

Background: Evidence is lacking to explain how septin function is controlled during the cell cycle.
Results: Phosphorylation of SEPT9 by Cdk1 creates a binding site for Pin1 that is required for cytokinesis.
Conclusion: Septin interaction with Pin1 is controlled by Cdk1 and is necessary for timely cytokinesis.
Significance: Improper cell division results in cancer, in which both SEPT9 and Pin1 have been implicated.
Keywords: CDK (Cyclin-dependent Kinase), Cytokinesis, Cytoskeleton, GTPase, Phosphorylation, Pin1, Proline Isomerase, Septin
Abstract
Precise cell division is essential for multicellular development, and defects in this process have been linked to cancer. Septins are a family of proteins that are required for mammalian cell division, but their function and mode of regulation during this process are poorly understood. Here, we demonstrate that cyclin-dependent kinase 1 (Cdk1) phosphorylates septin 9 (SEPT9) upon mitotic entry, and this phosphorylation controls association with the proline isomerase, Pin1. Both SEPT9 and Pin1 are critical for mediating the final separation of daughter cells. Expression of mutant SEPT9 that is defective in Pin1 binding was unable to rescue cytokinesis defects caused by SEPT9 depletion but rather induced dominant-negative defects in cytokinesis. However, unlike SEPT9 depletion, Pin1 was not required for the accumulation of the exocyst complex at the midbody. These results suggest that SEPT9 plays multiple roles in abscission, one of which is regulated by the action of Cdk1 and Pin1.
Introduction
Septins are filament-forming GTPases required for cell division in diverse organisms from yeast to man (1). They were first identified in yeast during screens for cell cycle mutations and septin mutations lead to cytokinesis defects. All septins contain a GTP-binding domain and variable N and C termini, the latter of which often contains a coiled coil. In yeast, seven genes encode septins, five that are expressed during vegetative growth and two that are expressed only during sporulation (2). Septins exist in complexes that are composed of ordered non-polar arrays of multiple different septins (3, 4). In yeast, septins Cdc3, Cdc10, Cdc11, and Cdc12 form an octameric complex, and the order was determined to be Cdc11-Cdc12-Cdc3-Cdc10-Cdc10-Cdc3-Cdc12-Cdc11 through visualization of tagged proteins by electron microscopy (3).
In humans, 13 genes encode septins, and their diversity is further amplified by alternative splicing giving rise to additional N and C termini. The 13 genes can be grouped, based on sequence similarity, into four subgroups. The SEPT2 subgroup consists of septins 1, 2, 4, and 5; the SEPT6 subgroup consists of septins 6, 8, 10, 11, and 14; and the SEPT3 subgroup consists of septins 3, 9, and 12, whereas SEPT7 represents a unique subgroup (5). The crystal structure of a complex composed of the recombinant mammalian septins 2, 6, and 7 revealed that as in yeast, mammalian septins are also precisely ordered (4). Recent studies have revealed that in cell lines, human septins, similar to those of yeast, also form octameric complexes with SEPT9 at the terminal position (6, 7). Hence, a human septin complex is ordered SETP9-SEPT7-SEPT6-SEPT2-SEPT2-SEPT6-SEPT7-SEPT9, with a member of each subgroup represented at two places in the octamer.
Among mammalian septins, SEPT9 undergoes the most alternative splicing, with five alternative N termini and three alternative C termini (8). Coordinated expression of different SEPT9 isoforms seems to be critical as alteration in SEPT9 expression patterns has been linked to cancer (9–11) and mutations in the SEPT9 gene have been associated with hereditary neuralgic amyotrophy (12). In HeLa cell lysates, SEPT9_i1-SEPT9_i3 co-migrate as a single band, whereas SEPT9_i4 migrates more quickly. The function of the N terminus is not known, but we recently showed that only the longest three SEPT9 N-terminal variants (SEPT9_i1-SEPT9_i3) could rescue cytokinesis phenotypes caused by SEPT9 depletion, whereas shorter forms (SEPT9_i4) resulted in a dominant-negative abscission defect (13), indicating that the N termini of different isoforms control unique functions.
Analysis of the N termini of SEPT9 reveals few structural features that would explain this isoform specificity, but two recent high throughput phosphorylation screens identified a threonine residue within the N-terminal region of the long forms of SEPT9, but lacking from the short forms, as a putative mitotic phosphorylation site (14, 15). This raises the possibility that phosphorylation of the N-terminal region of SEPT9_i1-SEPT9_i3 may be important for their role during cell division. Although regulation of mammalian septin function by kinases during the cell cycle has been speculated for some time (16), it has never been demonstrated experimentally.
We therefore considered the possibility that the N termini of SEPT9 might be an important region controlling septin function during cytokinesis. Here, we show that SEPT9 is phosphorylated by cyclin-dependent kinase 1 (Cdk1)4 at Thr-24 and that this phosphorylation creates a binding site for the peptidylprolyl isomerase Pin1. We also show that Pin1, similar to SEPT9, is required for abscission in human cells, and its depletion causes phenotypes similar to knockdown of SEPT9, controlling a previously uncharacterized step in abscission.
EXPERIMENTAL PROCEDURES
Cell Culture
HeLa cells were cultured as described (17). Cells were arrested in mitosis by adding 50 ng/ml nocodazole (Sigma) to the culture medium and incubating for ∼16 h. For Cdk1 inhibition, 25 μm roscovitine (Sigma) was added for the final 3 h of nocodazole treatment. SF21 cells were cultured in suspension in a 1:1 mixture of Grace's media and Sf-900II SFM (Invitrogen) at room temperature.
Plasmids
Human SEPT9_v3 (accession no. NM_006640) and SEPT9_v4 (accession no. NM_001113494.1) were cloned into pFastBacHTA using standard methods. Threonine 24 in SEPT9_v3 was mutated to alanine via site-directed mutagenesis. Both SEPT9_v3 and SEPT9_v3 T24A were then subcloned into a modified version of pEGFPC. GST-Pin1 and GST-Pin1Y23A were described previously (18). Human Polo-like kinase-1 (Plk1) cDNA was obtained from ATCC (IMAGE 3854860), and the Polo-box domain (PBD) (19) was cloned into pGEX-6P. H538A and K540M mutations were introduced by site-directed mutagenesis, generating a Plk1 PBD mutation (which cannot bind to phosphorylated ligands (20)).
Transfections
GFP-tagged constructs were transfected into HeLa cells using FuGENE 6 (Roche Applied Science) according to the manufacturer's instructions. Protein expression was permitted for ∼24 h before treatment with or without nocodazole.
Immunoprecipitation
Cells were lysed in Triton X-100 lysis buffer (30 mm HEPES, pH 7.5, 100 mm NaCl, 1 mm EGTA, 1% Triton X-100, 20 mm NaF) with additional phosphatase (1 mm sodium orthovanadate, 100 nm okadaic acid, and 100 nm calyculin A) and protease inhibitors. 1–4 μg of 10C10 (13) or GFP (Invitrogen) antibody was added to ∼1 mg of lysate and incubated at 4 °C with constant mixing for at least 1 h. After washing with Triton X-100 lysis buffer, 30–50 μl of protein A Sepharose (Sigma) or protein G Sepharose (GE Healthcare) was added to the antibody-lysate mixture and incubated at 4 °C with constant mixing for at least 1 h. The beads were then washed three times, resuspended in SDS-PAGE loading buffer containing DTT (see Fig. 4E) or N-ethylmaleimide (see Fig. 1, A, B, E, and F, and Fig. 5, A and B), and subjected to Western blotting.
FIGURE 4.

