

Background: Prion proteins may adopt multiple aggregate conformations, known as strains.
Results: Kosmotropic and chaotropic anions exhibit opposite effects on aggregation kinetics and favor different strains.
Conclusion: Both prion nucleation kinetics and prevailing strain patterns strongly depend on ionic composition of the aggregation mixture.
Significance: Ionic composition is shown to be a critical determinant in the generation of prion strains.
Keywords: Amyloid, Protein Aggregation, Protein Conformation, Translation Release Factors, Yeast, Chaotrope, Hofmeister Series, Kosmotrope
Abstract
Ordered, fibrous, self-seeding aggregates of misfolded proteins known as amyloids are associated with important diseases in mammals and control phenotypic traits in fungi. A given protein may adopt multiple amyloid conformations, known as variants or strains, each of which leads to a distinct disease pattern or phenotype. Here, we study the effect of Hofmeister ions on amyloid nucleation and strain generation by the prion domain-containing fragment (Sup35NM) of a yeast protein Sup35p. Strongly hydrated anions (kosmotropes) initiate nucleation quickly and cause rapid fiber elongation, whereas poorly hydrated anions (chaotropes) delay nucleation and mildly affect the elongation rate. For the first time, we demonstrate that kosmotropes favor formation of amyloid strains that are characterized by lower thermostability and higher frangibility in vitro and stronger phenotypic and proliferation patterns effectively in vivo as compared with amyloids formed in chaotropes. These phenomena point to inherent differences in the biochemistry of Hofmeister ions. Our work shows that the ionic composition of a solution not only influences the kinetics of amyloid nucleation but also determines the amyloid strain that is preferentially formed.
Introduction
Amyloidoses are disorders marked by the deposition of fibrous protein aggregates (amyloids) within an organism (1, 2). In mammals, these disorders include widespread sporadic or familial neurodegenerative diseases such as Alzheimer and Parkinson disease. Infectious amyloids, termed “prions,” cause transmissible spongiform encephalopathies or prion diseases, which include Creutzfeldt-Jakob disease in humans and “mad cow” disease in cattle (for review, see Refs. 1, 3, and 4). Not all amyloidoses are pathological, however. For example, amyloid of Pmel17 protein, formed in the mammalian melanocyte, is biologically beneficial (5). In yeast and fungi, there is an ever growing list of prions that control specific phenotypic traits; some of these prions may also have pathological effects (6–9). Amyloids are also formed spontaneously in the preparations of protein- or peptide-based drugs upon storage that decreases drug efficiency and may increase immunogenicity (10).
The basic scheme of amyloid formation appears to be the same for all amyloidogenic proteins, mammalian or fungal. The process typically originates with a change in the secondary structure of the protein, resulting in an increased proportion of β-sheets (11). For many, maybe most (12) proteins, such a conversion can occur in vitro under extreme solution conditions (high temperature and low pH), as in the cases of lysozyme, transthyretin, and insulin (13–16). Prion proteins, however, spontaneously misfold under physiological conditions. Regardless of how the conformers were induced, the β-sheet-rich peptides aggregate to form nuclei. Solution conditions play a great role in determining the rate of nucleation and the structure of the nucleus (17–19); however, this relationship is not yet clearly understood. After nucleation, other peptides of the same amino acid sequence are recruited to the aggregate in a unidirectional manner, analogous to “one-dimensional crystallization.”
Two main factors dictate the folding state, nucleation, and aggregation propensity of proteins: the primary sequence and environmental conditions (20–22). The high primary sequence specificity of amyloid propagation has been clearly demonstrated through mutational studies, construction of synthetic prion, and species barrier studies (23–28). However, the mechanism of a curious phenomenon in prion biology where a given peptide can misfold into a variety of distinct amyloid structures, each leading to a distinct transmissible or inheritable phenotype (29–31), remains unclear. The question arises as to whether environmental factors, such as pH, co-solutes, shear, temperature, and ionic strength, can elicit these different aggregation states giving rise to different prion “strains” or “variants.”
Prion strains have been extensively studied for the yeast prion protein Sup35p, the protein determinate of the [PSI+] prion (11, 31–36). In vitro studies typically employ a 253-residue, amyloid-forming fragment of Sup35p, called Sup35NM. Sup35NM has been shown to predominantly form different strains of [PSI+] when nucleation occurs at different temperatures (17, 37–40). Amyloids formed in vitro at 4 °C (Sc4) and 37 °C (Sc37) preferentially induced “strong” and “weak” [PSI+] strains, respectively, when transfected into Saccharomyces cerevisiae (37). These different [PSI+] strains were shown to form at different rates (38), through different pathways (17), and create distinct structures (39). Notably, phenotypically strong strains are characterized by less stable physical structure and shorter amyloid core region; the opposite is true of phenotypically weak strains. This is due to the fact that efficient prion propagation in vivo occurs via a chaperone-mediated fragmentation of aggregates that generates new seeds. Amyloids that are physically more stable are less efficiently fragmented and therefore are phenotypically weak (6).
