

Background: Amyloid precursor protein (APP) and ADAM10, main α-secretase involved in generation of secreted APP (sAPPα), are overexpressed in pancreatic cancer.
Results: Inhibition of APP processing by ADAM10 prevents anchorage independent growth and survival of cancer cells.
Conclusion: Inhibition of sAPPα generation enhances chemotherapeutic potential of gemcitabine.
Significance: Supplementing established pancreatic cancer therapy regimen with sAPPα inhibition will significantly improve the efficacy of treatment.
Keywords: ADAM ADAMTS, Amyloid Precursor Protein, Pancreatic Cancer, Protein Secretion, Secretases
Abstract
Pancreatic adenocarcinoma or pancreatic cancer is often diagnosed at a very late stage at which point treatment options are minimal. Current chemotherapeutic interventions prolong survival marginally, thereby emphasizing the acute need for better treatment options to effectively manage this disease. Studies from different laboratories have shown that the Alzheimer disease-associated amyloid precursor protein (APP) is overexpressed in various cancers but its significance is not known. Here we sought to determine the role of APP in pancreatic cancer cell survival and proliferation. Our results show that pancreatic cancer cells secrete high levels of sAPPα, the α-secretase cleaved ectodomain fragment of APP, as compared with normal non-cancerous cells. Treatment of cells with batimastat or GI254023X, inhibitors of the α-secretase ADAM10, prevented sAPPα generation and reduced cell survival. Additionally, inhibition of sAPPα significantly reduced anchorage independent growth of the cancer cells. The effect of batimastat on cell survival and colony formation was enhanced when sAPPα downregulation was combined with gemcitabine treatment. Moreover, treatment of batimastat-treated cells with recombinant sAPPα reversed the inhibitory effect of the drug thereby indicating that sAPPα can indeed induce proliferation of cancer cells. Down-regulation of APP and ADAM10 brought about similar results, as did batimastat treatment, thereby confirming that APP processing is important for growth and proliferation of these cells. These results suggest that inhibition of sAPPα generation might enhance the effectiveness of the existing chemotherapeutic regimen for a better outcome.
Introduction
Pancreatic cancer is the fourth leading cause of cancer-related deaths in the United States and shows poor prognosis and a survival rate of less than 5% (1). This is attributed mainly to the asymptomatic progression of the disease with aggressive growth and resistance of the cancer cells to conventional chemotherapy and radiation (1, 2). At present, the nucleoside analog, gemcitabine, is the prescribed standard drug for disease management despite the dismal survival rates (3, 4). Several other compounds such as 5-FU, Irinotecan, and premetrexed were tested as potential chemotherapeutic agents but they did not increase the overall survival of patients as compared with treatment with gemcitabine alone (5–8). Therefore, much of the focus in the field of pancreatic cancer research involves identification of new molecular targets for development of more effective drugs and drug combinations.
Recent studies have shown that the transmembrane protein, amyloid precursor protein (APP),2 is overexpressed in pancreatic cancer (9, 10). APP is the major protein implicated in the pathogenesis of Alzheimer disease (AD) mainly due to the deposition of plaque-forming amyloid β (Aβ) oligomers (11, 12). APP is cleaved by secretases to produce fragments with different biological functions (13) (Fig. 1A). Proteolysis can follow the amyloidogenic or non-amyloidogenic pathway based on the secretases that cleave APP. The amyloidogenic pathway involves sequential cleavage of APP by β-secretase, BACE, and γ-secretase to generate secreted APPβ (sAPPβ), pathogenic Aβ and intracellular APP domain, AICD (14, 15). In the non-amyloidogenic pathway, no Aβ is formed. Instead, APP is cleaved by α-secretase and γ-secretase to produce secreted APPα (sAPPα), the P3 fragment, and AICD (16). These two pathways are mutually exclusive and it has been suggested that cleavage of APP by α-secretase, ADAM10 (a disintegrin and metalloprotease domain 10), may down-regulate Aβ generation and have beneficial effects on AD pathology (Fig. 1A) (17).
FIGURE 1.
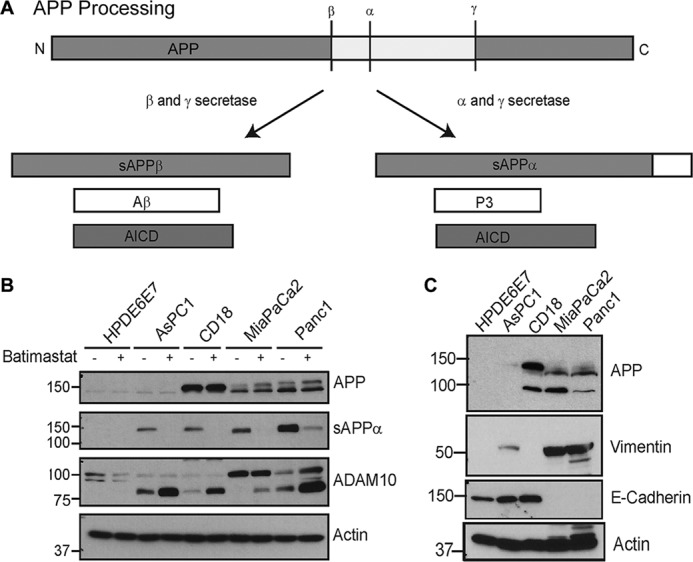
Pancreatic cancer cells show enhanced APP expression and processing. A, schematic showing APP processing by β- and γ-secretases, which follows the amyloidogenic pathway to produce Aβ, sAPPβ (secreted β-secretase cleaved APP), and AICD (APP intracellular domain) or the non-amyloidogenic processing by α and γ-secretases to generate sAPPα, P3 (small fragment generated by α and γ-secretase cleavage of APP), and AICD. B, pancreatic cancer cell lines AsPC1, CD18, MiaPaCa2, and Panc1 were compared with normal pancreatic epithelial cells, HPDE6E7, for expression and secretion of sAPPα and expression of ADAM10 in the presence or absence of batimastat. C, analysis of epithelial and mesenchymal markers in different pancreatic cancer cell lines. Vimentin is shown in panel 2 and E-cadherin in panel 3. Actin was used as loading control.
Although ADAM17, another member of the ADAM family of proteases, has been shown to cleave APP to generate sAPPα, studies have shown that ADAM10 is the most physiologically relevant α-secretase that constitutively cleaves APP at this particular site (16, 18–20). In addition to APP, ADAM10 has been shown to cleave other substrates such as Notch, cadherins, β-catenin, etc. (21–23). Several studies have indicated that up-regulation of ADAM10 might be beneficial in AD pathology, not only because preferential cleavage of APP by ADAM10 inhibits the amyloidogenic proteolysis of APP and Aβ generation, but also because the sAPPα fragment has antiapoptotic and neuroprotective effect on cells such that it can counter the degenerative effects mediated by Aβ (24, 25). In the case of cancer, however, molecules that promote proliferation and growth are undesirable and therapy involves the down-regulation of the levels or activity of such molecules. ADAM10 has been found to be up-regulated in pancreatic cancer and shown to promote migration and invasion of cancer cells (26). In addition, studies in epithelial cells have shown that secreted APP at nanomolar concentrations enhances cell proliferation and migration (27). Based on these findings, we hypothesize that ADAM10-mediated generation of sAPPα may enhance growth and proliferation of pancreatic cancer cells.
