

Background: Ricin A chain (RTA) uses the ribosomal stalk to access the sarcin/ricin loop (SRL).
Results: Arginine residues at the interface of RTB are critical for RTA to bind to the stalk and to stimulate depurination of the SRL.
Conclusion: Stalk binding stimulates depurination by orienting RTA toward the SRL.
Significance: We propose a model that describes how RTA accesses the SRL.
Keywords: Protein Synthesis, Ribosomal RNA (rRNA), Ribosomes, RIP, Toxins, Ribosome-inactivating Protein, Ricin
Abstract
Ricin inhibits protein synthesis by depurinating the α-sarcin/ricin loop (SRL). Ricin holotoxin does not inhibit translation unless the disulfide bond between the A (RTA) and B (RTB) subunits is reduced. Ricin holotoxin did not bind ribosomes or depurinate them but could depurinate free RNA. When RTA is separated from RTB, arginine residues located at the interface are exposed to the solvent. Because this positively charged region, but not the active site, is blocked by RTB, we mutated arginine residues at or near the interface of RTB to determine if they are critical for ribosome binding. These variants were structurally similar to wild type RTA but could not bind ribosomes. Their Km values and catalytic rates (kcat) for an SRL mimic RNA were similar to those of wild type, indicating that their activity was not altered. However, they showed an up to 5-fold increase in Km and up to 38-fold decrease in kcat toward ribosomes. These results suggest that the stalk binding stimulates the catalysis of ribosome depurination by RTA. The mutated arginines have side chains behind the active site cleft, indicating that the ribosome binding surface of RTA is on the opposite side of the surface that interacts with the SRL. We propose that stalk binding stimulates the catalysis of ribosome depurination by orienting the active site of RTA toward the SRL and thereby allows docking of the target adenine into the active site. This model may apply to the translation factors that interact with the stalk.
Introduction
Ricin derived from the castor bean seeds is a type 2 ribosome-inactivating protein (RIP),2 composed of a catalytically active A subunit (RTA) covalently linked to a cell-binding B subunit (RTB) through a disulfide bond. Because of its potent toxicity, ricin is classified as a category B bioterrorism agent (1). Currently, no United States Food and Drug Administration-approved vaccines or therapeutics exist to protect against ricin, Shiga toxins (Stxs), or any other RIP. RTB binds to glycoproteins or glycolipids on the cell surface and facilitates transport of RTA into the cell. RTA enters the cytosol after the disulfide bond between RTA and RTB is reduced (2) and cleaves a specific adenine (A4324 in rat, A3027 in yeast and A2660 in bacteria) from the α-sarcin/ricin loop (SRL) of the large rRNA (3). The depurination of the SRL inhibits protein synthesis at the elongation step (4–7). Depurination of A2660 of the bacterial SRL has been shown to inhibit the GTPase activity of elongation factor G (8). However, another study showed that a point mutation at A2660 did not affect GTP hydrolysis but affected anchoring of elongation factor G to the ribosome during translocation (9). Although very toxic to eukaryotic cells, ricin holotoxin is not active in cell-free protein translation systems (5, 10, 11). RTA is activated only after it is separated from RTB by reduction of the disulfide bond (10). It is not known why RTA in the holotoxin is inactive despite the fact that it has the same conformation as free RTA with its active site cleft exposed on the surface (12).
Although the SRL is nearly universally conserved, the depurination activity of RTA differs among ribosomes isolated from different organisms. RTA is most active toward mammalian ribosomes and less active against plant and yeast ribosomes, and it cannot depurinate bacterial ribosomes (6, 13). RTA can bind to 60 S rat liver ribosomes but not ribosomes from Escherichia coli (14, 15). However, RTA shows similar activity when rRNA is used as the substrate, although the activity against rat rRNA is about 5 orders of magnitude lower than against intact rat ribosomes (6). In contrast to ricin, other RIPs, such as Stx and pokeweed antiviral protein, are equally active on eukaryotic and prokaryotic ribosomes (16). These studies suggested that ricin and related RIPs may require docking to ribosomal proteins to maintain an optimal depurination rate (17). We showed that RTA binds to a component of the large ribosomal subunit known as the ribosomal P-protein stalk to depurinate the SRL in yeast (18, 19) and in human cells (20). RTA interacts directly with isolated in vivo assembled P-protein stalk complexes from yeast (21).
The ribosomal stalk is a lateral protuberance of the large ribosome subunit, which recruits elongation factor 2 and other GTPase factors to the ribosome and stimulates factor-dependent GTP hydrolysis during translation (22, 23). In eukaryotes, it forms a pentameric structure, consisting of a P0 protein, which anchors two P1-P2 heterodimers (24, 25). The unique feature of all P-proteins is the C-terminal 11 amino acids, which are identical in all eukaryotes and are probably involved in direct interaction with the translation factors (26–28). The mechanism of their interaction with the translation factors is not well understood. Previous studies showed that trichosanthin (TCS), a single chain RIP (29), and the A1 chain of Shiga toxin 1 (Stx1) (30, 31) interact with the conserved CTD fragment of P0, P1, and P2. A recent solution structure of the full-length P1/P2 heterodimer showed a helical N-terminal domain and an unstructured C-terminal tail, which is required for the depurination activity of TCS (32). The structure of a peptide corresponding to the last 11 amino acids of the stalk proteins in a complex with TCS has been determined (33). According to this structure, the acidic amino acids at the amino end of the peptide interact with the positively charged Lys173, Arg174, and Lys177 of TCS, whereas the hydrophobic part of the carboxyl end of the peptide is inserted into a hydrophobic pocket of TCS (33). The amino acids that interact with P2 protein are located in a different region of the maize RIP than in TCS and differ in primary sequence and electrostatic distribution (34). It has been suggested that the ability to interact with the stalk arose independently by convergent evolution (35). Kinetic analysis of binding showed that five identical C termini of the stalk proteins increase the association rate of the interaction between RTA and the stalk (21). Moreover, RTA may undergo a conformational change upon depurination (36). These results suggest that the interaction of RTA with the stalk is a dynamic process, which cannot be fully described by x-ray structure analysis.
Residues involved in ribosome binding of RTA have not been identified. Chemical modification analysis showed that RTA lost its activity in cell-free protein synthesis when only a few arginines were modified by phenylglyoxal and that this inactivation was reversible (37). Modification of arginines at positions 193, 196, 213, and 234/235, which are mainly located on the opposite side of the active site cleft, decreased the rate of depurination and the affinity for the ribosome without causing a detectable change in the conformation of the catalytic site (38). Deletion analysis showed that Arg193, Arg196, and Arg197 were important for the activity of RTA in vitro (39). However, chemical modification analysis could not distinguish between the roles of these residues in substrate binding versus catalytic activity and did not provide direct evidence for their involvement in substrate binding. Most of the surface residues of RTA were thought to be directly or indirectly involved in the interaction of RTA with ribosomes, and substitution of only a few residues was thought not to have a sufficient effect on the interaction (39).
In the present study, we made point mutations at arginines at or near the interface of RTA and RTB and determined the effect of these mutations on ribosome binding and catalytic activity of RTA. Our findings identify for the first time RTA residues critical for binding to the P-protein stalk but not for catalytic activity and show that the ribosome binding surface of RTA is on the opposite side of the active site cleft. These results provide novel information about the molecular action of RTA and may be applicable to translation factors that interact with ribosomes.
EXPERIMENTAL PROCEDURES
RIP Structure and Sequence Analysis
All crystallographic structures were downloaded from the Protein Data Bank (PDB) and are displayed with PyMOL (The PyMOL Molecular Graphics System, version 1.5.0.4, Schrödinger, LLC, New York).
RTA Mutagenesis
Point mutations at arginine residues were introduced into pre-RTA, which contains the mature RTA and the 35-residue N-terminal leader in pBluescript using PCR-based mutagenesis (40). Each mutation was confirmed by DNA sequence analysis. The wild type (NT849) and mutated RTA genes (NT1037 for G212E, NT1360 for R189A/R234A, NT1361 for R191A/R196A, and NT1362 for R193A/R235A) were cloned into the yeast expression vector, NT198 under the control of the GAL1 promoter, containing the LEU2 selectable marker. The Saccharomyces cerevisiae strain W303 (MATa ade2-1 trp1-1 ura3-1 leu2-3,112 his-3-11,15 can1-100) was transformed with each construct and the empty vector.
Cytotoxicity of RTA Variants in Yeast
To compare the cytotoxicity of each mutant with that of wild type RTA, yeast cells transformed with RTA constructs were grown overnight in synthetic medium with 2% glucose. The cells were spun down and then resuspended in synthetic medium containing 2% galactose to A600 of 0.3 to induce RTA expression. At different time points after induction, 50 μl of cells were sampled and normalized to A600 of 0.1. Four serial dilutions (1:10) were made, and 8 μl of each dilution were spotted on SD−Leu plates containing 2% glucose. To measure the growth rates, 50 μl of cells grown overnight were pipetted into each well of a 24-well plate containing 700 μl of SD−Leu medium with 2% galactose. After sealing the plates with a semipermeable membrane, they were incubated in a BioTeK Synergy 4 microplate reader (Winooski, VT) at 30 °C with constant shaking. The A600 was recorded every 30 min up to 30 h.
RTA Expression in Yeast
To analyze RTA expression, total protein was extracted from yeast as described by Zhang et al. (41). Yeast cells (A600 of 5) were harvested at 4 and 6 h postinduction (hpi) and were first suspended in 0.5 ml of 2 m lithium acetate for 5 min on ice. Next, they were spun down at 6000 × g and resuspended in 0.5 ml of 0.4 m NaOH for 5 min on ice. The cells were spun down again at 6000 × g and then neutralized with 0.5 ml of 100 mm Tris-HCl, pH 6.8. Cells were suspended in 100 μl of 2× SDS sample buffer and heated at 95 °C for 5 min. The extracts were spun at 16,000 × g for 10 min, and the supernatants were collected. The supernatants from A600 of 0.4 or A600 of 0.8 were separated on a 12% SDS-polyacrylamide gel. After transfer to nitrocellulose, RTA was detected with monoclonal antibody PB10 (42, 43). Monoclonal antibodies against phosphoglycerate kinase and dolichoylphosphate mannosyltransferase 1 (Dpm1) from Invitrogen were used as loading controls for cytosol and membranes, respectively.