Similar to temperature, the ionic composition of the solution has been shown to affect aggregation kinetics in a number of amyloid-forming proteins, including α-synuclein (41), mouse prion protein PrP (42, 43), PABPN1 (44), and Sup35NM (18); however, the effect of solution composition on amyloid structures and biological effects was not studied in detail. The present work relates the differences in aggregation kinetics of Sup35NM caused by different salts to the structures and propagation parameters of amyloids formed in respective conditions. We find striking differences between aggregates formed in the presence of kosmotropes (well hydrated anions) versus chaotropes (poorly hydrated anions). Overall, our data show for the first time that the ionic composition of the solution modulates the structural patterns of nucleated amyloids.
EXPERIMENTAL PROCEDURES
Sup35NM Purification and Polymerization
Sup35NM was heterologously produced in HMS174 (pLysS) Escherichia coli (Novagen) using a pET20b-SUP35NM-(His)6 expression vector as published previously (45). Purification on nickel-nitrilotriacetic acid resin was performed as described previously (18), and then the purified protein was methanol precipitated at −20 °C. Purified protein was stored at −80 °C in 80% methanol. To polymerize, Sup35NM was resuspended in PBS (pH 7.4) containing 0.5 m of a given sodium salt to a final concentration of 10 μm and allowed to rotate at 20 rpm at room temperature for 48 h. Amyloid samples were frozen at −80 °C until used.
Kinetic Assay
Solutions of thioflavin T (Sigma-Aldrich) were prepared daily in PBS. Aggregation experiments were conducted in quadruplicate with final thioflavin T and Sup35NM concentrations of 100 and 10 μm, respectively, and containing a sodium salt. Polymerization was initiated by shaking samples at 18 Hz linearly at 27 °C in a BioTek Instruments Synergy H4 hybrid multi-mode microplate reader (Winooski, VT). Fluorescent readings were taken every 15 min for 12 h using an excitation wavelength of 440 nm and emission wavelength of 480 nm. Data were modeled using a nonlinear MATLAB sigmoidal curve fitting program (see the supplemental information).
Thermostability Assays
Amyloid samples were centrifuged and washed with deionized water to remove residual salt. Amyloids were resuspended in 100× diluted PBS. For SDS-PAGE gel entry assay, aggregates were held for 15 min in a preheated thermal block and then run on an acrylamide gel. Densitometry analysis was performed using ImageJ software. For the dynamic light scattering (DLS)3 technique, resuspended amyloids were sonicated and then, in a quartz cuvette, subjected to a temperature ramp protocol in a Malvern Zetasizer Nano ZS90 (Worcestershire, UK). Samples were capped with silicon oil to prevent solvent evaporation. The protocol increased the temperature linearly by 1 °C, equilibrated the sample for 2 min, and then took a single size measurement for 1 min. For these size measurements, the z-average of the amyloid diffusion coefficients was obtained from a second-order cumulant fit to the scattering intensity autocorrelation function and translated into a hydrodynamic diameter via the Stokes-Einstein relation.
Semidenaturing Detergent-Agarose Gel Electrophoresis (SDD-AGE)
SDD-AGE experiments were conducted according to previously published protocol (46) except that 0.1% SDS was added to the transfer buffer.
Isoelectric Point Determination
Electrophoretic mobility measurements were conducted using a Malvern ZS90 Zetasizer. Monomeric Sup35NM was suspended in water and pH-adjusted using very dilute NaOH or HCl solutions. The isoelectric points (pI) were determined by finding the pH at which the electrophoretic mobility was zero (i.e. the protein carried no net charge). For Sup35NM, the pI was found at pH 5.3 (data not shown). This finding indicates that at our operating pH of 7.4, Sup35NM carries a negative charge.
Aggregate Imaging
Aggregates were first washed and resuspended in deionized water. For transmission electron microscopy (TEM) images, aggregates were adsorbed on Formvar carbon-coated 200 mesh copper grids by dropping 5 μl of protein solution onto the grid. Fibers were negatively stained using 2 μl of a 3% (w/v) uranyl acetate solution. After 60 s of staining, the sample was washed with deionized water and dried at 40 °C overnight. Images were acquired with a JEOL JEM 100CX II electron microscope operating at 100 kV. The microscope was equipped with a LaB6 filament and side-mounted 4.2 megapixel FLI CCD camera. Bright-field images were acquired at magnifications of 14,000–80,000× with a 300-μm second condenser aperture, 40-μm objective aperture, and “spot size” of 2. MaxIm DL software was used for imaging.
For atomic force microscopy (AFM) samples, 20 μl of aggregate solution was allowed to adsorb onto freshly cleaved mica for 20 min. The sample was then washed and dried at room temperature overnight. Images were acquired using a Veeco Dimension 3100 AFM (Plainview, NY) with a >10-nm AppNano silicon tip in tapping mode. Measurement of individual fibers was conducted offline using NanoScope Analysis software.
Transfection
In vitro generated amyloid was washed with deionized water, sonicated, and then transfected into yeast along with a URA3 marker plasmid using a protocol described by Tanaka et al. (37) with the following modifications. 1 m dithiothreitol was added separately to the SCE (1 m sorbitol, 100 mm sodium citrate, 10 mm EDTA, pH 5.8) buffer to a final concentration of 10 mm; PEG buffer was prepared with 44% (w/v) PEG 3350; and top agar concentration was 0.8%, and it was incubated at 42 °C to prevent solidification. Transfectants were transferred to −ura selective medium and then velveteen replica-plated onto YPD for phenotypic characterization of strains. Successful transfection experiments using sonicated amyloid produced 20–30% [PSI+] colonies among all transfectants analyzed. When nonsonicated amyloids were transfected, the transfection efficiency dropped to ∼10% (data not shown).