Here we have used different pancreatic cancer cell lines and examined the effect of inhibition of sAPPα generation on cancer cell proliferation and growth. Results presented here show that, as compared with an MMP inhibitor GM6001, compounds such as batimastat and GI254023X, which are more specific inhibitors of ADAM10, robustly inhibit cancer cell survival. At low concentrations, these drugs not only abolish generation of sAPPα, but also inhibit anchorage independent growth and cell proliferation in pancreatic cancer cells. This inhibitory effect is enhanced when ADAM10 inhibition is combined with gemcitabine treatment. These results indicate an important role of APP in promoting growth of pancreatic ductal adenocarcinomas (PDACs) and suggest that inhibitors of sAPPα generation may increase the efficacy of the current therapies used for treating pancreatic cancer.
EXPERIMENTAL PROCEDURES
Cell Lines
Pancreatic cancer cell lines CD18, MiaPaCa2, AsPC1, and Panc1 as well as normal pancreatic cell line, HPDE6E7, were either kindly provided by Dr. Srikumar Chellappan (Moffitt Cancer Research Center, Tampa, FL) or obtained from ATCC. These cell lines were maintained in either DMEM (GIBCO, catalogue no. 10569) supplemented with 10% fetal bovine serum (Atlanta Biologicals, catalogue no. S11050), and 1% penicillin-streptomycin or in the case of HPDE6E7 (normal immortalized pancreatic cell line), in Keratinocyte Serum-free Media (GIBCO, catalogue no. 10724-011) supplemented with bovine pituitary extract and recombinant epidermal growth factor. Phoenix cells were kindly gifted by Dr. Nicholas Woods (Monteiro Lab, Moffitt Cancer Research Center, Tampa, FL) and maintained in DMEM with FBS and antibiotics.
Plasmids, Reagents, and Transfections
C-terminal Myc-tagged ADAM10 in pRK5M vector was obtained from Derynck laboratory (Addgene). Untagged ADAM10 in pCDNA3 was kindly provided by Paul Saftig (Biochemisches Institut, Germany). Batimastat was purchased from Tocris Biosciences (catalogue no. 2961) and dissolved in DMSO at a stock concentration of 10 mm. Gemcitabine was kindly gifted by Dr. Srikumar Chellapan (Moffitt Cancer Center, Tampa, FL) and dissolved in DMSO at a stock concentration of 4.15 mg/ml. GM6001 was purchased from Enzo Life sciences (catalogue no. BML-EI300) and reconstituted in DMSO at a stock concentration of 25 mm. GI254023X was purchased from Okeanos Tech. Co., China and reconstituted in DMSO at a stock concentration of 20 mm. Recombinant secreted APP (sAPPα) was purchased from Sigma (catalogue no. S9564). Control siRNA, APP siRNA as well as ADAM10 siRNA (sc-37007, sc-29677, and sc-41410, respectively) were purchased from Santa Cruz Biotechnology. ADAM17 siRNA was purchased from Dharmacon, siGENOME SMARTPOOL (catalogue no. M-003453). Reagent for siRNA transfection was purchased from Santa Cruz Biotechnology (sc-29528).
For knockdown of APP, ADAM10, or ADAM17, siRNA to the respective mRNA were transfected into cells using protocol described by the manufacturer (Santa Cruz Biotechnology). Briefly, cells were plated to 70% confluency in 24-well dishes. In a sterile tube, desired concentration of control or target siRNA was diluted in 25 μl of OPTIMEM. In a separate tube, 2.5 μl transfection reagent was diluted in 25 μl of OPTIMEM. The contents of the two tubes were mixed together and incubated at room temperature for 15–45 min. Then, 150 μl of OPTIMEM was added to the mixture and overlaid on washed cells and incubated at 37 °C for 6 h. At the end of 6 h, equal volume of OPTIMEM containing 2× concentration of FBS was added to the cells and incubated overnight. Next day, the media was replaced with fresh regular medium containing 10% FBS and antibiotics. Cells were further incubated for 24 to 48 h at 37 °C and then collected for analysis.
For overexpression of proteins, transfections were performed using calcium phosphate transfection method. Cells were grown to 90% confluency in a 10 cm dish 24 h before transfection. Appropriate plasmids were mixed in 400 μl of water and added to 70 μl of 2.5 m calcium chloride. The entire mixture was added dropwise with bubbling to 500 μl of 2× HEPES Buffered Saline (16.4 g NaCl, 11.9 g HEPES acid, 0.21 g Na2HPO4 in 1 liter of water and pH adjusted to 7.05) and the resulting ∼1 ml solution was added gently to the media of the cells and incubated overnight. Next day, the media was aspirated and replaced with fresh media and cells were incubated further for 24 h at 37 °C. At the end of this period, efficiency of transfection was determined by resolving the cell lysates on SDS-PAGE and probing for protein expression of the transfected plasmids.
Western Blotting and Antibodies
Cells were scraped in tissue culture supernatant and the entire contents were transferred from the plate into tubes and centrifuged at 4 °C for 5 min 600 × g. 100 μl of the supernatant was boiled with Laemmli Buffer for 5 min. Cell pellet was washed once with PBS followed by lysis in 1× RIPA buffer (150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, and 100 mm Tris, pH 8.0) containing protease inhibitor mixture (Roche, catalogue no. 04693124001), 1 mm sodium fluoride, 1 mm sodium orthovanadate, and 1 mm PMSF. Protein estimation was performed against a BSA standard using the Coomassie Plus-Bradford Assay Reagent (Thermo Scientific, catalogue no. 23238). 20 μg of protein was loaded on a 12% SDS-PAGE gel and transferred to a PVDF membrane (Millipore, catalogue no. IPVH00010). Blots were blocked in 5% milk in TBST (TBS, 0.05% Tween 20) for 1 h and incubated overnight at 4 °C in primary antibody diluted in blocking solution. The blots were then washed three times with PBST (PBS, 0.05% Tween-20) for 10 min each and incubated in secondary antibody at room temperature for 2 h with gentle shaking. Subsequently, the blots were washed 5 times, 6 min each with PBST. Protein detection was achieved using Supersignal West Pico Luminol reagent (Thermo Scientific, catalogue no. 1856136). Primary antibodies used were rabbit polyclonal ADAM10 from Millipore (catalogue no. AB19026) at a dilution of 1:1000, 6E10 for APP from Covance (catalogue no. 39320) at a dilution of 1:1000, actin from Sigma (catalogue # A5316) at a dilution of 1:10,000, PARP antibody from Millipore (catalogue no. AB16661) at a dilution of 1:1000, ADAM17 antibody from Abcam (catalogue no. 2051) at a dilution of 1:3000, Na/K ATPase antibody from Santa Cruz (catalogue no. 21712) at a dilution of 1:1500, and anti-Myc antibody from Millipore (catalogue no. 05-724) at a dilution of 1:1000. Secondary antibodies were purchased from Thermo Scientific, Immunopure Donkey anti-Rabbit IgG-HRP (catalogue no. 31458) at 1:5000 dilution and Immunopure Goat anti-mouse IgG-HRP (catalogue no. 31430) at 1:5000 dilution.
