

Background: The contribution of endogenous norepinephrine (NE) to skeletal homeostasis is unclear.
Results: Bone forming cells, not only neurons, express the norepinephrine transporter (NET), and blockade of extracellular NE clearance causes alterations in bone homeostasis.
Conclusion: NE clearance by NET is a component of the homeostatic machinery by which sympathetic nerves and osteoblasts control bone remodeling.
Significance: NET blockers may increase fracture risk.
Keywords: Gene Knockout, Mouse, Nerve, Neurotransmitter Transport, Osteoblasts, ADHD, Bone Remodeling, Norepinephrine Transporter, Sympathetic Nervous System
Abstract
Changes in bone remodeling induced by pharmacological and genetic manipulation of β-adrenergic receptor (βAR) signaling in osteoblasts support a role of sympathetic nerves in the regulation of bone remodeling. However, the contribution of endogenous sympathetic outflow and nerve-derived norepinephrine (NE) to bone remodeling under pathophysiological conditions remains unclear. We show here that differentiated osteoblasts, like neurons, express the norepinephrine transporter (NET), exhibit specific NE uptake activity via NET and can catabolize, but not generate, NE. Pharmacological blockade of NE transport by reboxetine induced bone loss in WT mice. Similarly, lack of NE reuptake in norepinephrine transporter (Net)-deficient mice led to reduced bone formation and increased bone resorption, resulting in suboptimal peak bone mass and mechanical properties associated with low sympathetic outflow and high plasma NE levels. Last, daily sympathetic activation induced by mild chronic stress was unable to induce bone loss, unless NET activity was blocked. These findings indicate that the control of endogenous NE release and reuptake by presynaptic neurons and osteoblasts is an important component of the complex homeostatic machinery by which the sympathetic nervous system controls bone remodeling. These findings also suggest that drugs antagonizing NET activity, used for the treatment of hyperactivity disorders, may have deleterious effects on bone accrual.
Introduction
Bone remodeling is a tightly regulated process under the control of systemic hormones and paracrine/autocrine factors. The bone marrow environment is richly vascularized and innervated, and recent evidence support the existence of functional interactions between nerves and bone cells (1–3). Among this evidence, signaling via the β2-adrenergic receptor (β2AR)2 has emerged as critical. The β2AR is expressed in both bone mesenchymal and hematopoietic lineages (4–6). In osteoblasts, its expression is predominant among the nine α- and β-adrenergic receptor subtypes (α1A, α1B, α1D, α2A, α2B, α2C, β1, β2, and β3) (4, 7). Moreover, the β2AR in osteoblasts is functional, as demonstrated by the accumulation of cAMP and the activation of target genes, including Rankl, upon exogenous stimulation by NE or isoproterenol, a non-selective β adrenergic agonist (1, 8). β2AR activation in osteoblasts stimulates the formation of osteoclast via the induction of Rankl expression (1) and inhibits osteoblast proliferation via cyclin D1 and Clock genes (9). Genetic ablation of the β2AR, globally or specifically in osteoblasts, induces a high bone mass phenotype in mice (1, 10). Similarly, pharmacological blockade of βAR signaling using the non-selective βAR antagonist propranolol increases bone mass, whereas β2AR stimulation with various βAR agonists decreases it (11–13). As a whole, this body of knowledge strongly supports the importance and biological relevance of sympathetic nerves in the regulation of bone remodeling. However, these studies have mostly focused on post-synaptic βAR modulation and did not yet directly investigate the existence of presynaptic mechanisms regulating endogenous sympathetic outflow. Following sympathetic activation, 80–90% of the NE released in synaptic clefts is cleared by NE reuptake, the remaining extracellular NE diffusing into the circulation or being metabolized. The process of NE reuptake is mediated by the norepinephrine transporter (NET), a monoamine transporter and a member of the Na+/Cl−-dependent family of neurotransmitter transporters. NET controls the concentration of NE and duration of neurotransmission at synapses, and is located in the membrane of presynaptic neurons, although desipramine-sensitive [3H]NE uptake has been observed in astrocytes as well (14) and Net expression has been reported in several peripheral organs in embryos (15). NET is the target of drugs used for the treatment of depression and attention deficit hyperactivity disorder (ADHD), and of drugs of abuse, including cocaine and amphetamine. In this study, we investigated whether bone cells transport NE and therefore locally control NE extracellular levels in the skeleton, and if alteration in NE reuptake caused by NE transport inhibition or Net deficiency in mice has consequences on bone homeostasis.
EXPERIMENTAL PROCEDURES
Animals
C57BL/6J WT mice (Jackson Laboratory) were administered reboxetine at 15 mg/kg/day via subcutaneously implanted mini-osmotic pumps (Durect Corporation 0000298). Heterozygous Net+/− mice (genetic background C57BL/6J) (16) were mated to produce Net+/+ (wild-type) and Net−/− littermates. Chronic immobilization stress (CIS) was carried out 2 h daily by placing mice in 50-ml laboratory conical tubes perforated for adequate air supply. Murine body composition was measured using a Minispec Model mq7.5 (7.5 mHz) nuclear magnetic resonance (NMR) analyzer (Bruker Instruments). Tail suspension measurements were performed as previously described (17). All procedures were approved by the Institutional Animal Care and Use Committee at Vanderbilt University Medical Center.
Primary Cell Culture
Calvarial osteoblasts were isolated from 3–4-day-old neonatal mice. Bone marrow stromal cells (BMSCs) were isolated from 2–3-month-old adult mice. Differentiation was induced with 50 μg/ml of ascorbic acid. Spleen-derived osteoclasts were prepared from 2–3-month-old adult mice. Differentiation was induced with 30 ng/ml of macrophage colony-stimulating factor and 50 ng/ml of receptor activator of nuclear factor κB ligand. For alkaline phosphatase-positive colony forming units (cfu-AP) assays, BMSCs were isolated from long bones, plated at a density of 1 × 107 cells/10-cm plate. One week after plating the cells, culture medium was switched to osteogenic medium containing 50 μg/ml of ascorbic acid. Two weeks following plating, alkaline phosphatase staining was performed, and the number of cfu-AP colonies was counted manually.
Semi-quantitative and Quantitative Real-time RT-PCR
Semi-quantitative RT-PCR was performed using the following primers: Net (norepinephrine transporter): forward, 5′-caggcacctccattctgttt-3′, reverse, 5′-taggtgagcggcttgaagtt-3′, Tm = 60 °C; Dbh (dopamine β-hydroxylase): forward, 5′-gactcaactactgccggcacgt-3′, reverse, 5′-ctgggtgcacttgtctgtgcagt-3′, Tm = 60 °C; Th (tyrosine hydroxylase): forward, 5′-ccccacctggagtactttgtg-3′, reverse, 5′-cttgtcctctctggaactgc-3′, Tm = 60 °C; Mao (monoamine oxidase): forward, 5′-ggagaagcccagtatcacagg-3′, reverse, 5′-gaaccaagacattaattttgtattctgac-3′, Tm = 60 °C; Comt (catechol-O-methyltransferase): forward, 5′-agaccgctaccttccagaca-3′, reverse, 5′-tgagtggtcaggacttcacg-3′, Tm = 60 °C; Emt (extraneuronal monoamine transporter): forward, 5′ctgggtggtccctgagtctcc3′, reverse, 5′-tcccaggcgcatgacaagtcc-3′, Tm= 61 °C; Ocn (osteocalcin): forward, 5′-accctggctgcgctctgtctct-3′, reverse, 5′-gatgcgtttgtaggcggtcttca-3′, Tm= 62 °C; Ctr (calcitonin receptor): forward, 5′-tgctggctgagtgcagaaacc-3′, reverse, 5′-ggccttcacagccttcaggtac-3′, Tm = 62 °C; Hprt (hypoxanthine-guanine phosphoribosyltransferase): forward, 5′-gttgagagatcatctccacc-3′, reverse, 5′-agcgatgatgaaccaggtt-3′, Tm = 55 °C. Real-time PCR was performed using TaqMan® or SYBR Green gene expression assays. TaqMan probes/primers were from Applied Biosystems (Sost, Mm00470479_m1; Hprt1, Mm00446968_m1; Ucp1, Mm00494069_m1). SYBR Green primer sequences were: Ocn forward, 5′-accctggctgcgctctgtctct-3′ and reverse, 5′-gatgcgtttgtaggcggtcttca-3′; Net forward, 5′-caggcacctccattctgttt-3′ and reverse, 5′-taggtgagcggcttgaagtt-3′; b2 mg (β2-microglobulin): forward, 5′-ttctggtgcttgtctcactga-3′ and reverse, 5′-cagtatgttcggcttcccattc-3′. Specificity of SYBR Green amplifications was verified by the presence of a single peak on the dissociation curve.
