

Background: Cytochrome c oxidase reduces O2 coupled with proton pumping.
Results: A newly developed time-resolved infrared system reveals transient conformational changes in the proton-pumping pathway upon CO binding to CuB in the O2 reduction site.
Conclusion: CuB promotes proton collection and effective blockage of back-leak of pumping protons.
Significance: These critical findings in bioenergetics stimulate the new infrared approach for mechanistic investigation of any other protein function.
Keywords: Bioenergetics, Biophysics, Infrared Spectroscopy, Membrane Proteins, Respiratory Chain
Abstract
X-ray structural and mutational analyses have shown that bovine heart cytochrome c oxidase (CcO) pumps protons electrostatically through a hydrogen bond network using net positive charges created upon oxidation of a heme iron (located near the hydrogen bond network) for O2 reduction. Pumping protons are transferred by mobile water molecules from the negative side of the mitochondrial inner membrane through a water channel into the hydrogen bond network. For blockage of spontaneous proton back-leak, the water channel is closed upon O2 binding to the second heme (heme a3) after complete collection of the pumping protons in the hydrogen bond network. For elucidation of the structural bases for the mechanism of the proton collection and timely closure of the water channel, conformational dynamics after photolysis of CO (an O2 analog)-bound CcO was examined using a newly developed time-resolved infrared system feasible for accurate detection of a single C=O stretch band of α-helices of CcO in H2O medium. The present results indicate that migration of CO from heme a3 to CuB in the O2 reduction site induces an intermediate state in which a bulge conformation at Ser-382 in a transmembrane helix is eliminated to open the water channel. The structural changes suggest that, using a conformational relay system, including CuB, O2, heme a3, and two helix turns extending to Ser-382, CuB induces the conformational changes of the water channel that stimulate the proton collection, and senses complete proton loading into the hydrogen bond network to trigger the timely channel closure by O2 transfer from CuB to heme a3.
Introduction
Cytochrome c oxidase (CcO),4 the terminal oxidase of cellular respiration, reduces O2 to H2O. This process occurs at a site that includes an iron site (Fea3 of heme a3) and a copper site (CuB). The electron equivalents for O2 reduction are transferred from cytochrome c via the second copper (CuA) and iron (Fea of heme a) sites. In bovine CcO, the process is coupled with the pumping of protons from the negative side to the positive side of the mitochondrial inner membrane in a system (H-pathway) that includes a hydrogen bond network and a water channel operating in tandem (1). The protons being pumped are transferred by water molecules to the hydrogen bond network from the negative side of the mitochondrial inner membrane through the water channel by thermal motion of the protein. The active transport of protons to the positive side is driven through the hydrogen bond network by electrostatic repulsion between protons and the positive charges, created upon oxidation of Fea, which is located near the hydrogen bond network (1). The redox-coupled conformational changes of Asp-51 at the positive side end of the hydrogen bond network, revealed by x-ray structural analyses, indicate that Asp-51 functions as one of the proton-loading sites for proton pumping (or the proton exit of the H-pathway) (2).
The function of Asp-51 has been confirmed by the D51N mutation of bovine heart CcO (3). Several other mutations for the key residues of the H-pathway (4) as well as x-ray structural analyses at various oxidation and ligand-binding states (5–7) have established that bovine heart CcO pumps protons through the H-pathway. However, various key amino acid residues in the H-pathway are not well conserved. For example, Asp-51 is not conserved in bacterial and plant CcOs, although similar possible proton-conducting structures are detectable in their x-ray structures. Extensive mutational analyses of the bacterial possible proton-conducting structures corresponding to the bovine H-pathway did not show any significant influence on the proton pumping activity (8). Based on mutagenesis analyses, another proton-conducting pathway, the D pathway, has been proposed to transport protons for pumping as well as for the water formation in bacterial aa3 type CcOs (9). Furthermore, mutagenesis analyses for the other bacterial terminal oxidases (ba3 type) showed that protons are pumped through the third proton-conducting pathway, the K pathway (10). These results strongly suggest that the proton-pumping mechanism of terminal oxidases is not conserved completely. No experimental evidence against the proton pumping function of the bovine H-pathway has been reported thus far, although the H-pathway structures are not completely conserved.
The water channel of the H-pathway of bovine heart CcO includes water cavities, each containing at least one water molecule, and water pathways through which water molecules can be transferred by thermal motion of the protein. The largest water cavity, located near the junction point for entry to the hydrogen bond network, is detectable only when both Fea3 and CuB are in the reduced state during catalytic turnover. Significant narrowing of the water channel occurs upon elimination of the largest cavity. This greatly decreases the efficiency of water exchange and thus decreases the rate of entry of protons (supplied by mobile water molecules in the water channel) into the hydrogen bond network as well as backward leakage of protons from the hydrogen bond network. Therefore, this state is designated as the “closed state.”
In the catalytic cycle, the O2 reduction site in the fully reduced state [Fea32+,CuB1+] receives O2 and then sequentially receives four electron equivalent from cytochrome c. Each of the one-electron reduction processes is coupled with the pumping of one proton equivalent (11). The water channel is closed during these proton-pumping processes (1). Thus, four proton equivalents must enter the hydrogen bond network in the reduced state before O2 binds to Fea3 (1). Just before the water channel is opened, the hydrogen bond network is completely in the deprotonated state, because four protons are pumped in the preceding catalytic cycle under the blockage of the proton supply from the water channel. Upon opening the channel, the hydrogen bond network gets protonically equilibrated with the negative side space of the mitochondrial inner membrane through the water channel. The protonic equilibration is the driving force for proton collection of the hydrogen bond network from the negative side.
The relative location of heme a against the hydrogen bond network suggests that the overall direction of proton pumping, which is driven by the electrostatic repulsion between protons and the positive charges of heme a, is determined essentially by the timing of closure of the water channel. Thus, the timely closure of the channel is critical to ensure highly efficient energy transduction.
For efficient energy transduction, molecular machinery must be adapted for collection of the protons being pumped and for sensing of the protonation state of the hydrogen bond network, to close the water-channel immediately after complete protonation is attained. It is likely that the molecular machinery operates during the transition from the fully reduced state in the O2 reduction site to the O2-bound state. Thus, time-resolved infrared (TRIR) examination of reactions between the fully reduced enzyme and nonreducible O2 analogs, such as CO, NO, and cyanide, are expected to provide various important insights for the mechanism of proton collection and timely closure of the water channel.
Recent x-ray structural analyses indicate that the most prominent change occurring in the protein moiety upon binding of CO is a bulge structural formation at Ser-382 providing a new single unpaired main chain C=O group (7). However, because of strong IR absorption of solvent water (12), IR measurements of proteins in aqueous solution (i.e. under physiological conditions) with sufficient sensitivity for analysis of IR spectral changes due to a single peptide C=O band have remained technically challenging. We have developed a novel nanosecond TRIR system that provides performance sufficient for this purpose. The TRIR results of this study indicate that CuB is not only a simple electron donor to the bound O2 but also plays key roles in the efficient collection of protons used in the proton-pumping process and timely closure of the water channel by using a conformational relay system that connects the CuB site and the water channel.
EXPERIMENTAL PROCEDURES
Sample Preparation
CcO, purified from bovine heart muscle as described previously (13), was dissolved in H2O buffered with 100 mm sodium phosphate (pH 7.4), containing 0.2% (w/v) n-decyl-β-d-maltoside. The protein concentration was determined by absorption spectroscopy, using an extinction coefficient of Δϵ604–630 nm = 46.6 mm−1cm−1 for the fully reduced form (13). Absorption spectra were also recorded after the experiments to confirm sample integrity after exposure to laser irradiation.
Experimental Setup
A femtosecond mid-IR pulse (>10 μJ) with a spectral width of 350 cm−1 was produced by a difference frequency generator with an optical parametric amplifier (OPerA Solo, Coherent), which was pumped by the output of an integrated Ti:Sapphire oscillator/regenerative amplifier system, operating at 1 kHz (Micra-5 and Legend Elite-USP, Coherent). A dual-row detector array (2 × 64) of MCT elements (Infrared Systems Development) coupled to a spectrograph (TRIAX-190, HORIBA Jobin Yvon) was employed, and the signals due to probe and reference pulses were read out with a boxcar integrator system (FPAS-0144, Infrared Systems Development) on a single shot basis with 16-bit resolution. The wave number resolution was 2 cm−1.
The 25-ns, 532-nm output (0.15 mJ) of an Nd:YAG laser (Navigator I, Spectra Physics) at 1 kHz was used as a pump pulse, which gave 80% CO photolysis for the highest performance for minimization of the photochemical damage by the pump pulse. A timing jitter between the visible pump and mid-IR probe pulses was ±25 ns. The pump beam was modulated using a phase-locked chopper operating at 0.5 kHz, which allowed us to perform nearly simultaneous (1-ms interval) measurements of the pump-on and pump-off spectra.