Recombinant RTA Expression and Purification
Wild type RTA (NT1430) and RTA variants (NT1434 for G212E, NT1484 for R189A/R234A, and NT1414 for R193A/R235A) were cloned into the E. coli expression vector, pET28, with an N-terminal His10 followed by the mature RTA. Wild type RTA and His10-tagged enhanced green fluorescent protein (EGFP) were purified using nickel-nitrilotriacetic acid-agarose from Qiagen (Valencia, CA), and RTA variants were purified by the Northeast Biodefense Center protein expression core facility.
Proteinase Digestion
Glu-C and Tosylphenylalanyl Chloromethyl Ketone (TPCK)-treated trypsin were obtained from New England Biolabs (Ipswich, MA). N-α-p-Tosyl-l-lysine chloromethyl ketone (TLCK)-treated chymotrypsin was from Thermo Scientific (Rockford, IL). For Glu-C digestion, 3 μg of each RTA variant was digested with 0.15 μg of Glu-C (20:1) in 1× Glu-C buffer and in a total volume of 10 μl. Reactions were incubated at 25 °C for 5 h. For trypsin, 10 μg of each RTA variant was digested with 0.4 μg of TPCK-treated trypsin (25:1) in 1× digestion buffer. The reaction was incubated at 25 °C for 60 min. For chymotrypsin, 10 μg of each RTA variant was digested with 0.4 μg of TLCK-treated chymotrypsin (25:1) in buffer containing 100 mm Tris-HCl, pH 8.8, plus 2 mm CaCl2. Reactions were incubated at 25 °C for 30 min.
Circular Dichroism (CD) Spectroscopic Measurement of Recombinant Wild Type RTA and Arginine Variants
Purified RTAs were dialyzed against 50 mm phosphate buffer, pH 6.0, overnight at 4 °C. The solutions were then centrifuged or filtered to remove any precipitate that had accumulated. Protein concentrations were determined from the absorbance at 280 nm using an extinction coefficient of 26,300 m−1 cm−1 (see the ExPASy Web site) using an Aviv model 14 spectrophotometer. For the near-UV CD spectra (250–350 nm), the concentrations ranged from 1.3 × 10−5 to 3.3 × 10−5 m, depending on the mutant, whereas for the far-UV CD spectra, the concentrations were 2.69 × 10−6 to 4.09 × 10−6 m. The near-UV CD spectra were measured in a 1-cm cell, which was moved directly from the CD instrument to the absorption spectrophotometer for the concentration measurements. The far-UV CD spectra were measured in a 1-mm cell, and the concentration of the same sample was determined in a 1-cm cell. All CD measurements were conducted at 25 °C in an Aviv model 410 CD spectrometer. Protein samples for CD were scanned eight times and averaged. Similarly, the buffer base lines were scanned eight times and averaged. The averaged base-line spectra were then subtracted from the averaged sample spectra, converted to molar elipticity, and smoothed. The fractions of secondary structure were calculated from the far-UV spectra with the Dichroweb server (44, 45) using the method of Provencher and Glöckner (46).
Interaction of Ricin Holotoxin and RTA with Yeast Ribosomes
A Biacore T200 (GE Healthcare) was used to measure the interaction after immobilizing RTA and ricin holotoxin on a CM5 chip using amine coupling at 4234 and 7040 RU, respectively. EGFP was immobilized on the reference surface at 4588 RU as a control. The running buffer contained 10 mm Hepes, pH 7.6, 150 mm NaCl, 10 mm MgOAc, 50 μm EDTA, and 0.005% surfactant P20. Yeast ribosomes at 0.25, 0.5, 1, 2, 4, 8, and 16 nm were passed over each surface at 40 μl/min for 5 min. Dissociation was at the same flow rate for another 5 min. The surface was regenerated by injection of 500 mm KCl in the running buffer for 20 s at a flow rate of 50 μl/min between each binding cycle. The signal from the reference surface containing EGFP was subtracted from the signal from the sensor surface containing RTA and ricin holotoxin to correct for nonspecific binding. The interactions were measured at 25 °C, and the data were analyzed using Biacore T200 evaluation software.
Interaction of RTA Variants with Yeast Ribosomes and Ribosomal Stalk Pentamer
To examine the interaction of RTA variants with the stalk pentamer, N-terminal His10-tagged RTA variants were immobilized on an NTA chip of a Biacore T200 at 2300 RU, and the same amount of EGFP was immobilized on the reference channel as the control. Stalk pentamer from yeast (21) was passed over the surface at 60 μl/min as the analyte. Running buffer contained 10 mm Hepes, pH 7.4, 150 mm NaCl, 10 mm MgOAc, 50 μm EDTA, and 0.005% surfactant P20. To examine the interaction of RTA variants with ribosomes, each RTA variant was immobilized on an NTA chip at 2000 RU, and the same amount of EGFP was immobilized to the reference channel as the control. Monomeric yeast ribosomes were passed over the surface at 30 μl/min as the analyte. Running buffer was the same as described for the stalk interaction with additional 50 mm KCl.
Depurination of Yeast Ribosomes and RNA by Ricin Holotoxin or RTA in Vitro
Ricin holotoxin was purchased from Vector Laboratories (Burlingame, CA). For ribosome depurination in vitro, the reaction mixture contained 40 nm yeast ribosomes, 1× reaction buffer (10 mm Tris-HCl, pH 7.4, 60 mm KCl, and 10 mm MgCl2), with different concentrations of RTA or ricin with or without tris(2-carboxyethyl)phosphine hydrochloride (TCEP) varying from 0 to 40 nm in a total reaction of 100 μl. The reaction was started by adding RTA to the reaction mixture and incubating at 30 °C for 10 min. It was stopped by adding 100 μl of 2× RNA extraction buffer (50 mm Tris-HCl, pH 8.8, 240 mm NaCl, 20 mm EDTA, and 2% SDS). The RNA was extracted with phenol and then phenol/chloroform and precipitated with ethanol. The extent of depurination was quantified by quantitative RT-PCR (qRT-PCR) as described previously (47). For RNA depurination, purified yeast RNA (1 μg) was dissolved in 18 μl of reaction buffer containing 20 mm citrate, pH 5.0. The reaction was started by adding 2 μl of ricin with or without TCEP or RTA. The reaction was incubated at 37 °C for 30 min. RNA was precipitated with ethanol after phenol/chloroform extraction. The extent of depurination was quantified by qRT-PCR as described previously (47).
Expression and Purification of S. cerevisiae Adenine Phosphoribosyltransferase
His-tagged S. cerevisiae adenine phosphoribosyltransferase was expressed and purified as described previously with some modifications (48, 49). Briefly, S. cerevisiae adenine phosphoribosyltransferase was expressed in E. coli (B25) cells; grown in 37 °C in Luria broth (LB) medium supplemented with kanamycin (25 μg/ml), ampicillin (100 μg/ml), and 10 mm sodium phosphate (pH 7.5) to an A600 of 0.6; and then induced with 2 mm isopropyl β-d-thiogalactopyranoside and grown for 5 h. The cells were harvested by centrifugation at 3500 × g for 20 min at 4 °C. The cell pellet was suspended in buffer A (50 mm Tris (pH 7.4), 10% glycerol, 300 mm NaCl, 5 mm MgCl2, 5 mm sodium pyrophosphate, 5 mm 2-mercaptoethanol, 1 mm PMSF with complete protease inhibitor mixture (Roche Applied Science), and the cells were disrupted by ultrasonication followed by centrifugations at 30,000 × g for 30 min and 200,000 × g for 2 h at 4 °C. The supernatant was fractionated by ammonium sulfate precipitation to obtain the fraction between 60 and 75% saturation. The resulting pellet was dialyzed against buffer A containing 20 mm imidazole and loaded onto a 5-ml HisTrap FF crude affinity column using an AKTA Purifier (GE Healthcare). After washing the column with 100 ml of buffer A containing 20 mm imidazole, the protein was eluted with a linear elution gradient of buffer A containing 500 mm imidazole (0–100%). The fractions with the highest purity and specific activity were combined, concentrated, and dialyzed overnight against buffer A supplemented with 4 g/liter charcoal. Purified enzyme was estimated to be 100% homogeneous by analysis on denaturing SDS-PAGE.
Expression and Purification of Clostridium symbiosum Pyruvate Orthophosphate Dikinase
C. symbiosum pyruvate orthophosphate dikinase was expressed and purified as described previously with some modifications (49–51). Briefly, pyruvate orthophosphate dikinase was expressed in E. coli (JM101) cells, grown in 37 °C in LB medium supplemented with 12.5 μm tetracycline to an A600 of 1.4. The cells were harvested by centrifugation at 3500 × g for 20 min at 4 °C. The cell pellet was suspended in buffer A (20 mm imidazole (pH 6.8), 10% glycerol, 2.5 mm Na2EDTA, 2 mm dithiothreitol (DTT), 75 mm KCl, 1 mm PMSF with complete protease inhibitor mixture), and the cells were disrupted by ultrasonication followed by centrifugations at 30,000 × g for 30 min and 200,000 × g for 2 h at 4 °C. The supernatant was fractionated by ammonium sulfate precipitation to obtain the fraction between 65 and 85% saturation. The resulting pellet was dialyzed against buffer A and loaded onto a Resource S cation exchange column (1 ml) using an AKTA purifier. The flow-through fraction containing active pyruvate orthophosphate dikinase enzyme was dialyzed against buffer A and loaded onto a Resource Q anion exchange column (6 ml). After washing the column with 120 ml of buffer A, the protein was eluted with a linear elution gradient of 75–1000 mm KCl. The fractions with the highest purity and specific activity were combined, concentrated, and dialyzed overnight against buffer A. The final step was size exclusion chromatography performed on an AKTA purifier, equipped with a Superose 12 10/300 GL gel filtration column and Unicorn version 5.31 software. After size exclusion chromatography, the fractions with the highest purity and specific activity were combined, concentrated, and dialyzed overnight against buffer A supplemented with 4 g/liter charcoal. Purified enzyme was estimated to be 95% homogeneous by SDS-PAGE analysis. The aliquots were frozen in dry ice/ethanol and stored at −80 °C.
