

Background: Gcn5-related N-acetyltransferases (GNATs) are involved in small molecule and protein acetylation in all organisms.
Results: Crystallographic and biochemical characterization of PA4794 is shown, including identification of substrates and inhibitors.
Conclusion: PA4794 is a new bacterial C-terminal lysine protein acetyltransferase inhibited by cephalosporins.
Significance: PA4794 is the first identified acetyltransferase specific for C-terminal lysine; identified interactions with cephalosporins may be of clinical relevance.
Keywords: Antibiotics, Crystal Structure, Enzyme inhibitors, High Throughput Screening (HTS), Pseudomonas, GNAT, Gcn5-related N-Acetyltransferase, Acetyltransferase, Cephalosporin, Lysine Acetylation
Abstract
The Gcn5-related N-acetyltransferase (GNAT) superfamily is a large group of evolutionarily related acetyltransferases, with multiple paralogs in organisms from all kingdoms of life. The functionally characterized GNATs have been shown to catalyze the transfer of an acetyl group from acetyl-coenzyme A (Ac-CoA) to the amine of a wide range of substrates, including small molecules and proteins. GNATs are prevalent and implicated in a myriad of aspects of eukaryotic and prokaryotic physiology, but functions of many GNATs remain unknown. In this work, we used a multi-pronged approach of x-ray crystallography and biochemical characterization to elucidate the sequence-structure-function relationship of the GNAT superfamily member PA4794 from Pseudomonas aeruginosa. We determined that PA4794 acetylates the Nϵ amine of a C-terminal lysine residue of a peptide, suggesting it is a protein acetyltransferase specific for a C-terminal lysine of a substrate protein or proteins. Furthermore, we identified a number of molecules, including cephalosporin antibiotics, which are inhibitors of PA4794 and bind in its substrate-binding site. Often, these molecules mimic the conformation of the acetylated peptide product. We have determined structures of PA4794 in the apo-form, in complexes with Ac-CoA, CoA, several antibiotics and other small molecules, and a ternary complex with the products of the reaction: CoA and acetylated peptide. Also, we analyzed PA4794 mutants to identify residues important for substrate binding and catalysis.
Introduction
Pseudomonas aeruginosa is a ubiquitous, Gram-negative bacterium that is an opportunistic pathogen of humans, animals, and plants (1, 2). As a nosocomial pathogen, P. aeruginosa mainly infects immune-compromised patients afflicted with severe burns, AIDS, or cystic fibrosis and it exhibits an extraordinary ability to acquire resistance to antibiotics (1).
The Gcn5-related N-acetyltransferase (GNAT)2 superfamily is a large and diverse group of evolutionarily related acetyltransferases. Members of the GNAT superfamily have been identified in all kingdoms of life, with multiple paralogs found in many organisms. These enzymes have been shown to catalyze the transfer of an acetyl group from acetyl-coenzyme A (Ac-CoA) to the primary amine of a wide range of substrates, including glucosamine 6-phosphate, aminoglycoside antibiotics, spermine, spermidine, and other small molecules (3, 4).
Moreover, GNATs are involved in protein acetylation, including both N-terminal (Nα) acetylation of protein termini and Nϵ acetylation of internal lysine residues. As with many other post-translational modifications, the acetylation of lysine residues is specific and reversible, which gives it a tremendous regulatory potential. Although the functional role of protein acetylation in eukaryotes has long been studied, it was recently discovered that acetylation of proteins is common in bacteria as well (5, 6). It appears that acetylated proteins are involved in many aspects of prokaryotic cellular physiology, including carbohydrate and energy metabolism, nucleotide and amino acid metabolism, transcription, translation, cell differentiation, survival, and apoptosis, stress response and many others, as reviewed in Ref. 7. Although many proteins have been identified as being acetylated, comparatively little is known about the acetyltransferases that catalyze these reactions.
PA4794 is an uncharacterized GNAT from P. aeruginosa, which has been annotated as a putative protein, and is remotely similar to the RimI ribosomal protein acetyltransferase. We used semi-high throughput crystallographic and biochemical screening experiments to characterize the structure and activity of this protein and found that it selectively acetylates the Nϵ group of the C-terminal lysine of peptides. This suggests that PA4794 functions as a C-terminal lysine protein acetyltransferase. During our biochemical screening, we also determined that PA4794 could acetylate chloramphenicol. These results show that PA4794 is not only capable of acetylating proteins, but also small molecules, and is capable of both N- and O-acetylation. Additionally, we show that several cephalosporin antibiotics and other molecules are competitive inhibitors of PA4794. Here we present multiple structures of PA4794 complexed with a variety of ligands, including substrates, products, and inhibitors. All structures were determined at resolutions of 1.2 to 1.8 Å, which allowed us to characterize interactions between ligands and protein in detail. We also kinetically characterized the enzyme, which included inhibition studies for the identified complexes and determining the influence of selected point mutations on kinetic activity.
EXPERIMENTAL PROCEDURES
Cloning, Expression, and purification
The PA4794 gene was cloned into the p11 pET-derived expression vector. The vector encodes for a His6 tag followed by a spacer and a tobacco etch virus protease cleavage site on the N terminus of the expressed protein. The amino acid sequence Gly-His remains on the N terminus of the protein after cleavage of the tag with tobacco etch virus protease. The fusion protein was overexpressed in Escherichia coli BL21-RIL (DE3) cells (Stratagene). The cells were grown in LB at 37 °C to an A600 of ∼1.0 and protein expression was induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside. After induction, the cells were incubated overnight with shaking at 16 °C. The harvested cells were resuspended in binding buffer (500 mm NaCl, 5% glycerol, 50 mm Tris-HCl, pH 7.5, 5 mm imidazole) and lysed by sonication after the addition of cOmplete, EDTA-free Protease Inhibitor Cocktail (Roche Applied Science). The lysate was clarified by centrifugation (30 min at 17,000 × g) and applied to a metal chelate affinity column charged with Ni2+ (Qiagen) and pre-equilibrated in binding buffer. The resin with bound protein was washed with wash buffer (500 mm NaCl, 50 mm Tris-HCl, pH 7.5, 5% glycerol, and 30 mm imidazole) to remove weakly binding contaminants. The tagged protein was eluted from the column in elution buffer (500 mm NaCl, 5% glycerol, 50 mm Tris-HCl, pH 7.5, 250 mm imidazole), and the tag was then cleaved from the protein by treatment with recombinant His-tagged tobacco etch virus protease (8, 9) during dialysis to remove the imidazole. The cleaved protein was then separated from the cleaved His tag and the His-tagged protease by passing the mixture through a second Ni2+-chelate affinity column. The flow-through was then passed through a Superdex 200 column attached to an ÅKTA FPLC gel filtration system (GE Healthcare) in a buffer containing 10 mm Tris-HCl, pH 7.5, and 150 mm NaCl. After gel filtration, fractions containing the protein were pooled and concentrated to 9 mg/ml.
Site-directed Mutagenesis
We generated single-site mutants using the QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions. The wild-type PA4794 gene cloned into the p11 vector was used as a template for PCR amplifications to introduce the single mutation, and the purified single mutant plasmids were used as templates to introduce consecutive mutations. The presence of the introduced mutations was confirmed by DNA sequencing.
Crystallization
Tracking and analysis of the crystallization experiments were performed with the Xtaldb system (10). The crystals were grown using vapor diffusion and hanging drop setups. The crystallization drops were a 1:1 mixture of protein solution and the precipitant solution from the wells (2 m ammonium sulfate and 100 mm BisTris, pH 6.5, or 1.5 m ammonium sulfate and 0.1 m Tris-HCl, pH 8.5, in the case of the covalently bound CoA), in which crystals grew overnight at 16 °C. The complexes with ligands were obtained by soaking ligands into crystals of “unbound” (apo-form) PA4794. The final concentration of each ligand in the drop was 5–10 mm, and the soaks were allowed to stand for 4–10 days. 5 mm 2-Mercaptoethanol was added to the crystallization conditions for the CoA soak. Prior to data collection, each crystal was transferred to a solution containing a 2:1 mixture of precipitant solution and ethylene glycol and immediately cryo-cooled in liquid nitrogen.