The sample was housed in two CaF2 windows separated by a Teflon spacer and spun at 1300 rpm to ensure a fresh spot for each IR pulse transmission. The sample cell temperature was kept at 28.0 °C (<0.1 °C accuracy). To avoid water vapor effects, the optical setup was contained in a dry air-purged chamber with <5% humidity. The TRIR measurements were repeated five times to be accumulated and averaged. Each measurement was performed with 48-s data accumulation.
Data Analysis
The TRIR measurements here are the accumulated and averaged difference spectra against the spectrum before the photolysis (the spectrum of the CO-bound form). Because both the visible and mid-IR pulses were linearly polarized, the difference spectra were recorded with the visible pulses polarized parallel (ΔA‖) and perpendicular (ΔA⊥) to the mid-IR, and isotropically averaged spectra, ΔAiso = (ΔA‖ + 2ΔA⊥)/3, are presented to eliminate rotational relaxation effects (14, 15).
The TRIR difference spectra were analyzed by global fit (Igor Pro 5.0, WaveMetrics), where each band was assumed to have a Gaussian shape, and the time dependence of its amplitude was fitted by a single exponential rise or decay with the wave number and bandwidth fixed. Base-line fluctuation (30 μOD levels, estimated from the standard deviation of the ΔA value at 2000 cm−1) was linearly corrected before the fitting. Prior to setting Gaussian bands, a singular value decomposition analysis was applied as a preliminary search to extract the principal components of spectral changes (Igor Pro 5.0, WaveMetrics). The first principal component showed two major peaks near 1662 and 1670 cm−1 with no time evolution after the initial appearance. The second principal component had a peak/trough at 1655(+)/1666(−) cm−1 with a time constant of ∼2.2 μs as the prominent change. First, four Gaussian bands were set to reproduce the above features. Then, additional Gaussian bands sufficient for giving the fitting residual not exceeding the noise level (30 μOD) were set to reproduce each peak. The bumps in the fitting residual, at the peak edges which are likely to be induced by the Gaussian curve fitting, were ignored.
Structural Exploration for the Intermediate State of Helix X between the Ligand-free and CO-bound States
The atomic coordinates of the crystal structures of bovine CcO in the ligand-free and CO-bound forms were obtained from the Protein Data Bank (codes 2eij and 3ag2, respectively) (5, 7), and subunits 1–3 were used in the following calculations.
First, the interpolated coordinates between the two states were generated as c→ = a→ + λ(b→ − a→), where a→, b→, and c→ are the sets of the coordinates of the ligand-free form, the CO-bound form (note that the coordinates of the ligand was removed), and the generated interpolated states. λ is a parameter (the reaction coordinate), which takes values between 0 and 1 with each step of 0.01 (i.e. 0.01, 0.02, … , 0.99). Thus, 99 sets of the interpolated coordinates were created as the initial conformations of helix X for the present modeling.
The hydrogen (H) atoms were then added to all of these structures using the LEAP module of the AMBER 9 program package (16), and the positions of the added H atoms were relaxed by using the steepest descent energy minimization scheme. In all the following processes, the dielectric constant was set to 4.0. Next, the side chain atoms were optimized, and then all atoms were relaxed with the energy minimization. These calculations were performed using the SANDER module and the parm99sb force field parameter in the AMBER 9 program package (16). With respect to the transition metal-binding sites, i.e. CuA, CuB, heme a, and heme a3, the electrostatic potentials were obtained by density functional theory calculations, which were performed in our previous study (17), and the restrained electrostatic potential (RESP) charges were generated by using the ANTECHAMBER module of AMBER 9 (16, 18).
To explore the conformations between the substrate-free and CO-bound states, the amino acid residues of helix X (i.e. from Val-380 to Met-390) were moved, and the other residues were fixed in the following calculations. For each of the 99 structures generated above, the simulated annealing protocol was adopted for the extended conformational sampling. First, 1-ps molecular dynamics simulations involving distance constrains (as described below) were performed sequentially at temperatures of 150, 200, 300, 400, 500, 450, 400, 350, and finally 300 K. Here, the hydrogen bonds that are not relevant to the bulge-out moieties in the crystal structures were restrained by using the distance constrains of the backbone H atoms of amide groups and O atoms of carbonyl groups. The distance constrains were imposed with the use of a harmonic function, U(r→) = k(r→ − r→0)2, with a force constant (k) of 100 kcal/mol Å−2. Then, energy minimization was performed for each structure, without the distance constrains. Finally, the intermediate state was identified as the minimum energy conformation among the stationary energy states.
Because the root mean square deviation value between helix X in the two crystal structures used for the sampling (i.e. the initial and final structures of the calculations) is as small as 0.7 Å (for the heavy atoms), our present scheme can provide a fine-grained sampling enough to examine whether the time-resolved observations are corresponding to the conformational changes that were found in helix X in the crystal structures.
Exploration of Cavity in the Protein
To identify cavities within the protein, CAVER (19) was applied to the regions in the vicinity of helix X, with respect to the above-mentioned two crystal structures and the intermediate structure of helix X. The cavities that were identified as having radii lager than 1.2 Å were used to prepare visual models.
RESULTS
Performance of Our Newly Developed TRIR System Designed for TRIR Analysis of the Conformational Changes Occurring after Photolysis of Carbonmonoxy (CO)-bound CcO
The strong IR absorption of water is the largest hindrance for the IR analyses of proteins in aqueous solution. We developed a TRIR system designed to eliminate this hindrance. One of the key components of our system is a femtosecond IR laser as the strong light source. We take advantage of the brightness of the femtosecond pulse to ensure a sufficient number of photons (1014 photons/<100-fs pulse) to detect after transmission through aqueous solution. Although the femtosecond IR technology has been developed and employed so far to investigate ultrafast events (20–24), it has never been utilized for the current purpose, namely high sensitivity against the high background absorption.
The present nanosecond TRIR system achieved 30 μOD sensitivity against a background OD of 2 in only 48 s of data accumulation, as given in Fig. 1. Although a femtosecond TRIR system has been previously applied to a protein in H2O, the reported sensitivity was 100 μOD at best for the Amide-I region (25), partially because no reference pulse was used to compensate the pulse-to-pulse fluctuation of the light source in the reported system. Recently, a quantum cascade laser has emerged as another type of strong IR light source. However, it provided the sensitivity of a few hundreds of μOD at present (26, 27).
FIGURE 1.
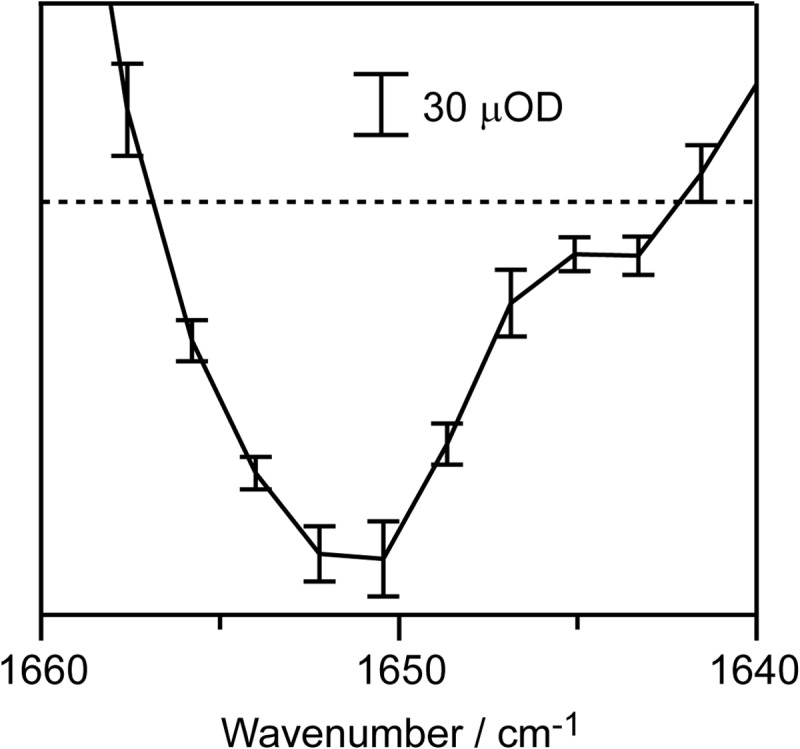
Experimental accuracy in a spectral region with the strong background absorption (OD of 2). The data shown are the TRIR difference spectra of CcO at 50 μs after CO photolysis in H2O buffer. Error bars represent the standard deviation of five independent experiments (each performed with 48-s data accumulation) on different days using different batches of CcO preparation. The protein concentration and optical path length were 0.68 mm and 13 μm, respectively.