The 10C10 epitope on SEPT9 is masked upon mitotic entry in a Pin1-dependent manner. A, HeLa cells were treated with control (Con) or Pin1 siRNA. SEPT9 was then immunoprecipitated with 10C10 from interphase (Inter) or mitotic HeLa cells. Precipitates were probed with a polyclonal SEPT9 antibody (left), and lysates were probed for Pin1and SEPT9 (right). B, quantification of the amount of SEPT9 present in the 10C10 immunoprecipitates shown in A. Data are expressed as a percentage of the interphase + control knockdown (ConKD) signal after normalizing to the amount of protein present in the lysate. Data are represented as mean ± S.E. (n ≥ 3); *, p < 0.01 (t test). WB, Western blot.
FIGURE 1.

SEPT9 is phosphorylated by Cdk1 at Thr-24 in a mitosis-specific manner. A, SEPT9_i3 is phosphorylated in a mitosis-specific manner. HeLa cells were transfected with a plasmid encoding GFP-SEPT9_i3. Cells were either left untreated, arrested in mitosis with nocodazole (Noco.) for 16 h, or arrested in mitosis and treated with the Cdk inhibitor roscovitine for the final 3 h of nocodazole treatment. Left, GFP-SEPT9_i3 was immunoprecipitated, and the precipitates were probed for pT-P and GFP. Right, lysates were probed for pT-P. Unsync, unsynchronized; rosc, roscovitine. B, mitotic phosphorylation of SEPT9_i3 occurs at Thr-24. GFP-SEPT9_i3 WT or GFP-SEPT9_i3 T24A was immunoprecipitated from mitotically arrested HeLa cells and probed for pT-P and GFP. C, Cdk1/cyclin B preferentially phosphorylates SEPT9_i3 over SEPT9_i4. SEPT9_i3 or SEPT9_i4 were incubated with Cdk1/cyclin B in the presence of radiolabeled ATP. Reactions were spotted on nitrocellulose, and radioactivity was counted using a scintillation counter. Data are expressed as a percentage of the counts per minute (cpm) obtained for SEPT9_i3 after normalizing to a no substrate control and are represented as mean ± S.E. (n = 3). D, Cdk1/cyclin B phosphorylates SEPT9_i3 at Thr-24. SEPT9_i3 and SEPT9_i3 T24A were subjected to kinase assays as described in C. Data are expressed as a percentage of the cpm obtained for SEPT9_i3 after normalizing to the SEPT9_i4 control and are represented as mean ± S.E. (n = 3). E, endogenous SEPT9 is phosphorylated during mitosis in a Cdk1-dependent manner. HeLa cells were arrested in mitosis with nocodazole and treated with or without roscovitine. Left, SEPT9 was immunoprecipitated, and the precipitates were probed for pT-P and SEPT9. Right, lysates were probed for pT-P. F, Cdk1 knockdown abrogates the mitotic phosphorylation of SEPT9. HeLa cells were treated with control or Cdk1 siRNA and arrested in mitosis with nocodazole. Left, SEPT9 was immunoprecipitated, and the precipitates were probed for pT-P and SEPT9. Right, lysates were probed for Cdk1, pT-P, and GAPDH. G, quantification of mitotic SEPT9 phosphorylation upon Cdk1 inhibition with roscovitine. Data are expressed as a percentage of the pT-P signal obtained upon control treatment after normalizing to the amount of SEPT9_i1–3 present in the immunoprecipitate and are represented as mean ± S.E. (n = 3). KD, knockdown; Con, control.
FIGURE 5.

Mapping the epitope of SEPT9 that is recognized by 10C10. A, 10C10 does not recognize SEPT9_i4. His-SEPT9_i1, His-SEPT9_i3, and His-SEPT9_i4 were purified from insect cells and subjected to Western blotting with His and 10C10 antibodies. B, schematic of the 139 amino acids at the N-terminal region of SEPT9 that are common between SEPT9_i1 and SEPT9_i3 and are absent from SEPT9_i4 (SEPT9 N-terminal). Constructs expressing peptides Epia, Epia-b, Epib, Epib-c, and Epic were generated. The Cdk1 phosphorylation site (Thr-24) is shown in red. C, lysates of induced bacteria expressing the various peptides were subjected to SDS-PAGE and stained with Coomassie. D, lysates of uninduced (U) and induced (I) bacteria expressing the various peptides were subjected to Western blotting with 10C10. Numbers to the right of the blot represent molecular mass standards in kDa. KD, knockdown.
Western Blotting
Western blotting was performed according to standard procedures using the following primary antibodies: SEPT9 (17), GAPDH (Millipore), GFP (Santa Cruz Biotechnology), Cdk1 (Calbiochem), Pin1 (Santa Cruz Biotechnology), Mpm-2 (Upstate), Thr(P)-Pro(P) (Cell Signaling), and FLAG (Sigma).
Protein Purification from Bacteria
Bacteria expressing the GST fusion protein of interest were inoculated in 5 ml of Luria broth containing 100 μg/ml ampicillin or Luria broth containing 100 μg/ml ampicillin and 50 μg/ml kanamycin (for BL21 Magic) and grown overnight at 37 °C (BL21 for GST-Pin1 and its variants, and BL21 Magic for GST-Plk1 and its variants). This starter culture was then diluted into 500 ml of Luria broth with ampicillin (or with ampicillin and kanamycin for BL21 Magic) and grown at 37 °C until the A600 nm reached 0.8–1. Expression was induced by adding isopropyl-β-d-thiogalactopyranoside as follows: 1 mm isopropyl-β-d-thiogalactopyranoside for 4 h at 30 °C for GST-Pin1 and its variants and 0.1 mm isopropyl-β-d-thiogalactopyranoside for 3 h at 37 °C for GST-Plk1 and its variants. Bacteria were centrifuged at 6000 × g for 10 min. For long term storage, bacterial pellets were frozen at −80 °C at this stage. Bacterial pellets were suspended in 20 ml of resuspension buffer (25 mm HEPES, pH 7.8, 100 mm NaCl, 5 mm MgCl2, 0.05% Tween 20, 1 mm DTT, and protease inhibitors) and lysed using a French press. Lysates were centrifuged at 15,000 × g for 10 min, and the supernatant was added to 1 ml of glutathione-agarose beads (50% slurry in resuspension buffer). This mixture was rotated end-over-end for at least 1.5 h. The beads were then washed three times with resuspension buffer (lacking protease inhibitors) and stored at 4 °C as a 50% slurry (in resuspension buffer) for up to 4 weeks. To elute GST-Pin1 for Far Western analysis, beads were first washed three times with 50 mm Tris, pH 8.0, 1 mm DTT, and then incubated with 1 ml of elution buffer (50 mm Tris, pH 8.0, 1 mm DTT, 20 mm reduced glutathione) for 1 h at 4 °C with constant mixing. The sample was dialyzed with 50 mm Tris, pH 8.0, 150 mm NaCl, 1 mm DTT, and frozen at −80 °C until use.