Western Blot
Western blot analyses were performed using anti-mouse NET (Mab Technologies NET05-2) or monoclonal mouse anti-β-actin antibody (Sigma A5441) and chemiluminescent detection (PerkinElmer Life Sciences).
[3H]NE Uptake Assay
Microcomputed Tomography Analysis
Three-dimensional microcomputed tomography analyses were performed using a Scanco μCT 40 system (Scanco Medical, Bassersdorf, Switzerland). Tomographic images were acquired at 55 kVp and 145 μA with an isotropic voxel size of 12 mm and at an integration time of 300 ms. To segment bone from non-mineralized tissue, a Gaussian noise suppression filter (σ = 0.8, support = 1) was used, and global thresholds were consistent across scans per anatomical site.
Biomechanical Test
Each femur was placed on the lower support points of a three-point bending fixture with the anterior side down and medial side forward. With the span between the lower supports set to 8 mm, the hydrated bones were loaded to failure at a rate of 3.0 mm/min using a servo-hydraulic, bench-top material testing system (Dynamight 8841, Instron, Canton, OH).
Histomorphometry
The bones were dehydrated and embedded undecalcified in methyl methacrylate. Histomorphometric measurements were performed using the Bioquant Image Analysis System (R&M Biometrics, Nashville, TN).
Bone Immunocytochemistry
Immunocytochemistry was performed in formalin-fixed long bones of neonatal mice using an antibody against NET (Mab Technologies NET05-2) or a non-immune IgG antibody, according to established protocols.
Urine DPD and Creatinine
Urinary deoxypyridinoline (DPD) and creatinine were measured by MicroVue DPD EIA Kit (Quidel Corporation 8007) and MicroVue Creatinine Assay Kit (Quidel Corporation 8009), respectively.
Statistics
All data are presented as mean ± S.E. Statistical analyses were performed using one-way analysis of variance for multiple comparisons followed by post hoc pairwise comparison with Bonferroni adjustment and unpaired two-tailed Student's t tests for two-group comparisons. For all analyses, p < 0.05 was considered significant.
RESULTS
Osteoblasts Express Genes Required for NE Transport and Catabolism
Although the presence and functionality of the β2AR in bone cells are well described, the homeostasis of NE within the skeleton remains unknown. Surprisingly, a significant amount of Net mRNA were detected in bone tissues by RT-PCR, suggesting that cells distinct from neurons within the skeleton may express Net (Fig. 1A). Using RNAs from cultures of primary bone cells as well as from clonal bone cell lines, Net mRNA transcripts were detected by RT-PCR in BMSCs, the mesenchymal progenitor cell line C3H10T1/2, rib-derived primary chondrocytes, and in calvaria-derived primary osteoblasts (POB) as well as in the homogenous osteoblastic cell line MC3T3, with higher expression levels in osteocalcin (Ocn)-positive differentiated osteoblasts (Fig. 1B). In addition, these analyses showed that these cells express Monoamine oxidase (Mao) and catechol-O-methyltransferase (Comt), two genes required for NE catabolism, but do not express Tyrosine hydroxylase (Th) and Dopamine β-hydroxylase (Dbh), two genes necessary for NE synthesis. Extraneuronal monoamine transporter (Emt) was detected in differentiated BMSCs only. Net expression was not detected in calcitonin receptor (Ctr)-positive primary osteoclast cultures prepared from spleen and differentiated in vitro with macrophage colony-stimulating factor and receptor activator of nuclear factor κB ligand.
FIGURE 1.
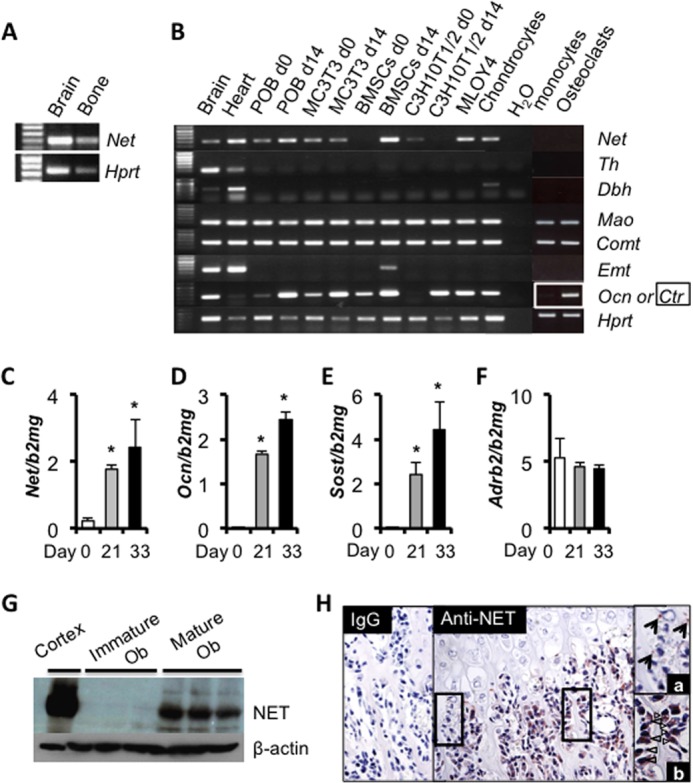
NET is expressed in differentiated osteoblasts. A and B, RT-PCR analysis of Net, Tyrosine hydroxylase (Th), Dopamine β-hydroxylase (Dbh), Monoamine oxidase (Mao), Catechol-O-methyltransferase (Comt), and Extraneuronal monoamine transporter (Emt). RNA extracts from brain and heart serve as positive controls. Osteocalcin (Ocn) is used as a marker for differentiated osteoblasts. Calcitonin receptor (Ctr) is used as a marker for differentiated osteoclasts. C–F, real-time quantitative PCR analysis of Net expression in undifferentiated (day 0) and differentiated (day 21 or day 33) primary calvarial osteoblasts. Ocn and Sclerostin (Sost) are used as markers for differentiated osteoblasts/osteocytes. Values are given as mean ± S.E., *, p < 0.05 versus day 0, n = 3. G, Western blot analysis of NET expression in undifferentiated (day 0) and differentiated (day 14) mouse calvarial primary osteoblasts. Protein extract from the cerebral cortex serves as a positive control. H, NET immunoreactivity (brown staining) detected by immunohistochemistry in nerves (a) and osteoblasts (b) in neonate tibia sections.
To further characterize the expression pattern of Net during differentiation of the osteoblast lineage, POB were isolated from mouse calvariae, differentiated in vitro with ascorbic acid for 21 or 33 days and Net gene expression was quantified by quantitative PCR. Net expression increased during osteoblast differentiation, with a pattern similar to that of Ocn and Sclerostin (Sost, a marker of matrix-embedded osteoblasts/osteocytes) expression (Fig. 1, C–E). In contrast, Adrb2 expression remained constant during differentiation (Fig. 1F). The expression of NET in differentiated osteoblasts was confirmed at the protein level by Western blot analysis using cultures of undifferentiated and differentiated POB (Fig. 1G). In addition, NET immunoreactivity was clearly detected in osteoblasts and cross-sections of nerves in long bone sections of neonatal mice (Fig. 1H). These results indicate that NET expression is not restricted to central and peripheral neurons, and suggest that differentiated osteoblasts have the potential to contribute to NE clearance.
