1 (NGNATR); light blue, switch-2 (DTPGFQ). C, multiple-sequence alignment of the motor domain (MD) of D. discoideum (Dd) myosin-2 and the motor domains of human (Hs) myosin-18A, mouse (Mm) myosin-18A, and D. melanogaster (Dm) myosin-18. Important myosin motor domain features are indicated and labeled. The gray shaded box marks Glu-459 (D. discoideum), which constitutes the salt bridge with Arg-238 in D. discoideum myosin-2.
The switch-2 region of myosin-18A displays an insertion of six residues, and the conserved glutamic acid in switch-2 is changed to glutamine (pink in Fig. 8, A and B). Previously, it has been shown that this residue plays a key role in the mechanism of chemo-mechanical coupling (36) and that mutation of this residue abolishes the cellular functions of myosin-2 in D. discoideum cells (19). Accordingly, the elongation of the switch-2 region in myosin-18A by a flexible linker (GGSARGA) and the disturbed salt bridge between switch-1 and switch-2 are possible structural determinants for the absent intrinsic ATPase activity of the motor domain.
Furthermore, the region preceding the SH2 domain contains a 29-residue surface loop extension (yellow in Fig. 8, A and B). This flexible loop is located near the entrance of the nucleotide binding pocket and comprises multiple serine residues. We observed biphasic ATP and ADP binding kinetics that may possibly be attributed to the existence of two structural states of this extended SH2 loop modulating nucleotide interactions and triggering slow and fast binding and release kinetics. This interpretation is supported by the fact that both slow and fast rates for ATP and ADP binding are dependent on nucleotide concentration. The biphasic nucleotide binding behavior has not been observed for the mouse myosin-18A S1 construct (4). Also, the overall nucleotide affinities of human M18A-MD are about 30 times higher than for the mouse isoform, and the ADP-mediated switching to full binding ability has not been described before. Because mouse and human isoforms of myosin-18A share complete amino acid sequence conservation in the SH2 loop extension, this is not the only determinant for the nucleotide binding properties of myosin-18A.
The CM-loop is located at the tip of the motor domain and involved in the actomyosin binding interface. Remarkably, in myosin-18A, the loop is considerably modified because it contains 14 additional amino acids (blue in Fig. 8A). This enables higher flexibility of the loop and alters the actin binding properties in comparison with generic myosin motor domains. A more flexible and elongated CM-loop might render the motor's actin affinity more independent from cleft closure and thereby permit high actin affinity that is less influenced by the nucleotide state of the active site than in other myosins.
The activation loop, which is part of the actin-myosin interface, is located within helix HR in the helix-loop-helix actin binding motif (orange in Fig. 8A) (37, 38). In human myosin-18A, this loop is elongated by 13 extra amino acids that introduce additional positive charges (three arginine residues instead of one in D. discoideum myosin-2), which interferes with regular activation by this loop (cf. Fig. 8C).
The actin affinities of the human myosin-18A motor domain in the absence and presence of nucleotide are 2 orders of magnitude stronger than for mouse myosin-18A S1 (Table 1). Accordingly, for the mouse isoform, no decoration of F-actin filaments could be observed by electron microscopic investigation. In contrast, M18A-MD shows the classic arrowhead decoration of F-actin filaments. The sequence comparison (Fig. 8C) of the actin binding regions (CM-loop and activation loop) displays only subtle differences between the two myosin isoforms. Nevertheless, in their study, Guzik-Lendrum et al. (4) utilized an S1 construct comprising a short N-terminal sequence preceding the motor domain and a neck region with bound essential and regulatory light chains. A previous study revealed differences for chicken skeletal muscle myosin S1 with wild-type versus truncated essential light chains in actin binding and ATPase activity, suggesting a direct interaction of the essential light chain N terminus with actin that is regulated by the SH3-like subdomain of myosin (39). Accordingly, one could speculate about a similar mechanism for myosin-18A, where the short N-terminal sequence of the mouse myosin-18A S1 construct used in the study of Guzik-Lendrum et al. (4) and the bound light chains modulate the actin (and maybe also nucleotide) affinity of myosin-18A.