Pulldown Analysis
Interphase cells were washed with TBS and harvested by scraping in Tris lysis buffer (50 mm Tris, pH 8.0, 140 mm NaCl, 10% glycerol, 1% Triton X-100, 1 mm DTT, 100 mm NaF) containing phosphatase (1 mm sodium orthovanadate, 100 nm okadaic acid) and protease (1 μg/ml leupeptin, 1 mm PMSF, 2 mm benzamidine, and 1 μg/ml pepstatin A) inhibitors. Fresh medium was added to mitotic cells, which were subsequently harvested by shake off, washed with TBS, and resuspended in Tris lysis buffer. Cells were then lysed by passing through a 27.5-gauge needle four to five times and rotated for 45 min at 4 °C. Lysates were clarified by centrifugation at 4000 × g for 5 min, and the supernatant was used for pulldown experiments. In some cases, 50 units/ml of calf intestinal phosphatase (New England Biolabs) was added to the lysate for 30 min at 30 °C prior to pulldown analysis. Equal amounts of glutathione-agarose beads complexed with GST, GST-Pin1, GST-Pin1Y23A (18), GST-Plk1 PBD, or GST-Plk1 PBD mutant were added to 0.5–1 mg of cell lysate and incubated for 1.5 h at 4 °C with continuous mixing. The beads were subsequently washed four times with Tris lysis buffer, resuspended in SDS-PAGE loading buffer, and subjected to Western blotting.
Far Western Analysis
HeLa cells were transfected with GFP-SEPT9_v3 or GFP-SEPT9_v3 T24A, and treated with 100 nm calyculin A (Sigma) for the final 30 min of nocodazole arrest. Exogenously expressed SEPT9 was immunoprecipitated as above, subjected to SDS-PAGE, and transferred to PVDF. After overnight incubation at 4 °C in PBS with 5% milk, 0.2% Tween 20, 1 mm DTT, and 1 mm PMSF, the membrane was treated with 10 μg/ml GST-Pin1 in PBS with 0.05% Tween 20, 1 mm DTT, and 1 mm PMSF for 2 h at 4 °C. Following extensive washing with PBST, GST-Pin1 bound to the membrane was detected by Western blotting.
Protein Purification from Insect Cells
SF21 cells were infected with baculovirus expressing His6-SEPT9_i3, His6-SEPT9_i4, or His6-SEPT9_i3 T24A and incubated for ∼6 days. Cells were lysed by a French press in isolation buffer (40 mm Tris, pH 7.4, 100 mm NaCl, 2 mm β-mercaptoethanol, 20% glycerol, and 25 mm imidazole) with protease inhibitors, and His-tagged protein was purified on nickel-nitrilotriacetic acid-agarose beads (Qiagen). Bound protein was eluted in 40 mm Tris, pH 7.4, 100 mm NaCl, 2 mm β-mercaptoethanol, 20% glycerol, and 500 mm imidazole and stored at −80 °C until use.
Kinase Assays
5 μl of 5× reaction buffer (40 mm MOPS, pH 7.0, 1 mm EDTA), 10 μl of ATP mixture (25 mm MgAc, 0.25 mm ATP, 1 μCi of [γ-32P]ATP (PerkinElmer Life Science) per 10 μl of mixture), and 2.5 μg of substrate were mixed to a final volume of 22.5 μl and warmed to 30 °C. Reactions were initiated by adding 10 ng of recombinant Cdk1/cyclin B (Upstate) in kinase dilution buffer (20 mm MOPS, pH 7.0, 1 mm EDTA, 5% glycerol, 0.01% Brij-35, 0.1% 2-mercaptoethanol, 1 mg/ml BSA). After incubating for 10 min at 30 °C with vortexing every minute, reactions were stopped by adding 5 μl of 3% phosphoric acid, spotted on MF-Membrane filters (Millipore), washed extensively with 0.75% phosphoric acid, and subjected to scintillation counting.
siRNA Treatment
siRNA for SEPT9 (13) (GCACGATATTGAGGAGAAA), Pin1 (21) (Pin1-1, GCCATTTGAAGACGCCTCG) and control siRNA (13) (GCAGCGACCATGAGTA-TCA) were obtained from Dharmacon. Cdk1 siRNA (AAGGGGTTCCTAGTACTGCAA) was obtained from Qiagen (hs_Cdc2_10 HP), as was the second Pin1 siRNA (Pin1-2, CCGCCAGAT TCTCCCTTAA; hs_PIN1_5 HP). Cells grown to 60–80% confluence in six-well plates were transfected with 120–240 pmol of double stranded siRNA using Lipofectamine 2000 (Invitrogen). Knockdown was achieved after at least 56 h for SEPT9 and Cdk1 and 72 h for Pin1. For siRNA treatment of stable cell lines, antibiotics were removed prior to transfection.
Immunofluorescence and Time-lapse Microscopy
This was performed as described previously (13).
Generation of SEPT9_i3 T24A Stable Cell Line
SEPT9_v3 T24A was FLAG-tagged and made siRNA-resistant by introducing three silent point mutations into the siRNA target sequence and was subsequently cloned into pRetroX Tight Pur (Clontech). A stable cell line that inducibly expresses siRNA-resistant SEPT9_i3 T24A was then generated as described (13).
Mapping of the 10C10 Epitope
The DNA sequences encoding amino acids 8–39 (Epia), 8–95 (Epia-b), 40–95 (Epib), 40–146 (Epib-c), and 96–146 (Epic) of SEPT9_i3 were amplified by PCR. Each sequence was cloned into His-Myc-SUMO pQE30 and transformed into BL21 magic cells. Bacteria expressing the fusion protein of interest were grown overnight in terrific broth containing ampicillin and kanamycin. 2.5 ml of this starter culture was diluted to 50 ml of terrific broth containing ampicillin and kanamycin and grown for 30 min at 37 °C. 100 μl of uninduced culture was collected, spun down, and resuspended in 50 μl of SDS-PAGE loading buffer containing DTT. The remaining culture was induced with 0.4 mm isopropyl-β-d-thiogalactopyranoside for 2.5 h. 50 μl of induced culture was collected, spun down, and resuspended in 50 μl of SDS-PAGE loading buffer containing DTT. 10 μl of each lysate was subjected to SDS-PAGE and Coomassie staining, whereas 5 μl of each sample was subjected to Western blotting with 10C10.
Statistical Analysis
Two-tailed Student's t tests were applied to determine statistical significance.
RESULTS
Cdk1 Phosphorylates Thr-24 in the N-terminal Region of SEPT9 in a Mitosis-specific Manner
We first addressed the possibility that SEPT9 may be regulated by phosphorylation during mitosis. Exogenously expressed GFP-SEPT9_i3 was immunoprecipitated from unsynchronized or nocodazole-arrested (hereafter called “mitotic”) HeLa cell lysates and probed with a phospho-Thr-Pro (pT-P) antibody (Fig. 1A). Although we did not detect phosphorylation of SEPT9 in unsynchronized cells, SEPT9 in mitotic cells was recognized by the pT-P antibody. Cdk1 phosphorylates at least 70 different proteins critical for both the initiation of and progression through mitosis (22). This kinase preferentially phosphorylates the motif (S/T)PX(K/R), where X is any amino acid (23). The phosphorylation screens mentioned above identified Thr-24 of SEPT9 (numbered according to SEPT9_i3) as a putative phosphorylation site (14, 15), and this residue matches the Cdk1 consensus motif perfectly. We therefore hypothesized that Cdk1 phosphorylates SEPT9 at Thr-24 during mitosis. Consistent with this notion, treatment of mitotic cells with the cyclin-dependent kinase inhibitor roscovitine completely abolished SEPT9 phosphorylation (Fig. 1A). Control total cell lysates (right panel) reveal the increase in pT-P levels in the synchronized cells that is reduced by roscovitine treatment. In addition, mutation of Thr-24 to alanine completely abolished SEPT9 phosphorylation detected by the pT-P antibody (Fig. 1B). In vitro kinase assays demonstrated that Cdk1 preferentially phosphorylates SEPT9_i3, but not SEPT9_i4, which lacks Thr-24 (Fig. 1C). Likewise, mutation of Thr-24 to alanine greatly decreased the in vitro phosphorylation of SEPT9_i3 (Fig. 1D), demonstrating that Cdk1 directly phosphorylates SEPT9 at Thr-24. Although the in vivo phosphorylation of SEPT9 (Fig. 1B) was completely abolished by the T24A mutation, the in vitro phosphorylation (Fig. 1D) was only decreased by ∼60%. This suggests that Cdk1 phosphorylates at least one other site in SEPT9 in vitro that is either not phosphorylated in vivo or is not recognized by the pT-P antibody (as would be the case with a pSer-Pro motif).