Active NE Transport via NET in Differentiated Osteoblasts
The function of NET in presynaptic nerve terminals is to recapture NE from the synaptic extracellular environment following action potential and NE release. To determine whether NET in differentiated osteoblasts serves a similar function as in nerves, [3H]NE uptake assays were performed using 3 independent bone cell types. [3H]NE uptake was readily detectable in CAD neuronal cells transfected with Net (used here as positive control), but could not be detected in undifferentiated MC3T3/E1 and C3H10T1/2 mesenchymal cells (Fig. 2A), in agreement with the undetectable level of the transporter in undifferentiated POB measured by Western blot (Fig. 1G). However, significant [3H]NE uptake was detected in differentiated osteoblasts, where NET is expressed (Figs. 1, C and G, and 2B). The specificity of this uptake activity was demonstrated by the significant reduction in NE uptake observed upon treatment of WT differentiated POB with the NET blocker desipramine (DMI) (Fig. 2B) and upon using differentiated BMSCs genetically deficient for Net (Fig. 2C). These results indicate that differentiated osteoblasts are equipped with a functional NE transporter and thus may contribute to skeletal NE clearance in vivo.
FIGURE 2.
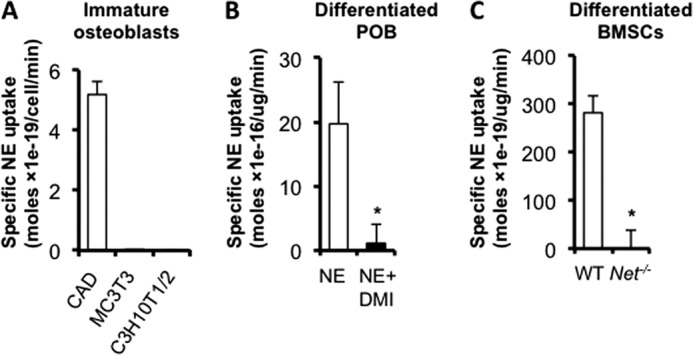
Differentiated osteoblasts transport NE. In vitro uptake assays using: A, non-differentiated osteoblastic cells (CAD: neuronal cell line stably expressing hNET, used as positive control); B, differentiated calvarial POB; and C, differentiated BMSCs. Values are given as mean ± S.E., *, p < 0.05, n = 3.
NET Pharmacological Blockade Reduces Bone Mass
To gain insights into the in vivo relevance of our in vitro findings, we treated WT mice chronically for 8 weeks with reboxetine (15 mg/kg/day), a selective NET blocker, using 28-day releasing subcutaneous mini-osmotic pumps. Pumps were changed once, 28 days following implantation of the first pump. To assess the long-term potency of this chronic treatment, tail suspension tests were performed 21 days following pump implantation, as NET blockade is known to have anti-depressant activity in this behavioral test (20). As expected, a significant reduction in mouse immobility was observed in both male and female reboxetine-treated mice compared with controls (Fig. 3, A and D) indicating that the drug remained active in the pumps. Two months of chronic reboxetine treatment significantly decreased bone mass in both femurs and lumbar vertebrae (Fig. 3, B and C), as assessed by three-dimensional micro-computed tomography (μCT) and two-dimensional static and dynamic bone histomorphometric analyses (Fig. 3G), but surprisingly did so in males predominantly (Fig. 3, B, C, E, and F). Bone formation was significantly decreased in reboxetine-treated males compared with vehicle control mice, as demonstrated by a significant decrease in osteoblast surface/bone surface, bone formation rate/bone surface, and mineral apposition rate (Fig. 3G). Reboxetine treatment induced a trend toward increased osteoclast number and a significant increase in urinary DPD/creatinine (an indicator of osteoclast activity) (control, 8.8 ± 0.6 nmol of DPD/mmol of creatinine versus reboxetine, 10.8 ± 0.5 nmol of DPD/mmol of creatinine, p < 0.05, n = 9). These results indicate that pharmacological blockade of NE transport in vivo induces deleterious effects on bone mass.
FIGURE 3.
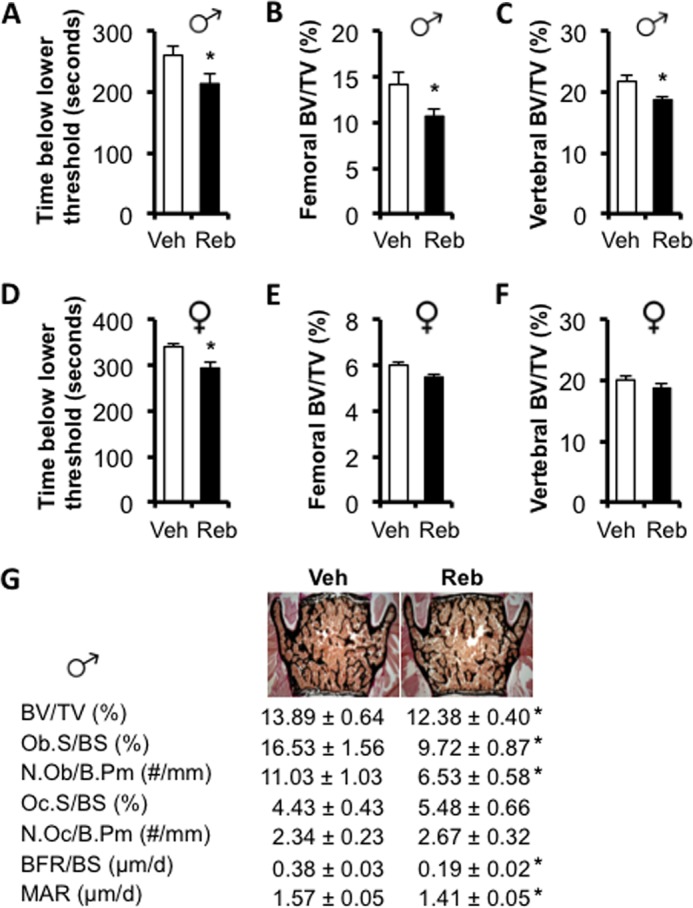
Chronic reboxetine treatment reduces bone mass. Behavioral tail suspension test in 14-week-old male (A) and female (D) mice treated with vehicle (Veh) or reboxetine (Reb). Micro-CT analyses of femoral (B and E) and vertebral (C and F) trabecular bone in male (B and C) and female (E and F) mice. G, histomorphometric analyses of vertebrae in 14-week-old male mice. ObS/BS, osteoblast surface/bone surface; N.Ob/B.Pm, number of osteoblast per bone perimeter; OcS/BS, osteoclast surface/bone surface; N.Oc/B.Pm, number of osteoclast per bone perimeter; BFR/BS, bone formation rate/bone surface; MAR, mineral apposition rate. Values are given as mean ± S.E., *, p < 0.05 versus vehicle, n = 8–10 per group.