Crystallographic Screening of Ligand Cocktails
Crystals of the apo-form of PA4794 were soaked with cocktail solutions, containing mixtures of several (usually 5–10) potential ligands simultaneously. Crystallographic screening of cocktails of several potential ligands has been shown to be useful in functional analyses of previously uncharacterized proteins (11). The cocktail components included representatives of different classes of small molecules to provide a wide range of potential substrates, cofactors, and inhibitors. The cocktail soaks showed that 4-methylumbelliferyl phosphate and the antibiotic cefmetazole bound to PA4794, so similar compounds were used in subsequent soaks. The ligand cocktails (Table 2) were prepared as aqueous solutions or suspensions with each component at a concentration of 100 mm. To minimize crystal damage, 0.3 μl of each cocktail was mixed with 0.7 μl of the mother liquor and then this mixture was gently combined with a 2-μl crystallization drop and incubated for 4–10 days. Based upon “hits” in the initial binding screen with the cocktails, additional compounds were selected for further study. Soaks with individual ligands were prepared in a similar way as the cocktail soaks, where the initial stock solutions contained 100 mm ligand in aqueous solution or suspension.
TABLE 2.
Composition of the cocktails used for crystals soaking
| Screen | Components |
|---|---|
| 1 | Paromomycin, tobramycin, apramycin, sisomicin, streptomycin, dihydrostreptomycin, amikacin, ribostamycin |
| 2 | Neomycin, gentamycin, kanamycin, kasugamycin, moxalactam, cefmetazol, hygromycin B, daunorubicin |
| 3 | α-d-Glucose, d-(+)-mannose, d-(−)-fructose, d-(+)-galactose, d-(+)-cellobiose, maltodextrin, l-(−)-fucose |
| 4 | d-(+)-Maltose, methyl α-d-glucopyranoside, n-octyl-β-d-glucopyranoside, n-dodecyl β-d-glucopyranoside, uridine-5′-diphosphoglucose, d-glucose 6-phosphate |
| 5 | n-Nonyl-β-d-glucopyranoside, n-hexyl β-d-glucopyranoside, n-dodecyl β-d-glucopyranoside, 4-nitrophenyl α-d-glucopyranoside, maltopentaose |
| 6 | Adenine, adenosine, 2′-deoxyadenosine 5′-monophosphate |
| 7 | O-Phosphorylethanolamine, guanine, guanosine, 2′-deoxyguanosine 5′-diphosphate, 2′-deoxyguanosine 5′-triphosphate |
| 8 | Cytosine, cytidine, 2′-deoxycytidine 5′-monophosphate, 2′-deoxyguanosine, dl-carnitine |
| 9 | Glycerol phosphate, uracil, uridine, uridine 5′-monophosphate, uridine 5′-diphosphate, 2′-deoxyuridine 5′-monophosphate, uridine-5′-diphosphoglucose |
| 10 | Thymine, thymidine, thymidine 5′-monophosphate |
| 11 | Coenzyme Q10, ergocalciferol, deoxycholic acid, (+)-α-tocopherol, 2′-deoxyinosine 5′-monophosphate |
| 12 | S-(5′-Adenosyl)-l-methionine, nicotinic acid adenine dinucleotide, biotin, nicotinic acid, folic acid, thiamine, 4-aminobenzoic acid |
| 13 | Riboflavin, riboflavin 5′-monophosphate, pyridoxal, pyridoxal 5′-phosphate, pyridoxamine |
| 14 | d-(+)-Xylose, l-rhamnose, N-acetyl-d-galactosamine, N-alcetylmuramic acid |
| 15 | N-acetyl-d-glucosamine, d-pantothenic acid |
| 16 | Sucrose, l-(+)-arabinose, d-sorbitol, d-(−)-fructose, d-(+)-trehalose, xylitol, d-mannitol, myo-inositol |
| 17 | Kasugamycin, geneticin |
| 18 | (1S,2S)-1-Amino-2-benzyloxycyclopentane, dl-kynurenine, 4-methylumbelliferyl phosphate |
| 19 | (1R,2R)-trans-2-Aminocyclohexanol, (1R,2R)-trans-2-AMinocyclopentanol, phosphocreatine |
| 20 | N-Acetyl-l-lysine, l-proline, d-cycloserine, O-phospho-dl-serine |
| 21 | Phosphinothricin, serotonin, l-methionine sulfoximine, l-methionine sulfone |
| 22 | Spermine, spermidine, d-glucosamine 6-phosphate, glycine, alanine |
| 23 | Arginine, lysine, cysteine, Gly-Gly-Gly, sodium acetate |
Other Ligand Screening
PA4794 was also screened by printed microarray for binding to a set of 465 glycans (Mammalian Printed Array version 4.1) by the Functional Glycomics Gateway, although no binding to any glycan in the array was detected. In an effort to identify a possible specific peptide substrate sequence for PA4794 we conducted phage display assays with a library of ∼109 randomized heptapeptides (New England Biolabs), but no binding of a specific peptide was detected under the conditions recommended by the manufacturer. However, in the Phage Display peptide library used, the peptides free N termini are exposed, but not the C termini, therefore binding of C-terminal residue could not be detected. In both cases it was necessary to use the His-tagged protein, and because PA4794 binds the His tag in the active site3 this may have influenced the results of the assays.
Data Collection, Structure Determination, and Refinement
Data collection for PA4794 was performed at beam lines 19-BM and 19-ID of the Structural Biology Center (12) at the Advanced Photon Source (APS), and at beam lines 21-ID-F and 21-ID-G of the Life Sciences Collaborative Access Team at the APS. Data were collected at a temperature of 100 K and processed with HKL-2000 (13). All structures were solved using HKL-3000 (14) coupled with MOLREP (15). The structures of PA4794 were determined by molecular replacement using the previously solved selenomethionine-substituted structure (PDB accession code 4M3S). Refinement was performed using HKL-3000 coupled with REFMAC5 (16), COOT (17, 18), and selected programs from the CCP4 package (19). The atom B-factors were refined either using Translation/Libration/Screw (TLS) groups assigned by the TLSMD server (20) or using anisotropic refinement. Validation of the structures was performed using MOLPROBITY (21) and ADIT (22). The 15 structures of PA4794 and its complexes were determined at resolutions varying from 1.2 to 1.8 Å. The models of all structures had reasonable stereochemistry. The coordinates, structure factors, and intensities were deposited in the PDB (PDB codes 4KUA, 3PGP, 4KUB, 4L89, 4L8A, 4KLV, 4KOT, 4KOW, 4KOX, 4KOV, 4KOU, 4KOY, 4KOR, 4KOS, and 4KLW). Statistics describing crystallographic data collection and refinement are summarized in supplemental Table S1.
Isothermal Titration Calorimetry
Isothermal titration calorimetry (ITC) measurements were performed at 25 °C using an iTC200 calorimeter (MicroCal). Preparations of purified protein were dialyzed against buffer containing 50 mm NaCl and 50 mm Tris, pH 7.5, overnight at 4 °C. The concentration of PA4794 used ranged from 55 to 456 μm, and the final concentrations of ligands used exceeded the protein concentration by a factor of 1.5 to 3. Data analysis was conducted with the Origin software (OriginLab).
Activity Screening and Measurement of Kinetic Activity
Because the PA4794 protein had not been characterized prior to this work, we subjected it to a previously described broad-substrate screen (23). When potential substrates were identified, a thorough characterization was performed to determine the kinetic parameters of the enzyme. The reaction conditions for the assay were slightly modified from the published broad-substrate screen protocol to substitute 50 mm Bicine, pH 9.0, for Tris-HCl, pH 8.0. Reactions were initiated with 2.6 μm enzyme (with the exception of C29A, which was 4.3 μm) and the reaction was allowed to proceed for 5 min at 37 °C. The reactions were stopped and measured as described previously (23). Substrate saturation curves were produced by holding the concentration of one substrate constant, while varying the concentrations of the other and vice versa. Inhibition curves were produced by varying the concentration of inhibitor, while holding the substrates at a constant concentration (0.5 mm Ac-CoA and 10 mm N-phenylacetyl-Gly-Lys (NPAcGK), respectively). The activity was the same regardless of whether the enzyme was preincubated with inhibitor or not. Data were fitted to a modified Hill equation: V = (Vo + (Vmax − Vo) [C]n)/(kn + [C]n), with Origin 8v1. V is the initial velocity, Vo is the velocity in the absence of substrate or inhibitor, Vmax is the maximal velocity, [C] is the concentration of substrate or inhibitor being varied, k is the concentration of substrate (S0.5) or inhibitor (I0.5) that produces half-maximal velocity, and n is the Hill coefficient. In an attempt to determine the preferential peptide sequence that the enzyme acetylates, a variety of synthesized peptides (Genescript) were screened for activity using 5 mm peptide and 0.5 mm Ac-CoA. One international unit of enzyme activity is defined as the amount of enzyme that produces 1 nmol of CoA per min in the described assay.