Ribosome Depurination Kinetics Assay
Monomeric ribosomes were isolated from W303 (MATa ade2-1 trp1-1 ura3-1 leu2-3,112 his-3-11,15 can1-100) as described previously (18, 19). The final pellet was resuspended in 10% glycerol, 10 mm Tris-HCl, pH 7.5, 5 mm MgCl2. Prior to use, the ribosome solution was passed through a G25 spin column to reduce AMP and ATP contamination as described (49). Depurination was measured by the continuous luminescence assay as described by Sturm and Schramm (49). RTA concentrations used were between 1 and 50 nm, depending on the activity of the RTA variants with increasing ribosome concentrations, as indicated. The reaction was set up in a 96-well plate in a total reaction volume of 50 μl. The reaction was started by adding RTA to the reaction mixture, and the luminescence intensity was recorded continuously for 10 min using a BioTek Synergy 4 microplate reader. The rates were determined from the linear region of the luminescence intensity. The RTA-independent rate of adenine generation was subtracted. Adenine standards covering the range of depurination were measured for each mutant. Complete depurination of ribosomes was assessed as described (49). The initial rate of adenine formation was calculated by converting luminescent rate (lumens/s) to enzymatic rate (pmol of adenine/min/pmol of enzyme) using the adenine standard curve. Kinetic parameters (kcat and Km) were calculated by fitting the initial rates to the Michaelis-Menten equation using the Sigma Enzyme Kinetics Module version 1.3 (Systat Software, Inc., San Jose, CA).
SRL Depurination Kinetics Assay
Stem-loop depurination was performed using a synthetic 10-mer SRL oligonucleotide (5′-rCrGrCrGrArGrArGrCrG-3′) purchased from Integrated DNA Technologies. The discontinuous luminescence assay described by Sturm and Schramm (49) was used. The reaction was performed at 37 °C in 50 μl in 10 mm potassium citrate-KOH and 1 mm EDTA at pH 4.0. RTA concentrations were 30–100 nm. The reaction was started by adding RTA to the reaction mixture. Samples were taken at 1-min intervals into 2× coupling buffer to stop the reaction, as described (49), for up to 5 min. Adenine standards were assayed for each sample using the same conditions. Luminescence intensity was measured using a BioTek Synergy 4 microplate reader. Kinetic parameters were calculated as described above for ribosomes.
RESULTS
Ricin Holotoxin Could Depurinate Free RNA but Not Ribosomes, and It Does Not Interact with Ribosomes
In free RTA, the active site residues (Glu177 and Arg180) and the putative rRNA binding residues (Arg213, Arg258, and Trp211), which are located on the edge of the active site, are exposed to the solvent (Fig. 1A). Similarly, in ricin holotoxin, the active site and the RNA binding residues are not covered by RTB, and they have similar conformations as in free RTA (Fig. 1B). Despite the exposed active site, ricin holotoxin does not inhibit translation in cell-free translation assays (5, 10, 11). To determine if this is due to lack of depurination, we examined the depurination activity of ricin holotoxin on yeast ribosomes and total RNA in the presence or absence of 1 mm TCEP, which reduces the disulfide bond between RTA and RTB. As shown in Fig. 1C, ricin holotoxin could depurinate total RNA similarly in the absence or presence of TCEP. In contrast, ricin holotoxin had very low activity on ribosomes in the absence of TCEP treatment (Fig. 1D). The depurination activity increased dramatically when 1 mm TCEP was included in the reaction buffer (Fig. 1D). Ricin holotoxin depurinated total RNA but not ribosomes in the absence of the reducing agent, suggesting that RTB blocks ribosome binding of RTA.
FIGURE 1.

Interaction of RTA and ricin holotoxin with ribosomes and depurination of total RNA and ribosomes. A, RTA crystal structure (PDB code 1RTC) oriented to show residues at the active site and residues involved in SRL binding. RTA is shown in green. Arg180 (R180; orange), Glu177 (E177; red), and Gly212 (G212; blue) are shown to indicate the active site area. Residues predicted to be involved in binding the SRL are shown in cyan. B, ricin holotoxin crystal structure (PDB code 2AAI) in a similar orientation as in A, to show that the SRL binding site and the active site are not blocked by RTB (yellow). C, depurination of RNA by ricin holotoxin. Ricin holotoxin was incubated with yeast total RNA (1 μg) in the presence or absence of 1 mm TCEP, and depurination was quantified using qRT-PCR (47). D, depurination of yeast ribosomes by ricin holotoxin. Ribosomes (40 nm) were treated with ricin holotoxin in the presence or absence of 1 mm TCEP, RNA was extracted, and depurination was quantified using qRT-PCR (47). E, interaction of ricin holotoxin and RTA with yeast ribosomes by Biacore analysis. RTA (dashed lines) and ricin (solid lines) were immobilized on a CM5 chip of a Biacore T200 using amine coupling at 4234 and 7040 RU, respectively. EGFP was immobilized on the reference surface at 4588 RU as a control. The ribosomes at 0.25, 0.5, 1, 2, 4, 8, and 16 nm were passed over each surface at 40 μl/min for 5 min. Dissociation was at the same flow rate for another 5 min. The rate constants and the equilibrium dissociation constant for RTA are shown based on fitting of the data with the heterogeneous ligand parallel reaction model using Biacore T200 evaluation software. Error bars, S.D.
To determine if the lack of depurination activity of the holotoxin toward ribosomes is due to the lack of ribosome binding, we immobilized ricin holotoxin and RTA on a CM5 chip of a Biacore T200 by amine coupling and analyzed their interaction with yeast ribosomes. As previously observed with His-tagged RTA captured on an NTA chip (18), RTA immobilized on a CM5 chip bound ribosomes, and the interaction signal increased with increasing ribosome concentrations (Fig. 1E). The interaction of immobilized RTA with ribosomes did not follow a single step binding model but was characterized best by the heterogeneous ligand parallel reaction model, which predicts two distinct types of interactions (AB1 and AB2) between RTA and ribosomes (18). The dissociation constants for both types of interactions were in the nanomolar range (Fig. 1E), as previously observed with His-tagged RTA captured on an NTA chip (18). In contrast to RTA alone, we did not observe any interaction between monomeric ribosomes and ricin holotoxin even at high ribosome concentrations (Fig. 1E), suggesting that the ribosome binding site of RTA is blocked in the holotoxin.
Double Mutations in Arginines at the Interface of RTB Reduce the Depurination Activity and Cytotoxicity of RTA in Yeast
The electrostatic character of the ribosomal surface has been shown to be responsible for the unusually high catalytic activity of ribotoxins and RIPs (52, 53). We previously showed that electrostatic interactions concentrate RTA on the ribosome surface and facilitate its interaction with the stalk (18). Because electrostatic interactions with the ribosome would require a positively charged surface on RTA, we mutated positively charged arginine residues, which are located at or near the interface of RTB (Fig. 2A). The arginines at positions 189, 191, 193, 196, 197, 234, and 235 were mutated to alanine individually. Following reductive separation of RTA from RTB, these arginines were exposed to the solvent and contributed to the positive surface charge in the carboxyl-terminal domain of RTA (Fig. 2B). These residues are located in a region similar to that of Lys173, Arg174, and Lys177 in TCS (33) and might be involved in the interaction of RTA with the stalk. None of the seven individual Arg to Ala mutations caused an obvious reduction in toxicity compared with wild type RTA in S. cerevisiae (data not shown). These results suggested that mutation of more than one arginine may be needed to reduce the toxicity of RTA. Therefore, we mutated two different arginines to alanine simultaneously and examined the toxicity of the variants in yeast. Double arginine mutations at R193A/R235A, R189A/R234A, and R191A/R196A reduced the toxicity of RTA compared with wild type (Fig. 2C). G212E, which was previously shown to reduce the depurination activity and cytotoxicity of RTA in yeast (40), was used as a positive control. R193A/R235A showed the greatest reduction in toxicity, whereas R189A/R234A and R191A/R196A showed a similar reduction, but they were more toxic than R193A/R235A (Fig. 2C). The doubling times correlated with the viability analysis. R193A/R235A had a doubling time of 6.4 ± 0.8 h compared with wild type RTA, which had a doubling time of 8.1 ± 1.3 h. R189A/R234A and R191A/R196A had doubling times of 8.0 ± 1.1 and 8.2 ± 1.0 h, respectively. In contrast, G212E had a doubling time of 4.6 ± 0.2 h, which was similar to the vector control (4.5 ± 0.1 h).
FIGURE 2.

Viability, RTA expression, and depurination activity of RTA variants in yeast. A, ricin holotoxin (PDB code 2AAI) oriented to show RTA residues involved in ribosome binding (magenta). Arg196 and Arg197 do not interact with RTB (yellow), but the presence of RTB prevents ribosomal access to these amino acids. A few of the residues at the SRL binding site (cyan) are visible on the right. The rest of RTA is green. B, RTA (PDB code 1RTC) oriented to show ribosome-binding residues. Residues at the ribosome binding site are in magenta. Only some of the residues at the putative SRL binding site are visible and are shown in cyan. C, viability of yeast expressing RTA variants. Yeast cells were transformed with wild type RTA or RTA variants under the GAL1 promoter. Cells carrying the empty vector were used as controls. Yeast cells were first grown in SD medium supplemented with 2% glucose and then transferred to SD medium supplemented with 2% galactose. At 0, 4, 6, and 8 h postinduction, a series of 10-fold dilutions were plated on media containing 2% glucose and grown at 30 °C for 2–3 days. D, immunoblot analysis of RTA expression in yeast at 4 hpi. Total protein extracted from equal amount of cells (A600 of 0.8) was loaded on the gel, except only half the amount of cells (A600 of 0.4) was used for G212E due to the higher expression of this variant in yeast (40). Monoclonal antibodies against phosphoglycerate kinase 1 (Pgk1) and dolichoylphosphate mannosyltransferase 1 (Dpm1) were used as loading controls for cytosol and membranes, respectively. E, ribosome depurination in yeast expressing arginine variants of RTA. Total RNA (375 ng) was used to quantify the relative level of depurination using qRT-PCR by the comparative ΔCT method (ΔΔCT) (47). The -fold increase in depurination in yeast expressing RTA variants compared with yeast harboring the empty vector is shown. Error bars, S.D.
