

Background: Phospholamban (PLB) regulates sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) activity and is thus a key regulator of cardiac contractility.
Results: We present the crystal structure of SERCA in complex with PLB at 2.8-Å resolution.
Conclusion: PLB stabilizes a divalent cation-free conformation of SERCA with collapsed Ca2+ binding sites. We call the structure E2-PLB.
Significance: The E2-PLB structure explains how PLB decreases Ca2+ affinity and depresses cardiac contractility.
Keywords: Calcium ATPase, Calcium Transport, Crystal Structure, Protein Cross-linking, Protein Crystallization, Sarcoplasmic Reticulum (SR), SERCA, Phospholamban, Sarcolipin
Abstract
P-type ATPases are a large family of enzymes that actively transport ions across biological membranes by interconverting between high (E1) and low (E2) ion-affinity states; these transmembrane transporters carry out critical processes in nearly all forms of life. In striated muscle, the archetype P-type ATPase, SERCA (sarco(endo)plasmic reticulum Ca2+-ATPase), pumps contractile-dependent Ca2+ ions into the lumen of sarcoplasmic reticulum, which initiates myocyte relaxation and refills the sarcoplasmic reticulum in preparation for the next contraction. In cardiac muscle, SERCA is regulated by phospholamban (PLB), a small inhibitory phosphoprotein that decreases the Ca2+ affinity of SERCA and attenuates contractile strength. cAMP-dependent phosphorylation of PLB reverses Ca2+-ATPase inhibition with powerful contractile effects. Here we present the long sought crystal structure of the PLB-SERCA complex at 2.8-Å resolution. The structure was solved in the absence of Ca2+ in a novel detergent system employing alkyl mannosides. The structure shows PLB bound to a previously undescribed conformation of SERCA in which the Ca2+ binding sites are collapsed and devoid of divalent cations (E2-PLB). This new structure represents one of the key unsolved conformational states of SERCA and provides a structural explanation for how dephosphorylated PLB decreases Ca2+ affinity and depresses cardiac contractility.
Introduction
Phospholamban (PLB),3 a single-span membrane protein of only 52 amino acids, is the principal membrane protein in the heart phosphorylated in response to β-adrenergic stimulation and a critical regulator of cardiac contractile strength (1). In the dephosphorylated state, PLB decreases cardiac contractility by inhibiting the activity of the sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA), an 110-kDa membrane protein with 10 membrane-spanning segments. PLB inhibits SERCA activity by decreasing Ca2+ affinity at its two Ca2+ binding sites (I and II) located in the SR membrane, thereby attenuating SR Ca2+ filling and contractile force development. Phosphorylation of PLB at Ser16 and Thr17 partially reverses PLB induced alterations in Ca2+ affinity, substantially augmenting contractility (2–4).
Chemical cross-linking was used previously to localize the PLB binding site on SERCA to a groove formed between transmembrane helices M2, M4, M6, and M9 of the Ca2+ pump (5–9). In conjunction with functional assays, these cross-linking studies led us to postulate that PLB binds to a unique Ca2+-free (E2) state of SERCA and decreases the Ca2+ affinity of the enzyme through direct effects on the Ca2+ binding sites (5). Recently, a gain-of-function, cross-linkable PLB mutant, PLB4 (N27A, N30C, L37A, V49G-PLB) (Fig. 1), was developed (10) and shown to bind to the Ca2+-ATPase with exceedingly high affinity, comparable with or even greater than that of thapsigargin (TG), the well known, irreversible Ca2+-ATPase inhibitor (11, 12). Here, we took advantage of PLB4 to stabilize the Ca2+-free state of SERCA and to crystallize it in complex with PLB. The 2.8-Å resolution structure, crystallized in nonyl maltoside, reveals a previously unresolved conformational state of the Ca2+ pump captured by PLB, which we define as E2-PLB. Importantly, the structure of E2-PLB is incompatible with Ca2+ (or Mg2+) binding to the enzyme, and confirms the hypothesis that mutually exclusive binding of PLB and Ca2+ to SERCA is the mechanism by which PLB decreases apparent Ca2+ affinity (5).
FIGURE 1.
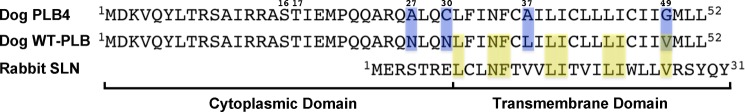
Amino acid sequences of PLB4 and WT-PLB (canine isoforms), and rabbit SLN. Cytoplasmic and transmembrane domains are shown as well as phosphorylated residues Ser16 and Thr17 of PLB. Mutations in PLB4 are shaded in blue. Amino acid residues common to both PLB and SLN are shaded in yellow.
EXPERIMENTAL PROCEDURES
Protein Preparation
SR vesicles sedimenting at 45,000 × g were isolated from rabbit back and hind leg muscles as described previously for cardiac SR vesicles (13). SR vesicle pellets were extracted with 0.6 m KCl, 20 mm MOPS (pH 7.0), resuspended in 0.25 m sucrose, 20 mm MOPS (pH 7.2), and stored frozen in small aliquots at −40 °C at a protein concentration of 30 mg/ml. Recombinant WT-PLB and PLB4 were expressed and purified from Sf21 insect cells using anti-PLB monoclonal antibody (2D12) affinity chromatography as previously described (14). Purified PLB was eluted from the column in 0.1% decyl maltoside or 0.01% dodecyl maltoside (Anatrace), which in control experiments were determined to be optimal detergents for co-crystallization of PLB with the solubilized Ca2+ pump. The purified, eluted PLB proteins were concentrated 100-fold with an Amicon concentrator, and then exhaustively dialyzed against 20 mm MOPS (pH 7.2), 20% glycerol, and 0.1% decyl maltoside or 0.01% dodecyl maltoside. The final working concentrations of PLB were 8–10 mg of protein/ml. PLB was stored frozen at −40 °C. Protein concentrations were determined by the Lowry method.
The Ca2+ pump suitable for crystallization was solubilized directly from SR vesicles without prior purification or extraction of SR vesicles with low concentrations of deoxycholate (15). Thawed SR vesicles were diluted 1:1 to a protein concentration of 15 mg/ml in buffer containing 2% nonyl maltoside (Anatrace), 20% glycerol, 100 mm MOPS (pH 7.0), 0.12 m sucrose, 80 mm KCl, 3 mm MgCl2, and 2.8 mm EGTA (final concentrations). The samples were allowed to stand for 7 min at room temperature, then ultracentrifuged at 4 °C at 100,000 × g for 15 min in a Beckman TLA 100.1 rotor. The supernatant was collected and PLB was added from the concentrated working solutions at a ratio of 0.14 mg of PLB/1.0 mg of solubilized SR vesicle protein, determined in control experiments to be a saturating concentration of PLB for inhibition of Ca2+-ATPase activity by lowering the apparent Ca2+ affinity. This amount of added PLB gave a molar ratio of PLB to SERCA of 2.9:1, as determined by quantitative immunoblotting (16). Final volumes of mother liquors were adjusted by addition of 20% glycerol to make the final EGTA concentration 2 mm and samples were stored at 4 °C. Ca2+-ATPase prepared by this method (in 2 mm EGTA) retained 95–100% of the initially solubilized activity for at least 3 weeks at 4 °C in the presence and absence of PLB (Fig. 2B). In pilot studies, the Ca2+ pump was solubilized from SR vesicles using other detergents, including C12E8 and octyl glucoside. Solubilization conditions were identical to that described above, using 2% detergent concentrations (Fig. 2B).
FIGURE 2.