Characterization of Prion Strains in Vivo
The S. cerevisiae strain GT17 (MATa ade1-14 (UGA) his3 leu2 trp1 ura3 [psi−] [pin−]) was used for all experiments (47). GT17 contains a nonsense mutation ade1-14 (UGA) in the ADE1 gene, which causes premature translational termination of Ade1 protein synthesis in [psi−] cells. As a result, [psi−] cells cannot grow on media lacking adenine and appear red on YPD media due to buildup of an intermediate in the adenine biosynthetic pathway (6). In [PSI+] cells, Sup35 protein is sequestered in aggregates, and read-through of the nonsense mutation occurs. Read-through results in proper Ade1p expression, allowing for growth on media lacking adenine and a white phenotype on YPD.
“Boiled Gel” Assay
To detect biochemical differences between prion strains, a boiled gel assay was used that allows the comparison of the proportions of soluble and polymerized fractions of the Sup35 protein (48). Monomeric protein can enter polyacrylamide, whereas aggregated protein remains trapped in the wells of the gel. After a short electrophoresis run, the wells were sealed with the new portion of polyacrylamide to prevent leakage, and the whole gel was boiled for 10 min to solubilize the trapped amyloid protein. Then, electrophoresis was resumed and followed by Western blot with the Sup35 antibody (kindly provided by D. Bedwell). Polymerized Sup35 is seen as the top band, whereas soluble Sup35 is seen as the bottom band.
RESULTS
Ion Specificity and Sup35NM Aggregation Kinetics
We first examined the effects of salt type and salt concentration on Sup35NM amyloid formation in vitro via a fluorescence assay using the amyloid-binding dye thioflavin T (49). A typical aggregation profile is presented in Fig. 1A and in the inset of Fig. 2A. The classical sigmoidal shape of these data is indicative of nucleation-dependent aggregation. Nucleation-dependent aggregation is characterized by three phases: lag phase, fiber elongation, and mature fibril (11). This behavior can be modeled empirically using a sigmoidal curve (50–52),
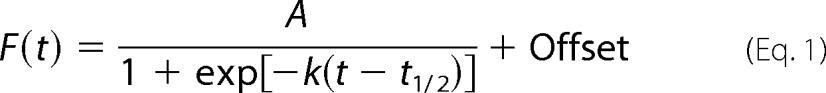 |
where A is the normalized amplitude, k is the fiber elongation rate constant, t is time, and t½ is the time at 50% aggregation. The lag time tlag is given by Equation 2.
 |
FIGURE 1.
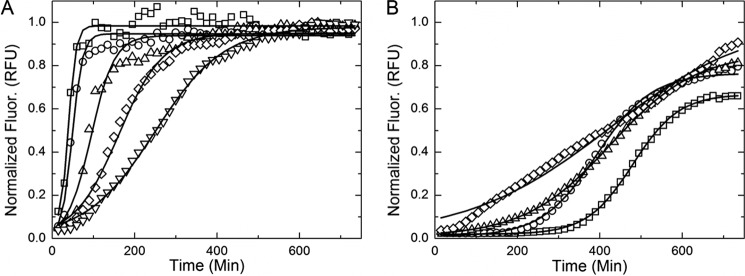
Examples of thioflavin T aggregation kinetic data and model fits for sulfate (A) and perchlorate (B). A and B, amyloid formation kinetics in the presence of sulfate (A) and perchlorate (B) are presented to show the contrasting effects of the ions on aggregation. In the presence of a strong kosmotrope (sulfate), the lag time decreases and aggregation rate increases with increasing salt concentration. The opposite is true of aggregation in a chaotropic solution (perchlorate). The solid black lines shown are nonlinear model fits described under “Results” (Equation 1).Salt molarities: 0.4 (□), 0.3 (○), 0.2 (▵), 0.1 (♢), and 0.025 (▿). MATLAB nonlinear regression models are shown in a solid black line. RFU, relative fluorescence units.
FIGURE 2.
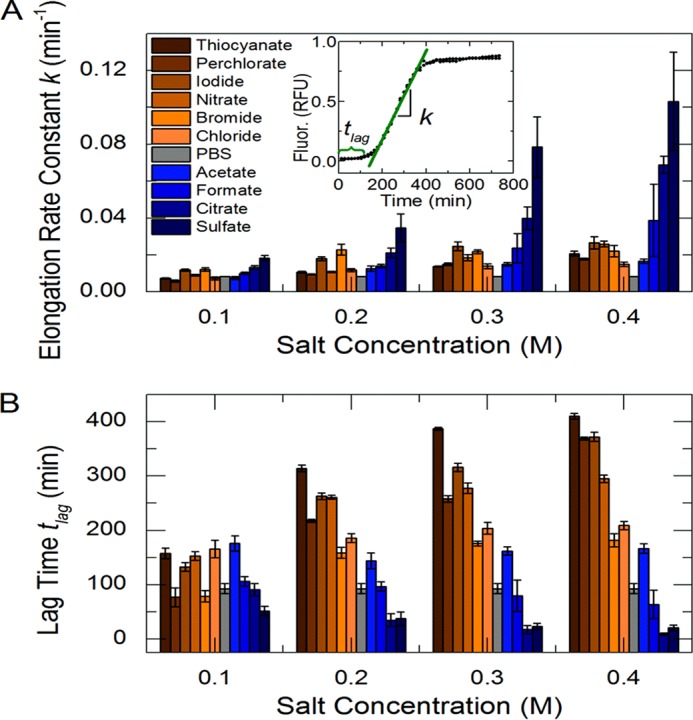
In vitro Sup35NM aggregation kinetics determined using thioflavin T fluorescence. A characteristic aggregation experiment and model parameter determination are shown in the inset to A. Chaotropes are shown in orange, and kosmotropes are shown in blue. The background buffer, PBS, without added salt is shown in gray. The elongation rate constants (A) and lag times (B) are shown over four salt concentrations. RFU, relative fluorescence units. Error bars indicate mean ± S.D.