The expression of arginine variants in yeast was examined by immunoblot analysis at 4 hpi. Total protein extracted from equal amounts of cells was loaded on the gel, except that only half the amount of cells was used for G212E due to the higher expression of this variant in yeast (40). The expression level of the variants correlated inversely with their toxicity (Fig. 2D). A lower level of protein expression was detected if the RTA variant was more toxic. R189A/R234A and R191A/R196A were expressed at a lower level than R193A/R235A, which was less toxic. G212E, which had the lowest toxicity, was expressed at the highest level, whereas wild type RTA, which had the highest toxicity, was expressed at the lowest level (Fig. 2D). The levels of expression at 6 hpi were similar to those at 4 hpi (data not shown).
To determine if the reduced cytotoxicity correlated with the reduced depurination activity in vivo, we examined the depurination activity of each variant using a previously described qRT-PCR assay (47). The time course of depurination was examined after expression of each RTA variant was induced using galactose. Samples were collected at 2, 3, 4, 6, and 8 h after galactose induction. Depurination reached the highest level at 4 or 6 hpi, depending on the type of mutation, and then it decreased as we have observed previously with other RTA variants (54). A low level of depurination was detected in yeast expressing wild type RTA even when cells were grown in glucose (at 0 h in Fig. 2E) due to leaky expression of RTA. The mutants did not show any depurination activity compared with the vector control at 0 h. At 2 hpi, the depurination level in yeast expressing R191A/R196A was about half that of yeast expressing wild type RTA and was still lower at 3 hpi (Fig. 2E). The depurination level of R189A/R234A was almost the same as that of wild type, whereas the depurination level of R193A/R235A was much lower than those of the other two mutants up to 4 hpi (Fig. 2E). The level of depurination at 2 hpi correlated with the relative toxicity of the arginine variants (Fig. 2C), indicating that arginine residues at the interface of RTA/RTB are critical for toxicity and depurination in yeast.
Recombinant RTA Variants Have Conformations Similar to That of Wild Type RTA
To determine if the reduced depurination activity and toxicity of the arginine variants are due to defects in ribosome binding, we expressed wild type RTA and R189A/R234A and R193A/R235A in E. coli and purified them to homogeneity. R191A/R196A could not be cloned into the E. coli expression vector, possibly because it was toxic to the bacteria. G212E, which had very low depurination activity and cytotoxicity in yeast (40), was also expressed in E. coli and purified to homogeneity.
To determine if the double mutations in Arg residues affected the structure of the RTA variants, we carried out partial digestion analysis of the recombinant proteins using three different proteinases: Glu-C, which cleaves at the carboxyl side of glutamic acid; TPCK-treated trypsin, which cleaves at the carboxyl side of arginine and lysine residues; and TLCK-treated chymotrypsin, which cuts on the carboxyl side of tyrosine, phenylalanine, tryptophan, and leucine residues. Surprisingly, Glu-C and trypsin did not digest wild type RTA or the mutants even after extended times (Fig. 3A). These results suggested that the peptide bonds at the potential cleavage sites are not on the surface and are therefore inaccessible to these two proteases. In contrast, chymotrypsin was able to digest RTA variants similarly as wild type RTA (Fig. 3A), suggesting that the RTA variants have conformations similar to that of wild type RTA.
FIGURE 3.

Partial digestion and far UV and near UV CD spectra of purified RTA variants. A, partial digestion of RTA variants with different proteinases. 3 μg of each RTA was digested with 0.15 μg of Glu-C (20:1) in 1× Glu-C buffer in a total volume of 10 μl at 25 °C for 5 h. 10 μg of RTA was digested with 0.4 μg of TPCK-treated trypsin (25:1) in 1× digestion buffer at 25 °C for 60 min. 10 μg of RTA was digested with 0.4 μg of TLCK-treated chymotrypsin (25:1) in buffer containing 100 mm Tris-HCl, pH 8.8, and 2 mm CaCl2 at 25 °C for 30 min. Shown are the far UV CD spectra (B) and near UV CD spectra (C) of RTA variants. The CD spectra of purified variants were determined at 25 °C in 50 mm phosphate buffer, pH 6.0, after overnight dialysis at 4 °C, followed by centrifugation or filtering. For the near-UV (250–350 nm), the concentrations ranged from 1.3 × 10−5 to 3.3 × 10−5 m depending on the variant, whereas for the far-UV, the concentrations were 2.69 × 10−6 to 4.09 × 10−6 m. Samples for CD were scanned eight times and averaged as described under “Experimental Procedures.”
To further investigate the structure of the variants compared with wild type RTA, we measured the circular dichroism (CD) spectra in both the far-UV (185–250 nm) and the near-UV (250–350 nm). The far-UV spectra are very similar in shape and magnitude (Fig. 3B), and the secondary structure content (Table 1) is practically identical to that of the crystallographic structure of RTA (PDB code 1RTC) with the slight exception of R189A/R234A and G212E. Compared with the crystal structure, ∼6–7% of these variants' helical residues and 2–3% of their non-repeating secondary structure (loop and turn residues) have been added to the β-strand content (Table 1). The close similarity of the four proteins to one another and to the crystallographic structure is strikingly shown in the near-UV CD spectra (Fig. 3C). Spectra in this region are diagnostic of tertiary structure, arising primarily from the side chain environments within the protein of tyrosine and tryptophan residues, of which RTA contains 14 and 1, respectively. Any tertiary structural change will affect the CD of these side chains through interactions with one another, with the peptide backbone, and with other side chains nearby (55). The near-UV CD spectra in Fig. 3C are identical within experimental error, indicating that there are no detectable tertiary structural differences and reinforcing the secondary structural analyses.
TABLE 1.
Secondary structure content of RTA variants based on far-UV CD analysis
| RTA variants | Percentages of residuesa |
||
|---|---|---|---|
| Helix | Strand | Other | |
| % | % | % | |
| X-ray structure (PDB code 1RTC) | 30.8 | 13.3 | 55.9 |
| Wild type | 34.2 | 13.5 | 52.4 |
| R189A/R234A | 23.7 | 23.2 | 53.1 |
| G212E | 24.8 | 21.7 | 53.6 |
| R193A/R235A | 32.2 | 13.1 | 54.6 |
Double Arginine Mutations Affect the Interaction of RTA with Ribosomes and with the Yeast Stalk Pentamer
To determine if the double arginine mutations affect the interaction of RTA with yeast ribosomes or with the isolated stalk pentamer from yeast (21), we examined their interaction by Biacore analysis. In order to detect even low levels of binding, His10-tagged wild type RTA or RTA variants G212E, R189A/R234A, and R193A/R235A were captured on the different channels of the NTA chip at a relatively high density (2300 RU). The same amount of His10-tagged EGFP was captured on the reference channel as a control. The yeast stalk pentamer was passed over the surface as the analyte (21). Wild type RTA and G212E bound the stalk pentamer similarly (Fig. 4A). The binding levels of stalk pentamer to wild type RTA and G212E reached 120 and 700 RU at pentamer concentrations of 1 and 10 nm, respectively. The binding signal could be detected even at a pentamer concentration of 0.1 nm (data not shown). Binding of R189A/R234A to the stalk pentamer was detected at 1 nm, and it reached around 60 RU at 10 nm. We could not detect binding of R193A/R235A to the stalk pentamer at stalk concentrations of 0.1, 1, or 10 nm (Fig. 4A).
FIGURE 4.
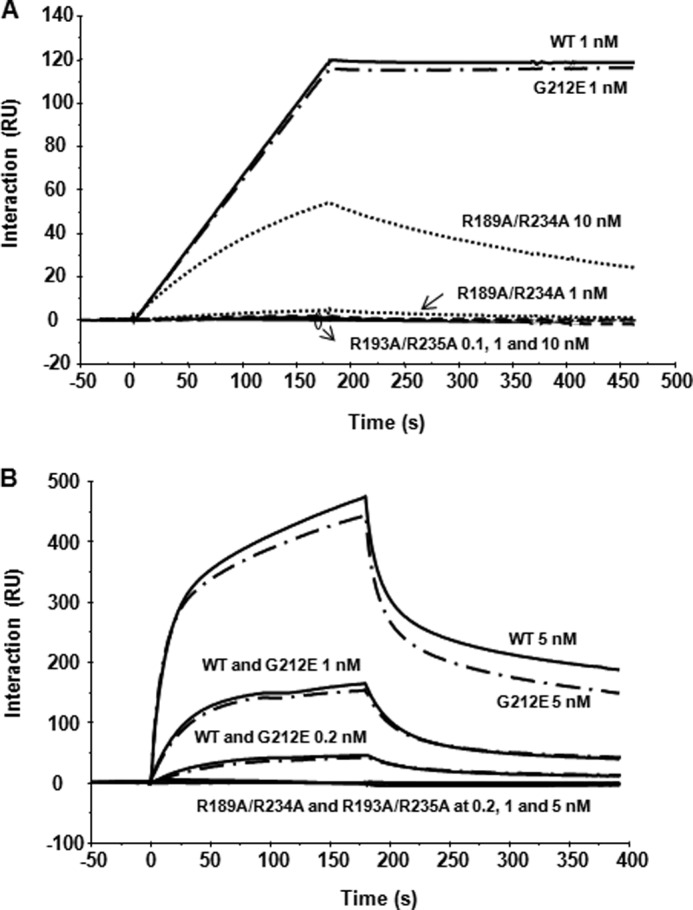
Interaction of RTA variants with the ribosomal stalk pentamer and with monomeric ribosomes. A, N-terminal His10-tagged RTA variants were captured on an NTA chip at 2300 RU, and the same amount of EGFP was captured on the reference channel as control. Isolated stalk pentamer was passed over the RTA surface at 0.1, 1, and 10 nm as the analyte. B, RTA variants were captured on an NTA chip at 2000 RU, and the same amount of EGFP was captured to the reference channel as a control. Yeast ribosomes at a concentration of 0.2, 1, and 5 nm were passed over the surface as the analyte.