The most widely used TRIR technique is step-scan Fourier transform (FT) IR (28–31). However, FTIR is not a suitable method for measuring strong absorbers. In FTIR, the IR lights in strong absorption regions (e.g. 1600–1700 cm−1) are spatially overlapped with those in weak absorption regions (e.g. 1800–1900 cm−1) for the sake of obtaining an interferogram and detected simultaneously with a single-channel detector, which prevents the use of the strong light source while avoiding detector saturation. Thus, the high accuracy (tens of μOD) of FTIR is normally achieved when the background absorption is lower than an OD of 1. Nevertheless, the best sensitivity was 100 μOD even using a system with the highest performance reported thus far (32) for the measurements at nanosecond time resolution. This is because the data acquisition in the step-scan procedure is in principle limited by the response speed of detector electronics, and nanosecond is near the upper limit of the response speed. Furthermore, long data accumulation (∼1 h or more) is usually required in step-scan FTIR to achieve the best sensitivity. The rapid data acquisition together with the high allowable background absorbance are critical for practical applications of the aqueous solution of proteins with high performance.
Although D2O exchange greatly decreases the background absorbance, especially in the Amide-I region, complete exchange is practically impossible. As a result, the experimental results obtained using a D2O-exchanged sample usually do not provide a straightforward interpretation. Furthermore, D2O exchange effects are often not simple, especially in proteins with proton transfer functions such as CcO (33). For such proteins, IR analyses in H2O are indispensable. Hydrated protein films are often successfully applied to stable proteins for reducing the strong water absorption (34). However, it is impossible to exclude the possibility of partial denaturation in the film especially for unstable proteins. Thus, we have developed an IR system for investigating aqueous (H2O) protein systems.
A single peptide C=O stretching band in the Amide-I region in the millimolar concentration range provides 260–1300 μOD using a light path of 13 μm, depending on the microenvironment of the group (35). A light path of 13 μm provides a background maximum OD of ∼2 in the mid-IR region (1200–2200 cm−1). Thus, the present system, which detects a 30 μOD difference against a background OD of 2 with nanosecond time resolution as described above, is suitable for use in obtaining TRIR measurements at sufficiently high resolution for analysis of the infrared behavior of a single peptide C=O group in the Amide-I region of proteins in H2O solution.
IR Spectral Changes after CO Photolysis of CO-bound Bovine Heart CcO
Difference spectra at various time points after photolysis against the spectrum before photolysis (the spectrum of the CO-bound form) are shown in Fig. 2. In this work, the intensity of the pump pulse was controlled to give 80% CO photolysis for the highest performance with minimization of photochemical damage.
FIGURE 2.
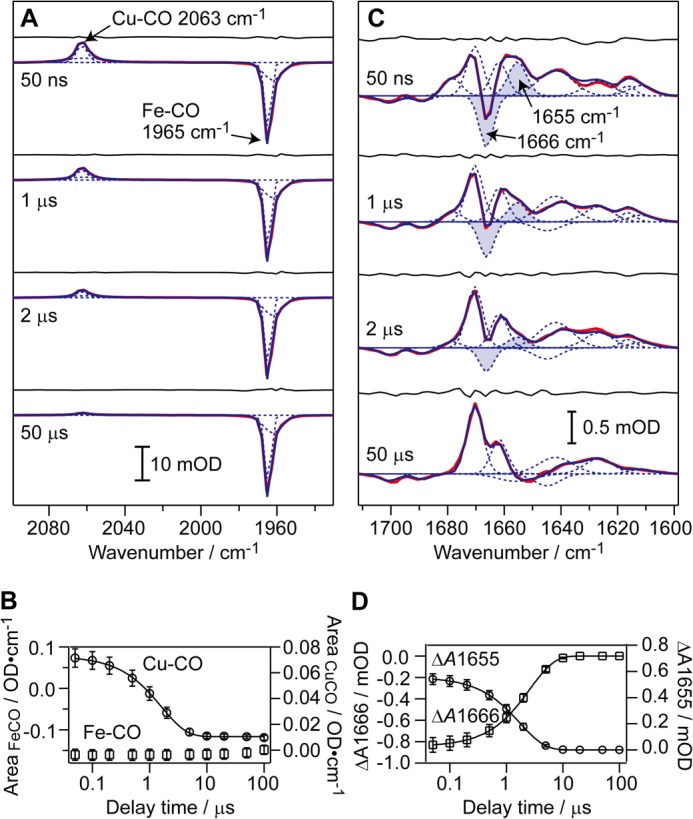
TRIR difference spectra of CcO in H2O buffer. Red, measured spectra; blue, fitted spectra, with each Gaussian component shown by a dotted curve. The fitting residual is also shown for each spectrum. The protein concentration and optical path length were 0.72 mm and 100 μm for A and 0.68 mm and 13 μm for C. Error bars represent the standard deviation of three (B) and five (D) independent experiments performed on different days using different batches of CcO preparation.
As indicated in Table 1, 30 bands are detectable between 2100 and 1500 cm−1. It should be noted that the kinetic behavior of these bands at this resolution, except for the carbonmonoxy CO stretch peaks for CuB-CO and Fea3-CO, has not been reported thus far. These bands can be classified in terms of the time scale of their appearance or decay into the following three types: (i) band appearance within 50 ns; (ii) band appearance or decay with a time constant of 0.7 ∼ 3 μs, and (iii) band appearance or decay with a time constant of 12–83 μs. The type i bands without further change after the initial rapid appearance are controlled by CO release from Fea3. The type ii bands are likely to be coupled with CO release from CuB, although the type iii bands appear following the process of the CO release from CuB. These results suggest that Fea3 and CuB control the conformations of different areas of the CcO protein.
TABLE 1.
Detected TRIR bands
| Wave number | Bandwidtha | Time constant | ΔIb | Δϵc |
|---|---|---|---|---|
| cm−1 | cm−1 | μs | mOD | m−1 cm−1 |
| 2063 | 10.0 (±0.1) | Rise <0.05 | 5.08 (±0.49) | 882 (±85) |
| Decay 1.6 (±0.1) | −4.65 (±0.39) | −807 (±68) | ||
| 2061 | 35.0 (±0.9) | Rise <0.05 | 1.33 (±0.17) | 231 (±30) |
| Decay 1.7 (±0.2) | −1.07 (±0.13) | −186 (±23) | ||
| 1965 | 5.9 (±0.0) | Rise <0.05 | −20.29 (±1.57) | −3523 (±273) |
| 1962 | 16.6 (±0.2) | Rise <0.05 | −5.23 (±0.48) | −908 (±83) |
| 1750 | 6.5 (±0.5) | Rise 15.7 (±6.6) | 0.06 (±0.00) | 19 (±0) |
| 1749 | 12.1 (±1.2) | Rise <0.05 | 0.28 (±0.01) | 90 (±3) |
| Decay 1.1 (±0.4) | −0.10 (±0.01) | −32 (±3) | ||
| 1745 | 11.1 (±0.4) | Rise <0.05 | −0.21 (±0.03) | −67 (±10) |
| Rise 15.6 (±9.9) | −0.04 (±0.01) | −13 (±3) | ||
| 1738 | 11.7 (±0.7) | Rise <0.05 | −0.08 (±0.01) | −26 (±3) |
| Decay 0.7 (±0.5) | 0.06 (±0.01) | 19 (±3) | ||
| 1701 | 11.4 (±0.4) | Rise <0.05 | −0.11 (±0.01) | −156 (±14) |
| 1689 | 9.7 (±0.3) | Rise <0.05 | −0.09 (±0.01) | −127 (±14) |
| 1678 | 10.6 (±0.4) | Rise <0.05 | 0.29 (±0.02) | 410 (±28) |
| Decay 1.9 (±0.8) | −0.29 (±0.02) | −410 (±28) | ||
| 1670 | 9.4 (±0.2) | Rise <0.05 | 0.79 (±0.07) | 1117 (±99) |
| Rise 2.0 (±0.2) | 0.31 (±0.08) | 438 (±113) | ||
| 1666 | 8.4 (±0.3) | Rise <0.05 | −0.85 (±0.07) | −1202 (±99) |
| Decay 2.6 (±0.3) | 0.85 (±0.07) | 1202 (±99) | ||
| 1662 | 9.5 (±0.1) | Rise <0.05 | 0.55 (±0.05) | 778 (±71) |
| 1656 | 23.1 (±4.4) | Rise 38.7 (±15.2) | −0.16 (±0.01) | −226 (±14) |
| 1655 | 10.6 (±0.2) | Rise <0.05 | 0.56 (±0.04) | 792 (±57) |
| Decay 1.8 (±0.2) | −0.56 (±0.04) | −792 (±57) | ||
| 1645 | 15.5 (±1.1) | Rise 0.7 (±0.2) | −0.19 (±0.02) | −269 (±28) |
| 1642 | 20.3 (±1.4) | Rise <0.05 | 0.41 (±0.02) | 580 (±28) |
| Decay 82.9 (±11.4) | −0.33 (±0.01) | −467 (±14) | ||
| 1627 | 16.5 (±1.0) | Rise <0.05 | 0.24 (±0.02) | 339 (±28) |
| 1616 | 8.6 (±0.5) | Rise <0.05 | 0.16 (±0.01) | 226 (±14) |
| Decay 14.3 (±2.7) | −0.16 (±0.01) | −226 (±14) | ||
| 1612 | 14.5 (±0.6) | Rise <0.05 | 0.17 (±0.01) | 240 (±14) |
| Decay 1.0 (±0.1) | −0.10 (±0.01) | −141 (±14) | ||
| 1592 | 10.6 (±0.1) | Rise <0.05 | −0.17 (±0.01) | −94 (±6) |
| 1577 | 16.8 (±0.1) | Rise <0.05 | −0.42 (±0.02) | −233 (±11) |
| Decay 2.3 (±0.6) | 0.20 (±0.00) | 111 (±1) | ||
| 1559 | 21.4 (±0.2) | Rise <0.05 | −0.52 (±0.02) | −289 (±11) |
| Decay 12.5 (±5.3) | 0.26 (±0.02) | 144 (±11) | ||
| 1549 | 15.2 (±0.5) | Rise 2.4 (±0.3) | 0.59 (±0.01) | 328 (±6) |
| 1545 | 10.6 (±0.1) | Rise <0.05 | −0.68 (±0.02) | −378 (±11) |
| 1538 | 11.2 (±0.0) | Rise <0.05 | 0.48 (±0.02) | 267 (±11) |
| Decay 3.0 (±0.4) | −0.48 (±0.02) | −267 (±11) | ||
| 1533 | 11.4 (±0.0) | Rise <0.05 | −1.17 (±0.02) | −650 (±11) |
| 1524 | 20.3 (±0.5) | Rise <0.05 | −0.59 (±0.02) | −328 (±11) |
| Decay 1.9 (±0.5) | 0.14 (±0.01) | 78 (±6) | ||
| 1509 | 15.5 (±0.3) | Rise <0.05 | 0.14 (±0.00) | 78 (±1) |
a Full width at half-maximum is given.