To determine whether endogenous SEPT9 undergoes mitotic phosphorylation, we immunoprecipitated SEPT9 from mitotic HeLa cells and probed with the pT-P antibody (Fig. 1E). As expected, endogenous SEPT9 was also recognized by this antibody, and treatment of mitotic cells with roscovitine completely abolished endogenous SEPT9 phosphorylation. Note that SEPT9_i4, which lacks this residue, was not recognized by the pT-P antibody. Total cell lysates (right panel) demonstrate the presence of other pT-P species and their reduction following roscovitine. To provide further evidence that Cdk1 is the kinase responsible for the mitotic phosphorylation of SEPT9 at Thr-24, we assessed the effect of Cdk1 depletion on SEPT9 phosphorylation. As shown in Fig. 1F, Cdk1 knockdown decreased the mitotic phosphorylation of SEPT9. pT-P levels in total cell lysates (right panel) were also reduced. Quantification of the effects of roscovitine on SEPT9 phosphorylation is shown in Fig. 1G. Collectively, our results demonstrate that Cdk1 phosphorylates SEPT9 at Thr-24 during mitosis and thus identify the first link between a mammalian septin and the cell cycle machinery. We cannot rule out the possibility that Cdk1 activates another kinase, which, in turn, phosphorylates SEPT9 at Thr-24, but given our in vitro data, we feel that this is unlikely.
Mitotic Phosphorylation of SEPT9 at Thr-24 Does Not Regulate Association with Plk1
We next sought to determine the functional significance of SEPT9 phosphorylation at Thr-24. A recent proteomic screen identified SEPT9 as a putative binding partner of Plk1, which has been shown to act as a regulator of cytokinesis (20, 24). Plk1 contains a PBD that binds to S(pS/pT)P motifs in target proteins (19). Because Thr-24 of SEPT9 resides in such a motif, we tested whether mitotic phosphorylation of SEPT9 regulates binding to Plk1. We performed GST pulldowns using recombinant GST-Plk1 PBD and mitotic lysate from HeLa cells that were transfected with a plasmid encoding GFP-SEPT9_i3. Blots were probed with SEPT9 antibody (Fig. 2, top panels) to detect SEPT9 interaction with Plk1 PBD and also the Mpm-2 antibody (Fig. 2, bottom panels) which recognizes the S(pS/pT)P consensus sequence recognized by PBDs and serves as a control to demonstrate pulldown of other PBD targets. SEPT9_i3 interacted with the PBD of Plk1, and this association was impaired upon mutation of residues in the PBD that are required for binding to phosphorylated ligands (Fig. 2A). Note that the many Mpm-2 positive bands were also unable to bind. However, mutation of Thr-24 to alanine had no effect on the ability of SEPT9_i3 to bind to the PBD of Plk1 (Fig. 2B). These results suggest that phosphorylation of SEPT9 at Thr-24 does not control association with Plk1.
FIGURE 2.
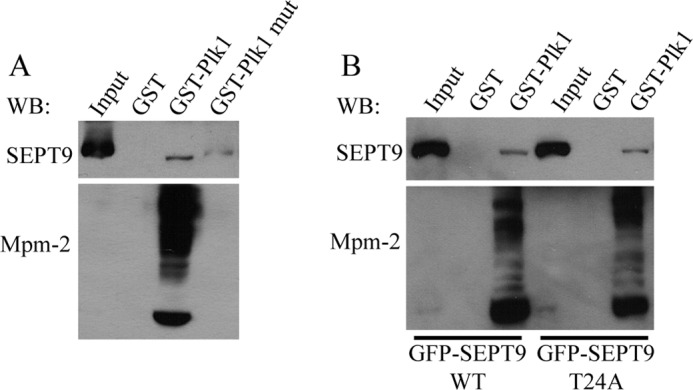
Mitotic phosphorylation of SEPT9 at Thr-24 does not regulate association with Plk1. A, the PBD of Plk1 interacts with SEPT9 in a phosphorylation-dependent manner. HeLa cells were transfected with a plasmid encoding GFP-SEPT9_i3 and arrested in mitosis with nocodazole. Lysates were incubated with glutathione-agarose beads complexed with GST, GST-Plk1 PBD, or GST-Plk1 PBD mutant (which cannot bind to phosphorylated ligands). Bound proteins were analyzed by Western blotting for SEPT9 or Mpm-2 (which recognizes the motif phospho-Ser/Thr-Pro and thus serves as a positive control). B, HeLa cells were transfected with plasmids encoding GFP-SEPT9_i3 or GFP-SEPT9_i3 T24A. Cells were arrested in mitosis and subjected to pulldown analysis as described in A. WB, Western blot.
Mitotic Phosphorylation of SEPT9 at Thr-24 Regulates Binding to Pin1
We identified the peptidyl-prolyl isomerase, Pin1, as a putative septin-interacting protein in a yeast two-hybrid screen (data not shown). Pin1 is a highly conserved enzyme consisting of two domains: an N-terminal WW domain and a C-terminal peptidyl-prolyl isomerase domain (25). The WW domain acts as a substrate recognition module by binding to specific phosphorylated Ser/Thr-Pro (phospho-Ser/Thr-Pro) motifs (26). The peptidyl-prolyl isomerase domain isomerizes the peptide bond between the pS/T and proline (27). This conformational change has been shown to have various effects on Pin1 substrates, such as regulation of function, stability, and/or localization (28, 29). Interestingly, Pin1 interacts with many of its substrates in a mitosis-specific manner (30), and has also been implicated in cancer (31–33).
We therefore hypothesized that Pin1 may interact with SEPT9 in response to mitotic phosphorylation at Thr-24 by Cdk1. To test this, we performed GST pulldowns using recombinant GST-Pin1 and unsynchronized or mitotic HeLa cell lysates. Pulldowns were blotted with antibody to SEPT9 (Fig. 2, top) and with Mpm-2 as a control (bottom) to prove that the Pin1 domains pulled down pT/S-P containing proteins. Although no interaction was observed between Pin1 and SEPT9 when unsynchronized lysates were used, SEPT9 in mitotic lysates associated with Pin1 (Fig. 3A). Note that SEPT9_i4 did not interact with Pin1. Ponceau S staining was performed to verify that equal amounts of each GST construct were used in the various pulldowns (data not shown). The SEPT9-Pin1 interaction was dependent on the WW domain of Pin1, as inactivation of the WW domain through a Y23A mutation (34) completely abolished the interaction (Fig. 3A). Treatment of lysates with calf intestinal phosphatase (Fig. 3B) or pretreatment of cells with roscovitine (Fig. 3C) prior to pulldown analysis greatly impaired the SEPT9-Pin1 association, arguing that the interaction is dependent on Cdk1-mediated phosphorylation.
FIGURE 3.