The biochemical characterization of modules that reside in the large N-terminal extension of human myosin-18Aα provides information on the molecular function of this myosin in the cellular context. Two previous studies proposed ATP-independent actin binding for the N-terminal subdomain of myosin-18Aα (5, 11). In both studies, immunoprecipitation from human cell line-derived cell lysates showed an interaction of the N-terminal extension of myosin-18A with actin filaments. In expanding these studies, we utilized purified bacterially expressed N-terminal subdomain constructs to define binding affinities and saturation ratios for F-actin. We confirm that the actin binding site resides within the KE-rich subdomain (amino acids 1–219 of human myosin-18Aα) with intermediate affinity and full binding ability. Although the isolated PDZ module does not interact with actin, it significantly strengthens the actin affinity of the N-terminal extension. Remarkably, the direct interaction of GOLPH3 with the PDZ module leads to changes in the actin binding properties, and this interplay acts as a modulator for the KE motif-mediated cytoskeleton interaction. Likewise, the affinity of the PDZ module with GOLPH3 is significantly enhanced by the presence of the KE-rich region (Fig. 7, C and D). This further confirms an interdependent connection between the neighboring functional domains KE and PDZ. The observed independence of the binding affinities from ATP confirms the assumption of Isogawa et al. (5) that this interaction is ATP-insensitive. The N-terminal extension of myosin-18A could therefore be used by the protein to cross-link F-actin filaments to higher order complexes in a GOLPH3 binding-regulated fashion.
We determined the ADP affinity of myosin-18A in the actin-bound state (KAD) to be ∼12 μm, which is in the range of reported physiological concentrations: 12 μm MgADP in brain and 6 μm MgADP in resting muscle or 43 μm MgADP in muscle after heavy exercise (40); 40 to 140 μm (relaxed/contracted) free ADP in the cytosol of smooth muscle cells (41, 42). Within the cytosol, local high ADP concentrations are expected in areas of high ATP turnover, such as myosin filaments or near membranes were ion pumps are located. Transiently increased ADP levels may facilitate strong actin binding of all four binding sites of a dimeric myosin-18A molecule, resulting in stable cross-linking of F-actin filaments or an attenuation of myosin filament contractility. However, in all possible nucleotide states, a fraction of the motor domain is strongly bound to the actin filaments. It is therefore likely that in cellular conditions, a release factor is needed to detach myosin-18A from the actin cytoskeleton to allow redistribution.
When feeding all equilibrium binding constants of M18A-MD for actin and ADP into Scheme 1, the product should equal 1. Using the values determined in this study, we obtain a product of 0.029. This discrepancy suggests the presence of at least one additional step in the interaction of M18A-MD with actin and/or ADP that is not resolved by our experiments.
For human myosin-18A, a number of confirmed or putative binding partners have been described (13, 14, 16, 43, 44). They can in principle act as modulators of myosin-18A function or stimulate enzymatic activity when bound to the protein. Furthermore, the presence of the complete N-terminal extension may be necessary for motor domain catalytic function as for example in myosin-3A the presence of the N-terminal kinase domain happens to modulate the kinetics of the motor domain (45, 46). Nevertheless, Guzik-Lendrum et al. (4) report nucleotide and actin binding but no significant ATPase activity for a recombinant mouse myosin-18Aα motor construct, which contains the complete N-terminal domain.
In an attempt to characterize the interaction of myosin-18A with GOLPH3, we were able to prove the direct binding of the phosphoprotein to the N-terminal extension of myosin-18A. Because the affinity is ionic strength-dependent, we suppose that charge-charge interactions are responsible for high affinity binding. We could show a direct effect of GOLPH3 on myosin-18A function because its binding to the PDZ module enhances the actin affinity of the N-terminal extension. Moreover, the GOLPH3·myosin-18A complex serves as a junction between the Golgi membrane and the cytoskeleton and may thus be highly regulated, presumably by other associated proteins.
The motor domain of myosin-18A appears to have evolved to serve as an actin cross-linker, whose activity is modulated in a nucleotide- and cargo-dependent manner. Direct actin binding to the motor domain involves generic actin binding motifs and conserved albeit attenuated communication pathways between nucleotide and actin binding regions. Actin binding to the N-terminal extension is modulated by binding of GOLPH3 and potentially other cargo molecules to the PDZ module. Non-muscle myosin-2A and myosin-18A have been shown to share the same essential and regulatory light chains (4). Moreover, they colocalize near the cell periphery in lamellar actomyosin bundles (14). Therefore, it is tempting to speculate that myosin-18A is part of these bipolar filaments acting as mediator between membrane and cytoskeleton components. Other potential roles of myosin-18A include a ratchet-like function, where the positive strain resulting from the productive interaction of non-muscle myosin-2A with actin pushes the molecule forward to the next actin binding site, where it snaps into place. In the same manner, strain-induced conformational changes can affect nucleotide and actin affinity, enabling myosin-18A to work as an efficient strain sensor within the contractile machinery.