Net Deficiency Results in Low Peak Bone Mass and Weakened Bone Mechanical Properties
The use of reboxetine to investigate the contribution of NE reuptake to bone remodeling has several advantages, including its clinical relevance and absence of developmental disturbances, but also drawbacks, including possible stimulation of other receptors and partial potency. Therefore, we embarked in the characterization of Net-deficient (Net−/−) mice through the analysis of multiple architectural, biomechanical, and histological parameters before and after sexual maturity. Net−/− mice weighed less than their wild-type (WT) littermates in both genders (Fig. 4, A and B). This decrease in body weight was not accompanied by changes in body fat or muscle mass over body weight (Fig. 5, A–D). However, a slight but significant reduction in femur length was detected at 3 months of age in male and female Net−/− mice compared with WT controls (Fig. 4, C and D). Distal femoral cancellous bone μCT analyses revealed a significant 15 and 24% decrease in femoral BV/TV in Net−/− mice at 3 and 6 months of age, respectively, compared with WT mice. A similar phenotype was observed in lumbar vertebrae, and it reached significance in males only (Fig. 6, A–D), similarly to what was observed in reboxetine-treated mice. The femoral mid-shaft of Net−/− mice was more slender and the medullary volume was smaller in comparison to WT mice (Table 1, 2). Similar to the trabecular compartment phenotypes, the difference in cortical bone structure (cross-sectional area and moment of inertia) was statistically significant in males only. Consistent with the alterations in bone structure, biomechanical testing indicated that the femurs from male Net−/− mice had weaker mechanical properties than WT littermates, as demonstrated by a significant 20% decrease in bone stiffness and peak force (Fig. 6, E and F).
FIGURE 4.
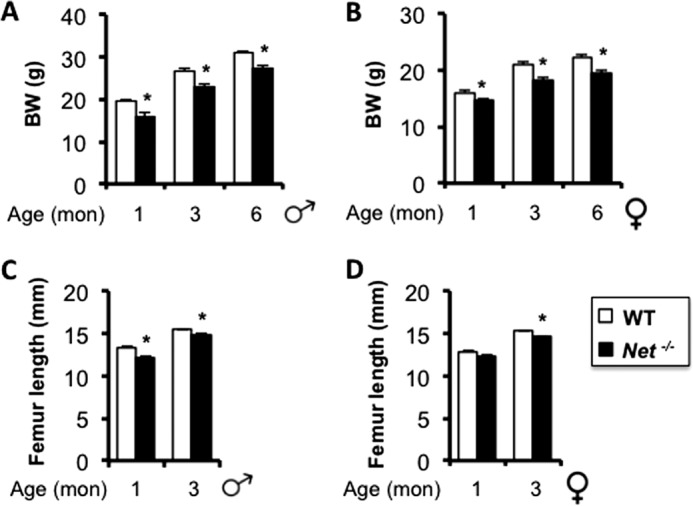
Net−/− mice are leaner and shorter than WT littermates. A and B, body weight (BW); C and D, femur length. Values are given as mean ± S.E., *, p < 0.05 versus WT, n > 8 per group.
FIGURE 5.
Normal body fat or muscle mass over body weight in 3-month-old Net−/− mice. A and B, males; C and D, females. Values are given as mean ± S.E., *, p < 0.05 versus WT, n > 8 per group.
FIGURE 6.
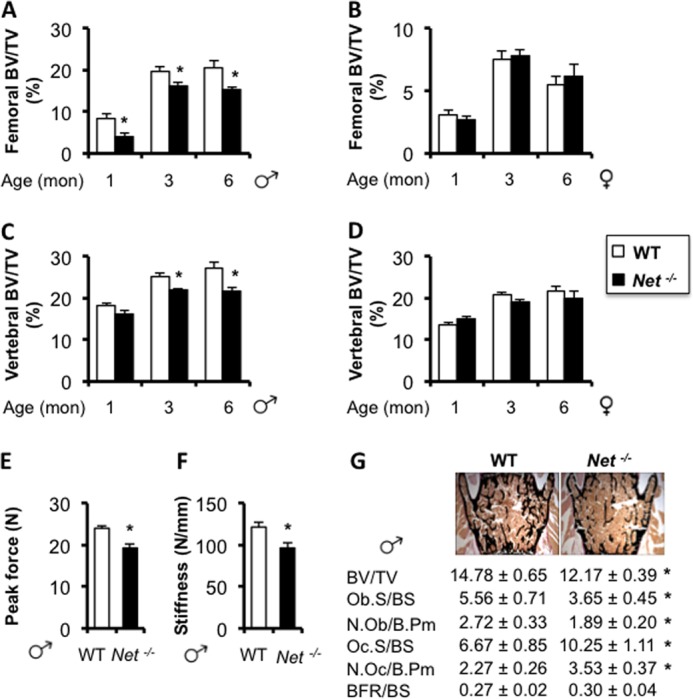
Net deficiency causes a low peak bone mass and reduced bone strength. Micro-CT analyses of femoral (A and B) and vertebral (C and D) trabecular bone in males (A and C) and females (B and D) at 1-, 3-, and 6-month-old ages (BV/TV: bone volume/tissue volume). Peak force (E) and stiffness (F) measured by biomechanical three-point bending tests of femurs from 3-month-old males. G, histomorphometric analyses of vertebrae in 6-month-old male mice. ObS/BS (%), osteoblast surface/bone surface; N.Ob/B.Pm (#/mm), number of osteoblast per bone perimeter; OcS/BS (%), osteoclast surface/bone surface; N.Oc/B.Pm (#/mm), number of osteoclast per bone perimeter; BFR/BS (μm/d), bone formation rate/bone surface. Values are given as mean ± S.E., *, p < 0.05 versus WT, n = 8 per group.
TABLE 1.
Bone cortical structural properties determined by μCT analysis of the femur mid-shaft from 3-month-old male mice (mean ± S.E., n ≥ 7)
| Property | Units | WT | Net−/− | p value |
|---|---|---|---|---|
| Ct.Tha | mm | 0.187 ± 0.003 | 0.181 ± 0.007 | 0.425 |
| Ma.V | mm3 | 1.42 ± 0.03 | 1.09 ± 0.03 | <0.0001 |
| Ct.Ar | mm2 | 0.867 ± 0.018 | 0.741 ± 0.038 | 0.006 |
| Imin | mm4 | 0.151 ± 0.006 | 0.106 ± 0.008 | 0.0005 |
| Slenderness | mm−1 | 7.25 ± 0.2 | 8.68 ± 0.4 | 0.004 |
a Ct.Th, cortical thickness; Ma.V, medullary volume; Ct.Ar, bone cross-sectional area; Imin, moment of inertia; Slenderness, ratio of femur length to total cross-sectional area (within periosteal perimeter).
TABLE 2.
Bone cortical structural properties determined by μCT analysis of the femur mid-shaft from 3-month-old female mice (mean ± S.E., n ≥ 7)
| Property | Units | WT | Net−/− | p value |
|---|---|---|---|---|
| Ct.Tha | mm | 0.16 ± 0.014 | 0.155 ± 0.012 | 0.442 |
| Ma.V | mm3 | 1.08 ± 0.08 | 0.96 ± 0.09 | 0.008 |
| Ct.Ar | mm2 | 0.646 ± 0.075 | 0.585 ± 0.054 | 0.076 |
| Imin | mm4 | 0.084 ± 0.014 | 0.073 ± 0.01 | 0.079 |
| Slenderness | mm−1 | 9.2 ± 0.66 | 9.81 ± 0.69 | 0.083 |
a Ct.Th, cortical thickness; Ma.V, medullary volume; Ct.Ar, bone cross-sectional area; Imin, moment of inertia; Slenderness, ratio of femur length to total cross-sectional area (within periosteal perimeter).
In agreement with the μCT results, we observed a significant (17%) reduction in bone volume/tissue volume (BV/TV) in L3/L4 lumbar vertebral bodies of Net−/− mice compared with control littermates using two-dimensional bone histomorphometry. Osteoclast surface/bone surface was significantly increased in Net−/− mice, whereas osteoblast surface/bone surface was significantly decreased. Osteoclast and osteoblast number/bone perimeter followed the same pattern (Fig. 6G). Bone formation rate, however, did not significantly differ between genotypes. These results indicate that NET is necessary for the acquisition of a normal peak bone mass in both appendicular and axial skeletons, and for optimal bone mechanical properties.