Effect of detergent solubilization of SERCA on apparent Ca2+ affinity (A) and Ca2+-ATPase stability over time (B). Mother liquors were prepared from rabbit skeletal SR vesicles in buffer containing 2% detergent concentrations and 2 mm EGTA as described under “Experimental Procedures.” A, apparent Ca2+ affinity (KCa) of the solubilized Ca2+-ATPase was determined immediately after membrane solubilization by measuring Ca2+ activation of ATP hydrolysis. Results are expressed as percentage of the maximal Ca2+-ATPase activity obtained for each detergent tested: NM (nonyl glucoside), OG (octyl glucoside), DM (decyl maltoside), and DDM (dodecyl maltoside). Control membranes (Con) were not treated with detergent. KCa values (μm) were 0.21 ± 0.005 (Con), 0.25 ± 0.02 (NM), 0.23 ± 0.02 (OG), 0.63 ± 0.02 (C12E8), 0.29 ± 0.01 (DM), and 0.40 ± 0.02 (DDM). Mean ± S.E. from four to eight determinations are shown. B, enzyme stability of SERCA was monitored after storage of the mother liquors in different detergents at 4 °C for the times indicated. At the designated times of storage, aliquots were taken for assay of Ca2+-ATPase activity (micromole of Pi/mg of protein/h) at saturating Ca2+ concentration (50 μm Ca2+). NM + PLB4 designates SERCA solubilized in NM reconstituted with PLB in DM, an optimal condition for crystal formation. Shown is one representative experiment, which was repeated at least three times for all the different detergents with similar results.
Crystallization
One day after the initial Ca2+-ATPase solubilization and addition of PLB, mother liquors were sedimented a second time by ultracentrifugation as described above. Hanging drops were made by mixing 1 μl of the sedimented mother liquors with 1 μl of reservoir solution (15% glycerol, 17% (w/v) PEG-2000, 200 mm NaOAc, and 5 mm β-mercaptoethanol) and crystals were grown by vapor diffusion at 4 °C. Single crystals appeared within 2 weeks and grew to a final size of 150 × 100 × 50 μm within 1 month. Crystals were mounted using nylon fiber loops and flash cooled in liquid nitrogen with no additional cryo-protectant.
Data Collection, Structure Solution, and Refinement
The x-ray diffraction data were collected at Beamline 19-ID operated by the Structural Biology Center at the Advanced Photon Source within Argonne National Laboratory. All diffraction data were collected at a wavelength of 0.979 Å from a single crystal at 100 K. The crystal was formed from PLB4 added in decyl maltoside. The diffraction data were integrated and scaled using the program package HKL3000 (17). The structure was solved by molecular replacement using the individual protein domains of SERCA (PDB code 2C8L) (18) as the search models. Solutions were found for the three cytoplasmic domains using Phaser (19), but no solution for the transmembrane region was obtained. The initial model was constructed from the three cytoplasmic domains and used to calculate initial electron density maps into which the individual transmembrane helices were manually fit using the program Coot (version 0.6.1 (20)) and the connectivity of the M4 and M5 helices to one of the cytoplasmic domains and the C-terminal transmembrane helix as points of reference. Helix M4 required fitting as two distinct sections and the connecting polypeptide was manually fit to the electron density in Coot. The structure was subjected to interative rounds of model building and refinement using the program Refmac5 (21) and included the use of TLS tensors (22) to model the anisotropy of the individual domains and PLB. In addition to SERCA and PLB, the final model includes one potassium ion and 2 non-covalently associated maltose molecules, for which the acyl chains were not visible in the electron density and hence have been modeled simply as maltose residues. Initial attempts to include a magnesium ion bound to site I in the transmembrane domain gave rise to a negative difference peak at 4.2 σ in the Fo − Fc electron density map at the position of the modeled magnesium ion. Based on this information, combined with the lack of a positive peak in the Fo − Fc electron density map greater than 2.7 σ when the model is refined in the absence of magnesium and the fact that magnesium ions are not required for PLB cross-linking, we conclude that the PLB-SERCA complex lacks metal ions bound to the transmembrane metal sites.
Ca2+-ATPase Assay
Ca2+-ATPase activity was determined colorimetrically by measuring inorganic phosphate release from ATP (16). 12 μg of solubilized Ca2+-ATPase protein (taken from mother liquors used for protein crystallization) were added to 1 ml of ATPase buffer containing 50 mm MOPS (pH 7.0), 3 mm MgCl2, 3 mm ATP, 100 mm KCl, 1 mm EGTA, 5 mm NaN3, and 3 μg/ml of A23187. Ionized Ca2+ concentrations were set by addition of CaCl2. Some reaction tubes in addition contained 60–80 μg of anti-PLB 2D12 antibody to reverse PLB inhibition of Ca2+-ATPase activity (23). Reactions were conducted at 37 °C and Ca2+-dependent activities are reported. KCa values designate Ca2+ concentrations at which Ca2+-ATPase is half-maximally activated.
Cross-linking
Cross-linking assays were conducted with solubilized Ca2+-ATPase protein reconstituted with PLB4 taken from mother liquors used for protein crystallization. The Ca2+-ATPase protein (12 μg) was added to 500 μl of buffer identical to that indicated above for determination of Ca2+-ATPase activity with omission of NaN3. Cross-linking was conducted with 1 mm KMUS for 2 min at room temperature. Reactions were stopped with 7.5 μl of gel loading buffer containing 15% SDS and 100 mm dithiothreitol. Samples were subjected to SDS-PAGE and immunoblotting with the anti-PLB antibody, 2D12, for detection of PLB cross-linked to SERCA1a (skeletal muscle isoform) (Fig. 3C). Cross-linking of the canine cardiac isoform of the Ca2+ pump (SERCA2a) to N30C-PLB or PLB4 co-expressed in insect cells was conducted identically as previously described (10) in 50 mm MOPS (pH 7.0), 100 mm KCl, and 1 mm Ca2+, EGTA buffer in the presence or absence of 3 mm MgCl2 (Fig. 6). Cross-linking was conducted with 1 mm KMUS for 1 min at room temperature. The results shown in Fig. 6 were conducted in the absence of ATP, however, similar results were obtained with inclusion of 3 mm ATP. Ki values indicate Ca2+ concentrations at which cross-linking is inhibited by 50%.
FIGURE 3.

Crystal structure and functional characterization of the PLB4-SERCA complex. A, a ribbon model of the complex between SERCA and PLB4. In SERCA the cytoplasmic headpiece consists of actuator (A) (gray), phosphorylation (P) (gold), and nucleotide-binding (N) (green) domains. The transmembrane domain helices of SERCA are colored cyan, except for M4, which is colored blue. The bound PLB4 molecules are colored magenta (chain B) and yellow (chain C) (Table 1). The figure was generated using PyMOL (The PyMOL Molecular Graphics System, version 1.5.0.4, Schrödinger, LLC). B, Ca2+-ATPase activity of nonyl maltoside-solubilized SERCA alone and after reconstitution with purified, solubilized WT-PLB or PLB4. Activity was measured in the presence and absence of the anti-PLB monoclonal antibody, 2D12. KCa values (μm) for Ca2+ activation of ATP hydrolysis were: no PLB, 0.26 ± 0.01; WT-PLB, 0.62 ± 0.03 and 0.35 ± 0.02 (+ 2D12); PLB4, 1.38 ± 0.07 and 0.53 ± 0.03 (+ 2D12). Mean ± S.E. from three to six determinations are shown. C, anti-PLB immunoblot showing the Ca2+ effect on cross-linking of PLB4 to SERCA with KMUS under the same conditions used for determination of Ca2+-ATPase activity. Ki value (μm) for Ca2+ inhibition of cross-linking was 1.60 ± 0.16. Mean ± S.E. from 6 determinations are shown. D, Coomassie Blue-stained gel showing SERCA solubilized from rabbit skeletal muscle SR membranes and WT-PLB and PLB4 purified in decyl maltoside (± boil). 10 μg of membrane protein were electrophoresed per gel lane.
FIGURE 6.