Cell Toxicity Assay
Thiazoyl Blue Tetrazolium Bromide (MTT) (Sigma, catalogue no. M2128) was used for the cytotoxicity assay. 10,000 cells were plated per well in a 24-well plate and allowed to attach overnight. The next day, 400 μl of complete medium containing 5 μm batimastat, 5 μm GI254023X, 100 ng/ml gemcitabine, 25 μm GM6001, or combinations of the drugs were added to the cells and incubated at 37 °C for 24 h. At the end of this period, MTT at a concentration of 1 mg/ml in complete media was added to the wells and incubated for an additional 3 h. Purple formazan crystals formed in viable cells were solubilized in isopropanol containing 4 mm hydrochloric acid and 0.1% Nonidet P-40 with vigorous shaking at room temperature for 20 min. Absorbance was measured at 540 nm to compare the intensity of color between control and drug-treated cells. Higher absorbance reflects more viable cells and less cell death. Each treatment was done in quadruplicates, and each experiment was performed at least three times.
Colony Formation Assay
To determine the effect of drugs on anchorage independent growth of cells, soft agar assays were performed. A 3% sterile agar solution was mixed with complete media at 45 °C to obtain a final concentration of 0.6%. 2 ml of this mixture was introduced in each well of a 12-well plate, which formed the base layer. For the top layer, 5,000 cells along with the appropriate drug concentrations were mixed with the 3% agar solution to obtain a final concentration of 0.3% agar and layered over the base layer in each well. Thus seeded, these cells will have the opportunity to grow and form colonies in the top layer of the soft agar. The wells were re-fed with drug-containing media once a week to prevent drying, and at the end of 2.5 weeks, the colonies were visualized by staining with MTT at a final concentration of 1 mg/ml for 5 h at 37 °C. Each treatment was performed in triplicate and repeated at least three times.
Zymography for Analysis of MMP Activity
Gelatin Zymogram gels (10%) were purchased from Novex Invitrogen (catalogue no. EC61755BOX). 7 μl of media (supernatant) from control or treated cells were incubated with 2× Sample Buffer (125 mm Tris-Cl, pH 8.0, 20% glycerol, 4% SDS, 0.05% Bromphenol Blue) at room temperature for 10 min and loaded on the gels. Tris-glycine running buffer was used to run the gels at a constant voltage of 125 V until the tracking dye reached the bottom of the gel. The gel was incubated in renaturing buffer (2.5% Triton X-100) for 30 min at room temperature with shaking. This was followed by equilibration of gel in developing buffer (50 mm Tris-Cl, pH 7.5, 0.2 m NaCl, 5 mm CaCl2, 0.02% Brij-35) for 30 min at room temperature with gentle shaking. After equilibration, the developing buffer was decanted and replaced with fresh developing buffer and incubated at 37 °C overnight. Next day, the gel was stained with Coomassie Blue R-250 (0.5% Coomassie Blue R-250, 20% methanol, and 10% acetic acid) for 30 min at room temperature. Gels were destained with destaining solution (50% methanol, 10% acetic acid), and clear bands of MMP activity were observed against the blue background.
Cell Surface Protein Isolation
To determine membrane association of ADAM10 and APP, cell surface protein isolation was performed using the Pierce Cell Surface Protein Isolation Kit following the manufacturer's protocol (Thermo Scientific, catalogue no. 89881). Briefly, drug treated and untreated cells were washed and incubated with Sulfo-NHS-SS-Biotin solution for 30 min at 4 °C to biotinylate cell surface proteins followed by addition of quencher to quench the reaction. Cells were harvested and lysed in Lysis Buffer with protease inhibitors. Cell lysate was centrifuged at 10,000 × g for 10 min at 4 °C, and clarified supernatant was collected. The supernatant was incubated with NeutrAvidin Agarose beads for 1 h at room temperature. This was followed by several washes of the beads with Wash Buffer supplied with the kit. At the end of the washes, biotinylated proteins bound to the NeutrAvidin beads were eluted by incubating the beads with SDS-PAGE sample buffer containing DTT for 1 h at room temperature. Eluted proteins were resolved on an SDS-PAGE gel and APP, ADAM10, and Na/K ATPase were detected by Western blot using appropriate antibody.
Deglycosylation Analysis
To determine the glycosylation status of ADAM10 in batimastat-treated cells, we performed experiments using a deglycosylation kit (catalogue no. 9PIV493) from Promega by following the manufacturer's protocol. The kit contains a mix of deglycosidases capable of removing both N- and O-linked glycans from proteins. Briefly, Phoenix cells were transfected with myc-ADAM10 and 48 h after transfection cells were treated with or without batimastat (5 μm) for 24 h and lysates were prepared in RIPA buffer. Samples were also prepared from MiaPaCa2 cells treated with or without 5 μm batimastat. 40 μg of protein from each sample was mixed with 10× denaturing solution to a final volume of 20 μl, and the mixture was boiled at 95 °C for 10 min. Samples were then incubated on ice for 5 min followed by addition of 10% Nonidet P-40, deglycosylation reaction buffer and the protein deglycosylation mix (PNGaseF, O-glycosidase, neuraminidase, β1–4 galactosidase, β-N-acetylglucosaminidase). Deglycosylation was carried out at 37 °C for 18 h, and the control as well as deglycosylated samples were analyzed by Western blot using C-terminal ADAM10 antibody.
RESULTS
Pancreatic Cancer Cells Express High Levels of APP and ADAM10
Pancreatic cancer cell lines CD18, MiaPaCa2, AsPC1, and Panc1 were compared with non-cancerous, immortalized HPDE6E7 pancreatic epithelial cells for APP and ADAM10 levels by Western blotting and it was found that most pancreatic cancer cells express higher levels of APP and ADAM10. These cells also showed higher levels of secreted APP, sAPPα, as compared with the normal HPDE6E7 (Fig. 1B). These findings are in agreement with previous reports (9, 26). This suggests an association of increased levels of APP and ADAM10 with pancreatic cancer. Treatment of cells with the ADAM10 inhibitor, batimastat, at 5 μm concentration inhibited the generation of sAPPα significantly (Fig. 1B, panel 2). In addition, ADAM10 blots showed an increase in the levels of a band around ∼80 kDa upon treatment with batimastat (Fig. 1B, panel 3), which appears to be an alternative form of ADAM10. In an attempt to characterize the properties of the different pancreatic cancer cells, we analyzed them for EMT (epithelial mesenchymal markers) such as E-cadherin and vimentin (Fig. 1C). Expression of E-cadherin suggested that AsPC1 and CD18 cell lines exhibit an epithelial phenotype similar to the normal immortalized HPDE6E7 cells. MiaPaCa2 and Panc1 cells expressed vimentin, which is an indicator of mesenchymal phenotype. This predicts that MiaPaCa2 and Panc1 cells will likely be more resistant to chemotherapy and more aggressive in their growth and proliferation properties as compared with CD18. AsPC1 seemed to express high levels of E-cadherin and low levels of vimentin, which suggests that it is mostly epithelial but has some degree of mesenchymal property.