The duration of the lag and fiber elongation phases varied significantly between ions and salt concentrations (Fig. 1). The rate of fiber elongation (Fig. 2A) was increased and the lag time (Fig. 2B) diminished in the presence of kosmotropes (shown in blue) in an ion-specific and concentration-dependent manner. Chaotropes (shown in orange) dramatically lengthened the lag time in an ion-specific and concentration-dependent fashion (Fig. 2B). Strong chaotropes or kosmotropes (shown as darkness of hue) accentuated these effects.
Chaotropes maintained the unfolded, monomeric form of Sup35NM, whereas kosmotropes promoted folding and thereby formation of the polymeric (aggregated) form. These results confirm the ostensibly “inverse” Hofmeister trend previously observed (18). Having established that ion-specific effects greatly affect aggregation rate, we examined the structural and biological effects of these differing rates.
Temperature Stability and Frangibility
We hypothesized that a correlation exists between the time aggregates take to form in different saline solutions and the compactness and stability of the resulting aggregate structure. This hypothesis is motivated by an analogous kinetics-structure relation known from the aggregation of isotropically interacting colloidal particles (53) and by the general notion that slower aggregation kinetics allows for the exploration of a larger configuration space and the realization of lower energy aggregate states. According to this expectation, a faster forming amyloid would contain more imperfect intermolecular bonds and have a less stable amyloid structure, as observed for Sup35NM amyloids preferentially formed at low temperature. On the other hand, a slow forming amyloid would create an ordered structure with well formed intermolecular bonds, allowing for a more robust amyloid core, as in Sup35NM amyloids preferentially formed at high temperature (37). We tested this prediction using two different techniques, both applied to amyloids produced in the presence of various salts (Fig. 3C). In both cases, amyloids were subjected to a temperature ramp, and either particle size was measured via DLS or the proportion of nonaggregated (monomeric) protein was determined based on its ability to enter an SDS-PAGE gel (gel entry assay). DLS detects a rapid growth of the protein size above a threshold temperature TD, which we attribute to the coagulation of at least partially denatured, colloidally unstable protein. This threshold temperature TD determined by DLS (Fig. 3B) correlates well with the temperature of disaggregation determined by gel entry assay (Fig. 3A). Observed data are consistent with a disruption of the highly ordered detergent-resistant structure of amyloid aggregates and their replacement by large detergent-sensitive agglomerates of partly denatured protein molecules with increased temperature. Moreover, despite slight numerical differences between the DLS-based and gel-based assays, caused by qualitative and conventional differences in the two techniques, both assays indicate that amyloids created in the presence of kosmotropes have lower thermostability than those formed more slowly in chaotropic conditions.
FIGURE 3.
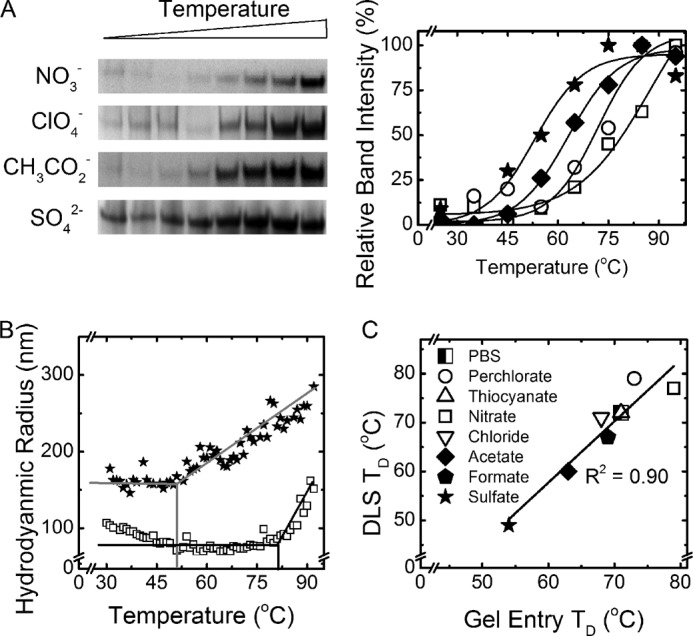
Analysis of the amyloid thermostability. A, examples of the gel entry assay are shown. B, examples of the DLS denaturation temperature ramp are shown. C, gel entry assay and DLS determined thermostability of amyloids. Chaotropes are shown with open icons, and kosmotropes are shown with closed icons.