Net−/− Mice Have Reduced Sympathetic Outflow but High Circulating Norepinephrine Levels
The increased osteoclast and decreased osteoblast number observed in Net−/− mice were reminiscent of what had been previously observed upon pharmacological activation of β2AR signaling in mice or rats receiving daily βAR agonist injections (1, 11, 21). These observations suggested that lack of NET leads to overt stimulation of the β2AR expressed in osteoblasts, leading to bone loss. However, independent studies have shown that NE content in nerve terminals in the heart and other sympathetically innervated organs were diminished in Net−/− mice, whereas NE serum levels were increased. These studies supported the notion that extracellular NE level increases and intracellular NE is depleted, in response to lack of NE reuptake and reduced repackaging (16). Consistent with these findings, we could detect a significant decrease in Uncoupling protein 1 (Ucp1) mRNA expression (used here as a readout for reduced sympathetic outflow to peripheral tissues) in brown adipose tissue from Net−/− mice (Fig. 7, A and B) and, most importantly, in bone NE content in 3-month-old Net−/− males and females compared with WT controls (Fig. 7, C and D). These results indicate that low sympathetic outflow is not systematically accompanied by a high bone mass phenotype and suggest that the bone loss observed in Net−/− mice may stem from a rise in serum NE levels resulting from the general lack of NE reuptake by NET.
FIGURE 7.
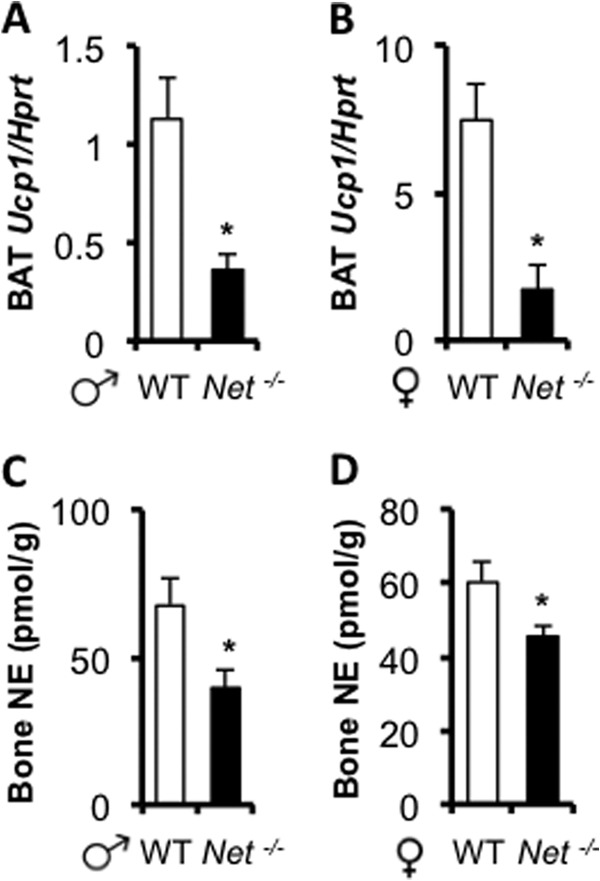
Low sympathetic outflow in Net−/− mice. A and B, brown adipose tissue (BAT) Ucp1 expression measured by quantitative PCR is decreased in Net−/− mice. C and D, Decreased NE content in Net−/− bones. Values are given as mean ± S.E., *, p < 0.05 versus WT, n ≥ 8.
Endogenous Sympathetic Activation by Chronic Immobilization Stress Causes Bone Loss Only When NE Reuptake Is Inhibited
Based on the above in vitro and in vivo observations, we hypothesized that endogenous sympathetic activation may have different repercussions on bone remodeling compared with the administration of exogenous pharmacological βAR agonists (which are not substrates for NET reuptake), and that NE reuptake by differentiated osteoblasts/osteocytes and/or neurons may contribute to the regulation of NE homeostasis and βAR signaling within the skeleton. To address this hypothesis, we subjected 2-month-old WT C57BL6J mice to CIS (daily) for 4 weeks, a time frame long enough to induce bone loss in several conditions such as ovariectomy (22) or in response to isoproterenol treatment (13). CIS has been widely used as a model of transient endogenous sympathetic activation and as an anxiety/depression-like model in mice (23). In contrast to what was observed using daily treatment with βAR agonists, a daily increase in endogenous NE serum levels induced by CIS (24) did not decrease bone volume compared with controls (no CIS), in both femurs and vertebrae, and in both genders (Fig. 8, A–D). These data distinguish the effect of exogenous pharmacological βAR agonists from endogenous NE on bone remodeling, and further support the existence of a homeostatic system that protects the skeleton from bone loss induced by sympathetic activation.
FIGURE 8.
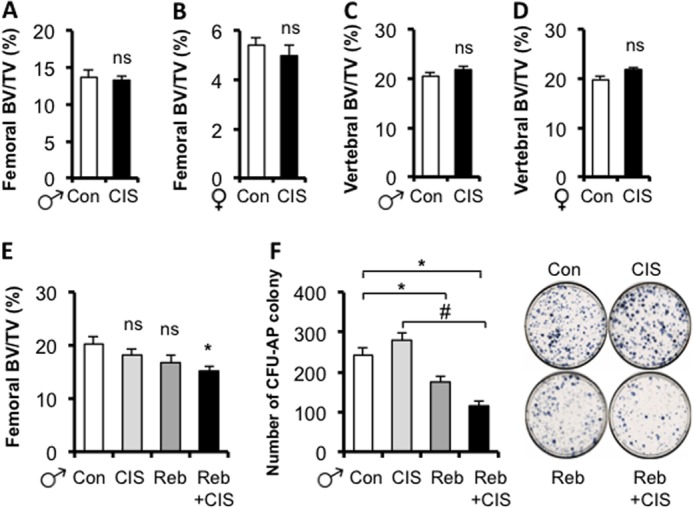
NET blockade is required for chronic immobilization stress-induced bone loss. A–D, CIS does not induce bone loss in 3-month-old WT mice. Micro-CT analyses of femoral (A and B) and vertebral (C and D) trabecular bones in males and females. E, CIS induces bone loss in male WT mice only when NE reuptake is inhibited. F, concurrent CIS and NET blockade reduces the number of bone marrow osteoprogenitors (assessed by the number of Cfu-AP colonies, right panel) compared with the control or CIS groups, following 14 days of in vitro differentiation in osteogenic condition. Values are given as mean ± S.E., *, p < 0.05 versus control; #, p < 0.05 versus CIS, n ≥ 8.
To further address whether NET plays a significant role in this homeostatic system, 5-week-old male WT mice were treated chronically with reboxetine, using subcutaneous mini-osmotic pumps 1 week before mice were subjected to CIS. Following 3 weeks of CIS with or without reboxetine infusion, femoral trabecular bone mass was quantified by μCT, and bone marrow stromal cells were extracted and grown under osteogenic conditions to assess the number of osteoprogenitor cells. As observed before, CIS alone did not induce a significant bone loss, nor did the 4-week-long treatment with reboxetine alone. However, NET blockade by reboxetine in mice subjected to CIS led to a significant 25% bone loss compared with vehicle-treated control mice (no CIS), and to a 16% bone loss (trend only) compared with CIS mice (Fig. 8E). Similarly, the number of bone marrow osteoprogenitor cells was not affected by CIS, but was significantly reduced upon reboxetine or CIS + reboxetine treatment, compared with control or CIS groups (Fig. 8F). These results suggest that concurrent CIS and NET blockade increases adrenergic signaling in bone and further support the notion that NE reuptake by NET is required to conserve a normal bone mass in conditions characterized by chronic sympathetic activation.