Batimastat Inhibits ADAM10 and Down-regulates sAPPα Generation
To confirm that batimastat is a better drug of choice for inhibition of ADAM10-mediated sAPPα generation, MiaPaCa2 and Panc1 cells were treated with 5 μm batimastat or with a general MMP inhibitor, GM6001, at 25 μm concentration for 24 h. Both cells and media (supernatant) were collected for Western blot analysis. Treatment with batimastat showed a more effective inhibition of sAPPα in the tissue culture supernatant as compared with GM6001 thereby indicating the specificity of batimastat against ADAM10 (Fig. 2A, top panel). Supernatants of MiaPaCa2 cells treated with either 5 μm batimastat or 25 μm GM6001 were analyzed using a gelatin zymogram to examine the activation of matrix metalloproteases. Batimastat did not inhibit the cleavage and formation of mature MMPs as effectively as GM6001 as a result of which the pro-MMP form at 92kDa (Fig. 2B) is clearly visible in GM6001-treated cells and not in batimastat-treated cells. This suggests that batimastat has very little effect on other MMPs. Cell lysates from the corresponding samples probed with anti-actin antibody served as the loading control for the zymography data (lower panel, Fig. 2B). Additionally, GI254023X, which is a more specific inhibitor of ADAM10 (28), was used to treat MiaPaCa2 cells, and its effect was compared with that of batimastat on these cells (Fig. 2C). Western blot analysis clearly indicates that the two drugs are equally effective in inhibiting the generation of sAPPα and promoting the formation of the ∼80 kDa ADAM10 form. Moreover, neither of the two drugs showed an effect on ADAM17 cleavage or levels, thereby suggesting that both drugs are specific inhibitors of ADAM10. Because of limited availability of the GI254023X compound, majority of our experiments were performed with batimastat for the purpose of inhibiting sAPPα.
FIGURE 2.

Batimastat inhibits ADAM10 and down-regulates sAPPα generation. A, MiaPaCa2 and Panc1 cells were treated with 5 μm batimastat or 25 μm GM6001. Tissue culture supernatant was blotted for sAPPα using 6E10 antibody, and cell extract was analyzed for ADAM10 levels (light and dark exposures of blot are shown). Actin was used as loading control. B, zymography was performed on a 10% gelatin zymogram with tissue culture supernatant of untreated MiaPaCa2 cells or those treated with either 5 μm batimastat or 25 μm GM6001. Cell lysates from the corresponding samples used for zymography were blotted for actin to ensure equal loading. C, MiaPaCa2 cells were treated with 5 μm batimastat or 5 μm GI254023X. Tissue culture supernatant was blotted for sAPPα using 6E10 antibody (top panel), and cell lysate was analyzed for ADAM10 (panel 2) and ADAM17 (bottom panel) levels. Actin was used as loading control (panel 3). D, MiaPaCa2 cells were treated with 5 μm batimastat, 100 ng/ml gemcitabine or both, and ADAM10 levels were analyzed by Western blotting. An antibody directed against the C terminus of ADAM10 was used to detect the different forms of ADAM10. Actin was used as loading control. E, phoenix cells were transfected with either untagged pcDNA3-ADAM10 or C-terminally Myc-tagged pRK5M-ADAM10 and cell lysates were used to detect the different forms of ADAM10 (light and dark exposures are shown). Myc antibody was used to detect the expression of Myc-tagged ADAM10 (myc, panel 4), and actin was used as loading control (actin, panel 3). Tissue culture supernatant was used to detect the levels of secreted sAPPα using 6E10 antibody (panel 5, sAPPα). F, sAPPα levels were quantified using ImageJ software from three independent experiments and normalized to actin. G, MiaPaCa2 and myc-ADAM10 transfected Phoenix cells were treated with or without 5 μm batimastat, samples were prepared in RIPA lysis buffer and deglycosylated following the manufacturer's protocol. Western blot analysis of samples was performed with ADAM10 C-terminal antibody. Actin was used as a loading control. H, ADAM10 levels in total cell lysate of Phoenix cells transfected with pcDNA3 or Myc-tagged pRK5 ADAM10 before immunoprecipitation with NeutrAvidin was detected by ADAM10 antibody. Actin was used as loading control. I, phoenix cells transfected with pcDNA3 or myc-tagged pRK5M-ADAM10 were analyzed using anti-Myc antibody for membrane bound ADAM10 fragments in the presence or absence of batimastat using biotinylation-based cell surface protein isolation (top and second panels). Na/K ATPase was used as loading control (third panel). 6E10 antibody was used to detect membrane-associated APP levels (bottom panel). J, to confirm that the ∼80 kDa band we observed in the pancreatic cancer cells is a form of ADAM10, we transfected MiaPaCa2 cells with a control or ADAM10 siRNA, performed biotinylation-based membrane isolation and analyzed the samples by Western blot using C-terminal ADAM10 antibody. Na/K ATPase was used as loading control (lower panel).
Upon analysis of the blots with an ADAM10 antibody, we noticed a slight increase in a band around ∼80 kDa with a corresponding decrease in a band around ∼60 kDa (Fig. 2D) in lysates from cells treated with batimastat. Such an effect was also observed in cells treated with GI254023X (Fig. 2C, panel 2) but was not so evident with GM6001 treatment (Fig. 2A, panels 2 and 3). Based on published reports (29, 30), the proenzyme form of ADAM10 migrates at ∼100 kDa on a SDS-PAGE gel and the metabolically active form at ∼60 kDa. Treatment with batimastat showed an increase in the levels of an ∼80 kDa form of ADAM10, which was associated with a decrease in sAPPα generation (Fig. 2D, top and middle panels). In addition to the ∼100 kDa and ∼60 kDa forms of ADAM10, an ∼85 kDa form of ADAM10, which is non-enzymatically active (31), has been reported. Thus, we believe that the ∼80 kDa fragment we observed in our studies could represent a non-enzymatically active form of ADAM10. The increase in ∼80 kDa ADAM10 fragment or decrease in sAPPα generation was not visible in either untreated or gemcitabine treated cells (Fig. 2D), confirming that the observed effect is specific to batimastat treatment. To study the batimastat-associated changes in ADAM10, Phoenix cells transfected with untagged or myc-tagged ADAM10 plasmid (Myc tag at C-terminal) were treated with or without batimastat for 18 h, and cell lysates were analyzed with ADAM10 antibody. Upon treatment of cells with batimastat, we observed an increase in the ∼80 kDa band and a decrease in the ∼60 kDa band as compared with untreated cells (Fig. 2E, panels 1 and 2). These two bands were recognized by the myc antibody (Myc tag at C-terminal end of ADAM10), which confirms that these are different forms of ADAM10 and contain the C-terminal end (Fig. 2E, panel 4). This also suggests that these fragments might be derived from the N-terminal cleavage of the proform. Increase in the levels of the ∼80 kDa band correlated with decrease in sAPPα generation (quantified and normalized to actin) (Fig. 2E, panel 5, and 2F). It was also noted, on several blots, that the ∼60 kDa active form of ADAM10 was visible at low levels or in some cases, undetectable, which suggests that this fragment might be short-lived or undergoing rapid turnover. Increase in level of the ∼80 kDa band could be attributed to the decreased generation of active form of ADAM10 (∼60 kDa, Fig. 2E, panels 1, 2, and 4) brought about by batimastat treatment. It is possible that the ∼80 kDa form of ADAM10 is an intermediate cleavage product generated during maturation, and treatment with batimastat prevents further processing