b Intensity change is shown.
c Molar extinction coefficient change is as follows: Δϵ = ΔI/([P]·0.8·l); where [P] is the protein concentration used in the experiment; l is the optical path length. The CO photolysis yield (0.8) was taken into account.
IR Spectral Changes in the CO Band Region
Positive CuB-CO and negative Fea3-CO peaks appear at 2063 and at 1965 cm−1, respectively, within 50 ns (Fig. 2A). The assignments of these bands have been given previously (36, 37). The CuB-CO species decays with a time constant of 1.6 ± 0.1 μs, with no recovery of the Fea3-CO species (Fig. 2B), consistent with previous reports (38–40). The integrated areas of these two peaks at 50 ns indicate stoichiometric transfer of CO from Fea3 to CuB upon photolysis.
1655(+)/1666(−) cm−1 Band Pair
The most prominent change in the Amide-I region is the appearance of a peak/trough at 1655(+)/1666(−) cm−1 within 50 ns (Fig. 2C). This feature vanishes with a time constant of 2.2 ± 0.3 μs (Fig. 2D). This signal is assignable to the Amide-I change of the bulge segment in the H-pathway, based on the wave number, intensity, and temporal behavior, as described below.
The only possible side chain that can give a signal at 1655/1666 cm−1 is the guanidino group of Arg (41). However, the contribution from this side chain is unlikely because the guanidino group shows two bands in the Amide-I spectral region: antisymmetric CN stretch with 1652–1695 cm−1 and symmetric CN stretch with 1614–1663 cm−1 (41) with similar intensity (300–500 m−1 cm−1). Their frequencies are known to be positioned higher by salt bridge formation (42). Thus, a salt bridge structural change upon photolysis of CO-bound CcO would provide a simultaneous two-band transition, which is not the present case. Thus, contribution of any guanidino group to the signal is unlikely
It has been reported that an α-helix with partial disorder provides an Amide-I signal with a higher-than-usual wave number (>1660 cm−1) (43, 44), consistent with the reasonable prediction that engagement of the peptide C=O with a hydrogen bond induces a lower wave number shift in the C=O stretching band. Thus, the 1666(−)/1655(+)-cm−1 band transition strongly suggests that bulge structures are eliminated by introduction of additional hydrogen bonds.
The intensity of the band transition suggests that the transition is induced by one C=O moiety or so, as revealed by the following data examinations. The averages of the peak and trough intensities and the positive and negative area intensities for the band transition at 1655(+)/1666(−) cm−1 induced by 0.68 mm CcO (placed in a cell with a path length of 13 μm) are 0.70 (±0.025) mOD and 5.0 (±0.15) mOD cm−1, respectively (under 80% photolysis), which correspond to 0.88 and 6.25 mOD cm−1 under complete photolysis conditions. The reported molar absorption coefficient of the Amide-I band is between 200 and 1000 m−1 cm−1 (35). Thus, the peak intensity of 0.88 mOD (995 OD m−1cm−1) for the band transition at 1655(+)/1666(−) cm−1 after complete photolysis, as described above, suggests that 1–5 C=O stretch bands are involved in the transition. On the other hand, the area intensity corresponds to 0.029% of the absolute area intensity of the Amide-I region (1610–1690 cm−1). Assuming that the absolute area intensity of the CcO sample (21.5 OD cm−1) is only due to Amide-I and that the area intensity of each peptide C=O moiety is independent of the microenvironment, the band area intensity of the transition is expected to be 0.6 of the single C=O stretching band intensity. The absolute spectrum in the 1610–1690-cm−1 region also includes various bands other than the peptide C=O stretching bands, such as bands arising from Arg and Tyr residues. Nevertheless, the experimental value of 0.6 supports the conclusion drawn from the peak intensity described above. Thus, the 1666(−)/1655(+) cm−1 band transition is most likely to be due to a transition in the stretching frequency of at least one C=O moiety.
X-ray structures of bovine CcO indicate that the structural transition related to the bulge conformation is detectable only in the segment extending from Val-380 to Ser-382 in helix X (the trans-membrane α-helix located between the planes of heme a3 and heme a). Ser-382 and Val-380 in the CO-bound and ligand-free reduced states, respectively, are in the bulge conformation of helix X (Fig. 3). Thus, upon CO photolysis, bulge elimination is detectable only at Ser-382. Therefore, the 1666(−)/1655(+) cm−1 band transition is conclusively assignable to bulge elimination at Ser-382. The transition indicates that an intermediate state in which Ser-382 is incorporated in helix X by forming a new hydrogen bond appears before the Val-380 bulge formation.
FIGURE 3.

Structural modeling of the intermediate form detected after photolysis of CO-Fea3. A, side view of the modeled structure of the intermediate form (center), compared with x-ray structures of the reduced (left) and CO-bound (right) forms. The location of the concerned helix structure in the overall proton pumping system in the reduced form is shown with a square. The red dotted surfaces (and gray portions in the left scheme) represent the water cavities identified as spaces with the radii greater than 1.2 Å. The green dotted lines indicate hydrogen bonds. The locations of water pathways are not given for simplicity. B, top stereo view of the modeled structure of the intermediate form (gray), superimposed with x-ray structures of the reduced (blue) and CO-bound (red) forms. The red circle indicates the location of the water cavity that is eliminated by Ser-382 upon CO binding.
The 1666 cm−1 negative band is assignable to the spectral change due to formation of hydrogen bonds to Ser-382 resulting in a lower wave number shift to give the 1655 cm−1 band. Then, the intermediate conformation was transformed to the ligand-free reduced form with the Val-380 bulge upon elimination of hydrogen bonds that exist in the intermediate state.
The segment from Val-380 to Ser-382 has one bulge C=O moiety and two α-helix C=O moieties in both the CO-bound and ligand-free reduced states, as shown in the reported x-ray structures (Fig. 3). Thus, the segment provides an essentially identical Amide-I band in both the states. Consistent with this expectation from the x-ray structures, the 1666(−)/1655(+)-cm−1 band disappears after CO release from CuB.
In the CO-bound form, the Ser-382 bulge feature eliminates the largest water cavity detectable in the ligand-free reduced form as given in Fig. 3 (7). However, the intermediate conformation in which Ser-382 is incorporated into helix X is likely to have a water cavity similar to the cavity detected in the ligand-free reduced state.
Other Spectral Changes in the Amide-I Region
Strong bands in the Amide-I region, other than the 1655(+)/1666(−) cm−1 band pair, are detectable as follows: the bands at 1670 and 1662 cm−1 appear within 50 ns. A 1678-cm−1 band also appears within 50 ns but shifts with the time constant of ∼2 μs to 1670 cm−1, which overlaps with the 1670-cm−1 band appearing within 50 ns (Fig. 2C and Table 1). These bands are likely to arise from CN stretch of Arg (41), C=O stretch of Asn/Gln (41), or C=O stretch of heme side chains (propionate or formyl group) (45–48). However, the conformational changes in these functional groups are too small to be detectable in the x-ray structural analyses at the highest resolution available at present (1.8 Å) (5, 7).