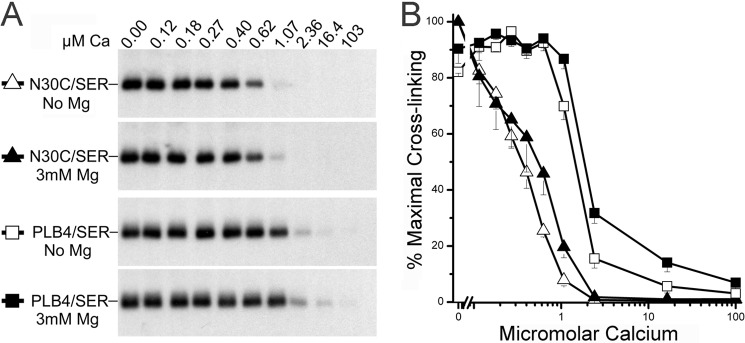
Lack of effect of Mg2+ on PLB cross-linking. A, autoradiograph showing cross-linking of N30C-PLB or PLB4 to SERCA2a expressed in insect cell membranes. Cross-linking was conducted with KMUS over a range of Ca2+ concentrations in the presence and absence of 3 mm MgCl2. B, quantified results plotted as percentage of maximal cross-linking determined at zero Ca2+ concentration. Mg2+ had no effect on maximal cross-linking. Ki values (μm) for Ca2+ inhibition of cross-linking of N30C-PLB to SERCA were 0.37 ± 0.05 and 0.46 ± 0.07 in the absence and presence of Mg2+, respectively. Ki values for Ca2+ inhibition of PLB4 cross-linking were 1.52 ± 0.06 and 1.70 ± 0.10, respectively. Mean ± S.E. from three to five determinations.
RESULTS AND DISCUSSION
Crystallization of the PLB-SERCA Complex
Crystallizing the novel Ca2+-free state of SERCA that binds PLB required that the solubilized enzyme be reconstituted with PLB in the absence of Ca2+ (presence of EGTA). However, the Ca2+-ATPase is extremely labile and prone to denaturation when solubilized without Ca2+ (24, 25), and the detergents typically used for SERCA solubilization/crystallization (C12E8) (15, 18, 26) and for PLB reconstitution with SERCA (C12E8 or octyl glucoside) (14, 27) had strong detrimental effects on Ca2+ affinity (Fig. 2A) and/or stability of the enzyme over time (Fig. 2B). For example, solubilizing SERCA in the absence of Ca2+ in C12E8 caused a 3-fold decrease in Ca2+ affinity and loss of enzyme activity with a t½ of ∼3 days at 4 °C. Octyl glucoside, on the other hand, did not affect Ca2+ affinity, but caused even more rapid denaturation of the enzyme, with a t½ of less than 1 day at 4 °C.
Consequently, a major challenge was to develop a new detergent system that would facilitate co-crystallization of SERCA reconstituted with PLB without denaturing the enzyme. The nonionic detergent nonyl maltoside was ultimately selected for solubilization of SERCA as it had little effect on the Ca2+ affinity of the enzyme (KCa = 0.25 versus 0.21 μm in the absence of detergent) (Fig. 2A), and preserved 90–100% of the solubilized Ca2+-ATPase activity for several weeks at 4 °C (Fig. 2B). SERCA solubilized in nonyl maltoside was then reconstituted with WT-PLB or PLB4 solubilized in decyl maltoside or dodecyl maltoside (shown with PLB4 solubilized in decyl maltoside in Fig. 2B), detergents that also preserved SERCA catalytic activity for weeks when stored at 4 °C. Ca2+-ATPase assays in the newly developed detergent system show that WT-PLB and PLB4 decreased the Ca2+ affinity of the solubilized enzyme by 2- and 5-fold, respectively, but had no significant effect on maximal ATPase activity measured at saturating Ca2+ concentrations (Fig. 3B). This alteration in the apparent Ca2+ affinity of SERCA is the hallmark of PLB inhibition (2, 3). As expected, addition of the 2D12 anti-PLB antibody (which mimics the effect of Ser16/Thr17 phosphorylation of PLB (23)) largely reversed SERCA inhibition by WT-PLB and PLB4 (Fig. 3B, shaded dotted lines).
Ca2+-dependent binding interactions between solubilized SERCA and PLB4 were also confirmed using chemical cross-linking. Cross-linking of residue N30C of PLB4 to Lys328 of SERCA with the heterobifunctional cross-linker KMUS (7, 10) was inhibited over the same Ca2+ concentration range (Ki = 1.6 μm) (Fig. 3C) as enzyme activation occurred (KCa = 1.4 μm) (Fig. 3B). This strongly suggests that PLB decreases the Ca2+ affinity of SERCA by disrupting Ca2+ binding (10) to the enzyme. Fig. 3 further confirms that normal protein-binding interactions between PLB and SERCA were maintained in the newly developed detergent system, recapitulating results previously observed with intact SR vesicles (16) or with ER membranes co-expressing the recombinant proteins (10). The remarkable stability of Ca2+-free SERCA solubilized in nonyl maltoside is reported here for the first time.
It should be mentioned that rabbit skeletal muscle SR vesicles were used as the source of the Ca2+ pumps in this study due to the high content of the ATPase protein in the SR membrane, making enzyme purification unnecessary (15) (Fig. 3D). (SERCA1a is highly homologous to the cardiac isoform, SERCA2a, and is regulated identically by PLB (6).) In addition, both WT-PLB and PLB4 prepared in alkyl mannosides were predominantly multimeric on SDS gels; WT-PLB was mostly pentameric, and PLB4 ran mostly as pentamers and tetramers. Boiling in 6% SDS prior to PAGE was required to destabilize these highly oligomeric forms of PLB (Fig. 3D), as observed previously with PLB purified in other detergents (14, 28).
Crystallization trials with SERCA solubilized in nonyl mannoside were conducted at pH 7.0 in the presence and absence of PLB4, nucleotides (ADP and ATP), and Ca2+. In the absence of Ca2+ (1 mm EGTA), SERCA crystals grew only in the presence of PLB4, and crystal growth was inhibited by increasing Ca2+ concentration, consistent with PLB stabilizing a Ca2+-free state of enzyme (5). Addition of ADP or ATP to the mother liquors appeared to improve crystal quality in some trials; however, no electron density corresponding to nucleotide bound to SERCA was resolved in any of these crystal forms (data not shown). Crystallization trials were also conducted with WT-PLB, but the naturally occurring protein proved inferior to PLB4 in promoting SERCA crystal growth, presumably because its lower affinity for SERCA relative to PLB4 (10) makes it less efficient at stabilizing the pump for crystallization.
PLB Binding Site on SERCA
The structure of E2-PLB at 2.83-Å resolution (Table 1) shows PLB4 as an α-helix bound within a groove formed between transmembrane helices M2, M4, M6, and M9 of SERCA (Fig. 3A, magenta helix). Electron density corresponding to residues 21 to 49 of the 52 amino acids comprising PLB (Fig. 1) was observed. The α-helix extends uninterrupted from residue 21 in the cytoplasm to residue 49 in transmembrane domain. The N-terminal 20 residues of PLB, which includes residues Ser16 and Thr17 that are subject to phosphorylation, are not visible in the electron density, but were, nonetheless, functional in the mother liquors as evidenced by the effects of the 2D12 antibody (Fig. 3B). (Western blots of the re-solubilized crystals demonstrated that the PLB in E2-PLB was completely intact; PLB was detected at the appropriate mobility by the 2D12 antibody, which recognizes residues 7–14 of PLB, and by the 1F1 antibody, which recognizes residues 1–5 of PLB.) Residues 50–52 of PLB, predicted previously to remain α helical (9), were also unobserved in the electron density. Consequently, it would appear that the central 29 residues of PLB form the majority of the stable inhibitory interactions with the Ca2+-ATPase. The E2-PLB structure confirms the location of the PLB binding site on the cardiac enzyme (SERCA2a) predicted previously by chemical cross-linking (5, 7–9). The distances between specific amino acid residues of PLB and SERCA observed in the E2-PLB structure were expected to be consistent with the distances estimated by cross-linking (5–9, 16); however, we were surprised by how closely the experimental values agreed with those determined from the structure (Table 2), highlighting the utility of cross-linkers as molecular rulers.