Pin1 interacts with SEPT9 upon mitotic phosphorylation at Thr-24 by Cdk1. A, Pin1 interacts with SEPT9 in a mitosis-specific manner via its WW domain. Unsynchronized or mitotic HeLa cell lysates were incubated with glutathione-agarose beads complexed with GST, GST-Pin1, or GST-Pin1Y23A (which has an inactivated WW domain). Bound proteins were analyzed by Western blotting for SEPT9 or Mpm-2 (which recognizes the motif phospho-Ser/Thr-Pro and thus serves as a positive control). B, the SEPT9-Pin1 interaction is phosphorylation-dependent. Mitotic HeLa cell lysates were incubated with or without calf intestinal phosphatase (CIP), and pulldowns were performed as described in A. C, the Pin1-SEPT9 interaction is dependent on Cdk1-mediated phosphorylation. Mitotic HeLa cells were treated with or without the Cdk1 inhibitor roscovitine prior to lysis, and pulldowns were performed as described in A. D, Thr-24 of SEPT9 is required for the association with Pin1. HeLa cells were transfected with plasmids encoding GFP-SEPT9_i3 or GFP-SEPT9_i3 T24A. Cells were arrested in mitosis and subjected to pulldown analysis as described in A. E, pulldown analysis was performed as described in D. The lower panel demonstrates that endogenous SEPT9_i1–3 are present in the GST-Pin1 pulldown upon transfection with either GFP-SEPT9 WT or GFP-SEPT9 T24A. F, Pin1 binds to SEPT9 directly via Thr-24. GFP-SEPT9_i3 or GFP-SEPT9_i3 T24A was immunoprecipitated from mitotically arrested HeLa cells and subjected to Far Western analysis using recombinant Pin1 as the probe. WB, Western blot.
To elucidate whether Pin1 binds SEPT9 at Thr-24, we first transfected HeLa cells with plasmids encoding GFP-SEPT9_i3 or GFP-SEPT9_i3 T24A, arrested the cells in mitosis, and performed pulldowns with GST-Pin1. Although GFP-SEPT9_i3 interacted with Pin1, the single T24A point mutation greatly inhibited the association, demonstrating that Thr-24 is critical for Pin1 binding (Fig. 3D). We performed a similar experiment to determine whether Pin1 directly binds to SEPT9 at Thr-24. GFP-SEPT9_i3 or GFP-SEPT9_i3 T24A was immunoprecipitated from mitotically arrested HeLa cells. Far Western analysis using recombinant Pin1 as a probe showed that Pin1 interacts directly with wild type SEPT9 but binds more poorly to the T24A mutant (Fig. 3E), even though endogenous SEPT9 is still detected in long exposures (bottom panel).
To evaluate an isomerization, we employed a monoclonal antibody called 10C10 that appears to be sensitive to N-terminal SEPT9 conformation (13). Intriguingly, this antibody consistently immunoprecipitates far more SEPT9 from unsynchronized cells than from mitotic cells, even though SEPT9 expression levels do not change upon mitotic entry (Fig. 4A). This raises the possibility that phosphorylation and/or isomerization of SEPT9 by Cdk1 and Pin1 (at onset on mitosis) may affect the ability of 10C10 to recognize SEPT9. To address this possibility, we first mapped the epitope of SEPT9 that is recognized by 10C10 (Fig. 5). Western blotting was performed to assess whether 10C10 can recognize purified His-SEPT9_i1, His-SEPT9_i3, and His-SEPT9_i4. As shown in Fig. 5A, 10C10 recognizes His-SEPT9_i1 and His-SEPT9_i3 but not His-SEPT9_i4. This demonstrates that the 10C10 epitope is somewhere within the 139 amino acids at the N-terminal region of SEPT9 that are common between SEPT9_i1 and SEPT9_i3 and are absent from SEPT9_i4 (Fig. 5B). To map the epitope further, we generated peptides corresponding to different fragments of this region (called Epia, Epia-b, Epib, Epib-c, and Epic; see Fig. 5, B and C) and assessed whether 10C10 can recognize these peptides by Western blotting (Fig. 5D). Only peptides containing Epic, which corresponds to amino acids 96 to 146 of SEPT9_i3, were recognized by 10C10. This shows that Thr-24 does not lie within the epitope of SEPT9 that is recognized by 10C10. Consequently, the inability of 10C10 to efficiently immunoprecipitate SEPT9 from mitotic cells is not simply a direct consequence of phosphorylation of SEPT9 at Thr-24.
We next depleted Pin1 (Fig. 4A) by siRNA, immunoprecipitated SEPT9 with 10C10 from unsynchronized and mitotic cells, and probed the immunoprecipitate with our polyclonal SEPT9 antibody (17). Consistent with previous work, cells treated with control and Pin1 siRNA had similar cell cycle profiles (21) and arrested in mitosis with similar efficiencies upon treatment with nocodazole (data not shown). Strikingly, Pin1 depletion significantly rescued the ability of 10C10 to immunoprecipitate SEPT9 from mitotic cells (Fig. 4, A and B). This demonstrates that the 10C10 epitope on SEPT9 is masked upon mitotic entry in a Cdk1- and Pin1-dependent manner, likely as a result of Pin1-mediated isomerization.
Pin1 Is also Important for Midbody Abscission
Given that SEPT9 plays an important role in midbody abscission (13), we next sought to elucidate whether Pin1 is also involved in this process. We first depleted Pin1 by siRNA (Fig. 6A) and assayed for defects in cytokinesis by immunofluorescence. Strikingly, Pin1-depleted cells exhibited a similar cytokinetic defect to that observed upon SEPT9 depletion. 19% ± 2% (S.E.) of Pin1-depleted cells remained joined by a midbody, compared with 7% ± 1% (S.E.) of the control cells (Fig. 6, B and C; t test, p < 0.005). Similar results were observed with a second siRNA targeting a different region of the Pin1 gene (Fig. 6C), arguing that this cytokinetic defect was not due to off-target effects of the siRNA. These results are consistent with previous observations (35) and suggest that Pin1 may also play a role in mediating midbody abscission.
FIGURE 6.

Pin1 is important for midbody abscission. A, efficiency of Pin1 depletion by siRNA. HeLa cells were treated with control siRNA or one of the two siRNAs targeting different regions of the Pin1 gene. Lysates were probed for Pin1 and GAPDH. Numbers to the right of the blots represent molecular mass standards in kilodaltons. B, representative example of cells attached by persistent midbodies following depletion of Pin1(Pin1-1 siRNA) (red, α-tubulin; blue, DNA). Scale bar represents 17 μm. C, quantification of effect of Pin1 depletion on cytokinesis. The percentage of cells exhibiting cytokinesis defects (multiple nuclei (Multinuc) or persistent midbodies) was determined upon treatment with control or Pin1 siRNA. The two Pin1 siRNAs were independently controlled so control siRNA results are shown separately. Data are represented as mean ± S.E. (n ≥ 300 cells from three or more independent experiments). Asterisks indicate differences between control and Pin1 knockdown cells; **, p < 0.005 (t test). D, division of HeLa cells after treatment with control siRNA. HeLa cells were transfected with control siRNA and randomly selected cells were followed through division by time-lapse microscopy. The time (in hours:minutes) since the beginning of DNA segregation is shown. Black arrows point to intact midbodies, whereas white arrows denote abscission. Scale bar represents 16 μm. E, Pin1 depletion causes defects in midbody abscission. HeLa cells were transfected with Pin1-1 siRNA and imaged as described in D. F, quantification of effect of Pin1 depletion on midbody abscission. The time from DNA segregation to midbody abscission was determined for each cell (n = 32 cells for control knockdown, n = 33 cells for Pin1-1 knockdown), and the cumulative percentage of cells that have abscised is plotted as a function of time. ConKD, control knockdown.