RESULTS
Overall Structure Analysis and Relationship to Other GNATs
The PA4794 gene, which encodes a polypeptide of 160 amino acids, was cloned into E. coli, overexpressed, purified, and crystallized. The PA4794 protein crystallized in the orthorhombic crystal system (space group P21212) with a monomer in the asymmetric unit. Gel filtration studies also suggest that PA4794 is a monomer in solution (data not shown). PA4794 is a member of the GNAT superfamily of acyltransferases, with a fold comprised of an N-terminal β-strand followed by three α-helices (although two helices are generally more common in GNATs (4)), three antiparallel β-strands, followed by an α-helix, a fifth β-strand, a fifth α-helix, and two β-strands. The seventh β-strand is positioned between strands β5 and β6 (Fig. 1). In contrast to some GNATs where the active site contains residues from both molecules of a dimer (24), the active site of PA4794 is located within a monomer.
FIGURE 1.

Overall structure of PA4794 and similarity to other GNATs. A, topological diagram showing the arrangement of secondary structural elements. α-Helices are shown as circles, β-strands are shown as triangles. Orientation of the triangles shows the orientation of the β-strands, with vertex of the triangle pointing up for the β-strand pointing toward the reader, and vertex of the triangle pointing down for opposite orientation of the β-strand. B, ribbon diagram colored from N to C, Ac-CoA is shown as sticks. C, ribbon diagrams of PA4794 and RimI. Fragments showing the largest differences are highlighted. D, superposition of PA4794 and related GNATs. Conservation of the Ac-CoA/CoA binding site and the flexibility of the substrate-binding site (indicated by a blue rectangle) are visible. CoA is shown as sticks.
PA4794 is classified as a member of the Pfam PF00583 family of acetyltransferases and COG0454 (histone acetyltransferase HPA2 and related acetyltransferases). The homologs of PA4794 with the highest sequence similarity were identified using a Blast search of the non-redundant (nr) database. The protein sequences that showed highest similarity to PA4794 are mainly from Pseudomonas and other proteobacteria (including the species Vibrio, Thioalkalivibrio, Photobacterium, Pelobacter, and Azospirillum), but homologs are also found in Bacteroides and Paenibacillus. PA4794 is the only structurally characterized representative of this group.
A structural similarity search of the PDB using DALI (25) reveals that the structure of PA4794 shows high structural similarity to several GNATs (root mean square deviation (r.m.s. deviations) values of 1.4–3.5 Å), but the sequence identity between these proteins is below 20%. In terms of sequence, the protein of known function and structure that is most similar to PA4794 is RimI. This GNAT is responsible for the Nα-acetylation of the ribosomal protein S18 (RimI from Salmonella typhimurium LT2, PDB code 2CNS; r.m.s. deviations 1.4 Å) (26). The other GNATs identified by DALI that showed high structural similarity to PA4794 included two proteins of unknown function from P. aeruginosa, PA2578 (PDB code 3OWC, r.m.s. deviations 3.1 Å) and PA4866 (1YVO; r.m.s. deviations 1.4 Å), phosphinothricin acetyltransferase from Agrobacterium tumefaciens (1YR0; r.m.s. deviations 3.5 Å), and yncA, a putative acetyltransferase from Salmonella typhimurium (3DR8; r.m.s. deviations 1.4 Å). In general, the structures of GNATs show high conservation in regions corresponding to the Ac-CoA binding site, but vary significantly in regions responsible for recognition and binding of their diverse substrates (Fig. 1D). The most pronounced differences between structures of PA4794 and the aforementioned proteins are observed in the first three α-helices. The first α-helix may be long and connected with the second canonical GNAT α-helix by a short loop as it is in RimI (Fig. 1C). In the structure of PA4794, the first α-helix is shorter and followed by an additional short α-helix in the region corresponding to the loop in other GNATs, and connected to the second of the canonical α-helices (Fig. 1C). The second canonical α-helix also shows high conformational diversity among the structures. The additional α-helix is relatively well conserved among the closest PA4794 homologs, but is not present in RimI.
Ac-CoA·CoA Complex
We determined structures of PA4794 in complexes with Ac-CoA and CoA, and we used ITC to measure the parameters of binding of Ac-CoA and CoA to PA4794 (Table 3). The binding of Ac-CoA and CoA by PA4794 (Fig. 2) is similar to what has been observed for other GNAT superfamily members (3, 4). The adenosine moiety of CoA is located on the surface of the protein, leaning against α4, but not making significant interactions with the protein molecule. The conserved “P-loop” between β4 and α3 coordinates the pyrophosphate moiety of CoA, which is bound mostly by main chain nitrogen atoms. In PA4794 the loop is composed of the sequence Arg88-Gly89-Leu90-Gly91-Val92-Ala93 (the conserved motif in GNATs is (Q/R)-X-X-G-X-(G/A)) and the specific residues are marked in bold (26, 27). Arg94, which is not conserved, also coordinates the pyrophosphate with its main chain nitrogen and the 3′-phosphate of the ribose with its side chain. As with other GNATs, Ac-CoA and CoA are bound in a characteristically bent conformation with a sharp flexion of the pantothenate moiety. The β4 and β5 strands splay apart, creating a V-shaped cleft in the β-sheet that provides a binding site for the pantetheine. This splay is characteristic of GNATs (3) and enables the pantetheine group to make hydrogen bonds with the main chain of β4, mimicking an antiparallel β-strand.
TABLE 3.
The effect of inhibitors on kinetics of PA4794, and binding affinities of PA4794 for inhibitors and cofactors
The inhibitory parameters for the compounds observed in the crystal structures of PA4794 are shown, specifically the I0.5 and fold-decrease in activity. These values were determined as described under “Experimental Procedures.” For the inhibition studies 7-aminocephalosporanic acid produced an interfering yellow color at higher concentrations; however, this interfering absorbance was accounted for by using proper background controls. We were unable to measure the inhibition kinetics for cephalosporin C because it is a zinc salt that readily hydrolyzes Ac-CoA in the absence of enzyme. In some cases only approximate values could be determined. The ITC-measured parameters of binding of each compound to PA4794, in the absence of other substrates, are also shown. ΔH, enthalpy change; ΔS, entropy change; Kd, dissociation constant; N, stoichiometry; T, temperature.
| Compound | I0.5 | Fold-decrease | Kd (ITC) | ΔH | −TΔS | n |
|---|---|---|---|---|---|---|
| μm | cal/mol | |||||
| 4-Methylumbelliferyl phosphate | 11.0 ± 0.4 | 8.5 | 165.3 ± 9.5 | −9138 ± 275 | 3963 | 1.00 ± 0.02 |
| Cefotaxime | 92.7 ± 5.1 | 10.3 | 3.5 ± 0.2 | −8714 ± 83 | 1268 | 1.07 ± 0.01 |
| Cefoxitin | >200 | 5.9 | 49.3 ± 2.9 | −13310 ± 583 | 7420 | 1.04 ± 0.03 |
| Cephalothin | >500 | 9.6 | 16.9 ± 1.2 | −10460 ± 279 | 3933 | 0.99 ± 0.02 |
| Cefuroxime | >500 | 2.8 | 176.7 ± 6.2 | −8280 ± 188 | 3159 | 0.98 ± 0.02 |
| Cefixime | >500 | 3.4 | Weak bindinga | |||
| Ammonium 2-(aminocarbonyl) benzoate | >500 | 4.0 | Weak bindinga | |||
| 7-Aminocephalosporanic acid | >1 mm | 1.5 | Weak bindinga | |||
| Cefmetazole | >1 mm | 1.5 | Weak bindinga | |||
| AcCoA | 3.8 ± 0.5 | −9475 ± 210 | 2077 | 1.09 ± 0.02 | ||
| CoA | 3.9 ± 0.2 | −11730 ± 90 | 4351 | 1.03 ± 0.01 | ||
a Binding of the compounds was too weak to reliably determine its parameters at achievable protein concentrations.