Amyloid frangibility was assessed by fractionating polymers by sizes using SDD-AGE after sonication. Amyloids formed in the presence of sulfate (ScS), a strong kosmotrope, were more readily fragmented by sonication than amyloids formed in perchlorate (ScP), a strong chaotrope (Fig. 4A). This result also confirms that ScS is physically less stable than ScP. Notably, nonsonicated ScP fibers could not enter the gel, whereas ScS fibers could, indicating that ScP fibers are larger than ScS fibers (Fig. 4B).
FIGURE 4.
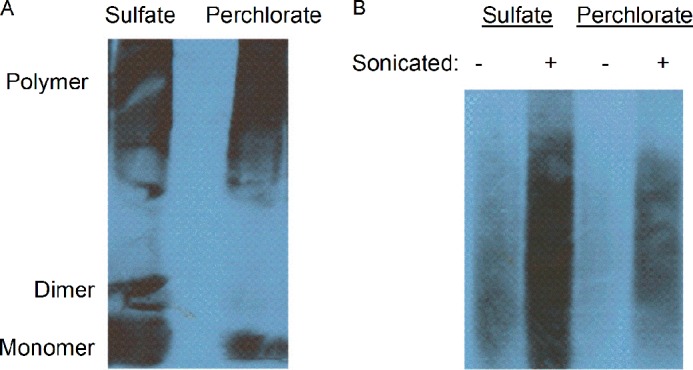
Comparison of the ScS (sulfate) and ScP (perchlorate) aggregates by SDD-AGE and sonication. A, sonication in the same conditions generates a large fraction of monomeric/dimeric protein in the sulfate-produced sample as compared with the perchlorate-produced sample. The images indicate higher frangibility of the sulfate-produced amyloids. B, amyloids generated in sulfate can enter the gel even without sonication, indicating the higher abundance of shorter fibers as compared with amyloids produced in perchlorate that cannot enter the gel in these conditions. After sonication, a larger amount of the sulfate-produced amyloid enters the gel as compared with perchlorate-produced amyloid. This confirms that the fibers formed in sulfate were more frangible than those formed in perchlorate. The gel in panel B was run for a longer time than the gel in panel A to resolve the upper limit of the polymer bands.
The differences in stability, like differences in kinetics (Fig. 2) between amyloids formed in the presence of intermediate Hofmeister ions, were less pronounced than for ScS and ScP (Fig. 3C). Ion-specific effects are more apparent when comparing ions at the extremes of the Hofmeister series, as exemplified by comparing ScS and ScP.
Microscopy Analysis of Amyloids Produced in Saline Solutions
TEM of Sup35NM amyloids formed in the presence of different salts revealed a systematic structural variation dependent on ionic composition. Amyloids formed in chaotropes were visibly more structured as compared with those formed in kosmotropes (Fig. 5, A–E). The most pronounced difference is seen by comparing the highly ordered helical fiber bundles of ScP aggregates (Fig. 5A) with the grainy ScS aggregates (Fig. 5E). The strong contrast between amyloids formed in chaotropes (Fig. 6F) and kosmotropes (Fig. 6G) was also confirmed by using AFM.
FIGURE 5.

Bright-field TEM images of negatively stained amyloid fibers. A–E, amyloids were formed in the presence of the following salts: perchlorate (A), nitrate (B), chloride (C), acetate (D), and sulfate (E).
FIGURE 6.

AFM analysis of amyloid fibers. A–E, AFM analysis of the Sup35NM amyloids formed in perchlorate (A), nitrate (B), chloride (C), acetate (D), and sulfate (E). Scale bar = 500 nm. Length distributions were fit to a log-normal probability density function, shown as a black curve: f(L, μ, σ) = (1/Lσ√2π) exp(−([ln(L) − μ2])/2σ2), where L, μ, and σ are fiber length, mean, and standard deviation, respectively (44). F and G, close-up images of amyloid formed in nitrate (F), a chaotrope, and sulfate (G), a kosmotrope, are shown to contrast their structures.
AFM was employed to measure average fiber lengths. All conditions produced roughly log-normal fiber length distributions (Fig. 6). For the fibers formed in chaotropes, such as perchlorate and nitrate (Fig. 6, A and B), the mean of the fiber length distribution was shifted to the right as compared with the distributions observed in kosmotropes such as acetate and sulfate (Fig. 6, C and D). This indicates that longer fibers are more abundant in chaotropes as compared with kosmotropes. Such a tendency agrees with the frangibility data discussed above.
Phenotypic Characterization of Prion Variants Caused by Ion Specificity
Next, we investigated whether the amyloid structures formed in different salt solutions would lead to different strains of the [PSI+] prion upon transfection into the yeast cells. Conversion of Sup35p into a prion ([PSI+]) form results in a defect of translation termination. [PSI+] strains cause termination defects of varying stringencies. These differences can be detected in a yeast culture bearing a reporter construct with a mutation generating a stop codon (UGA) in the middle of the ADE1 gene (ade1-14). Cells that contain a fully active (non-prion) form of Sup35p are denoted [psi−]; they do not grow on the medium lacking adenine (−Ade) and appear red on YPD medium due to accumulation of the intermediate of the adenine biosynthetic pathway. The degree of Sup35p inactivation depends on the prion variant or strain and leads to a spectrum of [PSI+] phenotypes ranging from white (strong [PSI+]) to dark pink (weak [PSI+]) (6). It is well established that phenotypically strong [PSI+] strains typically correspond to the physically less stable and shorter amyloids; the opposite is true of weak variants (38, 39). This is rationalized by the ability of frangible fibers to break and create more fiber ends at which soluble Sup35p can be quickly recruited to the fiber; conversely, more robust fibers create fewer fiber ends and therefore cannot immobilize and inactivate soluble Sup35p as efficiently in vivo. As a result, a strong correlation is observed between the amyloid frangibility and its phenotypic stringency or proliferation efficiency. Thus, we expected that smaller and frangible fibers assembled in the kosmotropes favoring rapid aggregation, such as ScS, would lead to phenotypically strong [PSI+] strains, whereas larger and more stable fibers, assembled as a result of slow aggregation in chaotropes such as ScP, would lead to phenotypically weak [PSI+].