DISCUSSION
The regulation of bone remodeling by sympathetic nerves has mainly been investigated by alteration in post-synaptic βAR signaling, pharmacologically or genetically (1, 8, 10–12, 25–33). This study, in contrast, addressed the contribution of endogenous sympathetic signaling and NE homeostasis to the control of bone remodeling. We show that differentiated osteoblasts, like sympathetic presynaptic neurons, can transport and catabolize NE, and thus may contribute to NE clearance within the richly vascularized bone marrow microenvironment. In addition, we report that pharmacological NE transport blockade by reboxetine induces bone loss, and that Net genetic ablation leads to a low peak bone mass. These findings indicate that the control of NE reuptake by NET is an integral part of the homeostatic system whereby bone remodeling is regulated. Along with independent studies suggesting a role of sympatho-inhibitory presynaptic α2ARs and endocannabinoids in the control of bone remodeling (4, 7, 34, 35), these findings also point to the existence of multiple endogenous regulatory pathways modulating bone remodeling via the control of both NE release and clearance. Last, these results suggest that drugs blocking NET activity, which are used for the treatment of depression and ADHD, may have a deleterious effect on the skeleton.
In Net−/− mice, the up-regulation of central NE extracellular levels and α2AR expression, and the subsequent increase in central sympatho-inhibition (36) leads to low sympathetic outflow but increased serum NE levels and low bone mass. Similarly, reduced sympathetic outflow is observed in patients and rats treated with the NET blockers reboxetine or desipramine (37–39). On the other hand, α2A/CAR−/− mice have elevated serum NE levels and increased sympathetic outflow associated with a high bone mass (4). The notion that conditions characterized by low sympathetic outflow systematically lead to bone gain is thus not correct. Collectively, these data warrant further investigations to understand how NE homeostasis in the central nervous system (CNS) and in the skeleton control bone accrual and maintenance. The deleterious effect of Net deficiency on bone mass indicates that NET is required for normal bone homeostasis and for the acquisition of normal bone size and peak bone mass. However, the global and non-inducible nature of this mouse model hampers the characterization of the precise mechanisms by which Net deficiency affects skeletal homeostasis. In addition, it remains unclear whether NET activity in CNS neurons or osteoblasts regulates bone remodeling. A number of observations from the analysis of Net−/− mice, however, provide initial mechanistic clues. First, although sympathetic outflow and NE tissue content are low in Net−/− mice, serum NE levels are increased due to NE spill over triggered by lack of NET activity (16). It is thus possible that the low bone mass of Net−/− mice is caused by increased circulating NE levels, which is in line with the increased bone resorption observed in patients with pheochromocytoma (40) and with the richly vascularized nature of the bone microenvironment. However, the observation that α2A/CAR−/− mice exhibit a high bone mass with elevated serum NE levels would argue against a major role of circulating NE in the regulation of bone remodeling, although a bone cell-autonomous role of the α2A/CAR suggested by this study or developmental phenotypes might explain the bone phenotype of these mutant mice (4). Second, Net−/− mice are characterized by increased central α2AR signaling (due to central accumulation of NE), low sympathetic outflow, and a low bone mass, whereas α2A/CAR−/− mice lacking α2AR signaling display a high bone mass with increased sympathetic outflow (4). Although these two models are limited by the global nature of the gene deletion that characterizes them, their analysis supports the hypothesis that increased central NE levels and central α2AR chronic stimulation causes bone loss. The relative contribution of α2A/CAR and NET in the CNS versus bone cells will thus need to be clarified to select optimal pharmacological αAR andβAR drugs that could increase bone mass. Third, a peripheral and extraneuronal role of NET in bone maintenance is supported by the rich vascularity of the bone microenvironment (hence high blood supply), the uptake of NE by differentiated osteoblasts in vitro, and the uptake of a norepinephrine analog in bone in rats in vivo (41). To what extent skeletal versus CNS or peripheral neuron NET activity contributes to the regulation of bone remodeling remains to be addressed. In addition, the gender specificity (male preferentially) of the bone phenotypes caused by genetic ablation of Net or NE transport blockade by reboxetine suggests that the function of NET may be linked to the biology of sexual hormones. This is in line with a number of clinical studies reporting gender differences in response to NET blockade. For instance, NET inhibition results in more pronounced changes in cardiac regulation in men than women (42), and is associated with higher frequency for sexual and genitourinary treatment-emergent adverse events in males than females (43). There is also evidence of gender difference in sympathetic nervous system regulation, which is in part controlled by the activity of NET in the CNS or in the periphery (44–47). Last, whereas administration of exogenous pharmacologic βAR agonists caused bone loss in multiple studies (8, 12, 13), daily endogenous sympathetic activation induced by mild chronic immobilization stress and the associated intermittent increase in sympathetic outflow and catecholamines did not lead to bone loss in nude immunodeficient mice (48) nor in C57BL6 mice (this study). It is only upon NET blockade that CIS and the associated increase in adrenergic signaling could induce bone loss. Collectively, these data are the first to dissociate the effect of exogenous pharmacologic βAR agonists from endogenous NE on bone remodeling and support the existence of homeostatic systems that protect the skeleton from bone loss induced by increased plasma NE levels and/or overt endogenous sympathetic activation.
From a clinical point of view, the inhibitory effect of chronic NET blockade by reboxetine on bone mass observed in this study lends credence to the hypothesis that children prescribed NET blockers may be at risk of reduced bone accrual during development and pubertal growth, suboptimal peak bone mass in young adults and premature fracture upon aging, because the peak bone mass attained in young adulthood is known as a predictor of later adult bone health (49, 50). ADHD is one of the conditions for which children are prescribed NET blockers. The prevalence of ADHD in the United States is 4.4%. It is more common in males (51) and tends to decrease with age (52). The most commonly prescribed medications include amphetamines (e.g. Adderall®), methylphenidate (e.g. Ritalin® and Concerta®) and atomoxetine (Straterra®), a selective norepinephrine reuptake inhibitor approved by the FDA for the treatment of ADHD in children older than 6 and adults. Atomoxetine is a non-stimulant drug prescribed to patients in case of non-response to other classes of drugs used for the treatment of ADHD, including stimulant drugs. It was put in the United States market in 2002 and possible deleterious effects on the skeleton were not expected and hence not scrutinized. Studies reporting the effect of NET-selective blockers on bone are scarce, include a very limited number of adult patients, and did not detect any significant effect on bone mineral density (53). Observational studies of the effect of NET blockers on fracture risk in patients with ADHD are also challenging as ADHD itself could be associated with increased fracture risk due to the risk-taking behavior associated with ADHD. Although the benefits of NET blockers use to treat ADHD remain undeniable, our observations support the need for assessing bone parameters in children and adults taking NET blockers.
Acknowledgments
We thank Dr. D. Robertson (Vanderbilt University Medical Center) for providing Net+/− mice, Dr. E. Garland and S. Lonce (Vanderbilt University Medical Center) for NE measurements, and Dr. G. Stanwood (Vanderbilt University Medical Center) for the behavioral assays.
This work was supported, in whole or in part, by National Institutes of Health, NIDDK Grant 5R01DK082471-03 and Grant UL1TR000445 from the Clinical and Translational Science Awards (CTSA).
- β2AR
- β2-adrenergic receptor
- NE
- norepinephrine
- NET
- norepinephrine transporter
- ADHD
- attention deficit hyperactivity disorder
- CIS
- chronic immobilization stress
- BMSC
- bone marrow stromal cell
- DPD
- deoxypyridinoline
- POB
- primary osteoblast
- BV/TV
- bone volume/tissue volume.