FIGURE 2.

Structures of PA4794 complexed with Ac-CoA (A), CoA (B), and covalently bound CoA (C); D, conformational change upon binding of AcCoA (blue, apo; gray, in complex with Ac-CoA).
Another characteristic feature of GNATs, also present in PA4794, is a “β-bulge” in β4 directly adjacent to the Ac-CoA binding site (3). The carbonyl of the acetyl group of Ac-CoA is stabilized by a hydrogen bond to the main chain amine of Met81, which is located just downstream of the β-bulge. Due to the lack of this stabilization in the complex with CoA, there is some flexibility of the β-mercaptoethylamine moiety, which was modeled in two alternate conformations. The complex of PA4794 with CoA, as crystallized in the initial conditions (1.5 m ammonium sulfate, 0.1 m Tris, pH 8.5) showed a conformational change in the protein with respect to the structure of unbound PA4794. In the apo-structure, as well as in the complex with Ac-CoA, the side chain of Cys29 faces away from the active site. Conversely, the complex with the CoA shows that the side chain is rotated into the active site pocket and CoA forms a covalent disulfide bond between its thiol sulfur and Cys29 of the protein. Covalent binding of the CoA to a Cys residue has also been observed for an S. typhimurium RimL acetyltransferase (28), and it has been suggested that the in vivo intracellular redox potential may regulate activity of the protein. The addition of 5 mm 2-mercaptoethanol to the crystallization conditions prevented the formation of the covalent link to CoA and resulted in a protein conformation similar to that of the PA4794·Ac-CoA complex (Fig. 2).
Upon binding of Ac-CoA and CoA to PA4794, there is a conformational change in the P-loop (residues 88–93), which coordinates the pyrophosphate moiety, and residues 120–130 of α4. There is a pronounced conformational change of Tyr128 when Ac-CoA is bound, and as described below, this residue is important for catalytic activity of PA4794 (Fig. 2).
Establishment of Acetyltransferase Activity of PA4794 and Determination of Substrates
In an effort to both confirm that PA4794 acts as an acetyltransferase and to determine possible substrates for the enzyme, we used a broad-substrate screen that was designed to identify potential substrates for functionally uncharacterized GNATs, as described in detail in Ref. 23. Using this screen, we determined that PA4794 acetylates l-lysine and some of its derivatives: thialysine, Nα-Ac-l-lysine, and N-phenylacetyl-Gly-Lys (NPAcGK). No acetylation was detected for Nϵ-acetyl-lysine, suggesting that PA4794 specifically catalyzes Nϵ-acetylation of lysine. The enzyme had a strong preference for the peptide NPAcGK, so this substrate was chosen for further detailed kinetic characterization of the enzyme. It should be noted that the screen also established that PA4794 acetylates chloramphenicol; however, in this work the focus will be on the probable protein acetylation function of PA4794. A subsequent manuscript will address the chloramphenicol acetylation activity of the enzyme.4
Structural-Functional Analysis of PA4794 in Complex with Substrates
Analysis of the hydrophobicity and electrostatic surface of PA4794 around the substrate-binding site shows that it is composed mainly of hydrophobic amino acids and is positively charged. We can also observe a small tunnel leading to the Ac-CoA binding site. To better understand the structure-function relationships of PA4794, we determined the structure of the ternary complex with CoA and acetylated NPAcGK peptide (Fig. 3A). The structure of the complex was obtained by soaking the crystals of apo-protein with substrates Ac-CoA and NPAcGK, but the resulting electron density clearly showed the products of the reaction: CoA and peptide acetylated on the Nϵ atom of the lysine. We were not able to observe electron density for NPAcGK when crystals of apo-protein were soaked with the peptide alone (i.e. without Ac-CoA). In the ternary complex, the acetylated NPAcGK peptide was modeled with two alternative conformations with the phenylacetyl moiety oxygen flipped. Due to the incomplete occupancy of the ligands, parts of the protein structure adopt two alternative conformations, approximately corresponding to the apo-form and Ac-CoA/CoA-bound structures, respectively.
FIGURE 3.
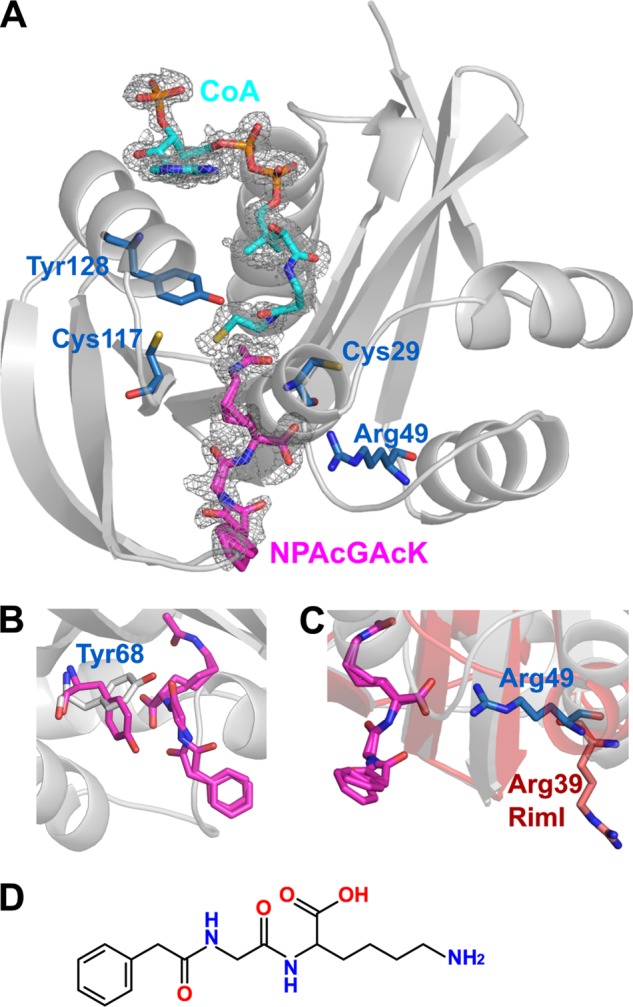
Structural features of the ternary complex. A, ternary complex structure of PA4794 with the reaction products CoA and Nϵ-acetylated NPAcGK bound. The residues thought to be important for substrate binding and activity are indicated. Fo − Fc omit electron density map of the bound ligands is presented (resolution 1.2 Å, σ = 2.5). B, conformational change of Tyr68 upon binding of substrate; the conformation found in apo-protein is gray and upon substrate binding is magenta. C, differences in the conformation of Arg49 in PA4794 (blue) compared with the analogous residue Arg39 in RimI (red). D, two-dimensional structure of NPAcGK.
The only residue that undergoes a significant conformational change upon binding NPAcGK is Tyr68. This residue participates in peptide binding (Fig. 3B) through hydrophobic interactions and hydrogen bonds with the main chain nitrogen of Gly in the peptide. The conformation of the Tyr128 in the ternary complex corresponds to its conformation in the apo-form not in the Ac-CoA-bound form. This shows that Tyr128 assumes its apo-form conformation right after the reaction occurs, but before the products are released. Most of the hydrogen bonding with the peptide is through the main chain of PA4794. The Nϵ atom of the C-terminal lysine of NPAcGK is stabilized by a hydrogen bond with the main chain oxygen of Ser116, which also undergoes a slight conformational change. The carbonyl of the Nϵ acetyl group produced after acetylation of NPAcGK is stabilized by a hydrogen bond with the main chain amine of Met81, which also stabilizes the acetyl moiety of Ac-CoA in the same manner prior to its transfer. The most distinct interaction in the ternary complex is hydrogen bonding of the C-terminal carboxyl group of the peptide with the Nω amines of the guanidine group of Arg49. The C-terminal carboxyl group of the peptide is also stabilized by a hydrogen bond with the main chain nitrogen of Asn80 and through a water molecule with Lys32. Main chain oxygens of the peptide interact with the main chain of PA4794 through water molecules. There are no substantial interactions with the phenyl moiety of the phenylacetylated peptide.