This expectation was tested by transfecting in vitro formed Sup35NM amyloids into S. cerevisiae cells (Fig. 7A). For simplicity, [PSI+] strains were divided into three classes: strong, intermediate, and weak (Fig. 7B). All three classes were curable by three passages on YPD medium containing 5 mm guanidine HCl (Fig. 7C), as typical of [PSI+] (54). Differences in the amount of Sup35p sequestered in aggregates between the representative [PSI+] strains of different classes were confirmed by Western blot (Fig. 7D).
FIGURE 7.
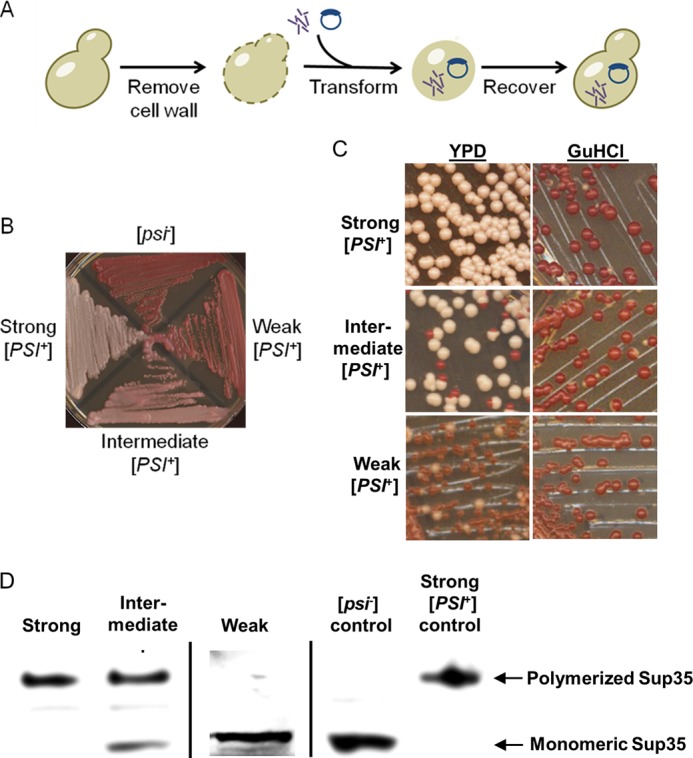
In vivo assay for in vitro generated prion strains. A, transfection experiments were conducted by enzymatically stripping the cell walls of the yeasts and then simultaneously transforming a marker plasmid and amyloid material. The yeast cells were recovered in the stabilizing medium, and resulting colonies were checked for phenotypes. B, [PSI+] strains were classified as strong, intermediate, or weak. C, these [PSI+] strains differed by frequencies of spontaneous mitotic loss of the [PSI+] phenotype, visualized as appearance of red ([psi−]) colonies after three passages on YPD medium, but were all curable after three passages on YPD medium containing 5 mm guanidine hydrochloride, an agent antagonizing prion propagation. D, boiled gel analysis demonstrates that cultures of strong [PSI+] strains contain essentially all Sup35 protein sequestered into an aggregated form (top band), whereas in intermediate strains, some Sup35 protein remains soluble, and in the weak strain, essentially all detectable Sup35 protein is soluble, apparently due to low propagation capabilities of aggregates and gross accumulation of the [psi−] cells. Standard [PSI+] and [psi−] cultures are shown as controls.
In line with our hypothesis, the fast-forming, frangible ScS amyloids primarily produced strong [PSI+] after transfection into yeast (Fig. 8, A and B), whereas the slowly forming, stable ScP amyloids predominantly produced weak [PSI+] (Fig. 8, A and C). A strain continuum was produced by transfected amyloid formed in anions that fall between perchlorate and sulfate; however, the proportion of strong prion isolates was steadily decreasing with an increase of chaotropicity. The differences in the proportions of strong and weak prion strains between the strong chaotrope (perchlorate) and strong kosmotrope (sulfate) were statistically significant (see the legend for Fig. 8). To compare the entire ensemble of phenotypes formed from ScS and ScP amyloids, we employed a “stringency rank” (SR), calculated as outlined below,
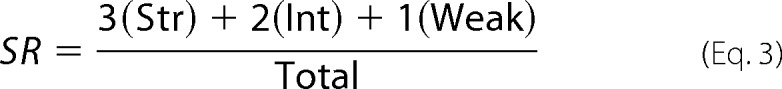 |
where Str, Int, and Weak are numbers of strong, intermediate, and weak [PSI+] colonies, and Total is a total number of [PSI+] colonies obtained in a given transfection experiment. Applying Equation 3 to the data for sulfate and perchlorate presented on Fig. 8, the SRs can be calculated as 2.55 and 1.47, respectively. Performing a Student's t test on these sample populations, we find that p < 0.0005, which provides strong evidence for these samples being statistically different.