To confirm these results, we followed HeLa cells treated with control (Fig. 6D) or Pin1 siRNA (Fig. 6E) through cell division by time-lapse microscopy. In cells treated with control siRNA, the midbody abscised an average of 3 ± 0.2 h (S.E.) after the onset of cytokinesis, and 97% of these cells successfully completed abscission within 5.5 h (Fig. 6F). Cells depleted of Pin1 exhibited defects in midbody abscission. Of the cells that successfully abscised, the average abscission time was 4.4 ± 0.4 h (S.E.) (Fig. 6F; t test, p < 0.005). However, only 64% of Pin1-depleted cells abscised within 5.5 h of cytokinesis onset. The remaining cells either underwent apoptosis after abscission failure or took longer than 6.25 h to abscise. (In some cases, imaging was terminated at this point.) This was somewhat surprising given that mice lacking Pin1 are viable (36), but the recent discovery of a second functional Pin1 isoform in mice (but not humans) suggests that redundancy may only exist in mice (37). In addition, detailed analysis of dividing mouse embryonic fibroblasts derived from Pin1 knock-out mice revealed that these cells take twice as long to abscise compared with wild type mouse embryonic fibroblasts (35). Therefore, it appears that Pin1 plays an important role in abscission.
The Pin1-SEPT9 Interaction Is Important for the Completion of Cytokinesis
Because both SEPT9 and Pin1 are important for midbody abscission, we sought to determine whether the SEPT9-Pin1 interaction is important for this process. To this end, we generated a stable cell line that expresses siRNA-resistant SEPT9_i3 T24A under the control of an inducible promoter and performed a similar rescue experiment to that described previously (13). This cell line exhibits low levels of leaky expression in the absence of induction, which is close to the level of endogenous SEPT9 expression (Fig. 7A). Hence, although we were able to induce robust expression by adding doxycycline, all subsequent experiments were performed without addition of the drug and relied on basal expression. We depleted endogenous SEPT9 from this cell line, which allowed us to determine whether this low expression of SEPT9_i3 T24A can rescue the cytokinetic defect seen upon SEPT9 depletion. As we showed previously, the accumulation of cells defective in midbody abscission caused by total SEPT9 depletion could be rescued by wild type SEPT9_i3, not SEPT9_i4 (13). Using the same approach, the basal expression of siRNA-resistant SEPT9_i3 T24A, similar to SEPT9_i4, caused severe cytokinetic defects, even in the presence of endogenous SEPT9 (Fig. 7C; compare control knockdown in the parent cell line to control knockdown in the SEPT9_i3 T24A cell line; t test, p < 0.005). Approximately half of these cells remained joined by midbodies to one or multiple cells, and/or had more than one nucleus (Fig. 7, B and C). Similar defects were observed in multiple independent clones, arguing that this is not a clone-specific phenomenon. These defects were not suppressed by depleting endogenous SEPT9 (Fig. 7C), suggesting that they are not simply the result of increased total SEPT9 levels. Furthermore, depletion of endogenous SEPT9 in the SEPT9_i3 T24A cell line did not cause an increase in cytokinesis defects. These results suggest that SEPT9_i3 T24A has a dominant negative effect and argue that the interaction between SEPT9 and Pin1 is important for the completion of cytokinesis.
FIGURE 7.

Expression of SEPT9_i3 T24A causes defects in cytokinesis. A, stable cell line inducibly expressing FLAG-SEPT9_i3 T24A was treated with increasing amounts of doxycycline, and expression was assayed by Western blotting with FLAG, SEPT9, and GAPDH antibodies. Note that FLAG-SEPT9_i3 T24A is expressed at low levels in the absence of doxycycline. B, expression of SEPT9_i3 T24A causes cytokinetic defects. Shown are representative examples of cells attached by persistent midbodies (in some cases to several cells) and (C) multinucleated cells following basal (uninduced) stable expression of siRNA-resistant SEPT9_i3 T24A (red, α-tubulin; blue, DNA). White arrows point to midbodies, which in some cases appear to have regressed. Scale bar represents 53 μm. D, Representative examples of the parent cell line used to generate the FLAG-SEPT9_i3 T24A cell line. Scale bar represents 53 μm. E, efficiency of SEPT9 depletion by siRNA. The parent and FLAG-SEPT9_i3 T24A cell lines were treated with control or SEPT9 siRNA. Lysates were probed for SEPT9 and GAPDH (all lysates were probed on the same blot). F, the parent and siRNA-resistant SEPT9_i3 T24A cell lines were treated with control or SEPT9 siRNA and assayed for defects in cytokinesis by immunofluorescence microscopy. Unresolved cytokinesis refers to cells exhibiting midbody attachment or multinucleation. Data are represented as mean ± S.E. (n ≥ 300 cells from 3 experiments); **, p < 0.005 (t test).
Pin1-mediated Isomerization of SEPT9 Is Not Important for Exocyst Recruitment to the Midbody
Because it has been demonstrated that SEPT9 mediates the recruitment of the exocyst complex to the midbody (13), we next assessed whether Pin1-mediated isomerization of SEPT9 is important for this process. Depletion of Pin1 by siRNA had no effect on the localization of the exocyst component Sec8 at the midbody (Fig. 8). In addition, Sec8 localization at the midbody appeared normal in the SEPT9_i3 T24A cell line (data not shown). This suggests that SEPT9 mediates recruitment of the exocyst complex to the midbody in a Pin1-independent manner and that Pin1 function is required for a distinct step in abcission.
FIGURE 8.

Pin1 is not important for the proper localization of the exocyst complex at the midbody. A, representative examples of Sec8 localization in cytokinetic cells following treatment with control or Pin1 siRNA. Scale bar represents 17 μm. Arrows point to the midbody. B, the percentage of cells exhibiting enrichment of Sec8 at the midbody was determined by immunofluorescence after treatment with control or Pin1 siRNA. Data are represented as mean ± S.E. (n ≥ 300 cells from three experiments). C, Sec8 fluorescence/pixel (arbitrary units) at the midbody was measured using ImageJ software after treatment with control or Pin1 siRNA. ConKD, control knockdown.
DISCUSSION
Given that mammalian septins undergo dramatic changes in localization and possibly function at the onset of cell division, it has long been speculated that they may be regulated by the cell cycle machinery that controls mitotic entry (16). However, experimental evidence supporting this hypothesis has been lacking. The work presented here describes the first link between a mammalian septin and a cell cycle-dependent kinase. Specifically, we have demonstrated that Cdk1 phosphorylates SEPT9 at Thr-24 at the onset of mitosis. This creates a S-pT-P motif, which is a perfect match to the consensus recognition motif of the Plk1 PBD (19). Although SEPT9 did interact with the PBD of Plk1, this association was not dependent on phosphorylation at Thr-24. Further work will be required to identify the Plk1 binding site in SEPT9 and elucidate the functional significance of this interaction. We have demonstrated that mitotic phosphorylation of SEPT9 at Thr-24 by Cdk1 creates a binding site for the WW domain of Pin1. Immunoprecipitation analysis revealed that the 10C10 epitope on SEPT9 is masked in a Pin1-dependent manner. This is consistent with the notion that Pin1 induces a conformational change in the N-terminal region of SEPT9 at the onset of mitosis, in response to phosphorylation at Thr-24 by Cdk1.