To investigate the hypothesis that the C-terminal position of the lysine is critical, we tested the activity of PA4794 with a Gly-Lys-Gly peptide. No activity was detected, suggesting that a free C-terminal carboxyl group on the lysine of the substrate is critical for binding and PA4794 cannot spatially accommodate larger substrates into the substrate-binding pocket. To confirm the key role of Arg49 in recognition and binding of the substrate C-terminal lysine, we mutated Arg49 to Gln. This mutation lead to a significant decrease in activity, likely due to the disruption of hydrogen bonding with the C-terminal carboxyl group of the peptide (Fig. 4). Arg49 is conserved in all of the closest homologs of PA4794, including RimI. Interestingly, when the structures of PA4794 and RimI are superimposed, the loops containing the corresponding arginines adopt substantially different conformations in the two proteins, causing the arginine to face in opposite directions in the two structures (Fig. 3C).
FIGURE 4.

Substrate saturation curves for Ac-CoA and NPAcGK. The specific activity is plotted as a function of Ac-CoA concentration at 10 mm NPAcGK (A) and as a function of NPAcGK concentration at 0.5 mm Ac-CoA (B) for wild-type and mutant proteins. The data are labeled as follows: wild-type (filled square), C29A (open circle), R49Q (filled triangle), C117A (open upside down triangle), C29A/C117A (filled diamond), N121A (open side triangle), Y128A (filled side triangle), and C29A/C117A/Y128A (open hexagon). Curves were produced and fitted as described under “Experimental Procedures.”
In an effort to determine preferred substrate peptide sequences for PA4794 we tested the ability of the enzyme to acetylate 33 different peptides with a C-terminal Lys residue. The peptide sequences were chosen based on their similarity to NPAcGK and an analysis of how the peptide binds in the structure of the ternary complex. This test also included some negative controls (i.e. Ac-GLKK and Ac-GRRK), peptides that structural analysis suggests would not bind effectively. We have detected some preferences, but the activity for all the peptides was much lower than that for NPAcGK (Fig. 5). The highest activity was observed for peptides that were similar to the original substrate: N-acetyl-Gly-Tyr-Gly-Lys and N-acetyl-Gly-Phe-Gly-Lys. We observed much lower activity for N-acetyl-Gly-Phe-Ala-Lys than for N-acetyl-Gly-Phe-Gly-Lys, indicating that Ala in the position adjacent to C-terminal Lys is much less preferred than the Gly. We observed some preference for small hydrophobic amino acids in the two positions adjacent to the C-terminal lysine residue, but were unable to conclusively determine a preferred substrate peptide sequence for the enzyme. We also tried soaking the peptides together with Ac-CoA into PA4794 crystals, but did not observe ordered electron density for any of the peptides.
FIGURE 5.
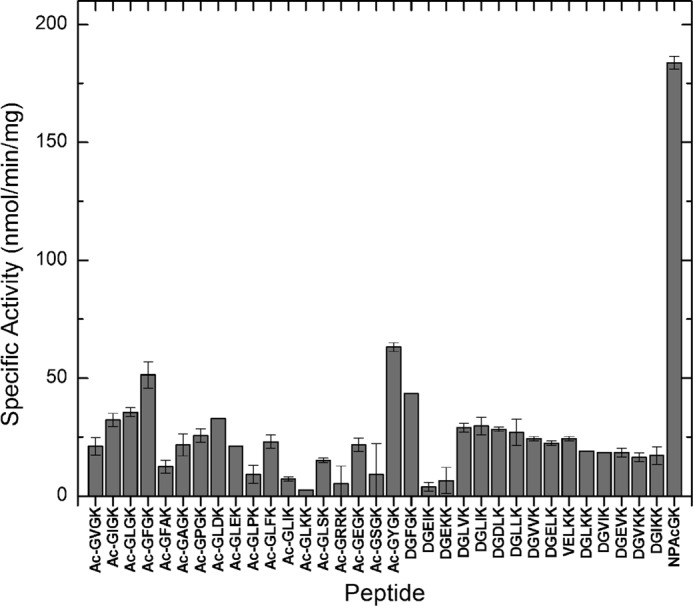
Peptide activity screening results. The ability of PA4794 to acetylate a variety of short peptides was tested as described in Ref. 23. The reaction mixture was composed of 0.5 mm Ac-CoA and 5 mm peptide and the activity was determined as described under “Experimental Procedures.”
Kinetic Activity and Mutants
Two main possible mechanisms have been proposed for acetyltransferase activity. In the direct transfer mechanism the acetyl group is transferred directly from Ac-CoA to the acceptor substrate. This involves formation of a ternary complex with enzyme, Ac-CoA, and the acceptor substrate where the primary amine performs nucleophilic attack on the carbonyl carbon of Ac-CoA. The second is a ping-pong mechanism where the acetyl group is transiently transferred to a Cys residue that functions as the nucleophile to form an acetyl-Cys enzyme intermediate. The acetyl group is subsequently transferred from the Cys to the acceptor substrate. The current consensus is that GNATs typically use a direct transfer mechanism (3).
To investigate the catalytic properties of PA4794, we selected and mutated residues that are suitably positioned in the structure for a role in catalysis (Fig. 3A). Because Tyr128 is positioned in the proximity of the sulfhydryl moiety of Ac-CoA and changes conformation upon binding of Ac-CoA, we prepared Y128F and Y128A mutants to test its function. Because there is no obvious residue that could function as a general base, and assuming that PA4794 relies only on general acid for catalysis, we would expect a substantial effect on its activity when Tyr128 is mutated. Indeed, both Y128F and Y128A mutants are inactive (Fig. 4), which substantiates the hypothesis that Tyr128 is critical for catalysis. It is likely that this residue functions as a general acid by donating a proton to the thiolate anion of CoA which is formed after the transfer of the acetyl group, but potentially could also serve as a nucleophile in the ping-pong mechanism. The residue is conserved among the most similar PA4794 homologs, with the exception of the homolog protein from Thioalkalivibrio sulfidophilus. The corresponding Tyr residue is also present and has been suggested to function as a general acid in RimI (26). Often, this residue is replaced by His in other PA4794 homologs with known structures (Fig. 6).
FIGURE 6.

Sequence alignment of selected GNAT homologous to PA4794. Cys29, Cys117, and Tyr128 are indicated with red dots, Arg49 with blue, and Asn121 with orange.
The primary amine of the acceptor substrate must be deprotonated to perform the direct nucleophilic attack, and due to the high pKa value of lysine it is likely that some mechanism of deprotonation is required (3). In many GNATs this involves an amino acid near the active site that can act as a general base. Usually it is a conserved Glu, Asp, or His, but there is no such residue suitably located in PA4794. Lack of an amino acid suitably placed to function as a general base suggests that PA4794 relies upon a different strategy for deprotonating the substrate. There is the possibility that water molecules act as a proton wire to transfer the proton out of the active site. We observe a well ordered water molecule that is coordinated by the main chain of Tyr28, Phe118, and the side chain of Asn121. The relatively low B-factor of this water molecule suggests it may play a role in catalysis by acting as an initial proton acceptor, and to investigate this we mutated Asn121 to Ala. This mutant showed a significant reduction in activity (Fig. 4). This Asn is also conserved in all PA4794 homologs analyzed in the sequence alignment (Fig. 6). Due to a slight conformational change of Asn121 upon binding of Ac-CoA, the distance from this water to Asn121 in the complex with Ac-CoA is 2.8 Å. However, in the ternary complex with CoA and the acetylated peptide it is 3.1 Å, and in the structure of the apo-form it is 3.5 Å, which suggests that the N121A mutation does not necessarily prevent binding of this water.