To examine the interaction of RTA variants with monomeric ribosomes, wild type RTA and variants were captured on different channels of an NTA chip at 2000 RU. Yeast ribosomes were passed over the surface as the analyte (18). Wild type RTA and G212E bound monomeric ribosomes similarly, except that G212E dissociated a little faster than wild type RTA. The binding levels of ribosomes to wild type RTA and G212E were 40, 150, and 450 RU at 0.2, 1, and 5 nm ribosome concentrations, respectively. We could not detect any ribosome binding for R189A/R234A and R193A/R235A even at a ribosome concentration of 5 nm (Fig. 4B). These results indicate that arginine mutations at or near the RTA/RTB interface disrupt the interaction of RTA with ribosomes. Although R189A/R234A showed weak binding to the stalk pentamer, we could not detect its interaction with ribosomes.
RTA Variants Depurinate Ribosomes at a Reduced Rate but Depurinate RNA at a Similar Rate as Wild Type RTA
To distinguish between the roles of the arginine residues in ribosome binding versus catalytic activity, we examined the depurination activity of the recombinant RTA variants using yeast ribosomes or RNA as a substrate. If arginine mutations affect either RNA binding or catalytic activity, depurination of both intact ribosomes and free RNA will be reduced. However, if the mutations affect only ribosome binding, but not RNA binding or catalytic activity, then depurination of intact ribosomes will be reduced, whereas depurination of RNA should not be affected. Because the conditions for ribosome depurination are different from RNA depurination for RTA (56), the depurination activity of arginine variants on each substrate was compared with wild type RTA. As shown in Fig. 5A, at a fixed ribosome concentration, the depurination levels increased with increasing RTA concentration. However, the depurination activity of R189A/R234A and G212E was reduced 20-fold compared with wild type RTA, whereas R193A/R235A had 50-fold lower activity compared with wild type. When total RNA was used as a substrate, R189A/R234A and R193A/R235A showed activity similar to that of wild type RTA, whereas the activity of G212E was about 5-fold lower (Fig. 5B).
FIGURE 5.

Depurination of ribosomes and total RNA by RTA variants in vitro. A, yeast ribosomes (40 nm) were incubated with different concentrations of RTA, varying from 0 to 40 nm. The extent of depurination was quantified by qRT-PCR as described previously (47). RTA concentrations are shown in log scale due to large differences in the activities of wild type RTA and arginine variants. The y axis shows the -fold change in depurination relative to the control samples without toxin treatment. B, total RNA (1 μg) was treated with different concentrations of RTA as described under “Experimental Procedures.” The extent of depurination was quantified by qRT-PCR as described previously (47). Error bars, S.D.
To confirm these results, we measured the kinetics of depurination, using an enzymatically coupled luminescence assay reported by Sturm and Schramm (49). This assay can quantify subpicomole levels of adenine by converting adenine to AMP by adenine phosphoribosyltransferase and AMP to ATP by pyruvate orthophosphate dikinase. The ATP is quantified by firefly luciferase (49). Wild type RTA catalyzed release of adenine from 80 S yeast ribosomes by causing a linear increase in luminescence in a continuous coupled assay. The initial rates were dependent on the ribosome concentration and fit to the Michaelis-Menten equation (Fig. 6A). The results from two independent measurements with different ribosome preparations are shown in Table 2. Wild type RTA depurinated 80S yeast ribosomes with an initial rate (kcat) of 19.2 ± 1.5 min−1 and a Km of 120 ± 18 nm, and its catalytic efficiency (kcat/Km) was 2.7 × 106 m−1 s−1. G212E, which could bind ribosomes similarly to wild type RTA by Biacore analysis (Fig. 4B), had a Km value (101 ± 10 nm) similar to that of wild type. In contrast, arginine variants had 4–5-fold higher Km values than wild type RTA, consistent with the Biacore data, which showed no detectable ribosome binding by the arginine variants (Fig. 4B). The initial rates (kcat) for G212E, R189A/R234A, and R193A/R235A were 96-, 9-, and 38-fold lower, respectively, than wild type RTA. The catalytic efficiency (kcat/Km) of G212E, R189A/R234A, and R193A/R235A was 71-, 35-, and 193-fold lower, respectively, than wild type RTA (Table 2). These results indicate that arginine mutations reduce the catalytic efficiency of RTA both by decreasing its ability to bind ribosomes (Fig. 4B) and by causing a decrease in the rate of depurination (Table 2). R193A/R235A, which could not bind ribosomes or ribosomal stalk, had a lower catalytic rate and catalytic efficiency toward ribosomes than R189A/R234A, which showed a low level of binding to the ribosomal stalk. G212E mutation did not affect ribosome binding but reduced the catalytic rate and catalytic efficiency toward ribosomes.
FIGURE 6.

Kinetic curve fits for catalysis of depurination of ribosomes and stem-loop RNA by RTA variants. A, Michaelis-Menten fits of ribosome depurination performed with the continuous assay described under “Experimental Procedures.” Wild type RTA was used at 1 nm, R189A/R234A at 10 nm, G212E at 30 nm, and R193A/R235A at 50 nm, all with increasing amounts of ribosomes as indicated. The RTA-independent rate of adenine generation was subtracted for each. Adenine standards covering the range of depurination were measured for each mutant. B, Michaelis-Menten fits of depurination of 10-mer loop performed with the discontinuous assay at pH 4.5 described under “Experimental Procedures.” Wild type RTA was used at 50 nm, R189A/R234A at 50 nm, G212E at 100 nm, and R193A/R235A at 50 nm. Adenine standards were measured for each under the same buffer conditions. Results from a representative experiment are shown.
TABLE 2.
Kinetic parameters of RTA variants on ribosome and on the stem-loop RNA
| RTA variants | Ribosomea |
Stem-loop RNAb |
||||
|---|---|---|---|---|---|---|
| kcat | Km | kcat/Km | kcat | Km | kcat/Km | |
| min−1 | nm | m−1 s−1 | min−1 | nm | m−1 s−1 | |
| Wild type | 19.2 ± 1.5 | 120 ± 18 | 2.7 × 106 | 3.7 ± 0.2 | 670 ± 56 | 9.3 × 104 |
| R189A/R234A | 2.2 ± 0.0 | 467 ± 1 | 7.8 × 104 | 2.3 ± 0.3 | 879 ± 29 | 4.4 × 104 |
| G212E | 0.23 ± 0.01 | 101 ± 10 | 3.8 × 104 | 1.1 ± 0.1 | 827 ± 70 | 2.2 × 104 |
| R193A/R235A | 0.50 ± 0.09 | 594 ± 128 | 1.4 × 104 | 2.7 ± 0.3 | 979 ± 133 | 4.7 × 104 |
a Ribosome substrate was assayed in continuous format.
b SRL mimic stem-loop RNA substrate was assayed in a discontinuous format.
To determine if ribosomal components affect the catalytic rates of the RTA variants, we examined the depurination kinetics using a 10-mer RNA stem-loop mimic of the SRL as a substrate (49). The activity of arginine variants on the stem-loop RNA should not be affected by ribosome or ribosomal stalk binding. Initial rate kinetics for the catalysis of stem-loop substrate was measured by a discontinuous luminescent assay for adenine release (49). Saturation curves, which fit well to the Michaelis-Menten equation, were obtained for wild type RTA and the RTA variants (Fig. 6B). The results from three independent measurements are shown in Table 2. Wild type RTA gave a kcat of 3.7 ± 0.2 min−1. The stem-loop RNA was depurinated by R189A/R234A and R193A/R235A at catalytic rates slightly lower than that of wild type RTA, whereas G212E had a 3-fold lower kcat than wild type RTA. The Km values of the arginine variants and G212E were slightly higher than wild type RTA. The kcat/Km values of the arginine variants for the stem-loop RNA were on the same order of magnitude (104) as wild type RTA (Table 2). In contrast, wild type RTA was ∼2 orders of magnitude more efficient against ribosomes than the arginine variants (Table 2). These results demonstrate that the double arginine mutations affect ribosome binding and catalytic activity of RTA toward ribosomes with minimal effect on RNA binding or catalytic activity against RNA.
DISCUSSION
Arginine Residues at the Interface of RTA and RTB Are Critical for the Interaction of RTA with Ribosomes and with the Stalk Pentamer
In the x-ray structure of ricin holotoxin, several arginine residues located at the interface of RTA and RTB are buried. To determine if these arginines are involved in the RTA-ribosome interaction, we mutated seven arginines, Arg189, Arg191, Arg193, Arg196, Arg197, Arg234, and Arg235, to alanine individually. We found that point mutations at any one of these arginines did not lead to an obvious reduction in cytotoxicity. It is possible that the effect due to each arginine residue is minimal, such that it does not show up as a reduction in cytotoxicity. To test this, we mutated more than one arginine at the same time and showed that at least two arginines had to be mutated to interrupt the interaction of RTA with the stalk pentamer and with intact ribosomes, such that the toxicity can be reduced. Similarly, single point mutations at K173A, R174A, and K177A reduced the interaction of TCS with the P2 protein, and triple mutations at K173/R174/K177 disrupted the interaction in vitro (29). We show here that the far-UV and the near-UV CD spectra for the arginine variants, R189A/R234A and R193A/R235A, were similar to those of wild type RTA, suggesting that they have similar secondary and tertiary structure. These mutations had a minimal effect on the catalytic rate of RTA when RNA was used as a substrate but reduced ribosome binding and the catalytic rate of RTA toward ribosomes. These results indicate that the decrease in toxicity was not due to the loss of catalytic properties but rather due to the loss of ribosome binding and provide the first in vivo evidence that ribosome binding is important for the catalysis of depurination and toxicity of RTA. Interestingly, monoclonal antibodies targeted against this arginine-rich region neutralize ricin in vitro and show protection in mice in vivo (42, 43), suggesting that this region is also important for the toxicity of ricin in an animal model.