We previously demonstrated that SEPT9_i3 is sufficient to drive cytokinesis in the absence of the other SEPT9 isoforms (13). In the present work, we show that SEPT9_i3 T24A (which cannot interact with Pin1) acts in a dominant-negative manner, causing severe defects in cytokinesis. Similar defects were observed previously upon expression of SEPT9_i4 (13), which lacks the Pin1 binding site. These results argue that the SEPT9-Pin1 interaction is important for the completion of cytokinesis and shed new light on how alterations in SEPT9 levels may contribute to carcinogenesis. Loss of SEPT9 function impairs the completion of cell division (13), thus potentially causing genomic instability, which could ultimately contribute to cancer (38). Increased expression of SEPT9_i4 (9, 11), which lacks the Pin1 binding site, could lead to cancer in a similar manner. Overexpression of SEPT9_i1 (10, 11), which contains the Pin1-binding site, could overwhelm Cdk1 and/or Pin1 and result in SEPT9 that is not isomerized by Pin1, which could have a similar effect. The finding that Pin1 is important for the cytokinetic function of SEPT9 is particularly interesting given the many links between alterations in Pin1 expression and cancer (33). Because variations in Pin1 levels would presumably directly affect the ability of SEPT9 to mediate abscission, it is possible that alterations in Pin1 expression could contribute to cancer through modulating SEPT9 activity. Future studies will be aimed at addressing this possibility.
As SEPT9 mediates the recruitment of the exocyst complex to the midbody (13), it seemed feasible that Pin1-mediated isomerization of SEPT9 may be important for this process. However, both depletion of Pin1 by siRNA and expression of SEPT9_i3 T24A had no effect on exocyst complex localization, suggesting that SEPT9 mediates recruitment of the exocyst complex to the midbody in a Pin1-independent manner. Future work will therefore focus on elucidating the mechanism by which Pin1-mediated isomerization of SEPT9 facilitates abscission. It is possible that isomerization of the N terminus of SEPT9 could unmask a binding site for some other protein, and this interaction may be important for abscission. SEPT9_i3 T24A would be unable to interact with this protein, as the binding site would remain masked upon mitotic entry. Consequently, SEPT9_i3 T24A would be expected to act in a dominant-negative manner, as was observed. SEPT9_i4 would lack the binding site for this protein altogether. As a result, SEPT9_i4 would also be expected to act in a dominant-negative manner, as shown previously (13).
Members of the septin family vary greatly in terms of the sequence and length of their N and C termini (16). In the crystal structure of the SEPT2/6/7 complex (4), the extreme N and C termini are disordered, and it remains unclear whether they are involved in septin-septin interactions. Consequently, the role of these regions is not understood. Based on the location of the Pin1-binding site near the extreme N terminus of SEPT9, it is tempting to speculate that the N-terminal region of septins may serve as an important regulatory domain. Consistent with this, phosphorylation near the extreme N terminus of mouse SEPT5 by Cdk5 regulates association with syntaxin-1 in the brain (39, 40). In addition, phosphorylation near the extreme C terminus of the yeast septin Cdc3p by Cdc28p is required for septin ring disassembly (41), suggesting that the C-terminal domain of septins may also serve a regulatory role. Because many septins, similar to SEPT9, have multiple isoforms with different termini, the various isoforms of a given septin may be differentially regulated.
Intriguingly, the above described septin phosphorylation sites also represent putative Pin1-binding sites. In addition, the yeast Pin1 homologue, Ess1 (42), demonstrates synthetic lethality with the yeast septin CDC12 (43). These studies therefore suggest that Pin1-mediated isomerization, in response to phosphorylation, may serve as an evolutionarily conserved mechanism of regulating septin function in a variety of cellular contexts.
Acknowledgments
We are grateful to all members of the Trimble and Kahr laboratories, Dr. B. Derry, and Dr. G. Brown for input and support.
This work was supported in part by the Canadian Cancer Society.
- Cdk1
- cyclin-dependent kinase 1
- Plk1
- Polo-like kinase-1
- pT-P
- phospho-Thr-Pro
- PBD
- Polo-box domain.
REFERENCES
- 1. Mostowy S., Cossart P. (2012) Septins: the fourth component of the cytoskeleton. Nat. Rev. Mol. Cell Biol. 13, 183–194 [DOI] [PubMed] [Google Scholar]
- 2. McMurray M. A., Thorner J. (2009) Reuse, replace, recycle. Specificity in subunit inheritance and assembly of higher-order septin structures during mitotic and meiotic division in budding yeast. Cell Cycle 8, 195–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bertin A., McMurray M. A., Grob P., Park S. S., Garcia G., 3rd, Patanwala I., Ng H. L., Alber T., Thorner J., Nogales E. (2008) Saccharomyces cerevisiae septins: supramolecular organization of heterooligomers and the mechanism of filament assembly. Proc. Natl. Acad. Sci. U.S.A. 105, 8274–8279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sirajuddin M., Farkasovsky M., Hauer F., Kühlmann D., Macara I. G., Weyand M., Stark H., Wittinghofer A. (2007) Structural insight into filament formation by mammalian septins. Nature 449, 311–315 [DOI] [PubMed] [Google Scholar]
- 5. Kinoshita M. (2003) Assembly of mammalian septins. J Biochem. 134, 491–496 [DOI] [PubMed] [Google Scholar]
- 6. Kim M. S., Froese C. D., Estey M. P., Trimble W. S. (2011) SEPT9 occupies the terminal positions in septin octamers and mediates polymerization-dependent functions in abscission. J. Cell Biol. 195, 815–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sellin M. E., Sandblad L., Stenmark S., Gullberg M. (2011) Deciphering the rules governing assembly order of mammalian septin complexes. Mol. Biol. Cell 22, 3152–3164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McIlhatton M. A., Burrows J. F., Donaghy P. G., Chanduloy S., Johnston P. G., Russell S. E. (2001) Genomic organization, complex splicing pattern and expression of a human septin gene on chromosome 17q25.3. Oncogene 20, 5930–5939 [DOI] [PubMed] [Google Scholar]
- 9. Burrows J. F., Chanduloy S., McIlhatton M. A., Nagar H., Yeates K., Donaghy P., Price J., Godwin A. K., Johnston P. G., Russell S. E. (2003) Altered expression of the septin gene, SEPT9, in ovarian neoplasia. J. Pathol. 201, 581–588 [DOI] [PubMed] [Google Scholar]
- 10. Gonzalez M. E., Peterson E. A., Privette L. M., Loffreda-Wren J. L., Kalikin L. M., Petty E. M. (2007) High SEPT9_v1 expression in human breast cancer cells is associated with oncogenic phenotypes. Cancer Res. 67, 8554–8564 [DOI] [PubMed] [Google Scholar]
- 11. Scott M., McCluggage W. G., Hillan K. J., Hall P. A., Russell S. E. (2006) Altered patterns of transcription of the septin gene, SEPT9, in ovarian tumorigenesis. Int. J. Cancer 118, 1325–1329 [DOI] [PubMed] [Google Scholar]