Surface of the Water Cavity Near Ser-382(OH) Group
The surface (or wall) of the cavity near the Ser-382(OH) group, defined by van der Waals radii of the atoms, includes only two negatively polarized atoms (peptide C=O) and one positively polarized atom (peptide N-H) (Fig. 4) (5). The rest of the wall is occupied by 20 nonpolarized carbon atoms, including –CH2–, –CH= of methine bridge of heme a porphyrin, and phenyl groups of Phe residues. The OH group of Ser-382 is located quite close to the wall of the cavity (3.4 Å from the cavity wall) but is not exposed to the cavity space. These structures provide a highly hydrophobic environment in the cavity. These polarized groups are likely to trap water molecules under the hydrophobic (low dielectric) environment inside this space. Furthermore, the hydrophobic environment would promote electrostatic interactions between any protonated water molecules inside the cavity and the polarized Ser-382(OH) group located close to the cavity wall in addition to the peptide C=O and N-H moieties, included in the wall as described above. Thus, these x-ray structures suggest that the protonation state of the water molecule trapped by the peptide N-H and C=O in the hydrophobic environment is electrostatically sensed by the Ser-382(OH) group. The Ser-382(OH) group in the reduced state migrates toward the cavity surface upon CO-binding to Fea3 to eliminate the cavity as shown in Fig. 3. Thus, in the intermediate state, the OH group is expected to be located closer to the wall of the cavity than in the ligand-free reduced state. Thus, the protons of a hydronium ion in the cavity would be stabilized significantly by interacting with the Ser-382(OH) group, to promote proton collection from the negative side of the mitochondrial membrane.
FIGURE 4.
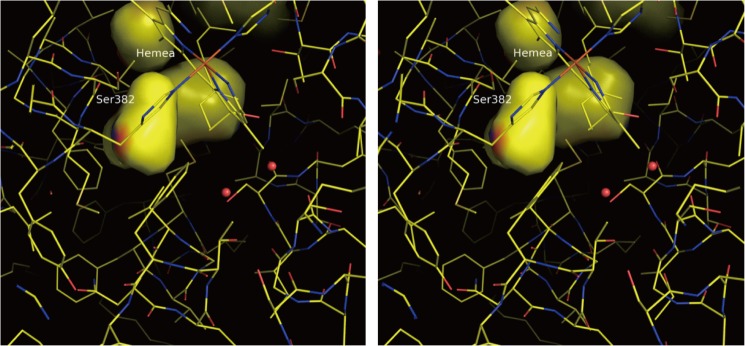
X-ray structure of the water cavity near Ser-382 in the fully reduced state. The stereo drawing is a view from the positive side perpendicular to the membrane surface. The water cavities are drawn on the surfaces calculated by the van der Waals radii of atoms exposed to the cavity spaces. The cavity near Ser-382 is located closest to the positive side among the four cavities seen in this figure. The red and blue areas on the cavity surface are due to the peptide C=O moieties of His-378 and Ser-382 and the peptide N-H of Met-383. The remainder of the surface is yellow and shows the location of nonpolar carbon atoms, including His-378 (Cβ), Ser-382 (Cβ), Met-383 (Cα, Cδ, and Cϵ), Val-386 (Cβ, Cδ1, and Cδ2), Phe-387 (Cδ2 and Cϵ2), Phe-425 (Cϵ1 and Cζ), Met-417(Cϵ), Val-421(Cβ and Cδ1), the heme a plane (2-methyl and a methine bridge), and the hydroxylfarnesylethyl group of heme a (C12, C13, and C14). The Ser-382(OH) group is located close to the cavity surface but does not form part of the cavity surface as described in the text.
Structural Modeling of the Conformational Changes in the Bulge, Revealed by the Present IR Analysis
Possible conformational changes occurring after CO photolysis were preliminarily explored by structural modeling combined with conformational sampling techniques as described under “Experimental Procedures.”
The energy of the system after the breakage of the Fe–CO bond (corresponding to photolysis) decreased almost monotonously in our calculation (i.e. barrierless) until the intermediate was formed. This is not contradictory to the time scale that was observed in the present experiment (< 50 ns). For the subsequent stage, the energy barrier between the intermediate and final states was estimated to be ∼10.2 kcal/mol through our calculations. The order of this value agrees well with that of the time scale observed in the present experiment, ∼2 μs. Here, we adopted the transition state theory to obtain the time scale that is corresponding to the energy barrier (49).
This preliminary analysis suggests that there is a transient stable structure in which Ser-382 in the bulge structure in the CO-bound form is incorporated into helix X to induce the formation of two additional hydrogen bonds without forming the Val-380 bulge (Fig. 3A). (The extensive theoretical analyses for this intermediate state are underway.) This structural change is consistent with the lower wave number shift from 1666 to 1655 cm−1 observed upon CO photolysis (Fig. 2C). The water channel is open in the intermediate state, as expected after interpretation of the results of the present IR analyses and the x-ray structures. However, it is apparent that the open conformation is different from the “open state” in the x-ray structure of the fully reduced CcO (Fig. 3A). This state is therefore designated as the “intermediate state”.
DISCUSSION
The respective time scales of CO dissociation from CuB, the 1666(−)/1655(+)-cm−1 band transition and the previously reported Fea3-His stretch resonance Raman shift (50), essentially coincide with each other (∼2 μs). Ser-382 and His-376 (the latter, the fifth ligand of heme a3) are located within the adjacent two turns of the α-helix of helix X (Fig. 3B). Furthermore, it has been shown that the CO stretch band of CO bound to CuB shifts from 2061 to 2040 cm−1 upon oxidation of Fea3 (51), suggesting that a significant interaction exists between CuB and Fea3 via the bound ligand. These results suggest the existence of a conformational relay system that includes CuB, CO (and thus O2), Fea3, His-376, a segment of two α-helix turns of helix X (from His-376 to Ser-382), and Ser-382.
The present TRIR analyses for CO flash photolysis of CcO indicate that the CuB site, upon O2 binding, induces conformational changes in the relay system to induce “intermediate” conformation in the water cavity. The location of Ser-382 closer to the cavity in the intermediate state than in the “open” state suggests higher proton affinity of the cavity in the former state (as described in Figs. 3 and 4). Thus, CuB upon O2 binding is expected to facilitate effective proton collection.
Ser-382(OH), which is located near the wall of the largest water cavity, is likely to sense the protonation state of the cavity, which is protonically equilibrated with the hydrogen bond network of the H-pathway. Conformational changes in Ser-382, upon sensing the protonation state, would stimulate the relay system to trigger a structural change in the O2 reduction site giving higher O2 affinity of Fea3 relative to CuB. Then, O2 binding to Fea3 triggers conformational changes in the relay system to eliminate the water cavity by forming the Ser-382 bulge, giving timely closure of the water channel.
Collection of four proton equivalents at once to the hydrogen bond network of the H-pathway is unlikely, because the water cavity does not have enough space for keeping four proton equivalents. Furthermore, existence of a possible O2 storage structure, located near the CuB site, as described below, suggests a reversible (or repetitive) O2 binding to CuB, coupled with the open to intermediate conformational transition in the cavity. These two structures (the narrow water cavity and the possible O2 storage structure) support the consecutive proton collection.
X-ray structures of bovine and bacterial CcOs indicate that a branch in the O2 pathway is detectable near the O2 reduction site between the two hemes (3, 52). The walls of both the branch and the O2 pathway are composed of highly hydrophobic residues. The branch also has enough space for O2 storage. No significant electron density peak is detectable in the interior spaces of the branch as well as the O2 pathway in the fully reduced state of CcO. However, it has been proposed that protons used in the proton-pumping process are transferred through the branch from Glu-242, assuming a water array inside the branch and the pathway (53, 54). Nevertheless, the structure of the branch, as described above, strongly suggests the O2 accepting function. Thus, the branch is expected to store the O2 molecule released from CuB to efficiently induce the repetitive formation of the intermediate state in the consecutive proton collection.
A more comprehensive description of the mechanisms for proton collection and timely closure of the water channel is given in Fig. 5. In the fully reduced CcO [Fea32+,CuB1+] under turnover conditions after the last proton-pumping step in the previous turnover, the water channel is in the open state, and the hydrogen bond network is fully deprotonated (Fig. 5A). In this conformation, CuB traps O2, which enters through the O2 pathway (55) (or from an O2 storage area located near the O2 reduction site (1)). The initial O2 binding to CuB and not to Fea3 is supported by the CO release from CuB without rebinding to Fea3 as revealed by TRIR analyses. The TRIR results indicate that Fea32+ before complete protonation of the hydrogen bond network of the H-pathway has essentially no affinity to O2.
FIGURE 5.