Because PA4794 lacks a key general base residue, it is possible the enzyme may act via a ping-pong mechanism rather than direct transfer. There are two cysteine residues in PA4794 (Cys29 and Cys117) and both are located within the active site close to the sulfhydryl group of CoA. Cys117 is conserved among the most similar PA4794 homologs and in the structure it faces toward the active site. Cys29 is not conserved and faces away from the active site. We wanted to test the possible role of these cysteine residues in the proton or the acetyl group transfer and examine the possibility that Cys29 and Cys117 have redundant roles in catalysis. To rule out the possibility that mutation of one cysteine could compensate for the other, we also prepared a double C29A/C117A mutant.
Compared with the wild-type protein, the C29A mutant shows similar activity and affinity for Ac-CoA when the NPAcGK peptide concentration is held constant (Fig. 4, Table 1); however, the Vmax decreases and the affinity for NPAcGK increases compared with the wild-type when the concentration of Ac-CoA is held constant (Fig. 4). On the other hand, the activity of the C117A mutant decreases significantly compared with wild-type protein regardless of which substrate is varied (Fig. 4, Table 1) suggesting that the effect of the C117A mutation is greater than that of the C29A. The double cysteine mutant behaves similarly to the C117A mutant, suggesting that the reduction in activity is mainly due to the C117A mutation. Because the double C29A/C117A mutation did not obliterate the activity of the enzyme and the presence of the two cysteine residues cannot compensate for the loss of Tyr128, it seems likely that PA4794 does not utilize the ping-pong mechanism as its primary mechanism when NPAcGK is the substrate. Additional double and triple mutants prepared to test if the enzyme could compensate for the loss of a residue suspected to be important catalytically (Y128A/C29A, Y128A/C117A, and Y128A/C29A/C117A) were also inactive. Like the lysine acetyltransferase PAT from Sulfolubus (29), it seems like PA4794 is also a relatively inefficient acetyltransferase.
TABLE 1.
Kinetic parameters. Kinetic parameters determined from the AcCoA substrate saturation curves (Fig. 4A) for wild-type (WT) and mutant PA4794 in the presence of 10 mm NPAcGK as described under “Experimental Procedures”
The NPAcGK substrate saturation curves did not reach saturation even at 25 mm peptide (Fig. 4B). Therefore, we could not determine accurate kinetic parameters for the NPAcGK substrate.
| Vmax | S0.5 | n | |
|---|---|---|---|
| nmol/min/mg | μm | Hill coefficient | |
| WT | 838 ± 57 | 232 ± 36 | 1.11 ± 0.11 |
| C29A | 1018 ± 70 | 466 ± 70 | 1.00 ± 0.06 |
| R49Q | 200 ± 7 | 52.2 ± 6.3 | 1.36 ± 0.22 |
| C117A | 172 ± 7 | 54.0 ± 7.1 | 1.42 ± 0.26 |
| C29A/C117A | 125 ± 6 | 57.0 ± 8.5 | 1.17 ± 0.19 |
| N121A | 279 ± 8 | 73.2 ± 6.2 | 1.16 ± 0.10 |
| Y128A | NDa | ND | ND |
| Y128F | ND | ND | ND |
| C29A/Y128A | ND | ND | ND |
| C117A/Y128A | ND | ND | ND |
| C29A/C117A/Y128A | ND | ND | ND |
a ND indicates that no activity for the mutants was detected.
The highest activity of the wild-type enzyme was observed at a pH of 9.0, which may indicate that the substrate requires deprotonation prior to catalysis. Although at this pH the cysteines are likely deprotonated and could theoretically act as a general base, the substrate will likely be deprotonated as well. To investigate the pH dependence of activity for PA4794, we tested the activity at pH 7.5 and observed about a 10-fold decrease for all variants of the enzyme (data not shown). Despite the reduced activity, the general kinetic behavior of the enzyme was the same as in pH 9.0, suggesting that the difference in the activity is due to the protonation state of the substrate rather than the cysteines. At pH 7.5 the C29A mutant shows activity that is even more similar to the wild-type protein, and further confirms its marginal functional relevance (data not shown).
Inhibitors Binding and Inhibition Studies
To determine whether PA4794 binds other small molecules that were not detected in the initial activity screen, we conducted a series of experiments in which we soaked crystals of unbound PA4794 with solutions of potential ligands. To reduce the number of required experiments, 112 compounds were combined into several cocktail solutions composed of molecules of different classes. If a hit was identified, the cocktail solutions were split into individual components and rescreened. The compounds used in the cocktails included previously identified GNAT substrates, antibiotics (because aminoglycoside antibiotics are known GNAT substrates), and various metabolites (for the cocktail compositions see Table 2). Determination of the structures of cocktail-soaked crystals revealed new electron density in the structures of two of them. Due to the comparatively high resolution of the soaked structures, the new electron density could be unambiguously assigned to 4-methylumbelliferyl phosphate and the antibiotic cefmetazole. To further explore the ligand specificity, other similar compounds were used in subsequent soaks. We tested an additional 30 compounds, of which we found well ordered electron densities for cefotaxime, cefalotin, cefuroxime, 7-aminocephalosporanic acid, cefmetazole, cefoxitin, cefixime, cephalosporin C, and ammonium 2-(aminocarbonyl)benzoate. The structures of all the bound compounds are presented in Fig. 8.
FIGURE 8.

Binding of inhibitors in the substrate-binding site. For the comparison, the NPAcGK is shown in the same orientation on the first panel. Fo − Fc omit electron density maps of bound ligands are presented (σ = 2.5). The α1b helix (residues 21–31) (indicated in cyan on the first panel) was removed from other panels for clarity. The cephalosporin core two-dimensional structure is shown on the last panel, R1 and R2 positions are indicated. 7-ACA, 7-aminocephalosporanic acid; Cephalosp. C, cephalosporin C; 2-(ac) benzoate, 2-(aminocarbonyl) benzoate; 4-MethUmbPh, 4-methylumbelliferyl phosphate.
The relatively high ligand concentrations used for the crystal soaks allowed us to detect ligand binding with both high and low affinity. The crystallographic determination of ligand-protein complexes was confirmed by ITC experiments, which allowed us to quantify the binding affinity of the ligands and to identify which compounds bound with the highest affinity. The strongest binding was observed for cefotaxime (Kd = 3.5 ± 0.2 μm), followed by cefalotin, cefoxitin, cefuroxime, and 4-methylumbelliferyl phosphate. The identified ligands were tested to determine whether they acted as inhibitors of NPAcGK acetylation. The binding and inhibition constants are given in Table 3. An example of ITC binding titrations and fitted isotherms for the compound with the highest affinity (Cefotaxime) and the lowest determined affinity (Cefuroxime) are presented in Fig. 7. The compound 4-methylumbelliferyl phosphate was the strongest inhibitor tested, and of the cephalosporins, the strongest inhibition was detected for the compound with the highest binding affinity (cefotaxime).
FIGURE 7.

Example ITC binding titrations and fitted isotherms. A, cefotaxime; B, cefuroxime.
The cephalosporins bind in the substrate-binding site of PA4794 and their conformation mimics the conformation of the acetylated peptide product (Fig. 8). Similar to interactions with the C-terminal lysine, the carboxyl group of the cephalosporin core hydrogen bonds with the side chain of Arg49 and the main chain nitrogen of Asn80. The fact that 7-aminocephalosporanic acid binds, but 7-aminodesacetoxycephalosporanic acid does not, indicates that the cephalosporin core alone is insufficient for binding. The best binding cephalosporins have a relatively small 3-(acetoxymethyl) or 3-(carbamoyloxymethyl) moiety in the R1 position (Fig. 8). This moiety is oriented toward the Ac-CoA binding site and much larger functional groups will not fit into this cavity.
In the complexes with cefotaxime and cefalotin, the compounds with the highest binding affinities (Table 3), the carbonyl oxygen of the 3-(acetoxymethyl) moiety is stabilized by a hydrogen bond to the main chain amine of Met81, as is the carbonyl of the acetyl group of Ac-CoA and the acetyl group of the product of the reaction. In the case of cefoxitin and cefuroxime, which have a 3-(carbamoyloxymethyl) moiety in the R1 position, the conformation is slightly different and the amino group of that moiety interacts with the main chain of the Ser116, which also stabilizes the Nϵ of the NPAcGK C-terminal lysine.