of this intermediate to generate the mature, active form of ADAM10. Since ADAM10 is a glycosylated protein, to determine whether the ∼80 kDa band is generated by deglycosylation we performed deglycosylation experiments using MiaPaCa2 cells and myc-ADAM10 transfected Phoenix cells, treated with or without batimastat. Results showed that all the three bands detected by ADAM10 (100, ∼80, and 60 kDa) are glycosylated and deglycosylation downshifts the molecular weights of each form of ADAM10 on an SDS-PAGE gel (Fig. 2G). Further, the deglycosylated proform of ADAM10 still migrated at a higher molecular weight than the ∼80 kDa form thus confirming that the ∼80 kDa fragment is not a deglysolyated form of the 100 kDa proform. In order to determine whether batimastat affects membrane localization of ADAM10, we used a biotinylation based cell surface protein isolation and analysis protocol (described under “Experimental Procedures”). Phoenix cells were used for this experiment due to their superior transfection ability. pcDNA3 or myc-ADAM10 transfected cells were treated with or without batimastat for 18 h. Cells were labeled, harvested, and processed following kit directions, and the cell surface proteins were resolved on SDS-PAGE. Blotting with anti-Myc antibody clearly showed an increase in the ∼80 kDa band in Myc-transfected, batimastat treated cells as compared with Myc-transfected, untreated cells (Fig. 2I, panels 1 and 2). For control, the same blot was probed for a cell surface protein, Na/K ATPase (Fig. 2I, panel 3). Additionally, we probed the samples with 6E10 antibody to detect APP (Fig. 2I, panel 4) and found that membrane associated APP levels were increased in response to batimastat treatment, which is to be expected since APP cleavage is reduced upon inhibition of ADAM10 function. To ensure ADAM10 expression in the transfected cells, a small amount of cell lysate collected prior to incubation with NeutrAvidin Agarose was resolved on SDS-PAGE and blotted with ADAM10 antibody (Fig. 2H). Actin was used as loading control. These results suggest that batimastat induces the formation of a membrane localized, non-enzymatically active form of ADAM10 with a corresponding decrease in the formation of the mature ADAM10 fragment. Further, to confirm that the 100, ∼80, and 60 kDa bands are derivatives of ADAM10, we downregulated ADAM10 in MiaPaCa2 cells, performed cell surface protein biotinylation and isolation of membrane proteins and examined the ADAM10 levels by Western blot. Results showed that ADAM10 siRNA transfected cells showed knockdown of all the three forms as compared with the siRNA control transfected cells confirming that the three observed bands are indeed different forms of ADAM10 (Fig. 2J). Na/K ATPase was used as loading control (Fig. 2J, lower panel).
Down-regulation of sAPPα by ADAM10 Inhibition Promotes Pancreatic Cancer Cell Death
CD18 and MiaPaCa2 cells were treated with different doses of batimastat (from 1.25 μm to 5 μm), and cell death was analyzed by cytotoxicity assay using MTT (data not shown). From the range of concentrations tested, it was found that 5 μm batimastat was most effective in causing cell death in CD18 cells when combined with 100 ng/ml gemcitabine as compared with either drug alone. The effect of these drugs was noted over a period of 24 h (Fig. 3A) and the same trend in cell death was observed at 48 h (data not shown). A similar effect was also observed in MiaPaCa2 cells (Fig. 3B). Moreover, the effect of the MMP inhibitor, GM6001, was not as dramatic as batimastat in this cell death assay (Fig. 3C) thereby confirming the specificity of batimastat for ADAM10. Additionally, GI254023X was compared with batimastat in a cytotoxicity assay and found to be equally effective in causing pancreatic cancer cell death as batimastat (Fig. 3, D and E). To determine whether the observed cell death was a result of apoptosis, CD18 and MiaPaCa2 cells treated with batimastat, gemcitabine, or both and were analyzed for cleaved PARP levels. Western blot analysis revealed PARP cleavage upon treatment with either drug and increased cleavage when treated with a combination of the drugs (Fig. 3, F and G). This indicates that apoptosis is likely the major mechanism by which these drugs cause cytotoxicity in cancer cells.
FIGURE 3.

Inhibition of sAPPα enhances the cytotoxic effect of gemcitabine on pancreatic cancer cells. A, 10,000 CD18 cells were plated in 24-well culture dishes and treated with 5 μm batimastat, 100 ng/ml gemcitabine, or a combination of both for 24 h. Cytotoxicity assay using MTT reagent was performed and samples analyzed at A540. B, 10,000 MiaPaCa2 cells were plated in 24-well culture dishes and treated with the indicated drugs, and the extent of cytotoxicity was determined using MTT reagent at A540. C, CD18 cells were plated in 24-well plates and treated with the indicated drugs for 24 h and cytotoxicity was measured using the MTT reagent at A540. Each experiment was performed more than three times and (*) indicates significant difference with p value less than 0.05 from an unpaired two-tailed Student's t test. D, 10,000 CD18 cells were plated in 24-well plates and treated with 5 μm batimastat, 5 μm GI254023X, 100 ng/ml gemcitabine, or combinations for 24 h. MTT reagent was used to determine cytotoxicity at A540. E, 10,000 MiaPaCa2 cells were plated in 24-well plates and treated with 5 μm batimastat, 5 μm GI254023X, 100 ng/ml gemcitabine or combinations for 24 h. MTT reagent was used to determine cytotoxicity at A540. F, CD18 and MiaPaCa2 cells were treated with 5 μm batimastat, 100 ng/ml gemcitabine or a combination of the drugs for 24 h, and cell lysates collected for analysis of PARP cleavage. The same blot was reprobed for actin, which served as a loading control. G, cleavage of PARP was quantified from three independent experiments and normalized to actin using the ImageJ software as represented by the bar graph. Each of the cytotoxicity and quantification experiments were performed more than three times, and (*)(#)(●)(■) indicate significant difference with p value less than 0.05 from an unpaired two-tailed Student's t test.
Down-regulation of APP or ADAM10, but Not ADAM17, Increases the Sensitivity of Pancreatic Cancer Cells to Gemcitabine
APP or ADAM10 gene expression was transiently knocked down in CD18 and MiaPaCa2 cells using siRNA as described under “Experimental Procedures.” The extent of knockdown was confirmed by Western blotting using APP (6E10) and ADAM10 antibodies (Fig. 4, A and B). 10,000 CD18 and MiaPaCa2 cells were plated in 24-well plates and transfected with control or target siRNA, and cell death was analyzed using MTT reagent. The results showed that the cytotoxicity caused by APP or ADAM10 siRNA was comparable to that of treatment with 5 μm batimastat (Fig. 4, C and D). To confirm that batimastat-induced cytotoxicity of pancreatic cancer cells is brought about via inhibition of ADAM10-mediated sAPPα generation, we performed the cytotoxicity assay with CD 18 and MiaPaCa2 cells (Fig. 4, E and F) in the presence of recombinant sAPPα. Control and batimastat-treated cells were incubated with 100 nm purified sAPPα for a period of 24 h. The effect of in vitro supplemented recombinant sAPPα protein on the cells was determined using MTT reagent. It was observed that treatment of cells with recombinant sAPPα was able to overcome the inhibitory effect of batimastat significantly thereby confirming our findings that inhibition of pancreatic cell proliferation is a result of inhibiting sAPPα generation from APP.