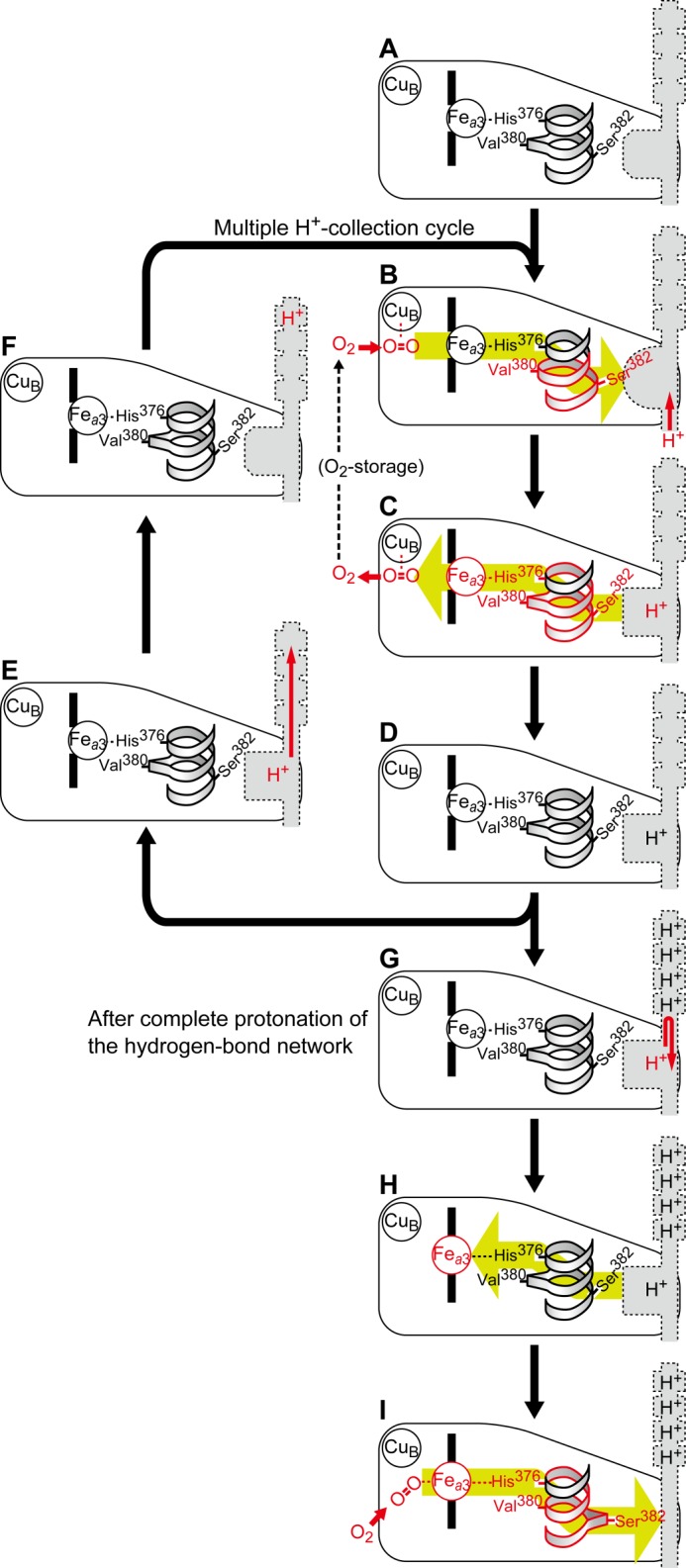
Schematic representation of the function of the conformational relay between CuB and Ser-382. The gray structures indicate a schematic representation of the hydrogen bond network and the water cavity detectable in the reduced state of the proton-pumping system. The green arrows indicate the direction of propagation of the conformational changes. The bulge conformation is indicated by the protruded shape of the helix ribbon. Three types of water cavity conformations are indicated by the shape of the water cavity. The CO-bound, fully reduced form used for the present experiments is the fully protonated CO-bound form that corresponds to I, because the hydrogen bond network is protonically equilibrated with the bulk aqueous phase before initiation of the CO photolysis experiments. Thus, the form that is generated after flash photolysis corresponds to the intermediate state (B) with the fully protonated hydrogen bond network. Then, a proton is taken up in the cavity, which releases CO from CuB. After that, the fully protonated and fully reduced form (H) is generated, which is ready to receive CO (and O2) and corresponds to the fully reduced form obtainable by reduction of the purified preparation.
The bound O2 triggers the conformational change in the water cavity with the relay system from CuB to Ser-382 to provide the intermediate conformation of the cavity (Fig. 5B). The conformational change from open to intermediate accelerates the rate of entry of a proton into the cavity from the negative side. Once a proton is incorporated into the cavity, the conformation of the cavity returns to the open state. The protonated open state (Fig. 5C) influences the O2-binding affinity of CuB through the relay system to trigger the release of O2 from CuB without rebinding to Fea3. The resulting protonated open state without O2 at CuB (Fig. 5D) is supported by the present TRIR results indicating the CO release from CuB (without rebinding to Fea3) coupled with the transition from the intermediate state to the open state of the cavity.
The cavity in the open state has weaker proton affinity than in the intermediate state. Thus, the proton in the cavity is readily taken up by the empty hydrogen bond network (Fig. 5E), thereby regenerating the deprotonated open state (Fig. 5F). The highly hydrophobic and fairly narrow structure of the cavity space (revealed by the x-ray structure, Fig. 4) indicates that the conformational change in the cavity upon deprotonation is a reasonable proposal. This state is ready to start another proton collection cycle by receiving O2 transferred from the O2 storage area (Fig. 5, B–D).
When the hydrogen bond network becomes saturated with 4 eq of protons, a proton in the cavity cannot be extracted by the hydrogen bond network (Fig. 5G). The increase in the protonation level is sensed by Ser-382, which triggers a conformational change at the Fea3 site using the relay system to increase the O2 affinity of the Fea3 site (Fig. 5H). For formation of the O2-bound form (Fig. 5I), the O2 affinity of CuB is lowered by the relay system as in the case of the O2 release step from CuB (Fig. 5C). In other words, CuB also contributes to the channel closure. The O2 binding eliminates the water cavity by the conformational changes in the relay system (by the Ser-382 bulge formation) (Fig. 5I) to close the water channel.
For complete protonation of the hydrogen bond network, it is critical that the protonated cavity induces the increase in O2 affinity of Fea3 at a controlled rate (Fig. 5, G and H). If the O2 affinity increase is faster than the rate of proton transfer from the cavity to the hydrogen bond network, the channel would close before complete protonation of the hydrogen bond network is attained. The kinetic requirement has not been experimentally proven, although various mechanisms are possible, for example, for control of the interaction between the water cavity and Ser-382.
The initial intermediate of the O2 reduction process by this enzyme is an oxygenated form (Fea32+-O2) as illustrated in Fig. 5I (1). The electron transfer process from cytochrome c via CuA and heme a is coupled with proton pumping (1, 11). The O2 reduction site in the oxygenated state does not receive electrons from cytochrome c, but it does after reduction of the bound O2, initiated by the electron transfer from CuB1+ (7). If the channel closure induced by the O2 binding to Fea3 is not sufficiently fast, the electron transfer to the O2 reduction site from cytochrome c, coupled with inefficient proton pumping in the open state of the water channel, would occur before the channel closure. To ensure that the O2 reduction occurs after the channel closure, CuB must sense the channel closure through the relay system (from Ser-382 to CuB) before the electron donation to the O2 at Fea32+. This sensing of the channel closure by CuB via the relay system is not included in Fig. 5 for the sake of simplicity.
After flash photolysis of CO-bound CcO, CO is transiently bound to CuB and released without rebinding to Fea3. The absence of CO rebinding after the CO release from CuB indicates that Fea3 has essentially no affinity for CO. The x-ray structures do not indicate the presence of any amino acid residue that could block the CO rebinding, as has been suggested (38). Thus, the affinity of Fea3 for CO is expected to be controlled by the coordination structure of the fifth ligand of heme a3, His-376, in the relay system. A subtle structural change in the coordination structure of the heme iron could greatly influence the ligand affinity as in the case of O2 affinity of hemoglobin (56, 57).
Except for the redox property of CuB as a single electron accepting site, the chemical properties (or functions) of the copper site have been essentially unknown, because the site is spectrally quite inert. In fact, electronic absorption of the site is completely masked by the strong absorption of the two hemes. The cupric state of CuB is EPR-silent because of the magnetic coupling with the ferric Fea3. This study has revealed a critical role of CuB in the proton pumping function of this enzyme for efficient proton collection and timely closure of the water channel. The role has never been proposed until this study, although CO binding to CuB was discovered by FTIR analysis 32 years ago (37).
The critical contribution of the newly developed TRIR system to the present unexpected findings is obvious. X-ray structural analysis of a protein is the most powerful for determination of the three-dimensional arrangements of atoms located in the functional site of the protein. However, the picture provided by an x-ray structure does not indicate the dynamic aspects of each of the atoms in the functional site. Thus, TRIR analyses of the functional site are indispensable for elucidation of the mechanism of any protein function. Our system provides a uniquely powerful strategy for elucidation of the mechanism of any protein function under physiological (i.e. aqueous) conditions.