TABLE 1.
Data collection and refinement statistics
| SERCA-PLB4 | |
|---|---|
| Data collection | |
| Space group | P212121 |
| Cell dimensions | |
| a, b, c (Å) | 61.59, 93.09, 316.4 |
| Cell angles (α, β, γ °) | 90.0, 90.0, 90.0 |
| Resolution (Å) | 50.0–2.83 (2.89–2.83) |
| Rmerge(%) | 8.4 (49.2) |
| I/σ〈I〉 | 13.2 (2.0) |
| Completeness (%) | 97 (97) |
| Redundancy | 5.5 (4.5) |
| Wilson B-value (Å2) | 88.5 |
| Refinement | |
| Resolution (Å) | 50.0–2.83 |
| No. reflections | 42925 |
| Rwork/Rfree(%) | 23.9/28.4 |
| No of atoms | |
| Protein | 7861 |
| Ligand/ion | 47 |
| B-factors (Å2) | |
| Serca (chain A) | 89.6 |
| PLB (chain B) | 140.2 |
| PLB (chain C) | 183.4 |
| Ligand/ion | 85.5 |
| Root mean square deviations | |
| Bond lengths (Å) | 0.009 |
| Bond angles (°) | 1.20 |
| Ramachandran plot | |
| Core | 88.9 |
| Allowed | 11.0 |
TABLE 2.
Distances between residues of PLB and SERCA determined by cross-linking (X-link) versus x-ray diffraction (crystal)
Cross-linking distances between residues of PLB and SERCA were determined after co-expression of selected mutants of PLB with SERCA2a (5, 7–9) or SERCA1a (6) in microsomal membranes, or with native PLB and SERCA2a in human cardiac SR vesicles (16). Cross-linking was conducted with homo- or heterobifunctional cross-linking reagents (5, 7–9, 16) or with copper phenanthrolene (6). The distances in the PLB4/SERCA crystal structure were determined by measuring from the β-carbon of the PLB residue to the closest side chain atom of the corresponding SERCA residue (native protein). For V49G in PLB4 the distance was measured from the α-carbon of Gly49.
| PLB | SERCA | Distance (Å) |
Ref. | |
|---|---|---|---|---|
| X-link | Crystal | |||
| N27C | Lys328 | 9–14 | 14 | 7 |
| Lys27 | Lys328 | 8 | 10–13a | 16 |
| N30C | Cys318 | 10 | 10 | 5 |
| N30C | Lys328 | 14–16 | 17 | 7 |
| L31C | T317C | 10 | 10 | 8 |
| V49C | V89C | 0–5 | 5 | 6, 9 |
| M50C | V89C | 10 | NDb | 9 |
| L52C | V89C | 5 | ND | 9 |
a The distance measurement between Lys27 and Lys328 was estimated by computational mutation of Ala27 to Lys and selecting side chain rotomers compatible with the local structure for both Lys27 and Lys328, then measuring the closest approaches for the side chains.
b ND, not determinable.
PLB Effects on the Ca2+ Binding Sites
The signature effect of PLB is its ability to decrease the apparent Ca2+ affinity of SERCA, and the physical basis for this functional effect is now revealed in the E2-PLB structure. The two high affinity Ca2+-binding sites (sites I and II) of SERCA, which are formed between transmembrane helices M4, M5, M6, and M8 near the cytoplasmic membrane interface (26), are both severely disrupted by PLB4 binding. Fig. 4A shows significant unraveling of the “M4 kink” induced by PLB4, and the side chain of the Ca2+ gating residue, Glu309, now faces away from the Ca2+ binding sites (Fig. 4B). The three main chain carbonyls on M4 contributing to site II are also displaced, making site II non-existent in our structure. Site I is largely collapsed and would not support coordination of either Ca2+ or Mg2+ in the conformation induced by PLB4 binding (Fig. 4B). The contact of PLB4 with helices M4 and M6 appears to close site I for metal occupancy because the side chains of Asn768, Glu771, Asn796, and Asp800 are all too close to accommodate the binding of a metal ion between their side chains and the peptide carbonyl oxygen of Ala305. In particular, the distance geometry of residues Asn768 and Asn796 suggests the orientation of their side chain amide groups are switched from the orientations compatible with metal ion coordination to support hydrogen bonding interactions with the peptide carbonyl oxygen of Ala305 and the side chain of Glu771 (Fig. 4B). Thus, with PLB4 bound to SERCA, the Ca2+ pump adopts a conformation that is incompatible with either Ca2+ or Mg2+ binding to sites I and II. The E2-PLB structure suggests that for the polar side chains and main chain carbonyls forming the Ca2+ binding sites to align properly, PLB4 must completely dissociate from its binding site in the groove between the M2, M4, M6, and M9. Conversely, close spacing of M2 and M9 in the Ca2+-bound structure of SERCA (E1·Ca2) (26) appears to be incompatible with PLB binding to the PLB binding site defined in our structure. This result confirms the cross-linking based theory of mutually exclusive binding of PLB and Ca2+ to SERCA (5–9), which has been controversial (29–31).
FIGURE 4.

Binding of PLB4 to the transmembrane domain of SERCA. A, a view of PLB4 bound to SERCA roughly orthogonal to that in Fig. 3A including the side chains of PLB4 interacting with the transmembrane domain; the color scheme is identical to Fig. 3A. B, a horizontal section of the SERCA transmembrane domain showing the interaction area of PLB4 in proximity of the Ca2+-binding sites, the color scheme is identical to Fig. 3A. Critical side chains are labeled and displayed, as are the nearby transmembrane helices. The dotted lines and numerical values indicate the distances between potential hydrogen bond donors and acceptors. The figure was generated using PyMOL.
In the E2-PLB structure, Asn34 of PLB appears to be an important and highly organized region of contact specifically perturbing the Ca2+ binding sites (Fig. 5A). Asn34 forms critical contacts with the main chain carbonyl oxygen of Gly801 (M6) and with the side chains of Thr805 (M6) and Gln108 (M2) of SERCA. Gln108, in turn, contacts the side chain of Thr317, which may influence the positioning of the important Ca2+-site II ligand, Glu309 (5, 8). The highly structured network of contacts with Asn34 of PLB appears to initiate a repositioning of residues 795–804 within the M6 helix; Asn796 now interacts with carbonyl oxygen of Ala305 and Ca2+ binding site I is collapsed (Fig. 4B). These findings highlight the key role of Asn34 in the mechanism of PLB inhibition, and explain how the N34A mutation of PLB causes loss of PLB function (32), and how the T795A, L802A, T805A, and F809A mutations in SERCA all partially attenuate PLB inhibition (33). Another SERCA mutation known to disable Ca2+ binding is G801V (34). Our structure provides a plausible explanation for this phenomenon because a side chain at position 801 would be impinged on by Val104 and reposition the M6 helix and its Ca2+ ligands, Asn796 and Glu800, in a manner similar to how Asn34 repositions the M6 helix and these same Ca2+ ligands to collapse the Ca2+ binding site upon PLB binding (Fig. 5A).
FIGURE 5.