Acknowledgments
We thank Michael Radke, Henning Grosskopf, Georg Adler-Gunzelmann, and Michal Stanczak for help and discussions.
This work was supported by Deutsche Forschungsgemeinschaft Grants MA 1081/19–1 (to D. J. M.) and RA 1781/1–1 (to S. R.), Fonds der Chemischen Industrie Grant 684052 (to E. B.), and the Max Planck Society (to S. R. and E. B.).
- PDZ
- PSD-95/Discs-Large/ZO-1
- TRITC
- tetramethylrhodamine isothiocyanate
- M18A-MD
- myosin-18A motor domain
- mant
- 2′/3′-O-(N-methyl-anthraniloyl)
- SH2 and SH3
- Src homology 2 and 3, respectively
- TCEP
- tris(2-carboxyethyl)phosphine
- CM-loop
- cardiomyopathy loop.
REFERENCES
- 1. Bloemink M. J., Geeves M. A. (2011) Shaking the myosin family tree. Biochemical kinetics defines four types of myosin motor. Semin. Cell Dev. Biol. 22, 961–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Odronitz F., Kollmar M. (2007) Drawing the tree of eukaryotic life based on the analysis of 2,269 manually annotated myosins from 328 species. Genome Biol. 8, R196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Furusawa T., Ikawa S., Yanai N., Obinata M. (2000) Isolation of a novel PDZ-containing myosin from hematopoietic supportive bone marrow stromal cell lines. Biochem. Biophys. Res. Commun. 270, 67–75 [DOI] [PubMed] [Google Scholar]
- 4. Guzik-Lendrum S., Heissler S. M., Billington N., Takagi Y., Yang Y., Knight P. J., Homsher E., Sellers J. R. (2013) Mammalian myosin-18A, a highly divergent myosin. J. Biol. Chem. 288, 9532–9548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Isogawa Y., Kon T., Inoue T., Ohkura R., Yamakawa H., Ohara O., Sutoh K. (2005) The N-terminal domain of MYO18A has an ATP-insensitive actin-binding site. Biochemistry 44, 6190–6196 [DOI] [PubMed] [Google Scholar]
- 6. Langer W., Sohler F., Leder G., Beckmann G., Seidel H., Gröne J., Hummel M., Sommer A. (2010) Exon array analysis using re-defined probe sets results in reliable identification of alternatively spliced genes in non-small cell lung cancer. BMC Genomics 11, 676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Walz C., Chase A., Schoch C., Weisser A., Schlegel F., Hochhaus A., Fuchs R., Schmitt-Gräff A., Hehlmann R., Cross N. C., Reiter A. (2005) The t(8;17)(p11;q23) in the 8p11 myeloproliferative syndrome fuses MYO18A to FGFR1. Leukemia 19, 1005–1009 [DOI] [PubMed] [Google Scholar]
- 8. Walz C., Haferlach C., Hänel A., Metzgeroth G., Erben P., Gosenca D., Hochhaus A., Cross N. C., Reiter A. (2009) Identification of a MYO18A-PDGFRB fusion gene in an eosinophilia-associated atypical myeloproliferative neoplasm with a t(5;17)(q33–34;q11.2). Genes Chromosomes Cancer 48, 179–183 [DOI] [PubMed] [Google Scholar]
- 9. Ussowicz M., Jaśkowiec A., Meyer C., Marschalek R., Chybicka A., Szczepański T., Haus O. (2012) A three-way translocation of MLL, MLLT11, and the novel reciprocal partner gene MYO18A in a child with acute myeloid leukemia. Cancer Genet. 205, 261–265 [DOI] [PubMed] [Google Scholar]
- 10. Mori K., Furusawa T., Okubo T., Inoue T., Ikawa S., Yanai N., Mori K. J., Obinata M. (2003) Genome structure and differential expression of two isoforms of a novel PDZ-containing myosin (MysPDZ) (Myo18A). J. Biochem. 133, 405–413 [DOI] [PubMed] [Google Scholar]
- 11. Mori K., Matsuda K., Furusawa T., Kawata M., Inoue T., Obinata M. (2005) Subcellular localization and dynamics of MysPDZ (Myo18A) in live mammalian cells. Biochem. Biophys. Res. Commun. 326, 491–498 [DOI] [PubMed] [Google Scholar]