Understanding of the functional mechanism of the H-pathway as the proton-pumping system has been improved significantly by this work. Proton pumping systems, including the D or K pathways instead of the H-pathway, have been proposed based on mutagenesis analyses for bacterial enzymes, as described in the Introduction. The present results do not improve the proposed mechanisms, including the K or D pathways, because the conformational changes in helix X provide no direct structural influence on either the K or D pathways. Furthermore, none of the two pathways is likely to pump protons in the bovine enzyme, consistent with a preliminary mutational result that indicates no involvement of the bovine D pathway in proton pumping (58). The H-pathway structures and functions are not conserved well between bovine and bacterial enzymes. Thus, it is not clear whether the present IR results are common between these enzymes.
This work was supported by a grant-in-aid from the Global Center of Excellence Program (to S. Y.), the Targeted Protein Research Program (to S. Y. and T. O.), Scientific Research Grant (A) 2247012 (to S. Y.), Young Scientists Grants (A) 23685040 and (B) 21750022 (to M. K.) and (B) 22770154 (to S. Y.), the Japanese Ministry of Education, Culture, Sports, Science and Technology, the Japan Science and Technology Agency, PRESTO and CREST.
- CcO
- cytochrome c oxidase
- TRIR
- time-resolved infrared.
REFERENCES
- 1. Yoshikawa S., Muramoto K., Shinzawa-Itoh K. (2011) Proton-pumping mechanism of cytochrome c oxidase. Annu. Rev. Biophys. 40, 205–223 [DOI] [PubMed] [Google Scholar]
- 2. Yoshikawa S., Shinzawa-Itoh K., Nakashima R., Yaono R., Yamashita E., Inoue N., Yao M., Fei M.J., Libeu C.P., Mizushima T., Yamaguchi H., Tomizaki T., Tsukihara T. (1998) Redox-coupled crystal structural changes in bovine heart cytochrome c oxidase. Science 280, 1723–1729 [DOI] [PubMed] [Google Scholar]
- 3. Tsukihara T., Shimokata K., Katayama Y., Shimada H., Muramoto K., Aoyama H., Mochizuki M., Shinzawa-Itoh K., Yamashita E., Yao M., Ishimura Y., Yoshikawa S. (2003) The low-spin heme of cytochrome c oxidase as the driving element of the proton-pumping process. Proc. Natl. Acad. Sci. U.S.A. 100, 15304–15309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shimokata K., Katayama Y., Murayama H., Suematsu M., Tsukihara T., Muramoto K., Aoyama H., Yoshikawa S., Shimada H. (2007) The proton pumping pathway of bovine heart cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A. 104, 4200–4205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Muramoto K., Hirata K., Shinzawa-Itoh K., Yoko-o S., Yamashita E., Aoyama H., Tsukihara T., Yoshikawa S. (2007) A histidine residue acting as a controlling site for dioxygen reduction and proton pumping by cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A. 104, 7881–7886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aoyama H., Muramoto K., Shinzawa-Itoh K., Hirata K., Yamashita E., Tsukihara T., Ogura T., Yoshikawa S. (2009) A peroxide bridge between Fe and Cu ions in the O2 reduction site of fully oxidized cytochrome c oxidase could suppress the proton pump. Proc. Natl. Acad. Sci. U.S.A. 106, 2165–2169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Muramoto K., Ohta K., Shinzawa-Itoh K., Kanda K., Taniguchi M., Nabekura H., Yamashita E., Tsukihara T., Yoshikawa S. (2010) Bovine cytochrome c oxidase structures enable O2 reduction with minimization of reactive oxygens and provide a proton-pumping gate. Proc. Natl. Acad. Sci. U.S.A. 107, 7740–7745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee H. M., Das T. K., Rousseau D. L., Mills D., Ferguson-Miller S., Gennis R. B. (2000) Mutations in the putative H-channel in the cytochrome c oxidase from Rhodobacter sphaeroides show that this channel is not important for proton conduction but reveal modulation of the properties of heme a. Biochemistry 39, 2989–2996 [DOI] [PubMed] [Google Scholar]
- 9. Konstantinov A. A., Siletsky S., Mitchell D., Kaulen A., Gennis R. B. (1997) The roles of the two proton input channels in cytochrome c oxidase from Rhodobacter sphaeroides probed by the effects of site-directed mutations on time-resolved electrogenic intraprotein proton transfer. Proc. Natl. Acad. Sci. U.S.A. 94, 9085–9090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chang H. Y., Hemp J., Chen Y., Fee J. A., Gennis R. B. (2009) The cytochrome ba3 oxygen reductase from Thermus thermophilus uses a single input channel for proton delivery to the active site and for proton pumping. Proc. Natl. Acad. Sci. U.S.A. 106, 16169–16173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bloch D., Belevich I., Jasaitis A., Ribacka C., Puustinen A., Verkhovsky M. I., Wikström M. (2004) The catalytic cycle of cytochrome c oxidase is not the sum of its two halves. Proc. Natl. Acad. Sci. U.S.A. 101, 529–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Venyaminov S. Yu., Prendergast F. G. (1997) Water (H2O and D2O) molar absorptivity in the 1000–4000 cm−1 range and quantitative infrared spectroscopy of aqueous solutions. Anal. Biochem. 248, 234–245 [DOI] [PubMed] [Google Scholar]
- 13. Mochizuki M., Aoyama H., Shinzawa-Itoh K., Usui T., Tsukihara T., Yoshikawa S. (1999) Quantitative reevaluation of the redox active sites of crystalline bovine heart cytochrome c oxidase. J. Biol. Chem. 274, 33403–33411 [DOI] [PubMed] [Google Scholar]
- 14. Ansari A., Szabo A. (1993) Theory of photoselection by intense light pulses. Influence of reorientational dynamics and chemical kinetics on absorbance measurements. Biophys. J. 64, 838–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ansari A., Jones C. M., Henry E. R., Hofrichter J., Eaton W. A. (1993) Photoselection in polarized photolysis experiments on heme proteins. Biophys. J. 64, 852–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Case D. A., Cheatham T. E., 3rd, Darden T., Gohlke H., Luo R., Merz K. M., Jr., Onufriev A., Simmerling C., Wang B., Woods R. J. (2005) The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kang J., Kino H., Tateno M. (2011) A theoretical investigation of the functional role of the axial methionine ligand of the CuA site in cytochrome c oxidase. Biochim. Biophys. Acta 1807, 1314–1327 [DOI] [PubMed] [Google Scholar]
- 18. Bayly C. I., Cieplak P., Cornell W., Kollman P. A. (1993) A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 97, 10269–10280 [Google Scholar]
- 19. Beneš P., Chovancová E., Kozlíková B., Pavelka A., Strnad O., Brezovský J., Šustr V., Klvaňa M., Szabó T., Gora A., Zamborský M., Biedermannová L., Medek P., Damborský J., Sochor J. (2010) CAVER 2.1, CaverSoft, Brno, Czech Republic [Google Scholar]
- 20. Locke B., Diller R., Hochstrasser R. M. (1993) Advances in Spectroscopy (Clark R. J. H., Hester R. E., eds) Vol. 21, Biomolecular Spectroscopy, Part B, pp. 1–47, Wiley, New York [Google Scholar]
- 21. Hamm P., Zurek M., Mäntele W., Meyer M., Scheer H., Zinth W. (1995) Femtosecond infrared spectroscopy of reaction centers from Rhodobacter sphaeroides between 100 and 1800 cm−1. Proc. Natl. Acad. Sci. U.S.A. 92, 1826–1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Slayton R. M., Anfinrud P. A. (1997) Time-resolved mid-infrared spectroscopy: methods and biological applications. Curr. Opin. Struct. Biol. 7, 717–721 [DOI] [PubMed] [Google Scholar]
- 23. Groot M. L., van Wilderen L. J., Di Donato M. (2007) Time-resolved methods in biophysics. 5. Femtosecond time-resolved and dispersed infrared spectroscopy on proteins. Photochem. Photobiol. Sci. 6, 501–507 [DOI] [PubMed] [Google Scholar]