Critical interactions at the interface between SERCA and PLB4. A, a horizontal section of the SERCA transmembrane domain centered near the key inhibitory residue Asn34 of PLB4. The color scheme is identical to Fig. 4A. Key residues at the interaction surface are labeled, as are the individual transmembrane helices in this region. The dotted line indicates inferred hydrogen bonding between donor and acceptor atoms and the numerical value indicates the distance between atoms potentially sharing hydrogens. B, a “peeled” surface view of the interaction surface between PLB4 and SERCA. For this figure, PLB4 was rotated 180 degrees in the vertical plane and translated to the right-hand side of this figure to expose the interacting surfaces and their chemical character. The color scheme utilizes “atom type” coloring (blue for nitrogen, red for oxygen, yellow for sulfur, and green for carbon atoms not involved in the contact surface between PLB4 and SERCA). Carbon atoms in amino acids residing either in PLB4 or SERCA that have at least one atom at the contact surface are colored gray. Amino acid residues that form the contact surface are labeled. C, the original figure-of-merit σA-weighted 2Fo − Fc (blue) and Fo − Fc (green) electron density maps for PLB4 bound to the transmembrane domain of SERCA were generated prior to their inclusion in the model, superimposed on the final refined coordinates for the complex. D, structural super-positioning of the PLB4-SERCA complex (cyan and magenta ribbons for SERCA and PLB4) with that of the Ca2+-free E2 complex stabilized by TG (green ribbons, RCSB code 1IWO (24)). The structures were super-imposed utilizing the Cα atoms of residues 750–990. Key residues involved in Ca2+ binding are labeled, as is Ile38 of PLB4.
Another functionally important residue of PLB, Leu31, is located one turn of the α-helix above Asn34, and only 3.7 Å away from Thr805 of SERCA. Leu31 fits in a hydrophobic pocket defined by the side chains of Leu802, Ala806, and Phe809 in M6, and Trp932 and Leu939 in M9 (Fig. 5, A and B). Like N34A, the L31A mutation completely disables PLB inhibition (32), whereas L31I preserves full functional activity (8). This suggests that hydrophobic interactions of the proper size between residue 31 of PLB and M6/M9 of SERCA are required for proper positioning of Asn34 and SERCA inhibition. The binding interface between PLB and SERCA is largely stabilized by hydrophobic interactions including Van der Waals contacts between carbon atoms (Fig. 5B, gray). The contact surface buries between 1100 and 1200 Å2 and about 40% of the available surface area of PLB4 between residues 21 and 49. There are only a few polar contacts, specifically between the amide nitrogen of Asn34 (blue) and the main chain carbonyl of Gly801 and the amide carbonyl of Gln108 and side chain of Thr805 (all in red, Fig. 5B). It is particularly interesting that the extensive polar contacts surrounding Asn34 takes place in the context of a particularly hydrophobic surface, which likely enhances their impact on SERCA function.
Manipulation of side chain contacts between PLB and SERCA can enhance as well as attenuate PLB inhibitory function, explaining the superinhibitory nature of PLB4. For example, the N27A mutation (35) near the cytoplasmic membrane interface of PLB would allow PLB4 to bind more tightly to SERCA because the smaller side chain of Ala fits more snugly into the hydrophobic cleft between Phe809 (M6) and Trp932 (M9), where the distances between the methyl side chain of Ala and the two aromatic side chains are only 3.6–3.9 Å (Fig. 5B). Ala substitution at Leu37 in the bilayer interior (32, 36) is likewise expected to attenuate steric clash between the long alkyl side chain of Leu37 and the hydrophobic side chains of Ile103 and Val104 at M2 (Fig. 5B). The more compact side chain of Ile37 appears to fit the available space within this hydrophobic pocket better than Leu37, which would also explain the superinhibitory effect of the L37I mutation (37). At the C-terminal end of PLB4 at the luminal membrane interface, residue 49 of PLB is very close to Val89 (M2) (Table 2) (6, 9). The smaller side chains of Ala or Gly at this position appears to reduce steric hindrance with Val89 (Fig. 5B), explaining the superinhibitory effects of the V49A and V49G mutations in PLB (9). Thus PLB4 inhibitory function is augmented by the additive effects of the N27A, L37A, and V49G mutations, each of which optimizes the packing interactions between PLB and hydrophobic residues in the Ca2+-ATPase. This observation also explains why PLB4 binds to SERCA with an affinity greater than that of TG (10), which binds with sub-nanomolar affinity to the Ca2+ pump (11, 12).
It should be pointed out that the above interpretation of how the point mutations in PLB4 would alter PLB interactions with the Ca2+-ATPase and enhance its inhibitory function are based upon the assumption that WT-PLB would stabilize the enzyme in a nearly identical state as that captured by PLB4. Indeed, the structure of SERCA with PLB4 bound suggests that the point mutations in PLB4 would have only localized structural affects relative to WT-PLB. Moreover, the distances between specific amino acids of PLB and SERCA determined by cross-linking are nearly identical, whether determined with PLB molecules of normal or superinhibitory strength, including cross-linking of WT-PLB in native human SR vesicles (16). The distances determined by cross-linking with the normal strength PLB mutants reported in Table 2 are in close agreement with the distances between the same residues observed in the PLB4/SERCA crystal structure. Therefore, the crystal structure of SERCA with WT-PLB bound is likely to be very similar to the PLB4 bound structure.
Multimeric Nature of PLB
PLB is a pentamer of identical monomers (28, 38, 39). It has been proposed for some time that there is a dynamic equilibrium between PLB pentamers and monomers in the SR membrane (40, 41), and that the PLB monomer is responsible for binding to SERCA and inhibiting it (32, 36). Residues Leu37, Ile40, Leu44, Ile47, and Leu51 of PLB form the Leu/Ile zipper (40) that stabilizes the non-inhibitory PLB pentamer (41); Ala substitution at any of these residues promotes pentamer dissociation, an increase in PLB monomers, and increased enzyme inhibition (32, 36, 37). It was also suggested that PLB has two distinct functional faces located on opposite sides of the transmembrane helix: the Leu/Ile zipper face, which when PLB is bound to SERCA points away from the enzyme and is involved only in PLB pentamer formation, and the inhibitory face, which comes in direct contact with the Ca2+-ATPase and mediates enzyme inhibition (32). Here we find that this idea is indeed correct for the Ile arm of the zipper; the heptad repeat of Ile zipper residues 40 and 47 is oriented away from the Ca2+ pump when the PLB monomer is bound to the enzyme (Figs. 4A and 5C, magenta). In contrast, on the Leu arm of the zipper, both Leu37 (mutated to Ala in PLB4) and Leu44 make direct contact with the Ca2+ pump at M2 (at Ile103 and Val104, and at Leu96, respectively) (Figs. 4A and 5B), consistent with the dual role of these two zipper residues in both pentamer formation and enzyme inhibition (37). At other positions along the transmembrane helix of PLB, residues Phe35, Ile38, Cys41, Leu42, Ile45, and Ile48 all make direct contact with non-polar residues in several transmembrane helices of SERCA (Fig. 5, B and C), contributing to the high binding affinity of the PLB monomer for the Ca2+-ATPase (10, 16).
It should be noted that the mobility forms of WT-PLB and PLB4 observed on SDS gels are quite different. Whereas WT-PLB ran predominately as pentamers with a small amount of monomers, all five mobility forms of PLB4 were observed (Fig. 3D). Prior to boiling in 6% SDS, pentamers and tetramers predominated with PLB4; after boiling in SDS, monomers and dimers were the main mobility forms. The significance of this observation became apparent when we discovered additional weak electron density embedded in the membrane near PLB4 in all of our datasets (Figs. 3A, 4A, and 5, A and C, yellow helices). The electron density is consistent with a 14-amino acid-long α-helix (yellow) that is in direct contact with the outer face of the PLB4 monomer directly bound to SERCA (magenta), but this second helix does not contact SERCA. The nature of the observed electron density for this second helical region (PLB (chain C) in Table 1) indicates significant conformational freedom, low occupancy, or both. This density was able to accommodate the PLB4 sequence, but was not compatible with the sarcolipin (SLN) sequence register, and PLB4 dimers were also present in our re-solubilized crystals run on SDS-PAGE (data not shown). Therefore, the extra helix is most likely attributable to a second PLB4 molecule bound to PLB but not physically in contact with SERCA. Residues Phe32, Cys36, Leu39, Ile40, Leu43, and Ile47 of PLB4 appear to be interacting with residues Cys30, Ile33, Cys36, Ala37, and Ile40 of the second PLB4 molecule, which includes the two Ile zipper residues (40 and 47) that help to stabilize PLB pentamers (Figs. 4A and 5C).