If the structures of the complexes with antibiotics, Ac-CoA, and acetylated peptide are superimposed, the distances between their acetyl groups are about or below 1 Å. Therefore the binding of cephalosporins may also influence the binding of Ac-CoA. The R2 position is oriented toward the surface of PA4794 and is coordinated mainly through water molecules. It is therefore less restrained in terms of its flexibility and the size of the moiety that can be accommodated. For most of the cephalosporins the moiety in this position is visible in the structures, with the exception of cephalosporin C, where it is exposed to the solvent. The absence of this moiety in the structure is likely due to its flexibility, but we cannot exclude the possibility of decomposition.
The ligand 4-methylumbelliferyl phosphate is a fluorogenic substrate for alkaline phosphatase. It also interacts with the side chain of Arg49 through its carbonyl oxygen, as well as with the main chain of Cys29 and Asn80 (Fig. 8). The Nϵ and Nω atoms of Arg141 and several water-mediated interactions coordinate the phosphate moiety. Although the compound is not a biologically relevant ligand for PA4794, it does inhibit PA4794 enzymatic activity. As a general trend, the inhibitors that bind to PA4794 do not induce substantial conformational changes.
For cefsulodin we observe well defined electron density for the moiety in the R2 position, but the large moiety at the R1 position seems to be decomposed. Weak or no electron density was observed for cefalonium, cefadroxil, cefoperazone, cefazolin, ceftriaxone, cefamandole, ceftiofur, cefapirin, cefaclor, 7-aminodesacetoxycephalosporanic acid, 3-aminophthalic acid, potassium clavulanate, and (S)-(−)-4-amino-2-hydroxybutyric acid. We also tested coumarin-3-carboxylic acid, 4-methylumbelliferone, l-leucine-7-amido-4-methylcoumarin hydrochloride, 7-amino-4-methyl-3-coumarinylacetic acid, linezolid, and anisomycin, but none of these molecules could be detected to bind to the protein.
DISCUSSION
The discovery that GNATs are involved in protein acetylation in bacteria, not just in eukaryotes, suggests that acetylation-based regulation may be an important process in prokaryotes as well. Reversible and specific acetylation of lysine residues has tremendous regulatory potential, and it is becoming clear that this process is involved in almost every aspect of cellular physiology. However, little is known about the enzymes catalyzing protein acetylation in bacteria, including their substrates. Using several screening approaches, we identified the prokaryotic protein PA4794 (a representative of an uncharacterized subgroup of GNATs) as an Nϵ-lysine acetyltransferase, and established that the enzyme specifically acetylates the Nϵ amino group of C-terminal lysines of substrate peptides (and presumably, proteins).
Crystal structures confirm that PA4794 has a characteristic GNAT-fold with a splayed V-cleft between the β-sheets that binds Ac-CoA and CoA. The Ac-CoA/CoA binds in a sharply bent conformation, which is also characteristic for GNATs, and induces a slight conformational change in the Ac-CoA/CoA binding regions of PA4794. Despite low sequence similarity, our structural analysis revealed that PA4794 shows structural similarity to RimI, which is the GNAT responsible for Nα-acetylation of the ribosomal protein S18. However, it seems that the closest homologs of PA4794 are specific for Pseudomonas and related species, and therefore its function may be species specific.
The substrate NPAcGK is an artificial sweetener and is thus not the biological substrate for PA4794. However, the structure of the complex with NPAcGK revealed its binding mode and allowed us to determine the critical importance of the free C-terminal carboxyl group in permitting the acetylation of lysine. Based on the structure of NPAcGK and other tested peptides, we determined the enzyme most likely prefers a (Phe/Tyr)-Gly-Lys C-terminal motif, although it should be noted that all possible combinations were not exhaustively tested. We analyzed the C termini of all P. aeruginosa sequences in the UniProt database, but did not find proteins with a Phe-Gly-Lys or Tyr-Gly-Lys C-terminal motif. However, a number of sequences of P. aeruginosa proteins end with Gly-Lys, often with a hydrophobic residue in the 3rd position from the C terminus. There is a possibility that if the full-length protein is a substrate for PA4794, there are more distant residues that are also recognized by PA4794, thus the acetylated C terminus might have a different sequence. There is also a possibility that PA4794 does not acetylate full-length proteins, but rather shorter peptides, such as the products of proteolysis. In that case, the exposed C-terminal lysine residue of the substrate might be an internal lysine residue. One plausible explanation for this would be to label hydrolyzed proteins for degradation. It has been shown previously that lysine acetylation might trigger protein degradation (30). Another possibility is that the substrate is not an endogenous Pseudomonas protein, but a host protein. One of the components of eukaryotic host defense systems is antimicrobial peptides or proteins, and due to the development of resistance against conventional antibiotics these have gained attention as possible treatment options for Pseudomonas infections (31–34).
Because new substrates for different GNATs are gradually being discovered, it is plausible that other substrates that have not been considered exist for PA4794 and should be investigated in further studies. Adding to the complexity of identifying physiological substrates, GNATs seem to be promiscuous and are capable of acetylating more than one class of substrates (23, 24, 35). Interestingly, the PA4794 enzyme can also acetylate chloramphenicol and this mechanism will be detailed in a subsequent publication.4 This suggests that PA4794 may acetylate proteins in addition to small molecules, and is therefore capable of both N- and O-acetylation.
Another important discovery is the inhibition of PA4794 by cephalosporins. The crystallographic determination of protein-antibiotic complexes shows that cephalosporins bind in the substrate-binding site and mimic the conformation of the product, and thus likely serve as competitive inhibitors. The crystallographic data, together with isothermal titration calorimetry experiments, allowed us to determine the binding parameters as well as the structural features of the best binders. If the biological function of PA4794 is to acetylate Pseudomonas proteins, it may play an important regulatory role in bacteria. Additionally, if its substrate is exogenous then its function may be defensive. Once the physiological function of PA4794 is known, our identification of the detailed interactions between PA4794 and cephalosporins may be of clinical relevance. Understanding PA4794-antibiotic interactions may also be useful in facilitating the design of optimal inhibitors.
Acknowledgments
We thank Dr. Igor Shumilin and Przemyslaw Porebski for valuable discussions and Dr. Matthew D. Zimmerman for critically reading the manuscript. Additionally, we thank Drs. Sergii Pshenychnyi and Izolda Popova at the Recombinant Protein Production Core at Northwestern University for expression and purification services. Some results shown in this report are derived from work performed at the Structural Biology Center Sector 19 and LS-CAT Sector 21 at the Advanced Photon Source. The Advanced Photon Source, an Office of Science User Facility, is operated for the United States Dept. of Energy (DOE) Office of Science by Argonne National Laboratory. Argonne is operated by University of Chicago Argonne, LLC, for the United States DOE Office of Biological and Environmental Research under contract DE-AC02–06CH11357. Use of LS-CAT Sector 21 was also supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor Grant 085P1000817.
This work was supported, in whole or in part, by National Institutes of Health grants from the NIAID, Dept. of Health and Human Services, under Contracts HHSN272200700058C and HHSN272201200026C (CSGID) and PSI:Biology NIGMS Grant U54 GM094585.

This article contains supplemental Table S1.
The atomic coordinates and structure factors (codes 4KUA, 3PGP, 4KUB, 4L89, 4L8A, 4KLV, 4KOT, 4KOW, 4KOX, 4KOV, 4KOU, 4KOY, 4KOR, 4KOS, and 4KLW) have been deposited in the Protein Data Bank (http://wwpdb.org/).
K. A. Majorek, M. L. Kuhn, M. Chruszcz, W. F. Anderson, and W. Minor, unpublished data.
K. A. Majorek, M. L. Kuhn, A. Miłaczewska, M. A. Ballicora, M. Chruszcz, T. Borowski, W. F. Anderson, and W. Minor, unpublished data.