FIGURE 4.

Cytotoxic effect of batimastat is brought about by inhibition of sAPPα generation. A, CD18 cells were transfected with siRNA to APP or ADAM10 and Western blot analysis was performed using 6E10 and ADAM10 antibodies to confirm knockdown of the respective genes. B, APP or ADAM10 were knocked down in MiaPaCa2 cells and Western blot was used to confirm knockdown. The effect of ADAM10 and APP knockdown in CD18 (C) and MiaPaCa2 (D) cells was compared with batimastat treatment in a cytotoxicity assay using MTT reagent at A540. CD18 (E) and MiaPaCa2 (F) cells were treated with 100 nm recombinant sAPPα alone or in combination with 5 μm batimastat for 24 h and changes in cell death was analyzed with the MTT reagent at A540. Each experiment was performed three times, and (*)(#)(**) indicates significant difference with p value less than 0.05 from an unpaired two-tailed Student's t test.
We also tested the effect of ADAM17 knockdown on sAPPα levels in MiaPaCa2 cells. Using different concentrations of siRNA, we were able to down-regulate the expression of ADAM17 effectively (Fig. 5A). However, ADAM17 knockdown did not have an effect on sAPPα levels (Fig. 5A, panel 2). MiaPaCa2 cells transfected with ADAM17 or ADAM10 siRNA and treated with or without batimastat were used to perform a cytotoxicity assay (Fig. 5B). While ADAM17 knockdown did not affect cell death unless treated with batimastat, there was no additional cell death in ADAM10 knockdown cells treated with batimastat as compared with untreated ADAM10 knockdown cells. This confirms the specificity of batimastat against ADAM10 as well as suggests that ADAM10-mediated generation of sAPPα is important for pancreatic cancer cell proliferation. Fig. 5C indicates that sAPPα down-regulation specifically correlates with ADAM10 knockdown and Fig. 5D indicates the extent of knockdown of ADAM10 and ADAM17 by the respective siRNAs. It is important to note that ADAM10 siRNA inhibits the levels of the ∼100 kDa full-length protein as well as the ∼80 kDa fragment suggesting that the latter is a cleaved form of ADAM10.
FIGURE 5.

ADAM10, and not ADAM17, plays a role in sAPPα-mediated pancreatic cancer cell proliferation. A, MiaPaCa2 cells were transfected with ADAM17 siRNA at different concentration starting from 20 nm to 60 nm and Western blot of cell lysates were performed to determine extent of ADAM17 knockdown (top panel). Actin was used as loading control (bottom panel). Cell supernatant was used to detect sAPPα level using 6E10 antibody. B, MiaPaCa2 cells transfected with control siRNA, ADAM17 siRNA or ADAM10 siRNA were treated with or without 5 μm batimastat for 24 h, and cell death was analyzed with the MTT reagent at A540. Each experiment was performed three times, and (*)(#) indicates significant difference with p value less than 0.05 from an unpaired two-tailed Student's t test. C, sAPPα levels in ADAM10 or ADAM17 knockdown cells were detected using 6E10 antibody. D, Western blot analysis was performed to determine levels of ADAM10 (left) or ADAM17 (right) in MiaPaCa2 cells transfected with ADAM10 or ADAM17 siRNA. Actin was used as loading control.
Inhibition of sAPPα Negatively Affects Anchorage-independent Growth of Pancreatic Cancer Cells
To study the effect of inhibiting sAPPα generation on cell proliferation and colony formation, soft agar assays were performed with CD18, MiaPaCa2, and Panc1 pancreatic cancer cells. This is a sensitive assay used for determination of drug toxicity. The experiment was performed as outlined under “Experimental Procedures.” 5,000 cells were seeded in soft agar for each treatment, which was performed in triplicates. In all the cell lines tested, fewer colonies were observed in batimastat-treated wells but least number of colonies were observed in wells containing a combination of batimastat and gemcitabine thereby suggesting a synergistic inhibitory effect of the two drugs on pancreatic cancer cell proliferation (Fig. 6). It was also found that as compared with CD18 (Fig. 6A, left and right panels), MiaPaCa2 (Fig. 6B) and Panc1 cells (Fig. 6C) required higher dosage of batimastat for effective reduction in colony formation probably because of their mesenchymal phenotype. Since colony formation in soft agar shows anchorage independent growth, and it may have implications in metastasis, the results from these studies suggest that down-regulation of sAPPα may reduce the metastatic potential of pancreatic cancer cells.
FIGURE 6.

Effect of sAPPα inhibition on colony formation by pancreatic cancer cells. A, 5,000 CD18 cells were seeded in soft agar and treated with 1 μm batimastat, 100 ng/ml gemcitabine or combination of both drugs (left panel in A) to study their effect on the ability of cells to form colonies over a period of 2 weeks. Soft agar assay performed with CD18 cells using 5 μm batimastat, 100 ng/ml gemcitabine or a combination of both drugs (right panel in A). Similarly, 5,000 MiaPaCa2 (B) or Panc1 (C) cells were seeded in soft agar and treated with 10 μm batimastat, 100 ng/ml gemcitabine, or a combination of the drugs to study their effect on colony formation. Treatment with each dose was performed in triplicate, and each experiment was performed three times.
DISCUSSION
Overexpression of the amyloid precursor protein in pancreatic cancer has been reported in a few studies (9, 10). It has also been shown that the alpha secretase-mediated cleavage of APP generates a fragment that may possess growth-promoting qualities (9). In brain, the function of sAPPα has been thought to be neuroprotective, specifically because its formation provides an alternate pathway of APP processing, which prevents the formation of Aβ peptides that are symptomatic of AD (32–34). Additionally, sAPPα has been shown to have anti-apoptotic and neuroprotective properties (34, 35). For that reason, there has been an interest in investigating the role of sAPPα in cancer. In this study, we show that APP cleavage contributes significantly to the proliferation of pancreatic cancer cells and that inhibition of ADAM10 blocks the formation of sAPPα from APP and enhances the sensitivity of cancer cells to gemcitabine. This was confirmed by cytotoxicity assays performed with combination of batimastat and gemcitabine, which showed increased cell death of pancreatic cancer cells as opposed to treatment with either drug alone. In vitro addition of recombinant sAPPα to batimastat-treated cells was able to reverse the inhibitory effects of the compound significantly in a cytotoxicity assay which confirms that sAPPα is indeed involved in proliferation and survival of pancreatic cancer cells. Both batimastat and GI254023X abolished the generation of the sAPPα fragment to similar extent, suggesting the effectiveness of these compounds on ADAM10 activity. Further investigation unraveled the possible mode of action of batimastat. Our results show that there is increased association of both APP and the ∼80 kDa fragment of ADAM10 with the membrane upon batimastat treatment of the cells. Regardless of the localization of the ∼80 kDa fragment the cells showed an inhibition of sAPPα generation suggesting that this fragment is an inactive form of ADAM10. The effect observed with batimastat was specific to ADAM10 as knockdown of ADAM17 did not affect sAPPα generation or induce cell death in pancreatic cancer cells. These studies therefore confirm that proliferation of the pancreatic cancer cells is specifically influenced by ADAM10-mediated cleavage of APP. In addition to the cytotoxic effect, batimastat-mediated inhibition of sAPPα generation was able to effectively inhibit anchorage independent growth of the pancreatic cancer cells on soft agar. This effect was more pronounced when batimastat was used in conjunction with gemcitabine, the current standard drug for chemotherapy in pancreatic cancer. Since anchorage independent growth is an indicator of the metastatic potential of cancer cells, these results imply that higher levels of sAPPα may contribute to the aggressiveness of tumors. Our studies show that sAPPα inhibition was able to greatly reduce the concentration of gemcitabine required to inhibit cancer cell growth, which suggests that combination therapy that includes inhibitor of sAPPα generation could substantially reduce gemcitabine-associated side effects in pancreatic cancer patients.