- 12. Guzik-Lendrum S., Nagy A., Takagi Y., Houdusse A., Sellers J. R. (2011) Drosophila melanogaster myosin-18 represents a highly divergent motor with actin tethering properties. J. Biol. Chem. 286, 21755–21766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hsu R.-M., Tsai M.-H., Hsieh Y.-J., Lyu P.-C., Yu J.-S. (2010) Identification of MYO18A as a novel interacting partner of the PAK2/βPIX/GIT1 complex and its potential function in modulating epithelial cell migration. Mol. Biol. Cell 21, 287–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tan I., Yong J., Dong J. M., Lim L., Leung T. (2008) A tripartite complex containing MRCK modulates lamellar actomyosin retrograde flow. Cell 135, 123–136 [DOI] [PubMed] [Google Scholar]
- 15. Heissler S. M., Manstein D. J. (2013) Nonmuscle myosin-2. Mix and match. Cell. Mol. Life Sci. 70, 1–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dippold H. C., Ng M. M., Farber-Katz S. E., Lee S.-K., Kerr M. L., Peterman M. C., Sim R., Wiharto P. A., Galbraith K. A., Madhavarapu S., Fuchs G. J., Meerloo T., Farquhar M. G., Zhou H., Field S. J. (2009) GOLPH3 bridges phosphatidylinositol-4-phosphate and actomyosin to stretch and shape the Golgi to promote budding. Cell 139, 337–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lehrer S. S., Kerwar G. (1972) Intrinsic fluorescence of actin. Biochemistry 11, 1211–1217 [DOI] [PubMed] [Google Scholar]
- 18. Diensthuber R. P., Müller M., Heissler S. M., Taft M. H., Chizhov I., Manstein D. J. (2011) Phalloidin perturbs the interaction of human non-muscle myosin isoforms 2A and 2C1 with F-actin. FEBS Lett. 585, 767–771 [DOI] [PubMed] [Google Scholar]
- 19. Furch M., Fujita-Becker S., Geeves M. A., Holmes K. C., Manstein D. J. (1999) Role of the salt-bridge between switch-1 and switch-2 of Dictyostelium myosin. J. Mol. Biol. 290, 797–809 [DOI] [PubMed] [Google Scholar]
- 20. Bagshaw C. R. (1975) The kinetic mechanism of the manganous ion-dependent adenosine triphosphatase of myosin subfragment 1. FEBS Lett. 58, 197–201 [DOI] [PubMed] [Google Scholar]
- 21. Taft M. H., Hartmann F. K., Rump A., Keller H., Chizhov I., Manstein D. J., Tsiavaliaris G. (2008) Dictyostelium myosin-5b is a conditional processive motor. J. Biol. Chem. 283, 26902–26910 [DOI] [PubMed] [Google Scholar]
- 22. Kurzawa S. E., Geeves M. A. (1996) A novel stopped-flow method for measuring the affinity of actin for myosin head fragments using microgram quantities of protein. J. Muscle Res. Cell. Motil. 17, 669–676 [DOI] [PubMed] [Google Scholar]
- 23. Nalavadi V., Nyitrai M., Bertolini C., Adamek N., Geeves M. A., Bähler M. (2005) Kinetic mechanism of myosin IXB and the contributions of two class IX-specific regions. J. Biol. Chem. 280, 38957–38968 [DOI] [PubMed] [Google Scholar]
- 24. Struchholz S., Elfrink K., Pieper U., Kalhammer G., Honnert U., Grützner A., Linke W. A., Liao W., Bähler M. (2009) Functional role of the extended loop 2 in the myosin 9b head for binding F-actin. J. Biol. Chem. 284, 3663–3671 [DOI] [PubMed] [Google Scholar]
- 25. Heissler S. M., Manstein D. J. (2011) Comparative kinetic and functional characterization of the motor domains of human nonmuscle myosin-2C isoforms. J. Biol. Chem. 286, 21191–21202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Senisterra G. A., Markin E., Yamazaki K., Hui R., Vedadi M., Awrey D. E. (2006) Screening for ligands using a generic and high-throughput light-scattering-based assay. J. Biomol. Screen. 11, 940–948 [DOI] [PubMed] [Google Scholar]
- 27. Duhr S., Braun D. (2006) Why molecules move along a temperature gradient. Proc. Natl. Acad. Sci. U.S.A. 103, 19678–19682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wienken C. J., Baaske P., Rothbauer U., Braun D., Duhr S. (2010) Protein-binding assays in biological liquids using microscale thermophoresis. Nat. Commun. 1, 100. [DOI] [PubMed] [Google Scholar]