REFERENCES
- 1. Elefteriou F., Ahn J. D., Takeda S., Starbuck M., Yang X., Liu X., Kondo H., Richards W. G., Bannon T. W., Noda M., Clement K., Vaisse C., Karsenty G. (2005) Leptin regulation of bone resorption by the sympathetic nervous system and CART. Nature 434, 514–520 [DOI] [PubMed] [Google Scholar]
- 2. Franquinho F., Liz M. A., Nunes A. F., Neto E., Lamghari M., Sousa M. M. (2010) Neuropeptide Y and osteoblast differentiation. The balance between the neuro-osteogenic network and local control. FEBS J. 277, 3664–3674 [DOI] [PubMed] [Google Scholar]
- 3. Lee N. J., Herzog H. (2009) NPY regulation of bone remodelling. Neuropeptides 43, 457–463 [DOI] [PubMed] [Google Scholar]
- 4. Fonseca T. L., Jorgetti V., Costa C. C., Capelo L. P., Covarrubias A. E., Moulatlet A. C., Teixeira M. B., Hesse E., Morethson P., Beber E. H., Freitas F. R., Wang C. C., Nonaka K. O., Oliveira R., Casarini D. E., Zorn T. M., Brum P. C., Gouveia C. H. (2011) Double disruption of α2A- and α2C-adrenoceptors results in sympathetic hyperactivity and high-bone-mass phenotype. J. Bone Miner. Res. 26, 591–603 [DOI] [PubMed] [Google Scholar]
- 5. Li H., Fong C., Chen Y., Cai G., Yang M. (2010) β-Adrenergic signals regulate adipogenesis of mouse mesenchymal stem cells via cAMP/PKA pathway. Mol. Cell. Endocrinol. 323, 201–207 [DOI] [PubMed] [Google Scholar]
- 6. Aitken S. J., Landao-Bassonga E., Ralston S. H., Idris A. I. (2009) β2-Adrenoreceptor ligands regulate osteoclast differentiation in vitro by direct and indirect mechanisms. Arch. Biochem. Biophys. 482, 96–103 [DOI] [PubMed] [Google Scholar]
- 7. Huang H. H., Brennan T. C., Muir M. M., Mason R. S. (2009) Functional α1- and β2-adrenergic receptors in human osteoblasts. J. Cell. Physiol. 220, 267–275 [DOI] [PubMed] [Google Scholar]
- 8. Takeda S., Elefteriou F., Levasseur R., Liu X., Zhao L., Parker K. L., Armstrong D., Ducy P., Karsenty G. (2002) Leptin regulates bone formation via the sympathetic nervous system. Cell 111, 305–317 [DOI] [PubMed] [Google Scholar]
- 9. Fu L., Patel M. S., Bradley A., Wagner E. F., Karsenty G. (2005) The molecular clock mediates leptin-regulated bone formation. Cell 122, 803–815 [DOI] [PubMed] [Google Scholar]
- 10. Kajimura D., Hinoi E., Ferron M., Kode A., Riley K. J., Zhou B., Guo X. E., Karsenty G. (2011) Genetic determination of the cellular basis of the sympathetic regulation of bone mass accrual. J. Exp. Med. 208, 841–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bonnet N., Benhamou C. L., Brunet-Imbault B., Arlettaz A., Horcajada M. N., Richard O., Vico L., Collomp K., Courteix D. (2005) Severe bone alterations under β2 agonist treatments. Bone mass, microarchitecture and strength analyses in female rats. Bone 37, 622–633 [DOI] [PubMed] [Google Scholar]
- 12. Bonnet N., Benhamou C. L., Beaupied H., Laroche N., Vico L., Dolleans E., Courteix D. (2007) Doping dose of salbutamol and exercise. Deleterious effect on cancellous and cortical bones in adult rats. J. Appl. Physiol. 102, 1502–1509 [DOI] [PubMed] [Google Scholar]
- 13. Kondo H., Togari A. (2011) Continuous treatment with a low-dose β-agonist reduces bone mass by increasing bone resorption without suppressing bone formation. Calcif. Tissue Int. 88, 23–32 [DOI] [PubMed] [Google Scholar]
- 14. Horvath G., Sutto Z., Torbati A., Conner G. E., Salathe M., Wanner A. (2003) Norepinephrine transport by the extraneuronal monoamine transporter in human bronchial arterial smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 285, L829–837 [DOI] [PubMed] [Google Scholar]
- 15. Ren Z. G., Pörzgen P. P., Youn Y. H., Sieber-Blum M. (2003) Ubiquitous embryonic expression of the norepinephrine transporter. Dev. Neurosci. 25, 1–13 [DOI] [PubMed] [Google Scholar]
- 16. Keller N. R., Diedrich A., Appalsamy M., Tuntrakool S., Lonce S., Finney C., Caron M. G., Robertson D. (2004) Norepinephrine transporter-deficient mice exhibit excessive tachycardia and elevated blood pressure with wakefulness and activity. Circulation 110, 1191–1196 [DOI] [PubMed] [Google Scholar]
- 17. Cryan J. F., Mombereau C., Vassout A. (2005) The tail suspension test as a model for assessing antidepressant activity. Review of pharmacological and genetic studies in mice. Neurosci. Biobehav. Rev. 29, 571–625 [DOI] [PubMed] [Google Scholar]
- 18. Hahn M. K., Mazei-Robison M. S., Blakely R. D. (2005) Single nucleotide polymorphisms in the human norepinephrine transporter gene affect expression, trafficking, antidepressant interaction, and protein kinase C regulation. Mol. Pharmacol. 68, 457–466 [DOI] [PubMed] [Google Scholar]
- 19. Hahn M. K., Steele A., Couch R. S., Stein M. A., Krueger J. J. (2009) Novel and functional norepinephrine transporter protein variants identified in attention-deficit hyperactivity disorder. Neuropharmacology 57, 694–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dziedzicka-Wasylewska M., Faron-Górecka A., Kuśmider M., Drozdowska E., Rogóz Z., Siwanowicz J., Caron M. G., Bönisch H. (2006) Effect of antidepressant drugs in mice lacking the norepinephrine transporter. Neuropsychopharmacology 31, 2424–2432 [DOI] [PubMed] [Google Scholar]
- 21. Pierroz D. D., Bonnet N., Bianchi E. N., Bouxsein M. L., Baldock P. A., Rizzoli R., Ferrari S. L. (2012) Deletion of β-adrenergic receptor 1, 2, or both leads to different bone phenotypes and response to mechanical stimulation. J. Bone miner. Res. 27, 1252–1262 [DOI] [PubMed] [Google Scholar]
- 22. Watkins M. P., Norris J. Y., Grimston S. K., Zhang X., Phipps R. J., Ebetino F. H., Civitelli R. (2012) Bisphosphonates improve trabecular bone mass and normalize cortical thickness in ovariectomized, osteoblast connexin43 deficient mice. Bone 51, 787–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Joo Y., Choi K. M., Lee Y. H., Kim G., Lee D. H., Roh G. S., Kang S. S., Cho G. J., Choi W. S., Kim H. J. (2009) Chronic immobilization stress induces anxiety- and depression-like behaviors and decreases transthyretin in the mouse cortex. Neurosci. Lett. 461, 121–125 [DOI] [PubMed] [Google Scholar]
- 24. Tjurmina O. A., Armando I., Saavedra J. M., Goldstein D. S., Murphy D. L. (2002) Exaggerated adrenomedullary response to immobilization in mice with targeted disruption of the serotonin transporter gene. Endocrinology 143, 4520–4526 [DOI] [PubMed] [Google Scholar]
- 25. Bonnet N., Benhamou C. L., Malaval L., Goncalves C., Vico L., Eder V., Pichon C., Courteix D. (2008) Low dose β-blocker prevents ovariectomy-induced bone loss in rats without affecting heart functions. J. Cell. Physiol. 217, 819–827 [DOI] [PubMed] [Google Scholar]
- 26. Bonnet N., Laroche N., Vico L., Dolleans E., Benhamou C. L., Courteix D. (2006) Dose effects of propranolol on cancellous and cortical bone in ovariectomized adult rats. J. Pharmacol. Exp. Ther. 318, 1118–1127 [DOI] [PubMed] [Google Scholar]
- 27. Kondo H., Nifuji A., Takeda S., Ezura Y., Rittling S. R., Denhardt D. T., Nakashima K., Karsenty G., Noda M. (2005) Unloading induces osteoblastic cell suppression and osteoclastic cell activation to lead to bone loss via sympathetic nervous system. J. Biol. Chem. 280, 30192–30200 [DOI] [PubMed] [Google Scholar]