Although PLB4 is largely pentameric or tetrameric when purified in non-denaturing detergents, the crystal structure clearly indicates that the PLB monomer forms the important contact surface between PLB4 and SERCA (Fig. 5C). No PLB4 pentamers or tetramers attached to SERCA were identified in any of the datasets. Therefore, a reversible equilibrium (32, 36) between pentamer-tetramers and monomer-dimers of PLB4 must exist to allow regulatable inhibition of SERCA activity over the range of Ca2+ concentrations depicted in Fig. 3, B and C. Consistent with this, at the molar ratio of PLB to SERCA used for crystal formation (2.9 to 1), there are sufficient dimers, but insufficient pentamers or tetramers, to allow complete inhibition of SERCA at the lower Ca2+ concentrations assayed (Fig. 3B). Pentamer formation by monomers released from SERCA thus appears to aid in complete reversal of enzyme inhibition at saturating Ca2+ concentration (Fig. 3, B and C), as recently demonstrated using intact SR vesicles from human hearts (16).
The structure is therefore consistent with there being an equilibrium between PLB-SERCA heterodimers, free PLB monomers, and PLB pentamers in the SR membrane, with reversible exchange of PLB protomers between the different oligomeric states. Even though PLB4 is strongly bound to the enzyme in the absence of Ca2+, there is still a reversible equilibrium with PLB continuously associating and dissociating from SERCA. During this rapid exchange, the collapsed Ca2+ binding sites can briefly re-form, providing an opportunity for a Ca2+ to bind to the enzyme. High Ca2+ concentrations shift the binding equilibrium toward PLB dissociation, which is aided by the fact that the free PLB molecules can oligomerize into stable homopentamers.
The Conformation of SERCA That Binds PLB
The structure of SERCA in the PLB4-SERCA complex is distinct from the previously determined structures of SERCA in the E2 (low Ca2+ affinity) or E1 (high Ca2+ affinity) states (26). The Ca2+-ATPase in our structure is globally similar to E1·Ca2 in the overall spacing of the transmembrane helices and in the open arrangement of the cytoplasmic headpiece (Fig. 3A). However, unlike E1·Ca2, the M1 helix in the PLB4-bound Ca2+ pump structure is positioned lower in the membrane, and is distinctly bent at Leu60. In this position, the “M1 sliding door” that controls the opening to the purported Ca2+ access channel is open, exposing a number of negatively charged residues that lead to the Ca2+ binding sties (26, 42). An open Ca2+ access channel, as observed in our structure, is a key feature of the enzyme in the E2 state (26, 42). In addition, the metal-free Ca2+ binding sites in the PLB4-SERCA complex more closely resemble E2 than E1 (24). PLB4 contacts with M4 and M6 appear to stabilize hydrogen bonding between residues Asn796, Asn768, Glu771, and the main chain carbonyl of Ala305 (Fig. 5D). Although the spatial arrangement is somewhat different, conceptually similar interactions are observed in the metal-free Ca2+ binding sites in the E2-TG structure (24) (Fig. 5D). The apparent hydrogen bonding at the Ca2+ binding sites is consistent with the enzyme being protonated; it is generally accepted that protons stabilize the empty Ca2+ binding sites in the E2 state (26, 42). Therefore, PLB stabilizes a structure in which the Ca2+ binding sites are collapsed and appear to be protonated, and we define this structure as E2-PLB.
While this manuscript was in preparation, two separate groups (Toyoshima et al. (43) and Winther et al. (44)) published crystal structures of Ca2+-free SERCA stabilized by very high concentrations of MgSO4 (40–75 mm) (referred to hereafter as E1·Mg (43)). The structures were nearly identical, and both had SLN bound. SLN is a 31-amino acid skeletal-muscle “homologue” of PLB with ambiguous function (Fig. 1). Because the contacts between SLN and SERCA observed in the E1·Mg structures were similar to PLB-SERCA interactions determined previously by cross-linking (5–9), both groups postulated that the E1·Mg state they crystallized is the same state that binds PLB. On a global scale, PLB and SLN appear to bind to similar conformations of the transmembrane domain of SERCA, albeit with distinct orientations of the cytoplasmic domains. However, at the molecular level, notable structural differences in their respective transmembrane domains support that PLB stabilizes a distinct conformation of SERCA. Specifically, the Ca2+ binding sites in the E1·Mg structures are partially formed and have a distinctly E1-like appearance. On the other hand, in the metal-free E2 state that binds PLB, the Ca2+ binding sites are completely collapsed with interactions more similar to E2 than E1. In contrast to claims in these two recent papers (43, 44), the effects of SLN on Ca2+-ATPase activity are different from those of PLB and remain controversial: no effect, Ca2+ affinity effects, Vmax effects, and uncoupling effects of SLN have all been reported (27, 45–47). In detailed comparative studies, two separate groups recently concluded that SLN and PLB regulate SERCA by completely different mechanisms (27, 45). In one study, SLN effects on Ca2+-ATPase activity were attributed to the unique C-terminal tail of SLN (containing Arg27 and Tyr31), which is not found in PLB (27). In the second study, SLN had no effect on Ca2+-ATPase activity, but instead uncoupled Ca2+ transport from ATP hydrolysis, and SLN continued to interact with SERCA in the presence of Ca2+ (45), in contrast to PLB, which dissociates from SERCA in the presence of Ca2+ (5). Therefore, unlike the recent SLN-bound E1·Mg structures, the PLB-induced structural changes in SERCA observed presently have clear mechanistic implications that are consistent with PLB effects on Ca2+ affinity (2).
The physiological significance of the E1·Mg state of SERCA is also worth discussion. In both studies mentioned above (43, 44), the structures were solved with the Ca2+-free enzyme solubilized in C12E8, a condition known to cause irreversible enzyme inactivation (24, 25) (Fig. 2). It seems likely that at the high concentrations of MgSO4 utilized, Mg2+ was acting as a surrogate for Ca2+, thereby preventing enzyme denaturation. However, whether the E1·Mg state occurs at physiological Mg2+ concentration is unknown, and the functional role of the putative state is unclear. Toyoshima et al. (43) concluded that E1·Mg is an obligate intermediate in the catalytic cycle of SERCA and that Mg2+ binding accelerates Ca2+ binding by reducing the energy required for formation of E1·Ca. On the other hand, Winther et al. (44) concluded the opposite, that Mg2+ binding to the “low affinity Mg2+ sites” actually retards SERCA activation by Ca2+.
Cross-linking of PLB to SERCA can be used to test whether Mg2+ is required for PLB binding, and whether Mg2+ affects SERCA Ca2+ affinity, as postulated by Toyoshima et al. (43) and Winther et al. (44). Using both N30C-PLB, which alters Ca2+ affinity like WT-PLB, and PLB4, which supershifts Ca2+ affinity, we found that physiological concentrations of Mg2+ (3 mm) had no effect on the extent of PLB cross-linking to SERCA, and only minor effects on Ki values for Ca2+ inhibition of PLB cross-linking, thus reflecting minimal effects of Mg2+ on Ca2+ affinity of the enzyme (Fig. 6). Clearly, E1·Mg is not the state required for PLB binding, as was recently proposed (43, 44). Moreover, based upon our crystal structure and cross-linking results (Fig. 6), we conclude that the E1·Mg state of SERCA obtained at 40–75 mm MgSO4 (43, 44) does not exist at a physiological Mg2+ concentration (∼3 mm).