- 29. Roy A., Kucukural A., Zhang Y. (2010) I-TASSER. A unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wu S., Skolnick J., Zhang Y. (2007) Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol. 5, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kliche W., Fujita-Becker S., Kollmar M., Manstein D. J., Kull F. J. (2001) Structure of a genetically engineered molecular motor. EMBO J. 20, 40–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zolkiewski M., Redowicz M. J., Korn E. D., Ginsburg A. (1996) Thermal unfolding of Acanthamoeba myosin II and skeletal muscle myosin. Biophys. Chem. 59, 365–371 [DOI] [PubMed] [Google Scholar]
- 33. Ponomarev M. A., Furch M., Levitsky D. I., Manstein D. J. (2000) Charge changes in loop 2 affect the thermal unfolding of the myosin motor domain bound to F-actin. Biochemistry 39, 4527–4532 [DOI] [PubMed] [Google Scholar]
- 34. Ponting C. P., Phillips C., Davies K. E., Blake D. J. (1997) PDZ domains. Targeting signalling molecules to sub-membranous sites. BioEssays 19, 469–479 [DOI] [PubMed] [Google Scholar]
- 35. Ranganathan R., Ross E. M. (1997) PDZ domain proteins. Scaffolds for signaling complexes. Curr. Biol. 7, R770–R773 [DOI] [PubMed] [Google Scholar]
- 36. Onishi H., Kojima S., Katoh K., Fujiwara K., Martinez H. M., Morales M. F. (1998) Functional transitions in myosin. Formation of a critical salt-bridge and transmission of effect to the sensitive tryptophan. Proc. Natl. Acad. Sci. U.S.A. 95, 6653–6658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Várkuti B. H., Yang Z., Kintses B., Erdélyi P., Bárdos-Nagy I., Kovács A. L., Hári P., Kellermayer M., Vellai T., Málnási-Csizmadia A. (2012) A novel actin binding site of myosin required for effective muscle contraction. Nat. Struct. Mol. Biol. 19, 299–306 [DOI] [PubMed] [Google Scholar]
- 38. Behrmann E., Müller M., Penczek P. A., Mannherz H. G., Manstein D. J., Raunser S. (2012) Structure of the rigor actin-tropomyosin-myosin complex. Cell 150, 327–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lowey S., Saraswat L. D., Liu H., Volkmann N., Hanein D. (2007) Evidence for an interaction between the SH3 domain and the N-terminal extension of the essential light chain in class II myosins. J. Mol. Biol. 371, 902–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Roth K., Weiner M. W. (1991) Determination of cytosolic ADP and AMP concentrations and the free energy of ATP hydrolysis in human muscle and brain tissues with 31P NMR spectroscopy. Magn. Reson. Med. 22, 505–511 [DOI] [PubMed] [Google Scholar]
- 41. Krisanda J. M., Paul R. J. (1983) Phosphagen and metabolite content during contraction in porcine carotid artery. Am. J. Physiol. 244, C385–C390 [DOI] [PubMed] [Google Scholar]
- 42. Khromov A., Somlyo A. V., Somlyo A. P. (1998) MgADP promotes a catch-like state developed through force-calcium hysteresis in tonic smooth muscle. Biophys. J. 75, 1926–1934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yang C.-H., Szeliga J., Jordan J., Faske S., Sever-Chroneos Z., Dorsett B., Christian R. E., Settlage R. E., Shabanowitz J., Hunt D. F., Whitsett J. A., Chroneos Z. C. (2005) Identification of the surfactant protein A receptor 210 as the unconventional myosin 18A. J. Biol. Chem. 280, 34447–34457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Matsui K., Parameswaran N., Bagheri N., Willard B., Gupta N. (2011) Proteomics analysis of the ezrin interactome in B cells reveals a novel association with Myo18aα. J. Proteome Res. 10, 3983–3992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dosé A. C., Ananthanarayanan S., Moore J. E., Burnside B., Yengo C. M. (2007) Kinetic mechanism of human myosin IIIA. J. Biol. Chem. 282, 216–231 [DOI] [PubMed] [Google Scholar]
- 46. Dosé A. C., Ananthanarayanan S., Moore J. E., Corsa A. C., Burnside B., Yengo C. M. (2008) The kinase domain alters the kinetic properties of the myosin IIIA motor. Biochemistry 47, 2485–2496 [DOI] [PubMed] [Google Scholar]