FIGURE 8.
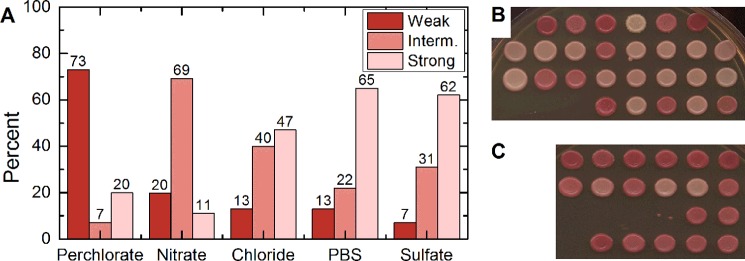
Phenotypic characterization of prion strains generated in the presence of various salts. A, distribution of weak (Weak, red filling), intermediate (Interm., medium pink), and strong (Strong, light pink) [PSI+] colonies in the samples obtained after transfection of yeast cells with Sup35NM amyloids obtained in the presence of various salts, as specified. B and C, patches from transfection of ScP (B) and ScS (C) are shown to illustrate the most divergent phenotype distributions. Proportions of strong [PSI+] colonies derived from transfection with amyloid formed in sulfate (ScS) and perchlorate (ScP) were statistically different with a p < 0.005. Proportions of weak [PSI+] between ScS and ScP were also statistically different with a p < 0.0001.
These results resemble data obtained for Sup35NM amyloids formed at different temperatures (37). We have repeated Sup35NM aggregation at different temperatures (4 and 37 °C) followed by transfection and confirmed that a variety of prion variants was generated at each temperature, with stronger strains more prevalent (but not exclusive) at lower temperature and weaker strains becoming more abundant at higher temperature (data not shown). Nonhomogeneity of each sample apparently originates from the fact that in vitro aggregation is initiated by multiple nucleation events in each case.
DISCUSSION
Our data confirm an inverse Hofmeister effect on Sup35NM aggregation, which agrees with our previous work (18) and with observations for other amyloidogenic proteins that allow this conclusion while not explicitly stating it (41, 42, 44). An inverse Hofmeister trend arises when a colloidal particle is positively charged and hydrophilic or when a particle is negatively charged and hydrophobic (55). We probably observe this trend because unfolded proteins are inherently hydrophobic (56), and at our operating pH, 7.4, Sup35NM carries a net negative charge (pI = 5.3) as determined by electrophoretic mobility measurements (see “Experimental Procedures”).
We hypothesize that the structural and biological differences observed between amyloids produced in kosmotropes versus chaotropes stem from their contrasting amyloid formation kinetics. Although the theory on why and how Hofmeister effects exist is still evolving, the notion that chaotropes adsorb onto the protein surface and kosmotropes preferentially interact with bulk water is generally accepted (55–57).
The adsorption of chaotropes to the surface of the proteins changes the topography and electrostatic landscape of the proteins. Given that amyloid formation is a site-specific, recognition-based process (23–27), such steric and/or electrostatic alterations likely hinder aggregation; however, resulting aggregates are characterized by higher order and more rigid structure. Apparently, the more robust amyloids form due to a selection pressure requiring a higher stringency of intermolecular interactions to overcome the anti-aggregation effect of chaotropic ions. This means that fewer collisions result in an aggregation event and that the aggregates that ultimately do form are structured and stable. Although chaotropes produce more ordered aggregates, their fiber lengths are generally longer than for fibers formed in kosmotropes. This is due to differences in frangibility, resulting in less efficient fragmentation of the chaotrope-derived fibers.
For aggregation in the presence of kosmotropes, we hypothesize that depletion interactions accelerate aggregation (58, 59). Depletion interaction arises when a solute molecule (a hydrated kosmotrope in this case) cannot fit between two colloidal particles (proteins). The absence or depletion of kosmotrope from the gap between colliding means that the osmotic pressure force associated with the solute no longer acts from within the gap but only on the outside and effectively pushes the two particles together. The more hydrated the ion and the higher the salt concentration, the greater the range and strength of this effect become (as seen in Fig. 2). As kosmotropes do not alter the surface of a protein but can be depleted from the gap, aggregates form faster, and intermolecular bonds need not be as precise and energetically favorable as in the case of aggregation in chaotropes. This leads to aggregates that nucleate rapidly, elongate quickly, and are less robust and less stable by comparison.
To explain how and why chaotropes and kosmotropes affect amyloid formation, we propose the following model. The bulk chaotropic solution, devoid of any protein, has lower cohesion as compared with kosmotropic solutions. Upon introducing protein to these solutions, there is a greater energetic penalty to creating an interface in kosmotropic solutions than in chaotropic ones. Therefore, aggregation in kosmotropes is expected to initiate at a higher free energy than in chaotropes. The energetic barrier to forming a nucleus in chaotropic solutions is greater than that of nucleating in kosmotropic solutions, for reasons described above. However, once a nucleus forms in chaotropic solutions, that structure is more highly ordered and stable, with lower free energy as compared with kosmotropic solutions.