The tertiary structures are similar between TCS and RTA. However, in plants, RTA is synthesized together with RTB as a single polypeptide chain. We show here that ricin holotoxin can depurinate total RNA from yeast but not yeast ribosomes, indicating that RTB blocks the access of RTA to the ribosome but not to RNA. Although RTA is toxic when expressed in plants, toxicity is reduced when RTA is co-expressed with RTB (57), suggesting that ribosomes are protected from RTA in the presence of RTB. Because TCS is a single chain RIP without a B chain, it is unclear how ribosomes are protected in TCS-producing plants.
We previously suggested a two-step binding model based on kinetic analysis of the interactions between RTA and ribosomes (18). According to this model, in the first step, non-stalk-specific electrostatic interactions (AB2 in Fig. 1E) increase the local concentration of RTA near the P-protein stalk and facilitate the second step, in which faster and more specific electrostatic interactions (AB1 in Fig. 1E) occur with the stalk complex (18). R193A/R235A and R189A/R234A could not bind intact ribosomes by Biacore analysis. Because the ribosome is about 120 times bigger than RTA (molecular mass of 30 kDa for RTA and 3600 kDa for the yeast ribosome), to reach the high depurination rate, RTA must have a very high local concentration near the ribosome (52, 53). We show here that the seven arginines at the RTA/RTB interface contribute to the positive surface charge, which allows RTA to interact with the ribosome via electrostatic interactions.
Our two-step model predicts that if RTA molecules do not concentrate on the ribosome and reach a very high local concentration near the stalk, they may not be recruited by the stalk. Although R189A/R234A showed a low level of interaction with the isolated stalk pentamer, it was not able to interact with the ribosome-bound stalk, possibly because it did not reach a high local concentration near the ribosome. Therefore, arginine residues in RTA at or near the interface of RTB are critical not only for the specific electrostatic interactions with the P-protein stalk, but also for the initial non-stalk-specific electrostatic interactions with the ribosome. These results provide further evidence for the two-step interaction model (18).
The Ribosome Binding Surface of RTA Is Opposite to the Surface That Contains the Active Site and Interacts with the rRNA
The crystal structure of RTA combined with point mutation analysis showed that Glu177 and Arg180 were critical for the catalytic activity (58, 59), whereas Tyr80, Tyr123, Asn209, and Trp211 contributed to binding to the target adenine (60, 61). The structures of small nucleotide ligands bound to RTA showed that Asn209 formed hydrogen bonds with the GAGA tetranucleotide of the SRL (62). Modeling analysis of the interaction of RTA with RNA suggested that Arg48, Arg134, Arg213, and Arg258 contribute significantly to the binding to the phosphate backbone of the rRNA (62, 63). Mutation analysis confirmed that these arginines located around the active site cleft play a role in the activity (64). Based on a combination of site-directed mutagenesis (60, 64), deletion (39), and computing simulation (63, 65) analysis, amino acids Arg48, Arg125, Arg134, Arg213, and Arg258, which are at the edge of the active site (Fig. 1A), were thought to be involved in rRNA binding. We show here that G212A binds ribosomes and ribosomal stalk similarly as wild type RTA but depurinates ribosomes and RNA at a lower catalytic rate than wild type RTA. Gly212 is located between Trp211, which is involved in binding to the target adenine (60, 61), and Arg213, which contributes to binding to the rRNA (62, 63) and is on the opposite side from the RTA/RTB contacts (Fig. 2B). Gly212 mutation affects catalytic activity rather than RNA binding. Consistent with this, deletion of Gly212–Arg213 produced an inactive RTA mutant (66). Arginine residues, which are shown here to interact with the ribosome and the stalk, are on the RTA-RTB interface, which is away from the area that contains the active site and is predicted to bind to the SRL. Therefore, RTA appears to use one side of the protein to interact with the ribosome and the P-protein stalk and the opposite side for binding to the SRL.
Stalk Binding Stimulates the Catalysis of Ribosome Depurination by RTA
Although arginine variants depurinated the stem-loop RNA at a similar rate as wild type RTA, their catalytic rate toward ribosomes was considerably lower. The kcat/Km ratios of the arginine variants for the stem-loop RNA were of the same magnitude as wild type RTA. However, wild type RTA was roughly 2 orders of magnitude more efficient against ribosomes than the arginine variants. Both arginine variants had similar activity toward the stem-loop RNA, but R189A/R234A had over 5-fold higher kcat/Km than R193A/R235A toward ribosomes. R189A/R234A showed a low level of interaction with the stalk. Based on these results, we conclude that stalk binding stimulates the catalysis of depurination of the rRNA by bringing RTA to the SRL on the ribosome. We propose a model, which describes how RTA accesses the SRL (Fig. 7). The stalk proteins interact with the stalk binding side of RTA, which is blocked by RTB in the holotoxin. The flexible hinge of the P-proteins delivers the properly oriented RTA to the SRL through a conformational change and correctly positions A3027 of the SRL at the active site of the stalk-bound RTA. This mechanism allows RTA to establish the specific contacts with the backbone of the SRL necessary for depurination. RTA has a domain that is thought to undergo a conformational change upon depurination (36). This conformational change may allow the stalk complex to disconnect from RTA. This model explains why RTA depurinates intact ribosomes much better than the free rRNA, how it selects the SRL instead of any other stem loop structure on the ribosome, and how it dissociates from the stalk. Because other RIPs, such as Stx1 (30, 31) and TCS (29, 33), also bind to the stalk to depurinate the SRL and contain basic residues at similar positions, the orientation of the active site toward the SRL by stalk binding may be a general mechanism applicable to other RIPs that interact with the stalk. Consistent with this model, recent results indicate that the level of depurination by TCS depends on the length of the hinge region within the C-terminal tail of the P-proteins (32).
FIGURE 7.

Model showing how RTA accesses the SRL. S. cerevisiae 26 S rRNA (PDB code 3U5H) and 60 S subunit (PDB code 3U5I) are indicated as light gray and dark gray colors, respectively. The fitted schematic structure of P0 fragment complexed with the N-terminal domain of P-proteins (PDB code 3A1Y) from Archaea is depicted as yellow and green, respectively. One CTD domain of a P-protein is shown as a gray line attached to the RTA molecule. The yeast 60 S subunit is oriented to show A3027 (red) of the SRL (cyan) and RTA (PDB code 1RTC) oriented to show R180 (orange) at the active site, the putative SRL binding site (cyan), and the stalk binding site (magenta). In step 1, RTA binds to the C-terminal tail of a P-protein using arginine residues located on the stalk binding surface. In step 2, the flexible hinge of the P-protein orients the active site of RTA toward the SRL and properly positions A3027 of the SRL into the active site of RTA. This mechanism enables RTA to establish the specific contacts with the backbone of the SRL necessary to hydrolyze the N-glycosidic bond of A3027. An enlarged image of the interaction is shown in the inset.
The translational GTPases, such as elongation factors, bind to a similar region of the ribosome as RIPs (26, 28). The GTPase activity of the translational GTPases increase markedly upon binding to the ribosome, and the ribosome plays an active role in catalysis (67). The GTPase binding site includes the SRL, the L11 protein, rRNA, and the stalk proteins, L12 (28). The mechanism used by the ribosome to activate GTP hydrolysis by translational GTPases is not clear. The conserved ribosome binding site between the translational GTPases and RTA suggests that translational GTPases may access the SRL by a similar mechanism. Stalk binding may correctly position the translational GTPases for binding to the ribosome and for activation of GTP hydrolysis.
The Rate of Depurination Determines the Cytotoxicity of RTA
Comparison of the RTA expression level, the depurination rate, and toxicity suggested a negative correlation between RTA expression and cytotoxicity. Although depurination reached the highest level at 4 hpi in yeast, cytotoxicity was more closely related to the rate of depurination than the absolute level of depurination. R189A/R234A, which depurinated ribosomes at a faster rate than R193A/R235A, was more toxic than R193A/R235A, whereas G212E, which had the lowest depurination rate, had the lowest toxicity. A low rate of depurination allows RTA to accumulate with time because cells do not die. Therefore, RTA variants such as G212E, which have lower a depurination rate, are expressed at a higher level. A high depurination rate induces cell death. Therefore, very little RTA accumulates in yeast expressing highly active RTA. These results suggest that the depurination rate, rather than the absolute level of depurination, determines cell survival. We show here that the depurination rate depends on how well RTA can interact with the stalk proteins, which is determined by the local concentration of RTA on the ribosome.
Although R189A/R234A and R193A/R235A did not bind ribosomes in vitro, they could depurinate ribosomes in vivo. The level of depurination in yeast expressing R189A/R234A was similar to that of wild type, suggesting that inside cells, the inability to bind ribosomes is at least partially compensated. Because RTA has to reach the ribosome through electrostatic interactions in vitro, arginine mutations, which reduce the surface charge, would affect these interactions. However, in vivo, cytosolic chaperones and even ribosomes are thought to be involved in the process of refolding of RTA (68, 69). Therefore, chaperone- or ribosome-mediated refolding of RTA may help the arginine variants to access ribosomes more efficiently inside cells.