In conclusion, we have determined the crystal structure of PLB4 bound to a unique metal-free E2 state of SERCA, one of the critical outstanding conformational states of the Ca2+-ATPase remaining to be solved (5, 10). Interpreted in light of the extensive biochemical and physiological studies on PLB (2, 3), the new SERCA structure has allowed us to explain how through direct effects on Ca2+ affinity, dephosphorylated PLB inhibits SERCA activity and decreases contractility of the heart. Co-crystallization of PLB4 with SERCA was achieved using a newly developed detergent system specifically designed to maintain SERCA catalytic activity and normal regulation by PLB after solubilization in the absence of Ca2+. Using this new system, it should be possible to crystallize SERCA with other forms of PLB, including WT-PLB, and ultimately to resolve the binding site for the N terminus of PLB on the Ca2+ pump, which has remained problematical (6, 7, 48). The N-terminal domain of PLB is of particular physiological interest because β-adrenergic receptor-stimulated phosphorylation of PLB at Ser16 and Thr17 is fundamental to the mechanism by which PLB regulates myocardial contractility (1, 4).
Acknowledgments
Results shown in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by UChicago Argonne, LLC, for the US Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357. We thank Stephan Ginell and Marianne Cuff for assistance at SBC-CAT 19ID. We also thank Glen Schmeisser.
This work was supported, in whole or in part, by National Institutes of Health Grants R37-HL049428 (to L. R. J.), and R01-AA18123 and R01-DK79887 (to T. D. H.). Thomas D. Hurley holds significant financial equity in SAJE Pharma, LLC. However, none of the work described in this study is related to, based on, or supported by the company.
- PLB
- phospholamban
- SR
- sarcoplasmic reticulum
- SERCA
- sarco(endo)plasmic reticulum Ca2+-ATPase
- SERCA1a
- fast skeletal muscle isoform of SERCA
- SERCA2a
- cardiac isoform of SERCA
- WT-PLB
- wild-type PLB
- PLB4
- N27A, N30C, L37A, V49G-PLB
- 2D12
- anti-PLB monoclonal antibody
- M
- transmembrane domain
- E1
- high Ca2+-affinity conformation of Ca2+-ATPase
- E2
- low Ca2+ affinity conformation of Ca2+-ATPase
- decyl maltoside
- n-decyl-β-d-maltopyranoside
- nonyl maltoside
- n-nonyl-β-d-maltopyranoside
- dodecyl maltoside
- n-dodecyl-β-d-maltopyranoside
- C12E8
- octaethylene glycol monododecyl ether
- octyl glucoside
- octyl β-d-glucopyranoside
- KCa
- Ca2+ concentration required for half-maximal effect
- KMUS
- N-κ-maleimidoundecanoyl-oxysulfosuccinimide ester
- SLN
- sarcolipin
- TG
- thapsigargin.
REFERENCES
- 1. Lindemann J. P., Jones L. R., Hathaway D. R., Henry B. G., Watanabe A. M. (1983) β-Adrenergic stimulation of phospholamban phosphorylation and Ca2+-ATPase activity in guinea pig ventricles. J. Biol. Chem. 258, 464–471 [PubMed] [Google Scholar]
- 2. Simmerman H. K., Jones L. R. (1998) Phospholamban. Protein structure, mechanism of action, and role in cardiac function. Physiol. Rev. 78, 921–947 [DOI] [PubMed] [Google Scholar]
- 3. Young H. S., Stokes D. L. (2004) The mechanics of calcium transport. J. Membr. Biol. 198, 55–63 [DOI] [PubMed] [Google Scholar]
- 4. Wegener A. D., Simmerman H. K., Lindemann J. P., Jones L. R. (1989) Phospholamban phosphorylation in intact ventricles. Phosphorylation of serine 16 and threonine 17 in response to β-adrenergic stimulation. J. Biol. Chem. 264, 11468–11474 [PubMed] [Google Scholar]
- 5. Jones L. R., Cornea R. L., Chen Z. (2002) Close proximity between residue 30 of phospholamban and cysteine 318 of the cardiac Ca2+ pump revealed by intermolecular thiol cross-linking. J. Biol. Chem. 277, 28319–28329 [DOI] [PubMed] [Google Scholar]
- 6. Toyoshima C., Asahi M., Sugita Y., Khanna R., Tsuda T., MacLennan D. H. (2003) Modeling of the inhibitory interaction of phospholamban with the Ca2+ ATPase. Proc. Natl. Acad. Sci. U.S.A. 100, 467–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen Z., Stokes D. L., Rice W. J., Jones L. R. (2003) Spatial and dynamic interactions between phospholamban and the canine cardiac Ca2+ pump revealed with use of heterobifunctional cross-linking agents. J. Biol. Chem. 278, 48348–48356 [DOI] [PubMed] [Google Scholar]
- 8. Chen Z., Stokes D. L., Jones L. R. (2005) Role of leucine 31 of phospholamban in structural and functional interactions with the Ca2+ pump of cardiac sarcoplasmic reticulum. J. Biol. Chem. 280, 10530–10539 [DOI] [PubMed] [Google Scholar]
- 9. Chen Z., Akin B. L., Stokes D. L., Jones L. R. (2006) Cross-linking of C-terminal residues of phospholamban to the Ca2+ pump of cardiac sarcoplasmic reticulum to probe spatial and functional interactions within the transmembrane domain. J. Biol. Chem. 281, 14163–14172 [DOI] [PubMed] [Google Scholar]
- 10. Akin B. L., Chen Z., Jones L. R. (2010) Superinhibitory phospholamban mutants compete with Ca2+ for binding to SERCA2a by stabilizing a unique nucleotide-dependent conformational state. J. Biol. Chem. 285, 28540–28552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lytton J., Westlin M., Hanley M. R. (1991) Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J. Biol. Chem. 266, 17067–17071 [PubMed] [Google Scholar]
- 12. Sagara Y., Fernandez-Belda F., de Meis L., Inesi G. (1992) Characterization of the inhibition of intracellular Ca2+ transport ATPases by thapsigargin. J. Biol. Chem. 267, 12606–12613 [PubMed] [Google Scholar]
- 13. Jones L. R., Besch H. R., Jr., Watanabe A. M. (1978) Regulation of the calcium pump of cardiac sarcoplasmic reticulum. Interactive roles of potassium and ATP on the phosphoprotein intermediate of the (K+,Ca2+)-ATPase. J. Biol. Chem. 253, 1643–1653 [PubMed] [Google Scholar]
- 14. Reddy L. G., Jones L. R., Cala S. E., O'Brian J. J., Tatulian S. A., Stokes D. L. (1995) Functional reconstitution of recombinant phospholamban with rabbit skeletal Ca2+-ATPase. J. Biol. Chem. 270, 9390–9397 [DOI] [PubMed] [Google Scholar]
- 15. Sørensen T. L., Olesen C., Jensen A. M., Møller J. V., Nissen P. (2006) Crystals of sarcoplasmic reticulum Ca2+-ATPase. J. Biotechnol. 124, 704–716 [DOI] [PubMed] [Google Scholar]
- 16. Akin B. L., Jones L. R. (2012) Characterizing phospholamban to sarco(endo)plasmic reticulum Ca2+-ATPase 2a (SERCA2a) protein binding interactions in human cardiac sarcoplasmic reticulum vesicles using chemical cross-linking. J. Biol. Chem. 287, 7582–7593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 18. Jensen A. M., Sørensen T. L., Olesen C., Møller J. V., Nissen P. (2006) Modulatory and catalytic modes of ATP binding by the calcium pump. EMBO J. 25, 2305–2314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 22. Painter J., Merritt E. A. (2006) Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D Biol. Crystallogr. 62, 439–450 [DOI] [PubMed] [Google Scholar]