- 28. Sato T., Arai M., Goto S., Togari A. (2010) Effects of propranolol on bone metabolism in spontaneously hypertensive rats. J. Pharmacol. Exp. Ther. 334, 99–105 [DOI] [PubMed] [Google Scholar]
- 29. Wang T. M., Hsu J. F., Jee W. S., Matthews J. L. (1993) Evidence for reduced cancellous bone mass in the spontaneously hypertensive rat. Bone Miner. 20, 251–264 [DOI] [PubMed] [Google Scholar]
- 30. Gotoh M., Mizuno K., Ono Y., Takahashi M. (2005) High blood pressure, bone-mineral loss and insulin resistance in women. Hypertens. Res. 28, 565–570 [DOI] [PubMed] [Google Scholar]
- 31. Graham S., Hammond-Jones D., Gamie Z., Polyzois I., Tsiridis E., Tsiridis E. (2008) The effect of β-blockers on bone metabolism as potential drugs under investigation for osteoporosis and fracture healing. Expert. Opin. Investig. Drugs 17, 1281–1299 [DOI] [PubMed] [Google Scholar]
- 32. Reid I. R. (2008) Effects of β-blockers on fracture risk. J. Musculoskelet. Neuronal Interact. 8, 105–110 [PubMed] [Google Scholar]
- 33. Bonnet N., Gadois C., McCloskey E., Lemineur G., Lespessailles E., Courteix D., Benhamou C. L. (2007) Protective effect of β blockers in postmenopausal women. Influence on fractures, bone density, micro and macroarchitecture. Bone 40, 1209–1216 [DOI] [PubMed] [Google Scholar]
- 34. Cuscito C., Colaianni G., Tamma R., Greco G., Dell'Endice S., Yuen T., Sun L., Zaidi M., Di Benedetto A., Zallone A. (2011) Adrenergic stimulation decreases osteoblast oxytocin synthesis. Ann. N.Y. Acad. Sci. 1237, 53–57 [DOI] [PubMed] [Google Scholar]
- 35. Tam J., Trembovler V., Di Marzo V., Petrosino S., Leo G., Alexandrovich A., Regev E., Casap N., Shteyer A., Ledent C., Karsak M., Zimmer A., Mechoulam R., Yirmiya R., Shohami E., Bab I. (2008) The cannabinoid CB1 receptor regulates bone formation by modulating adrenergic signaling. FASEB J. 22, 285–294 [DOI] [PubMed] [Google Scholar]
- 36. Gilsbach R., Faron-Górecka A., Rogóz Z., Brüss M., Caron M. G., Dziedzicka-Wasylewska M., Bönisch H. (2006) Norepinephrine transporter knockout-induced up-regulation of brain α2A/C-adrenergic receptors. J. Neurochem. 96, 1111–1120 [DOI] [PubMed] [Google Scholar]
- 37. Tank J., Schroeder C., Diedrich A., Szczech E., Haertter S., Sharma A. M., Luft F. C., Jordan J. (2003) Selective impairment in sympathetic vasomotor control with norepinephrine transporter inhibition. Circulation 107, 2949–2954 [DOI] [PubMed] [Google Scholar]
- 38. Carson R. P., Diedrich A., Robertson D. (2002) Autonomic control after blockade of the norepinephrine transporter. A model of orthostatic intolerance. J. Appl. Physiol. 93, 2192–2198 [DOI] [PubMed] [Google Scholar]
- 39. Bertram D., Barrès C., Cheng Y., Julien C. (2000) Norepinephrine reuptake, baroreflex dynamics, and arterial pressure variability in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 279, R1257–1267 [DOI] [PubMed] [Google Scholar]
- 40. Veldhuis-Vlug A. G., El Mahdiui M., Endert E., Heijboer A. C., Fliers E., Bisschop P. H. (2012) Bone resorption is increased in pheochromocytoma patients and normalizes following adrenalectomy. J. Clin. Endocrinol. Metab. 97, E2093–2097 [DOI] [PubMed] [Google Scholar]
- 41. Lin S. F., Fan X., Yeckel C. W., Weinzimmer D., Mulnix T., Gallezot J. D., Carson R. E., Sherwin R. S., Ding Y. S. (2012) Ex vivo and in vivo evaluation of the norepinephrine transporter ligand [11C]MRB for brown adipose tissue imaging. Nucl. Med. Biol. 39, 1081–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schroeder C., Adams F., Boschmann M., Tank J., Haertter S., Diedrich A., Biaggioni I., Luft F. C., Jordan J. (2004) Phenotypical evidence for a gender difference in cardiac norepinephrine transporter function. Am. J. Physiol. Regul. Integr. Comp. Physiol. 286, R851–856 [DOI] [PubMed] [Google Scholar]
- 43. Camporeale A., Day K. A., Ruff D., Arsenault J., Williams D., Kelsey D. K. (2013) Profile of sexual and genitourinary treatment-emergent adverse events associated with atomoxetine treatment. A pooled analysis. Drug Saf. 36, 663–671 [DOI] [PubMed] [Google Scholar]
- 44. Hinojosa-Laborde C., Chapa I., Lange D., Haywood J. R. (1999) Gender differences in sympathetic nervous system regulation. Clin. Exp. Pharmacol. Physiol. 26, 122–126 [DOI] [PubMed] [Google Scholar]
- 45. Hart E. C., Charkoudian N., Wallin B. G., Curry T. B., Eisenach J. H., Joyner M. J. (2009) Sex differences in sympathetic neural-hemodynamic balance. Implications for human blood pressure regulation. Hypertension 53, 571–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Moldovanova I., Schroeder C., Jacob G., Hiemke C., Diedrich A., Luft F. C., Jordan J. (2008) Hormonal influences on cardiovascular norepinephrine transporter responses in healthy women. Hypertension 51, 1203–1209 [DOI] [PubMed] [Google Scholar]
- 47. Heal D. J., Bristow L. M., Hurst E. M., Elliott J. M., Buckett W. R. (1989) Sex-related differences in central adrenergic function and responsiveness to repeated administration of desipramine or electroconvulsive shock. Br. J. Pharmacol. 97, 111–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Campbell J. P., Karolak M. R., Ma Y., Perrien D. S., Masood-Campbell S. K., Penner N. L., Munoz S. A., Zijlstra A., Yang X., Sterling J. A., Elefteriou F. (2012) Stimulation of host bone marrow stromal cells by sympathetic nerves promotes breast cancer bone metastasis in mice. PLoS Biol. 10, e1001363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Johnston C. C., Jr., Longcope C. (1990) Premenopausal bone loss. A risk factor for osteoporosis. N. Engl. J. Med. 323, 1271–1273 [DOI] [PubMed] [Google Scholar]
- 50. Matkovic V., Jelic T., Wardlaw G. M., Ilich J. Z., Goel P. K., Wright J. K., Andon M. B., Smith K. T., Heaney R. P. (1994) Timing of peak bone mass in Caucasian females and its implication for the prevention of osteoporosis. Inference from a cross-sectional model. J. Clin. Investig. 93, 799–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kessler R. C., Adler L., Barkley R., Biederman J., Conners C. K., Demler O., Faraone S. V., Greenhill L. L., Howes M. J., Secnik K., Spencer T., Ustun T. B., Walters E. E., Zaslavsky A. M. (2006) The prevalence and correlates of adult ADHD in the United States. Results from the National Comorbidity Survey Replication. Am. J. Psychiatry 163, 716–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Simon V., Czobor P., Bálint S., Mészáros A., Bitter I. (2009) Prevalence and correlates of adult attention-deficit hyperactivity disorder. Meta-analysis. Br. J. Psychiatry 194, 204–211 [DOI] [PubMed] [Google Scholar]
- 53. Ziere G., Dieleman J. P., van der Cammen T. J., Hofman A., Pols H. A., Stricker B. H. (2008) Selective serotonin reuptake inhibiting antidepressants are associated with an increased risk of nonvertebral fractures. J. Clin. Psychopharmacol. 28, 411–417 [DOI] [PubMed] [Google Scholar]