In summary, we demonstrate here that arginine residues that are blocked by RTB in ricin holotoxin are critical for RTA to bind to ribosomes. The ribosome binding surface of RTA is on the opposite side of the surface that contains the active site and the putative SRL binding site. These findings support a mechanism whereby the interaction of RTA with the P-protein stalk orients the active site of RTA toward the SRL and places it in correct orientation for binding to the target adenine. The properly oriented RTA is able to establish the specific contacts necessary to hydrolyze a single N-glycosidic bond from among 4000 stem loops in the 28 S rRNA. The stalk binding stimulates the catalysis of ribosome depurination by RTA by this mechanism and provides a basis for the exquisite specificity of RTA. This model may be applicable to other RIPs and to translational GTPases, which interact with the P-protein stalk to gain access to the SRL.
Acknowledgments
We acknowledge Drs. Vern Schramm and Emmanuel Burgos for reagents, including the expression vectors for adenine phosphoribosyltransferase and pyruvate orthophosphate dikinase, and valuable advice for measuring the kinetics of depurination; Dr. Karen Chave (Northeast Biodefense Center Protein Synthesis Core, U54-AI057158-Lipkin) for purification of RTA variants from E. coli; Dr. Marek Tchórzewski for providing the purified stalk pentamer from yeast; and Dr. Nicholas Mantis for providing PB10 monoclonal RTA antibody. We also thank Thong Vo for mutating the arginine residues and Maria Montano for the initial screening of the arginine variants.
This work was supported, in whole or in part, by National Institutes of Health Grant AI072425 (to N. E. T.). This work was also supported by a grant from the New Jersey Agriculture Experiment Station (to P. C. K.).
- RIP
- ribosome-inactivating protein
- SRL
- α-sarcin/ricin loop
- RTA
- ricin toxin A chain
- RTB
- ricin toxin B chain
- EGFP
- enhanced green fluorescent protein
- TCEP
- tris(2-carboxyethyl)phosphine hydrochloride
- TPCK
- tosyl phenylalanyl chloromethyl ketone
- TLCK
- N-α-p-tosyl-l-lysine chloromethyl ketone
- Stx
- Shiga toxin
- TCS
- trichosanthin
- PDB
- Protein Data Bank
- hpi
- hours postinduction
- qRT-PCR
- quantitative RT-PCR
- RU
- resonance units.
REFERENCES
- 1. Audi J., Belson M., Patel M., Schier J., Osterloh J. (2005) Ricin poisoning. A comprehensive review. JAMA 294, 2342–2351 [DOI] [PubMed] [Google Scholar]
- 2. Spooner R. A., Watson P. D., Marsden C. J., Smith D. C., Moore K. A., Cook J. P., Lord J. M., Roberts L. M. (2004) Protein disulphide-isomerase reduces ricin to its A and B chains in the endoplasmic reticulum. Biochem. J. 383, 285–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Endo Y., Mitsui K., Motizuki M., Tsurugi K. (1987) The mechanism of action of ricin and related toxic lectins on eukaryotic ribosomes. The site and the characteristics of the modification in 28 S ribosomal RNA caused by the toxins. J. Biol. Chem. 262, 5908–5912 [PubMed] [Google Scholar]
- 4. Montanaro L., Sperti S., Stirpe F. (1973) Inhibition by ricin of protein synthesis in vitro. Ribosomes as the target of the toxin. Biochem. J. 136, 677–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Olsnes S., Refsnes K., Pihl A. (1974) Mechanism of action of the toxic lectins abrin and ricin. Nature 249, 627–631 [DOI] [PubMed] [Google Scholar]
- 6. Endo Y., Tsurugi K. (1988) The RNA N-glycosidase activity of ricin A-chain. The characteristics of the enzymatic activity of ricin A-chain with ribosomes and with rRNA. J. Biol. Chem. 263, 8735–8739 [PubMed] [Google Scholar]
- 7. Sperti S., Montanaro L., Mattioli A., Stirpe F. (1973) Inhibition by ricin of protein synthesis in vitro. 60 S ribosomal subunit as the target of the toxin. Biochem. J. 136, 813–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Clementi N., Chirkova A., Puffer B., Micura R., Polacek N. (2010) Atomic mutagenesis reveals A2660 of 23S ribosomal RNA as key to EF-G GTPase activation. Nat. Chem. Biol. 6, 344–351 [DOI] [PubMed] [Google Scholar]
- 9. Shi X., Khade P. K., Sanbonmatsu K. Y., Joseph S. (2012) Functional role of the sarcin-ricin loop of the 23S rRNA in the elongation cycle of protein synthesis. J. Mol. Biol. 419, 125–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lugnier A. A., Creppy E. E., Le Meur M. A., Gerlinger P., Dirheimer G. (1977) Reciprocal interactions of ricin from Ricinus communis L. seeds with eukaryote ribosomes. FEBS Lett. 76, 166–172 [DOI] [PubMed] [Google Scholar]
- 11. Battelli M. G. (2004) Cytotoxicity and toxicity to animals and humans of ribosome-inactivating proteins. Mini Rev. Med. Chem. 4, 513–521 [DOI] [PubMed] [Google Scholar]
- 12. Mlsna D., Monzingo A. F., Katzin B. J., Ernst S., Robertus J. D. (1993) Structure of recombinant ricin A chain at 2.3 Å. Protein Sci. 2, 429–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cawley D. B., Hedblom M. L., Houston L. L. (1979) Protection and rescue of ribosomes from the action of ricin A chain. Biochemistry 18, 2648–2654 [DOI] [PubMed] [Google Scholar]
- 14. Honjo E., Watanabe K., Tsukamoto T. (2002) Real-time kinetic analyses of the interaction of ricin toxin A-chain with ribosomes prove a conformational change involved in complex formation. J. Biochem. 131, 267–275 [DOI] [PubMed] [Google Scholar]
- 15. Hedblom M. L., Cawley D. B., Houston L. L. (1976) The specific binding of ricin and its polypeptide chains to rat liver ribosomes and ribosomal subunits. Arch. Biochem. Biophys. 177, 46–55 [DOI] [PubMed] [Google Scholar]
- 16. Suh J. K., Hovde C. J., Robertus J. D. (1998) Shiga toxin attacks bacterial ribosomes as effectively as eucaryotic ribosomes. Biochemistry 37, 9394–9398 [DOI] [PubMed] [Google Scholar]
- 17. Tumer N. E., Li X. P. (2012) Interaction of ricin and Shiga toxins with ribosomes. Curr. Top. Microbiol. Immunol. 357, 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li X. P., Chiou J. C., Remacha M., Ballesta J. P., Tumer N. E. (2009) A two-step binding model proposed for the electrostatic interactions of ricin A chain with ribosomes. Biochemistry 48, 3853–3863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chiou J. C., Li X. P., Remacha M., Ballesta J. P., Tumer N. E. (2008) The ribosomal stalk is required for ribosome binding, depurination of the rRNA and cytotoxicity of ricin A chain in Saccharomyces cerevisiae. Mol. Microbiol. 70, 1441–1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. May K. L., Li X. P., Martínez-Azorin F., Ballesta J. P., Grela P., Tchórzewski M., Tumer N. E. (2012) The P1/P2 proteins of the human ribosomal stalk are required for ribosome binding and depurination by ricin in human cells. FEBS J. 279, 3925–3936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li X. P., Grela P., Krokowski D., Tchórzewski M., Tumer N. E. (2010) Pentameric organization of the ribosomal stalk accelerates recruitment of ricin a chain to the ribosome for depurination. J. Biol. Chem. 285, 41463–41471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wahl M. C., Möller W. (2002) Structure and function of the acidic ribosomal stalk proteins. Curr. Protein Pept. Sci. 3, 93–106 [DOI] [PubMed] [Google Scholar]
- 23. Gonzalo P., Reboud J. P. (2003) The puzzling lateral flexible stalk of the ribosome. Biol. Cell 95, 179–193 [DOI] [PubMed] [Google Scholar]
- 24. Grela P., Krokowski D., Gordiyenko Y., Krowarsch D., Robinson C. V., Otlewski J., Grankowski N., Tchórzewski M. (2010) Biophysical properties of the eukaryotic ribosomal stalk. Biochemistry 49, 924–933 [DOI] [PubMed] [Google Scholar]
- 25. Guarinos E., Santos C., Sánchez A., Qiu D. Y., Remacha M., Ballesta J. P. (2003) Tag-mediated fractionation of yeast ribosome populations proves the monomeric organization of the eukaryotic ribosomal stalk structure. Mol. Microbiol. 50, 703–712 [DOI] [PubMed] [Google Scholar]
- 26. Nomura N., Honda T., Baba K., Naganuma T., Tanzawa T., Arisaka F., Noda M., Uchiyama S., Tanaka I., Yao M., Uchiumi T. (2012) Archaeal ribosomal stalk protein interacts with translation factors in a nucleotide-independent manner via its conserved C terminus. Proc. Natl. Acad. Sci. U.S.A. 109, 3748–3753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Santos C., Ballesta J. P. (1995) The highly conserved protein P0 carboxyl end is essential for ribosome activity only in the absence of proteins P1 and P2. J. Biol. Chem. 270, 20608–20614 [DOI] [PubMed] [Google Scholar]
- 28. Diaconu M., Kothe U., Schlünzen F., Fischer N., Harms J. M., Tonevitsky A. G., Stark H., Rodnina M. V., Wahl M. C. (2005) Structural basis for the function of the ribosomal L7/12 stalk in factor binding and GTPase activation. Cell 121, 991–1004 [DOI] [PubMed] [Google Scholar]
- 29. Chan D. S., Chu L. O., Lee K. M., Too P. H., Ma K. W., Sze K. H., Zhu G., Shaw P. C., Wong K. B. (2007) Interaction between trichosanthin, a ribosome-inactivating protein, and the ribosomal stalk protein P2 by chemical shift perturbation and mutagenesis analyses. Nucleic Acids Res. 35, 1660–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McCluskey A. J., Poon G. M., Bolewska-Pedyczak E., Srikumar T., Jeram S. M., Raught B., Gariépy J. (2008) The catalytic subunit of Shiga-like toxin 1 interacts with ribosomal stalk proteins and is inhibited by their conserved C-terminal domain. J. Mol. Biol. 378, 375–386 [DOI] [PubMed] [Google Scholar]