- 24. Treuffet J., Kubarych K. J., Lambry J. C., Pilet E., Masson J. B., Martin J. L., Vos M. H., Joffre M., Alexandrou A. (2007) Direct observation of ligand transfer and bond formation in cytochrome c oxidase by using mid-infrared chirped-pulse upconversion. Proc. Natl. Acad. Sci. U.S.A. 104, 15705–15710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. van Wilderen L. J., van der Horst M. A., van Stokkum I. H., Hellingwerf K. J., van Grondelle R., Groot M. L. (2006) Ultrafast infrared spectroscopy reveals a key step for successful entry into the photocycle for photoactive yellow protein. Proc. Natl. Acad. Sci. U.S.A. 103, 15050–15055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Waegele M. M., Gai F. (2010) Infrared study of the folding mechanism of a helical hairpin: Porcine PYY. Biochemistry 49, 7659–7664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nagarajan S., Taskent-Sezgin H., Parul D., Carrico I., Raleigh D. P., Dyer R. B. (2011) Differential ordering of the protein backbone and side chains during protein folding revealed by site-specific recombinant infrared probes. J. Am. Chem. Soc. 133, 20335–20340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Palmer R. A., Manning C. J., Rzepiela J. A., Widder J. M., Chao J. L. (1989) Time-resolved spectroscopy using step-scan Fourier transform interferometry. Appl. Spectrosc., 43, 193–195 [Google Scholar]
- 29. Uhmann W., Becker A., Taran C., Siebert F. (1991) Time-resolved spectroscopy FT-IR absorption spectroscopy using a step-scan interferometer. Appl. Spectrosc. 45, 390–397 [Google Scholar]
- 30. Kötting C., Gerwert K. (2005) Proteins in action monitored by time-resolved FTIR spectroscopy. ChemPhysChem 6, 881–888 [DOI] [PubMed] [Google Scholar]
- 31. Radu I., Schleeger M., Bolwien C., Heberle J. (2009) Time-resolved methods in biophysics. 10. Time-resolved FTIR difference spectroscopy and the application to membrane proteins. Photochem. Photobiol. Sci. 8, 1517–1528 [DOI] [PubMed] [Google Scholar]
- 32. Magana D., Parul D., Dyer R. B., Shreve A. P. (2011) Implementation of time-resolved step-scan Fourier transform infrared (FT-IR) spectroscopy using a kHz repetition rate pump laser. Appl. Spectrosc 65, 535–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Salomonsson L., Faxén K., Adelroth P., Brzezinski P. (2005) The timing of proton migration in membrane-reconstituted cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A. 102, 17624–17629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kandori H. (2000) Role of internal water molecules in bacteriorhodopsin. Biochim. Biophys. Acta 1460, 177–191 [DOI] [PubMed] [Google Scholar]
- 35. Venyaminov S. Yu, Kalnin N. N. (1990) Quantitative IR spectrophotometry of peptide compounds in water (H2O) solutions. II. Amide absorption bands of polypeptides and fibrous proteins in α-, β-, and random coil conformations. Biopolymers 30, 1259–1271 [DOI] [PubMed] [Google Scholar]
- 36. Yoshikawa S., Choc M. G., O'Toole M. C., Caughey W. S. (1977) An infrared study of CO binding to heart cytochrome c oxidase and hemoglobin A. J. Biol. Chem. 252, 5498–5508 [PubMed] [Google Scholar]
- 37. Alben J. O., Moh P. P., Fiamingo F. G., Altschuld R. A. (1981) Cytochrome oxidase (a3) heme and copper observed by low-temperature Fourier transform infrared spectroscopy of the CO complex. Proc. Natl. Acad. Sci. U.S.A. 78, 234–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Einarsdóttir O., Dyer R. B., Lemon D. D., Killough P. M., Hubig S. M., Atherton S. J., López-Garriga J. J., Palmer G., Woodruff W. H. (1993) Photodissociation and recombination of carbonmonoxy cytochrome oxidase: Dynamics from picoseconds to kiloseconds. Biochemistry 32, 12013–12024 [DOI] [PubMed] [Google Scholar]
- 39. Dyer R. B., Peterson K. A., Stoutland P. O., Woodruff W. H. (1994) Picosecond infrared study of the photodynamics of carbonmonoxy-cytochrome c oxidase. Biochemistry 33, 500–507 [DOI] [PubMed] [Google Scholar]
- 40. Koutsoupakis C., Pinakoulaki E., Stavrakis S., Daskalakis V., Varotsis C. (2004) Time-resolved step-scan Fourier transform infrared investigation of heme-copper oxidases: implications for O2 input and H2O/H+ output channels. Biochim. Biophys. Acta 1655, 347–352 [DOI] [PubMed] [Google Scholar]
- 41. Barth A. (2007) Infrared spectroscopy of proteins. Biochim. Biophys. Acta 1767, 1073–1101 [DOI] [PubMed] [Google Scholar]
- 42. Braiman M. S., Briercheck D. M., Kriger K. M. (1999) Modeling vibrational spectra of amino acid side chains in proteins: effects of protonation state, counterion, and solvent on arginine C-N stretch frequencies. J. Phys. Chem. B 103, 4744–4750 [Google Scholar]
- 43. Torii H., Tasumi M. (1992) Model calculations on the amide-I infrared bands of globular proteins. J. Chem. Phys. 96, 3379–3387 [Google Scholar]
- 44. Torii H., Tasumi M. (1992) Application of the three-dimensional doorway-state theory to analyses of the amide-I infrared bands of globular proteins. J. Chem. Phys. 97, 92–98 [Google Scholar]
- 45. Babcock G. T. (1988) in Biological Applications of Raman Spectroscopy (Spiro T. G., ed) Vol. 3, pp. 293–346, Wiley, New York [Google Scholar]
- 46. Behr J., Hellwig P., Mäntele W., Michel H. (1998) Redox dependent changes at the heme propionates in cytochrome c oxidase from Paracoccus denitrificans: direct evidence from FTIR difference spectroscopy in combination with heme propionate 13C labeling. Biochemistry 37, 7400–7406 [DOI] [PubMed] [Google Scholar]
- 47. Hellwig P., Grzybek S., Behr J., Ludwig B., Michel H., Mäntele W. (1999) Electrochemical and ultraviolet/visible/infrared spectroscopic analysis of heme a and a3 redox reactions in the cytochrome c oxidase from Paracoccus denitrificans: separation of heme a and a3 contributions and assignment of vibrational modes. Biochemistry 38, 1685–1694 [DOI] [PubMed] [Google Scholar]
- 48. Behr J., Michel H., Mäntele W., Hellwig P. (2000) Functional properties of the heme propionates in cytochrome c oxidase from Paracoccus denitrificans: evidence from FTIR difference spectroscopy and site-directed mutagenesis. Biochemistry 39, 1356–1363 [DOI] [PubMed] [Google Scholar]
- 49. Siegbahn P. E., Blomberg M. R. (2007) Energy diagrams and mechanism for proton pumping in cytochrome c oxidase. Biochim. Biophys. Acta 1767, 1143–1156 [DOI] [PubMed] [Google Scholar]
- 50. Findsen E. W., Centeno J., Babcock G. T., Ondrias M. R. (1987) Cytochrome a3 hemepocket relaxation subsequent to ligand photolysis from cytochrome oxidase. J. Am. Chem. Soc. 109, 5367–5372 [Google Scholar]
- 51. Okuno D., Iwase T., Shinzawa-Itoh K., Yoshikawa S., Kitagawa T. (2003) FTIR detection of protonation/deprotonation of key carboxyl side chains caused by redox change of the CuA-heme a moiety and ligand dissociation from the heme a3-CuB center of bovine heart cytochrome c oxidase. J. Am. Chem. Soc. 125, 7209–7218 [DOI] [PubMed] [Google Scholar]
- 52. Qin L., Liu J., Mills D. A., Proshlyakov D. A., Hiser C., Ferguson-Miller S. (2009) Redox dependent conformational changes in cytochrome c oxidase suggests a gating mechanism for proton uptake. Biochemistry 48, 5121–5130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Brzezinski P., Larsson G. (2003) Redox-driven proton pumping by heme-copper oxidases. Biochim. Biophys. Acta 1605, 1–13 [DOI] [PubMed] [Google Scholar]
- 54. Wikström M., Verkhovsky M. I., Hummer G. (2003) Water-gated mechanism of proton translocation by cytochrome c oxidase. Biochim. Biophys. Acta 1604, 61–65 [DOI] [PubMed] [Google Scholar]
- 55. Shinzawa-Itoh K., Aoyama H., Muramoto K., Terada H., Kurauchi T., Tadehara Y., Yamasaki A., Sugimura T., Kurono S., Tsujimoto K., Mizushima T., Yamashita E., Tsukihara T., Yoshikawa S. (2007) Structures and physiological roles of 13 integral lipids of bovine heart cytochrome c oxidase. EMBO J. 26, 1713–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nagai K., Kitagawa T., Morimoto H. (1980) Quaternary structures and low frequency molecular vibrations of haems of deoxy and oxyhaemoglobin studied by resonance Raman scattering. J. Mol. Biol. 136, 271–289 [DOI] [PubMed] [Google Scholar]
- 57. Matsukawa S., Mawatari K., Yoneyama Y., Kitagawa T. (1985) Correlation between the iron-histidine stretching frequencies and oxygen affinity of hemoglobins. A continuous strain model. J. Am. Chem. Soc. 107, 1108–1113 [Google Scholar]
- 58. Aminaka R., Itoh M., Shimokata K., Katayama Y., Tsukihara T., Yoshikawa S., Shimada H. (2012) Mutational analyses of D-pathway of bovine heart cytochrome c oxidase suggest that the pathway does not transfer the pumping protons. Biochim. Biophys. Acta 1817, S104 [Google Scholar]