While our studies provide a novel concept of APP cleavage inhibition as a potential chemotherapeutic option for management of pancreatic cancer, the mechanism by which sAPPα promotes pancreatic cancer growth is still not known. Some studies have indicated that sAPPα might promote proliferation of non-neuronal cells by modulating transcription factors such as NFκB (35) while others have suggested that it might exhibit proliferative qualities in neural progenitor cells and mesenchymal stem cells by regulating the MAP kinase pathway (36). It could also be involved in metastasis as it has been shown that secreted APP promotes proliferation and migration of cells (27). Whether sAPPα promotes pancreatic cancer growth through similar mechanisms of transcription factor regulation or oncogenic pathway modulation is yet to be determined. It will also be useful to determine whether APP or its metabolites possesses oncogenic functions. These studies will help us fully understand the mechanisms that govern the progression of pancreatic cancer and hopefully provide us with novel methods to effectively combat this disease.
Acknowledgments
The support of the core facilities of the Moffitt Cancer Center is greatly acknowledged.
This work was supported, in whole or in part, by NIA, National Institutes of Health Grant 1R21AG031429-01A2 and Alzheimer's Association Grant IIRG-08-90842 (to J. P.)
- APP
- amyloid precursor protein
- AD
- Alzheimer disease
- Aβ
- amyloid β
- sAPP
- secreted APP
- ADAM
- a disintegrin and metalloprotease domain
- PDAC
- pancreatic ductal adenocarcinoma
- AICD
- APP intracellular domain.
REFERENCES
- 1. Jemal A., Siegel R., Xu J., Ward E. (2010) Cancer statistics, 2010. CA Cancer J. Clin. 60, 277–300 [DOI] [PubMed] [Google Scholar]
- 2. Jemal A., Siegel R., Ward E., Hao Y., Xu J., Thun M. J. (2009) Cancer statistics, 2009. CA Cancer J. Clin. 59, 225–249 [DOI] [PubMed] [Google Scholar]
- 3. Burris H. A., 3rd, Moore M. J., Andersen J., Green M. R., Rothenberg M. L., Modiano M. R., Cripps M. C., Portenoy R. K., Storniolo A. M., Tarassoff P., Nelson R., Dorr F. A., Stephens C. D., Von Hoff D. D. (1997) Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J. Clin. Oncol. 15, 2403–2413 [DOI] [PubMed] [Google Scholar]
- 4. Berlin J., Benson A. B., 3rd. (2010) Chemotherapy: Gemcitabine remains the standard of care for pancreatic cancer. Nat. Rev. Clin. Oncol. 7, 135–137 [DOI] [PubMed] [Google Scholar]
- 5. Berlin J. D., Catalano P., Thomas J. P., Kugler J. W., Haller D. G., Benson A. B., 3rd. (2002) Phase III study of gemcitabine in combination with fluorouracil versus gemcitabine alone in patients with advanced pancreatic carcinoma: Eastern Cooperative Oncology Group Trial E2297. J. Clin. Oncol. 20, 3270–3275 [DOI] [PubMed] [Google Scholar]
- 6. Rocha Lima C. M., Green M. R., Rotche R., Miller W. H., Jr., Jeffrey G. M., Cisar L. A., Morganti A., Orlando N., Gruia G., Miller L. L. (2004) Irinotecan plus gemcitabine results in no survival advantage compared with gemcitabine monotherapy in patients with locally advanced or metastatic pancreatic cancer despite increased tumor response rate. J. Clin. Oncol. 22, 3776–3783 [DOI] [PubMed] [Google Scholar]
- 7. Oettle H., Richards D., Ramanathan R. K., van Laethem J. L., Peeters M., Fuchs M., Zimmermann A., John W., Von Hoff D., Arning M., Kindler H. L. (2005) A phase III trial of pemetrexed plus gemcitabine versus gemcitabine in patients with unresectable or metastatic pancreatic cancer. Ann. Oncol. 16, 1639–1645 [DOI] [PubMed] [Google Scholar]
- 8. Van Cutsem E., Verslype C., Grusenmeyer P. A. (2007) Lessons learned in the management of advanced pancreatic cancer. J. Clin. Oncol. 25, 1949–1952 [DOI] [PubMed] [Google Scholar]
- 9. Hansel D. E., Rahman A., Wehner S., Herzog V., Yeo C. J., Maitra A. (2003) Increased expression and processing of the Alzheimer amyloid precursor protein in pancreatic cancer may influence cellular proliferation. Cancer Res. 63, 7032–7037 [PubMed] [Google Scholar]
- 10. Venkataramani V., Rossner C., Iffland L., Schweyer S., Tamboli I. Y., Walter J., Wirths O., Bayer T. A. (2010) Histone deacetylase inhibitor valproic acid inhibits cancer cell proliferation via down-regulation of the alzheimer amyloid precursor protein. J. Biol. Chem. 285, 10678–10689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lewis D. A., Higgins G. A., Young W. G., Goldgaber D., Gajdusek D. C., Wilson M. C., Morrison J. H. (1988) Distribution of precursor amyloid-beta-protein messenger RNA in human cerebral cortex: relationship to neurofibrillary tangles and neuritic plaques. Proc. Natl. Acad. Sci. U.S.A. 85, 1691–1695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Perry G., Lipphardt S., Mulvihill P., Kancherla M., Mijares M., Gambetti P., Sharma S., Maggiora L., Cornette J., Lobl T. (1988) Amyloid precursor protein in senile plaques of Alzheimer disease. Lancet 2, 746. [DOI] [PubMed] [Google Scholar]
- 13. De Strooper B., Annaert W. (2000) Proteolytic processing and cell biological functions of the amyloid precursor protein. J. Cell Sci. 113, 1857–1870 [DOI] [PubMed] [Google Scholar]
- 14. Vassar R., Bennett B. D., Babu-Khan S., Kahn S., Mendiaz E. A., Denis P., Teplow D. B., Ross S., Amarante P., Loeloff R., Luo Y., Fisher S., Fuller J., Edenson S., Lile J., Jarosinski M. A., Biere A. L., Curran E., Burgess T., Louis J. C., Collins F., Treanor J., Rogers G., Citron M. (1999) β-Secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741 [DOI] [PubMed] [Google Scholar]
- 15. Gandy S., Caporaso G., Buxbaum J., Frangione B., Greengard P. (1994) APP processing, A β-amyloidogenesis, and the pathogenesis of Alzheimer's disease. Neurobiol. Aging 15, 253–256 [DOI] [PubMed] [Google Scholar]