- 23. Chen Z., Akin B. L., Jones L. R. (2007) Mechanism of reversal of phospholamban inhibition of the cardiac Ca2+-ATPase by protein kinase A and by anti-phospholamban monoclonal antibody 2D12. J. Biol. Chem. 282, 20968–20976 [DOI] [PubMed] [Google Scholar]
- 24. Toyoshima C., Nomura H. (2002) Structural changes in the calcium pump accompanying the dissociation of calcium. Nature 418, 605–611 [DOI] [PubMed] [Google Scholar]
- 25. Montigny C., Arnou B., Champeil P. (2010) Glycyl betaine is effective in slowing down the irreversible denaturation of a detergent-solubilized membrane protein, sarcoplasmic reticulum Ca2+-ATPase (SERCA1a). Biochem. Biophys. Res. Commun. 391, 1067–1069 [DOI] [PubMed] [Google Scholar]
- 26. Toyoshima C. (2008) Structural aspects of ion pumping by Ca2+-ATPase of sarcoplasmic reticulum. Arch. Biochem. Biophys. 476, 3–11 [DOI] [PubMed] [Google Scholar]
- 27. Gorski P. A., Glaves J. P., Vangheluwe P., Young H. S. (2013) Sarco(endo)plasmic reticulum calcium ATPase (SERCA) inhibition by sarcolipin is encoded in its luminal tail. J. Biol. Chem. 288, 8456–8467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jones L. R., Simmerman H. K., Wilson W. W., Gurd F. R., Wegener A. D. (1985) Purification and characterization of phospholamban from canine cardiac sarcoplasmic reticulum. J. Biol. Chem. 260, 7721–7730 [PubMed] [Google Scholar]
- 29. Mueller B., Karim C. B., Negrashov I. V., Kutchai H., Thomas D. D. (2004) Direct detection of phospholamban and sarcoplasmic reticulum Ca-ATPase interaction in membranes using florescence resonance energy transfer. Biochemistry 43, 8754–8765 [DOI] [PubMed] [Google Scholar]
- 30. Li J., Bigelow D. J., Squier T. C. (2004) Conformational changes within the cytoplasmic portion of phospholamban upon release of Ca-ATPase inhibition. Biochemistry 43, 3870–3879 [DOI] [PubMed] [Google Scholar]
- 31. Bidwell P., Blackwell D. J., Hou Z., Zima A. V., Robia S. L. (2011) Phospholamban binds with differential affinity to calcium pump conformers. J. Biol. Chem. 286, 35044–35050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kimura Y., Kurzydlowski K., Tada M., MacLennan D. H. (1997) Phospholamban inhibitory function is activated by depolymerization. J. Biol. Chem. 272, 15061–15064 [DOI] [PubMed] [Google Scholar]
- 33. Asahi M., Kimura Y., Kurzydlowski K., Tada M., MacLennan D. H. (1999) Transmembrane helix M6 in sarco(endo)plasmic reticulum Ca2+-ATPase forms a functional interaction site with phospholamban. Evidence for physical interactions at other sites. J. Biol. Chem. 274, 32855–32862 [DOI] [PubMed] [Google Scholar]
- 34. Andersen J. P., Vilsen B., MacLennan D. H. (1992) Functional consequences of alterations to Gly-310, Gly-770, and Gly-801 located in the transmembrane domain of the Ca2+-ATPase of sarcoplasmic reticulum. J. Biol. Chem. 267, 2767–2774 [PubMed] [Google Scholar]
- 35. Kimura Y., Asahi M., Kurzydlowski K., Tada M., MacLennan D. H. (1998) Phospholamban domain Ib mutations influence functional interactions with the Ca2+-ATPase isoform of cardiac sarcoplasmic reticulum. J. Biol. Chem. 273, 14238–14241 [DOI] [PubMed] [Google Scholar]
- 36. Autry J. M., Jones L. R. (1997) Functional co-expression of the canine cardiac Ca2+ pump and phospholamban in Spodoptera frugiperda (Sf21) cells reveals new insights on ATPase regulation. J. Biol. Chem. 272, 15872–15880 [DOI] [PubMed] [Google Scholar]
- 37. Cornea R. L., Autry J. M., Chen Z., Jones L. R. (2000) Reexamination of the role of the leucine/isoleucine zipper residues of phospholamban in inhibition of the Ca2+ pump of cardiac sarcoplasmic reticulum. J. Biol. Chem. 275, 41487–41494 [DOI] [PubMed] [Google Scholar]
- 38. Wegener A. D., Jones L. R. (1984) Phosphorylation-induced mobility shift in phospholamban in sodium dodecyl sulfate-polyacrylamide gels. Evidence for a protein structure consisting of multiple identical phosphorylatable subunits. J. Biol. Chem. 259, 1834–1841 [PubMed] [Google Scholar]
- 39. Simmerman H. K., Collins J. H., Theibert J. L., Wegener A. D., Jones L. R. (1986) Sequence analysis of phospholamban. Identification of phosphorylation sites and two major structural domains. J. Biol. Chem. 261, 13333–13341 [PubMed] [Google Scholar]
- 40. Simmerman H. K., Kobayashi Y. M., Autry J. M., Jones L. R. (1996) A leucine zipper stabilizes the pentameric membrane domain of phospholamban and forms a coiled-coil pore structure. J. Biol. Chem. 271, 5941–5946 [DOI] [PubMed] [Google Scholar]
- 41. Cornea R. L., Jones L. R., Autry J. M., Thomas D. D. (1997) Mutation and phosphorylation change the oligomeric structure of phospholamban in lipid bilayers. Biochemistry 36, 2960–2967 [DOI] [PubMed] [Google Scholar]
- 42. Bublitz M., Musgaard M., Poulsen H., Thøgersen L., Olesen C., Schiøtt B., Morth J. P., Møller J. V., Nissen P. (2013) Ion pathways in the sarcoplasmic reticulum Ca2+-ATPase. J. Biol. Chem. 288, 10759–10765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Toyoshima C., Iwasawa S., Ogawa H., Hirata A., Tsueda J., Inesi G. (2013) Crystal structures of the calcium pump and sarcolipin in the Mg2+-bound E1 state. Nature 495, 260–264 [DOI] [PubMed] [Google Scholar]
- 44. Winther A. M., Bublitz M., Karlsen J. L., Møller J. V., Hansen J. B., Nissen P., Buch-Pedersen M. J. (2013) The sarcolipin-bound calcium pump stabilizes calcium sites exposed to the cytoplasm. Nature 495, 265–269 [DOI] [PubMed] [Google Scholar]
- 45. Sahoo S. K., Shaikh S. A., Sopariwala D. H., Bal N. C., Periasamy M. (2013) Sarcolipin protein interaction with sarco(endo)plasmic reticulum Ca2+ ATPase (SERCA) is distinct from phospholamban protein, and only sarcolipin can promote uncoupling of the SERCA pump. J. Biol. Chem. 288, 6881–6889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Odermatt A., Becker S., Khanna V. K., Kurzydlowski K., Leisner E., Pette D., MacLennan D. H. (1998) Sarcolipin regulates the activity of SERCA1, the fast-twitch skeletal muscle sarcoplasmic reticulum Ca2+-ATPase. J. Biol. Chem. 273, 12360–12369 [DOI] [PubMed] [Google Scholar]
- 47. Lytton J., Westlin M., Burk S. E., Shull G. E., MacLennan D. H. (1992) Functional comparisons between isoforms of the sarcoplasmic or endoplasmic reticulum family of calcium pumps. J. Biol. Chem. 267, 14483–14489 [PubMed] [Google Scholar]
- 48. James P., Inui M., Tada M., Chiesi M., Carafoli E. (1989) Nature and site of phospholamban regulation of the Ca2+ pump of sarcoplasmic reticulum. Nature 342, 90–92 [DOI] [PubMed] [Google Scholar]