- GNAT
- Gcn5-related N-acetyltransferase
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- ITC
- isothermal titration calorimetry
- PDB
- Protein Data Bank
- Bicine
- N,N-bis(2-hydroxyethyl)glycine
- NPAcGK
- N-phenylacetyl-Gly-Lys.
REFERENCES
- 1. Driscoll J. A., Brody S. L., Kollef M. H. (2007) The epidemiology, pathogenesis and treatment of Pseudomonas aeruginosa infections. Drugs 67, 351–368 [DOI] [PubMed] [Google Scholar]
- 2. Rahme L. G., Stevens E. J., Wolfort S. F., Shao J., Tompkins R. G., Ausubel F. M. (1995) Common virulence factors for bacterial pathogenicity in plants and animals. Science 268, 1899–1902 [DOI] [PubMed] [Google Scholar]
- 3. Dyda F., Klein D. C., Hickman A. B. (2000) GCN5-related N-acetyltransferases. A structural overview. Annu. Rev. Biophys. Biomol. Struct. 29, 81–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vetting M. W., S. de Carvalho L. P., Yu M., Hegde S. S., Magnet S., Roderick S. L., Blanchard J. S. (2005) Structure and functions of the GNAT superfamily of acetyltransferases. Arch. Biochem. Biophys. 433, 212–226 [DOI] [PubMed] [Google Scholar]
- 5. Yu B. J., Kim J. A., Moon J. H., Ryu S. E., Pan J. G. (2008) The diversity of lysine-acetylated proteins in Escherichia coli. J. Microbiol. Biotechnol. 18, 1529–1536 [PubMed] [Google Scholar]
- 6. Zhang J., Sprung R., Pei J., Tan X., Kim S., Zhu H., Liu C. F., Grishin N. V., Zhao Y. (2009) Lysine acetylation is a highly abundant and evolutionarily conserved modification in Escherichia coli. Mol. Cell. Proteomics 8, 215–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hu L. I., Lima B. P., Wolfe A. J. (2010) Bacterial protein acetylation. The dawning of a new age. Mol. Microbiol. 77, 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kapust R. B., Tözsér J., Fox J. D., Anderson D. E., Cherry S., Copeland T. D., Waugh D. S. (2001) Tobacco etch virus protease. Mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 14, 993–1000 [DOI] [PubMed] [Google Scholar]
- 9. Kapust R. B., Waugh D. S. (2000) Controlled intracellular processing of fusion proteins by TEV protease. Protein Expr. Purif. 19, 312–318 [DOI] [PubMed] [Google Scholar]
- 10. Zimmerman M. D., Chruszcz M., Koclega K. D., Otwinowski Z., Minor W. (2005) The Xtaldb system for project salvaging in high-throughput crystallization. Acta Crystallogr. A 61, 178–179 [Google Scholar]
- 11. Shumilin I. A., Cymborowski M., Chertihin O., Jha K. N., Herr J. C., Lesley S. A., Joachimiak A., Minor W. (2012) Identification of unknown protein function using metabolite cocktail screening. Structure 20, 1715–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rosenbaum G., Alkire R. W., Evans G., Rotella F. J., Lazarski K., Zhang R. G., Ginell S. L., Duke N., Naday I., Lazarz J., Molitsky M. J., Keefe L., Gonczy J., Rock L., Sanishvili R., Walsh M. A., Westbrook E., Joachimiak A. (2006) The Structural Biology Center 19ID undulator beamline. Facility specifications and protein crystallographic results. J Synchrotron Radiat. 13, 30–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Otwinowski Z., Minor W. (1997) in Methods Enzymology: Macromolecular Crystallography (Carter C. W., Jr., Sweet R. M., eds) Part A, pp. 307–326, Academic Press, New York [Google Scholar]
- 14. Minor W., Cymborowski M., Otwinowski Z., Chruszcz M. (2006) HKL-3000, the integration of data reduction and structure solution. From diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 62, 859–866 [DOI] [PubMed] [Google Scholar]
- 15. Vagin A., Teplyakov A. (1997) MOLREP. An automated program for molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 30, 1022–1025 [Google Scholar]
- 16. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 18. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Collaborative Computational Project, Number 4 (1994) The CCP4 suite. Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 20. Painter J., Merritt E. A. (2006) TLSMD web server for the generation of multi-group TLS models. J. Appl. Crystallogr. 39, 109–111 [Google Scholar]
- 21. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B., 3rd, Snoeyink J., Richardson J. S., Richardson D. C. (2007) MolProbity. All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yang H., Guranovic V., Dutta S., Feng Z., Berman H. M., Westbrook J. D. (2004) Automated and accurate deposition of structures solved by x-ray diffraction to the Protein Data Bank. Acta Crystallogr. D 60, 1833–1839 [DOI] [PubMed] [Google Scholar]
- 23. Kuhn M. L., Majorek K. A., Minor W., Anderson W. F. (2013) Broad-substrate screen as a tool to identify substrates for bacterial Gcn5-related N-acetyltransferases with unknown substrate specificity. Protein Sci. 22, 222–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vetting M. W., Magnet S., Nieves E., Roderick S. L., Blanchard J. S. (2004) A bacterial acetyltransferase capable of regioselective N-acetylation of antibiotics and histones. Chem. Biol. 11, 565–573 [DOI] [PubMed] [Google Scholar]
- 25. Holm L., Rosenström P. (2010) Dali server. Conservation mapping in 3D. Nucleic Acids Res. 38, W545–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vetting M. W., Bareich D. C., Yu M., Blanchard J. S. (2008) Crystal structure of RimI from Salmonella typhimurium LT2, the GNAT responsible for Nα-acetylation of ribosomal protein S18. Protein Sci. 17, 1781–1790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Neuwald A. F., Landsman D. (1997) GCN5-related histone N-acetyltransferases belong to a diverse superfamily that includes the yeast SPT10 protein. Trends Biochem. Sci. 22, 154–155 [DOI] [PubMed] [Google Scholar]
- 28. Vetting M. W., de Carvalho L. P., Roderick S. L., Blanchard J. S. (2005) A novel dimeric structure of the RimL Nα-acetyltransferase from Salmonella typhimurium. J. Biol. Chem. 280, 22108–22114 [DOI] [PubMed] [Google Scholar]
- 29. Brent M. M., Iwata A., Carten J., Zhao K., Marmorstein R. (2009) Structure and biochemical characterization of protein acetyltransferase from Sulfolobus solfataricus. J. Biol. Chem. 284, 19412–19419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lin S., Tsai S. C., Lee C. C., Wang B. W., Liou J. Y., Shyu K. G. (2004) Berberine inhibits HIF-1α expression via enhanced proteolysis. Mol. Pharmacol. 66, 612–619 [PubMed] [Google Scholar]
- 31. Sajjan U. S., Tran L. T., Sole N., Rovaldi C., Akiyama A., Friden P. M., Forstner J. F., Rothstein D. M. (2001) P-113D, an antimicrobial peptide active against Pseudomonas aeruginosa, retains activity in the presence of sputum from cystic fibrosis patients. Antimicrob. Agents Chemother. 45, 3437–3444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Naghmouchi K., Le Lay C., Baah J., Drider D. (2012) Antibiotic and antimicrobial peptide combinations. Synergistic inhibition of Pseudomonas fluorescens and antibiotic-resistant variants. Res. Microbiol. 163, 101–108 [DOI] [PubMed] [Google Scholar]
- 33. Malmsten M., Kasetty G., Pasupuleti M., Alenfall J., Schmidtchen A. (2011) Highly selective end-tagged antimicrobial peptides derived from PRELP. PLoS One 6, e16400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Izadpanah A., Gallo R. L. (2005) Antimicrobial peptides. J. Am. Acad. Dermatol. 52, 381–390; quiz 391–392 [DOI] [PubMed] [Google Scholar]
- 35. Kim C., Villegas-Estrada A., Hesek D., Mobashery S. (2007) Mechanistic characterization of the bifunctional aminoglycoside-modifying enzyme AAC(3)-Ib/AAC(6′)-Ib′ from Pseudomonas aeruginosa. Biochemistry 46, 5270–5282 [DOI] [PubMed] [Google Scholar]