- 31. McCluskey A. J., Bolewska-Pedyczak E., Jarvik N., Chen G., Sidhu S. S., Gariépy J. (2012) Charged and hydrophobic surfaces on the a chain of shiga-like toxin 1 recognize the C-terminal domain of ribosomal stalk proteins. PLoS One 7, e31191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee K. M., Yusa K., Chu L. O., Wing-Heng Yu C., Oono M., Miyoshi T., Ito K., Shaw P. C., Wong K. B., Uchiumi T. (2013) Solution structure of human P1*P2 heterodimer provides insights into the role of eukaryotic stalk in recruiting the ribosome-inactivating protein trichosanthin to the ribosome. Nucleic Acids Res., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Too P. H., Ma M. K., Mak A. N., Wong Y. T., Tung C. K., Zhu G., Au S. W., Wong K. B., Shaw P. C. (2009) The C-terminal fragment of the ribosomal P protein complexed to trichosanthin reveals the interaction between the ribosome-inactivating protein and the ribosome. Nucleic Acids Res. 37, 602–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang Y., Mak A. N., Shaw P. C., Sze K. H. (2010) Solution structure of an active mutant of maize ribosome-inactivating protein (MOD) and its interaction with the ribosomal stalk protein P2. J. Mol. Biol. 395, 897–907 [DOI] [PubMed] [Google Scholar]
- 35. Lapadula W. J., Sanchez-Puerta M. V., Ayub M. J. (2012) Convergent evolution led ribosome inactivating proteins to interact with ribosomal stalk. Toxicon 59, 427–432 [DOI] [PubMed] [Google Scholar]
- 36. Dai J., Zhao L., Yang H., Guo H., Fan K., Wang H., Qian W., Zhang D., Li B., Wang H., Guo Y. (2011) Identification of a novel functional domain of ricin responsible for its potent toxicity. J. Biol. Chem. 286, 12166–12171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Watanabe K., Funatsu G. (1986) Involvement of arginine residues in inhibition of protein synthesis by ricin A-chain. FEBS Lett. 204, 219–222 [DOI] [PubMed] [Google Scholar]
- 38. Watanabe K., Dansako H., Asada N., Sakai M., Funatsu G. (1994) Effects of chemical modification of arginine residues outside the active site cleft of ricin A-chain on its RNA N-glycosidase activity for ribosomes. Biosci. Biotechnol. Biochem. 58, 716–721 [DOI] [PubMed] [Google Scholar]
- 39. Kitaoka Y. (1998) Involvement of the amino acids outside the active-site cleft in the catalysis of ricin A chain. Eur. J. Biochem. 257, 255–262 [DOI] [PubMed] [Google Scholar]
- 40. Li X. P., Baricevic M., Saidasan H., Tumer N. E. (2007) Ribosome depurination is not sufficient for ricin-mediated cell death in Saccharomyces cerevisiae. Infect. Immun. 75, 417–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang T., Lei J., Yang H., Xu K., Wang R., Zhang Z. (2011) An improved method for whole protein extraction from yeast Saccharomyces cerevisiae. Yeast 28, 795–798 [DOI] [PubMed] [Google Scholar]
- 42. O'Hara J. M., Neal L. M., McCarthy E. A., Kasten-Jolly J. A., Brey R. N., 3rd, Mantis N. J. (2010) Folding domains within the ricin toxin A subunit as targets of protective antibodies. Vaccine 28, 7035–7046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. O'Hara J. M., Yermakova A., Mantis N. J. (2012) Immunity to ricin. Fundamental insights into toxin-antibody interactions. Curr. Top. Microbiol. Immunol. 357, 209–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Whitmore L., Wallace B. A. (2008) Protein secondary structure analyses from circular dichroism spectroscopy. Methods and reference databases. Biopolymers 89, 392–400 [DOI] [PubMed] [Google Scholar]
- 45. Lees J. G., Miles A. J., Wien F., Wallace B. A. (2006) A reference database for circular dichroism spectroscopy covering fold and secondary structure space. Bioinformatics 22, 1955–1962 [DOI] [PubMed] [Google Scholar]
- 46. Provencher S. W., Glöckner J. (1981) Estimation of globular protein secondary structure from circular dichroism. Biochemistry 20, 33–37 [DOI] [PubMed] [Google Scholar]
- 47. Pierce M., Kahn J. N., Chiou J., Tumer N. E. (2011) Development of a quantitative RT-PCR assay to examine the kinetics of ribosome depurination by ribosome inactivating proteins using Saccharomyces cerevisiae as a model. RNA 17, 201–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shi W., Tanaka K. S., Crother T. R., Taylor M. W., Almo S. C., Schramm V. L. (2001) Structural analysis of adenine phosphoribosyltransferase from Saccharomyces cerevisiae. Biochemistry 40, 10800–10809 [DOI] [PubMed] [Google Scholar]
- 49. Sturm M. B., Schramm V. L. (2009) Detecting ricin. Sensitive luminescent assay for ricin A-chain ribosome depurination kinetics. Anal Chem. 81, 2847–2853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang H. C., Ciskanik L., Dunaway-Mariano D., von der Saal W., Villafranca J. J. (1988) Investigations of the partial reactions catalyzed by pyruvate phosphate dikinase. Biochemistry 27, 625–633 [DOI] [PubMed] [Google Scholar]
- 51. Goss N. H., Evans C. T., Wood H. G. (1980) Pyruvate phosphate dikinase. Sequence of the histidyl peptide, the pyrophosphoryl and phosphoryl carrier. Biochemistry 19, 5805–5809 [DOI] [PubMed] [Google Scholar]
- 52. Korennykh A. V., Correll C. C., Piccirilli J. A. (2007) Evidence for the importance of electrostatics in the function of two distinct families of ribosome inactivating toxins. RNA 13, 1391–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Korennykh A. V., Piccirilli J. A., Correll C. C. (2006) The electrostatic character of the ribosomal surface enables extraordinarily rapid target location by ribotoxins. Nat. Struct. Mol. Biol. 13, 436–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yan Q., Li X. P., Tumer N. E. (2012) N-Glycosylation does not affect the catalytic activity of ricin a chain but stimulates cytotoxicity by promoting its transport out of the endoplasmic reticulum. Traffic 13, 1508–1521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kahn P. C. (1979) The interpretation of near-ultraviolet circular dichroism. Methods Enzymol. 61, 339–378 [DOI] [PubMed] [Google Scholar]
- 56. Chen X. Y., Link T. M., Schramm V. L. (1998) Ricin A-chain. Kinetics, mechanism, and RNA stem-loop inhibitors. Biochemistry 37, 11605–11613 [DOI] [PubMed] [Google Scholar]
- 57. Frigerio L., Vitale A., Lord J. M., Ceriotti A., Roberts L. M. (1998) Free ricin A chain, proricin, and native toxin have different cellular fates when expressed in tobacco protoplasts. J. Biol. Chem. 273, 14194–14199 [DOI] [PubMed] [Google Scholar]
- 58. Schlossman D., Withers D., Welsh P., Alexander A., Robertus J., Frankel A. (1989) Role of glutamic acid 177 of the ricin toxin A chain in enzymatic inactivation of ribosomes. Mol. Cell. Biol. 9, 5012–5021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Frankel A., Welsh P., Richardson J., Robertus J. D. (1990) Role of arginine 180 and glutamic acid 177 of ricin toxin A chain in enzymatic inactivation of ribosomes. Mol. Cell. Biol. 10, 6257–6263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ready M. P., Kim Y., Robertus J. D. (1991) Site-directed mutagenesis of ricin A-chain and implications for the mechanism of action. Proteins 10, 270–278 [DOI] [PubMed] [Google Scholar]
- 61. Kim Y., Robertus J. D. (1992) Analysis of several key active site residues of ricin A chain by mutagenesis and x-ray crystallography. Protein Eng. 5, 775–779 [DOI] [PubMed] [Google Scholar]
- 62. Monzingo A. F., Robertus J. D. (1992) X-ray analysis of substrate analogs in the ricin A-chain active site. J. Mol. Biol. 227, 1136–1145 [DOI] [PubMed] [Google Scholar]
- 63. Olson M. A., Cuff L. (1999) Free energy determinants of binding the rRNA substrate and small ligands to ricin A-chain. Biophys. J. 76, 28–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Marsden C. J., Fülöp V., Day P. J., Lord J. M. (2004) The effect of mutations surrounding and within the active site on the catalytic activity of ricin A chain. Eur. J. Biochem. 271, 153–162 [DOI] [PubMed] [Google Scholar]
- 65. Olson M. A. (1997) Ricin A-chain structural determinant for binding substrate analogues. A molecular dynamics simulation analysis. Proteins 27, 80–95 [DOI] [PubMed] [Google Scholar]
- 66. Morris K. N., Wool I. G. (1992) Determination by systematic deletion of the amino acids essential for catalysis by ricin A chain. Proc. Natl. Acad. Sci. U.S.A. 89, 4869–4873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Voorhees R. M., Schmeing T. M., Kelley A. C., Ramakrishnan V. (2010) The mechanism for activation of GTP hydrolysis on the ribosome. Science 330, 835–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Argent R. H., Parrott A. M., Day P. J., Roberts L. M., Stockley P. G., Lord J. M., Radford S. E. (2000) Ribosome-mediated folding of partially unfolded ricin A-chain. J. Biol. Chem. 275, 9263–9269 [DOI] [PubMed] [Google Scholar]
- 69. Spooner R. A., Lord J. M. (2012) How ricin and Shiga toxin reach the cytosol of target cells. Retrotranslocation from the endoplasmic reticulum. Curr. Top. Microbiol. Immunol. 357, 19–40 [DOI] [PubMed] [Google Scholar]