- 12. Kuhlenbäumer G., Hannibal M. C., Nelis E., Schirmacher A., Verpoorten N., Meuleman J., Watts G. D., De Vriendt E., Young P., Stögbauer F., Halfter H., Irobi J., Goossens D., Del-Favero J., Betz B. G., Hor H., Kurlemann G., Bird T. D., Airaksinen E., Mononen T., Serradell A. P., Prats J. M., Van Broeckhoven C., De Jonghe P., Timmerman V., Ringelstein E. B., Chance P. F. (2005) Mutations in SEPT9 cause hereditary neuralgic amyotrophy. Nat. Genet. 37, 1044–1046 [DOI] [PubMed] [Google Scholar]
- 13. Estey M. P., Di Ciano-Oliveira C., Froese C. D., Bejide M. T., Trimble W. S. (2010) Distinct roles of septins in cytokinesis: SEPT9 mediates midbody abscission. J. Cell Biol. 191, 741–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Beausoleil S. A., Villén J., Gerber S. A., Rush J., Gygi S. P. (2006) A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 24, 1285–1292 [DOI] [PubMed] [Google Scholar]
- 15. Dephoure N., Zhou C., Villén J., Beausoleil S. A., Bakalarski C. E., Elledge S. J., Gygi S. P. (2008) A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 105, 10762–10767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hall P. A., Russell S. E. (2004) The pathobiology of the septin gene family. J. Pathol. 204, 489–505 [DOI] [PubMed] [Google Scholar]
- 17. Surka M. C., Tsang C. W., Trimble W. S. (2002) The mammalian septin MSF localizes with microtubules and is required for completion of cytokinesis. Mol. Bio. Cell 13, 3532–3545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Messenger M. M., Saulnier R. B., Gilchrist A. D., Diamond P., Gorbsky G. J., Litchfield D. W. (2002) Interactions between protein kinase CK2 and Pin1. Evidence for phosphorylation-dependent interactions. J. Biol. Chem. 277, 23054–23064 [DOI] [PubMed] [Google Scholar]
- 19. Elia A. E., Cantley L. C., Yaffe M. B. (2003) Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science 299, 1228–1231 [DOI] [PubMed] [Google Scholar]
- 20. Lowery D. M., Clauser K. R., Hjerrild M., Lim D., Alexander J., Kishi K., Ong S. E., Gammeltoft S., Carr S. A., Yaffe M. B. (2007) Proteomic screen defines the Polo-box domain interactome and identifies Rock2 as a Plk1 substrate. EMBO J. 26, 2262–2273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xu Y. X., Manley J. L. (2007) The prolyl isomerase Pin1 functions in mitotic chromosome condensation. Mol Cell 26, 287–300 [DOI] [PubMed] [Google Scholar]
- 22. Malumbres M., Barbacid M. (2005) Mammalian cyclin-dependent kinases. Trends Biochem. Sci. 30, 630–641 [DOI] [PubMed] [Google Scholar]
- 23. Songyang Z., Blechner S., Hoagland N., Hoekstra M. F., Piwnica-Worms H., Cantley L. C. (1994) Use of an oriented peptide library to determine the optimal substrates of protein kinases. Curr. Biol. 4, 973–982 [DOI] [PubMed] [Google Scholar]
- 24. Petronczki M., Glotzer M., Kraut N., Peters J. M. (2007) Polo-like kinase 1 triggers the initiation of cytokinesis in human cells by promoting recruitment of the RhoGEF Ect2 to the central spindle. Dev. Cell 12, 713–725 [DOI] [PubMed] [Google Scholar]
- 25. Lu K. P., Hanes S. D., Hunter T. (1996) A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature 380, 544–547 [DOI] [PubMed] [Google Scholar]
- 26. Ranganathan R., Lu K. P., Hunter T., Noel J. P. (1997) Structural and functional analysis of the mitotic rotamase Pin1 suggests substrate recognition is phosphorylation dependent. Cell 89, 875–886 [DOI] [PubMed] [Google Scholar]
- 27. Yaffe M. B., Schutkowski M., Shen M., Zhou X. Z., Stukenberg P. T., Rahfeld J. U., Xu J., Kuang J., Kirschner M. W., Fischer G., Cantley L. C., Lu K. P. (1997) Sequence-specific and phosphorylation-dependent proline isomerization: a potential mitotic regulatory mechanism. Science 278, 1957–1960 [DOI] [PubMed] [Google Scholar]
- 28. Lu P. J., Wulf G., Zhou X. Z., Davies P., Lu K. P. (1999) The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature 399, 784–788 [DOI] [PubMed] [Google Scholar]
- 29. Ryo A., Nakamura M., Wulf G., Liou Y. C., Lu K. P. (2001) Pin1 regulates turnover and subcellular localization of β-catenin by inhibiting its interaction with APC. Nat. Cell Biol. 3, 793–801 [DOI] [PubMed] [Google Scholar]
- 30. Shen M., Stukenberg P. T., Kirschner M. W., Lu K. P. (1998) The essential mitotic peptidyl-prolyl isomerase Pin1 binds and regulates mitosis-specific phosphoproteins. Genes Dev. 12, 706–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bao L., Kimzey A., Sauter G., Sowadski J. M., Lu K. P., Wang D. G. (2004) Prevalent overexpression of prolyl isomerase Pin1 in human cancers. Am. J. Pathol. 164, 1727–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wulf G. M., Ryo A., Wulf G. G., Lee S. W., Niu T., Petkova V., Lu K. P. (2001) Pin1 is overexpressed in breast cancer and cooperates with Ras signaling in increasing the transcriptional activity of c-Jun towards cyclin D1. EMBO J. 20, 3459–3472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yeh E. S., Means A. R. (2007) PIN1, the cell cycle and cancer. Nat. Rev. Cancer 7, 381–388 [DOI] [PubMed] [Google Scholar]
- 34. Lu P. J., Zhou X. Z., Shen M., Lu K. P. (1999) Function of WW domains as phosphoserine- or phosphothreonine-binding modules. Science 283, 1325–1328 [DOI] [PubMed] [Google Scholar]
- 35. van der Horst A., Khanna K. K. (2009) The peptidyl-prolyl isomerase Pin1 regulates cytokinesis through Cep55. Cancer Res. 69, 6651–6659 [DOI] [PubMed] [Google Scholar]
- 36. Fujimori F., Takahashi K., Uchida C., Uchida T. (1999) Mice lacking Pin1 develop normally, but are defective in entering cell cycle from G0 arrest. Biochem. Biophys. Res. Commun. 265, 658–663 [DOI] [PubMed] [Google Scholar]
- 37. Zhu J. X., Dagostino E., Rejto P. A., Mroczkowski B., Murray B. (2007) Identification and characterization of a novel and functional murine Pin1 isoform. Biochem. Biophys. Res. Commun. 359, 529–535 [DOI] [PubMed] [Google Scholar]
- 38. Fujiwara T., Bandi M., Nitta M., Ivanova E. V., Bronson R. T., Pellman D. (2005) Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 437, 1043–1047 [DOI] [PubMed] [Google Scholar]
- 39. Amin N. D., Zheng Y. L., Kesavapany S., Kanungo J., Guszczynski T., Sihag R. K., Rudrabhatla P., Albers W., Grant P., Pant H. C. (2008) Cyclin-dependent kinase 5 phosphorylation of human septin SEPT5 (hCDCrel-1) modulates exocytosis. J. Neurosci. 28, 3631–3643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Taniguchi M., Taoka M., Itakura M., Asada A., Saito T., Kinoshita M., Takahashi M., Isobe T., Hisanaga S. (2007) Phosphorylation of adult type Sept5 (CDCrel-1) by cyclin-dependent kinase 5 inhibits interaction with syntaxin-1. J. Biol. Chem. 282, 7869–7876 [DOI] [PubMed] [Google Scholar]
- 41. Tang C. S., Reed S. I. (2002) Phosphorylation of the septin cdc3 in g1 by the cdc28 kinase is essential for efficient septin ring disassembly. Cell Cycle 1, 42–49 [PubMed] [Google Scholar]
- 42. Hanes S. D., Shank P. R., Bostian K. A. (1989) Sequence and mutational analysis of ESS1, a gene essential for growth in Saccharomyces cerevisiae. Yeast 5, 55–72 [DOI] [PubMed] [Google Scholar]
- 43. Davierwala A. P., Haynes J., Li Z., Brost R. L., Robinson M. D., Yu L., Mnaimneh S., Ding H., Zhu H., Chen Y., Cheng X., Brown G. W., Boone C., Andrews B. J., Hughes T. R. (2005) The synthetic genetic interaction spectrum of essential genes. Nat. Genet. 37, 1147–1152 [DOI] [PubMed] [Google Scholar]