- 16. Lammich S., Kojro E., Postina R., Gilbert S., Pfeiffer R., Jasionowski M., Haass C., Fahrenholz F. (1999) Constitutive and regulated α-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc. Natl. Acad. Sci. U.S.A. 96, 3922–3927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Prinzen C., Trümbach D., Wurst W., Endres K., Postina R., Fahrenholz F. (2009) Differential gene expression in ADAM10 and mutant ADAM10 transgenic mice. BMC Genomics 10, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kuhn P. H., Wang H., Dislich B., Colombo A., Zeitschel U., Ellwart J. W., Kremmer E., Rossner S., Lichtenthaler S. F. (2010) ADAM10 is the physiologically relevant, constitutive α-secretase of the amyloid precursor protein in primary neurons. EMBO J. 29, 3020–3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lopez-Perez E., Zhang Y., Frank S. J., Creemers J., Seidah N., Checler F. (2001) Constitutive α-secretase cleavage of the β-amyloid precursor protein in the furin-deficient LoVo cell line: involvement of the pro-hormone convertase 7 and the disintegrin metalloprotease ADAM10. J. Neurochem. 76, 1532–1539 [DOI] [PubMed] [Google Scholar]
- 20. Buxbaum J. D., Liu K. N., Luo Y., Slack J. L., Stocking K. L., Peschon J. J., Johnson R. S., Castner B. J., Cerretti D. P., Black R. A. (1998) Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated α-secretase cleavage of the Alzheimer amyloid protein precursor. J. Biol. Chem. 273, 27765–27767 [DOI] [PubMed] [Google Scholar]
- 21. Kohutek Z. A., diPierro C. G., Redpath G. T., Hussaini I. M. (2009) ADAM-10-mediated N-cadherin cleavage is protein kinase C-α-dependent and promotes glioblastoma cell migration. J. Neurosci. 29, 4605–4615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maretzky T., Reiss K., Ludwig A., Buchholz J., Scholz F., Proksch E., de Strooper B., Hartmann D., Saftig P. (2005) ADAM10 mediates E-cadherin shedding and regulates epithelial cell-cell adhesion, migration, and β-catenin translocation. Proc. Natl. Acad. Sci. U.S.A. 102, 9182–9187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. van Tetering G., van Diest P., Verlaan I., van der Wall E., Kopan R., Vooijs M. (2009) Metalloprotease ADAM10 is required for Notch1 site 2 cleavage. J. Biol. Chem. 284, 31018–31027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vincent B., Govitrapong P. (2011) Activation of the α-secretase processing of AβPP as a therapeutic approach in Alzheimer's disease. J. Alzheimers Dis. 24, 75–94 [DOI] [PubMed] [Google Scholar]
- 25. Postina R., Schroeder A., Dewachter I., Bohl J., Schmitt U., Kojro E., Prinzen C., Endres K., Hiemke C., Blessing M., Flamez P., Dequenne A., Godaux E., van Leuven F., Fahrenholz F. (2004) A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J. Clin. Invest. 113, 1456–1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gaida M. M., Haag N., Günther F., Tschaharganeh D. F., Schirmacher P., Friess H., Giese N. A., Schmidt J., Wente M. N. (2010) Expression of A disintegrin and metalloprotease 10 in pancreatic carcinoma. Int. J. Mol. Med. 26, 281–288 [DOI] [PubMed] [Google Scholar]
- 27. Schmitz A., Tikkanen R., Kirfel G., Herzog V. (2002) The biological role of the Alzheimer amyloid precursor protein in epithelial cells. Histochem. Cell Biol. 117, 171–180 [DOI] [PubMed] [Google Scholar]
- 28. Hundhausen C., Misztela D., Berkhout T. A., Broadway N., Saftig P., Reiss K., Hartmann D., Fahrenholz F., Postina R., Matthews V., Kallen K. J., Rose-John S., Ludwig A. (2003) The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood 102, 1186–1195 [DOI] [PubMed] [Google Scholar]
- 29. Arima T., Enokida H., Kubo H., Kagara I., Matsuda R., Toki K., Nishimura H., Chiyomaru T., Tatarano S., Idesako T., Nishiyama K., Nakagawa M. (2007) Nuclear translocation of ADAM-10 contributes to the pathogenesis and progression of human prostate cancer. Cancer Science 98, 1720–1726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McCulloch D. R., Akl P., Samaratunga H., Herington A. C., Odorico D. M. (2004) Expression of the disintegrin metalloprotease, ADAM-10, in prostate cancer and its regulation by dihydrotestosterone, insulin-like growth factor I, and epidermal growth factor in the prostate cancer cell model LNCaP. Clinical Cancer Research 10, 314–323 [DOI] [PubMed] [Google Scholar]
- 31. Zimmermann M., Gardoni F., Marcello E., Colciaghi F., Borroni B., Padovani A., Cattabeni F., Di Luca M. (2004) Acetylcholinesterase inhibitors increase ADAM10 activity by promoting its trafficking in neuroblastoma cell lines. J. Neurochem. 90, 1489–1499 [DOI] [PubMed] [Google Scholar]
- 32. Esch F. S., Keim P. S., Beattie E. C., Blacher R. W., Culwell A. R., Oltersdorf T., McClure D., Ward P. J. (1990) Cleavage of amyloid beta peptide during constitutive processing of its precursor. Science 248, 1122–1124 [DOI] [PubMed] [Google Scholar]
- 33. Jäger S., Leuchtenberger S., Martin A., Czirr E., Wesselowski J., Dieckmann M., Waldron E., Korth C., Koo E. H., Heneka M., Weggen S., Pietrzik C. U. (2009) α-secretase mediated conversion of the amyloid precursor protein derived membrane stub C99 to C83 limits Aβ generation. J. Neurochem. 111, 1369–1382 [DOI] [PubMed] [Google Scholar]
- 34. Furukawa K., Sopher B. L., Rydel R. E., Begley J. G., Pham D. G., Martin G. M., Fox M., Mattson M. P. (1996) Increased activity-regulating and neuroprotective efficacy of α-secretase-derived secreted amyloid precursor protein conferred by a C-terminal heparin-binding domain. J. Neurochem. 67, 1882–1896 [DOI] [PubMed] [Google Scholar]
- 35. Mattson M. P. (1997) Cellular actions of β-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol. Rev. 77, 1081–1132 [DOI] [PubMed] [Google Scholar]
- 36. Demars M. P., Bartholomew A., Strakova Z., Lazarov O. (2011) Soluble amyloid precursor protein: a novel proliferation factor of adult progenitor cells of ectodermal and mesodermal origin. Stem Cell Res. Ther. 2, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]