Transfection experiments confirm that amyloids formed in chaotropes and kosmotropes indeed produce different sets of [PSI+] strains. Both thermostability and transfection assays reveal a clear analogy between amyloids produced in different salts (present work) and at different temperatures (37–39). At low temperature, strong kosmotropes induce amyloids that are poorly thermostable, frangible, and preferentially generate the phenotypically strong [PSI+] variants upon transfection. The opposite is true for amyloids produced in strong chaotropes or at high temperature. As this and previous works have shown (60, 61), Sup35NM is able to create a number of distinct fiber types, which alludes to fiber conformations as the determinant of strains (37).
We hypothesize that chaotropic anions sterically alter the prion recognition domain and encourage an unfolded intermediate conformation, thus slowing aggregation and forcing the most energetically favorable intermolecular bonds to be selected. In contrast, kosmotropes accelerate aggregation through structural forces (depletion interactions, crowding, and preferential hydration), thus promoting a folded conformation. In the case of an amyloidogenic/prionogenic protein, this favors prion formation because an alternative (prion) fold, once formed, exhibits a high stability and is able to reproduce itself. The disparate kinetics gave rise to varied fiber morphologies, as observed using TEM and AFM imaging. Thermostability and frangibility assays indicated the differences in the fiber robustness and stability, and finally, transfection experiments proved that the varied structures induce different strains. To conclude, our work demonstrates that ion composition of the solution determines the amyloid formation rate, which in turn has a strong bearing on fiber morphology, prion strains, and infectivity. We further corroborate the assertion that the form and function of prion aggregates are interconnected.
More generally, this work supports the notion that environmental conditions are a strong driver of prion variants. The rate of fiber formation, whether it is influenced by temperature or ion composition, is the salient determinant of aggregate size, compactness/order, stability, and infectivity. Our data may be relevant to understanding amyloid formation both in vivo (as local gradients of ions exist in living systems) and in vitro (e.g. in the preparations of protein- or peptide-based drugs).
Acknowledgments
We thank J. T. Park, R. M. Clairmont, G. P. Newnam, N. V. Hud, D. A. Kiktev, and A. V. Romanyuk for technical assistance and fruitful discussion, and D. Bedwell for antibodies.
This work was supported, in whole or in part, by the subaward (to Y. O. C.) on National Institutes of Health Grant R01GM093294. This work was also supported by National Science Foundation (NSF) Grant MCB-1024854 (to Y. O. C.), NSF Industry/University Cooperative Research Centers (I/UCRC) Program Center for Pharmaceutical Development (CPD) Grant 0969003 (to A. S. B. and S. H. B.), the Graduate Assistance in Areas of National Need (GAANN) fellowship through the United States Department of Education (to J. R. and K. L. B.), and the Petite Undergraduate Scholar program through Medtronics (to H. K.).
We dedicate this work to Peter Ziemendorf, Dr. Mélanie Hall, and Dr. Gabriel Rubin for imparting their quality.

This article contains supplemental information about MATLAB model code.
- DLS
- dynamic light scattering
- SDD-AGE
- semidenaturing detergent-agarose gel electrophoresis
- TEM
- transmission electron microscopy
- AFM
- atomic force microscopy
- ScS and ScP
- S. cerevisiae Sup35NM amyloids formed in the presence of sulfate and perchlorate, respectively.
REFERENCES
- 1. Eisenberg D., Jucker M. (2012) The amyloid state of proteins in human diseases. Cell 148, 1188–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Prusiner S. B. (1991) Molecular biology of prion diseases. Science 252, 1515–1522 [DOI] [PubMed] [Google Scholar]
- 3. Prusiner S. B. (1998) Prions. Proc. Natl. Acad. Sci. U.S.A. 95, 13363–13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Weissmann C. (2004) The state of the prion. Nat. Rev. Microbiol. 2, 861–871 [DOI] [PubMed] [Google Scholar]
- 5. Fowler D. M., Koulov A. V., Alory-Jost C., Marks M. S., Balch W. E., Kelly J. W. (2006) Functional amyloid formation within mammalian tissue. PLoS Biol. 4, e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liebman S. W., Chernoff Y. O. (2012) Prions in yeast. Genetics 191, 1041–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McGlinchey R. P., Kryndushkin D., Wickner R. B. (2011) Suicidal [PSI+] is a lethal yeast prion. Proc. Natl. Acad. Sci. U.S.A. 108, 5337–5341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wickner R. B., Edskes H. K., Bateman D., Kelly A. C., Gorkovskiy A. (2011) The yeast prions [PSI+] and [URE3] are molecular degenerative diseases. Prion 5, 258–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wickner R. B., Edskes H. K., Shewmaker F., Nakayashiki T., Engel A., McCann L., Kryndushkin D. (2007) Yeast prions: evolution of the prion concept. Prion 1, 94–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chiti F., Dobson C. M. (2006) Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366 [DOI] [PubMed] [Google Scholar]
- 11. Shorter J., Lindquist S. (2005) Prions as adaptive conduits of memory and inheritance. Nat. Rev. Genet. 6, 435–450 [DOI] [PubMed] [Google Scholar]
- 12. Dobson C. M. (2002) Protein-misfolding diseases: Getting out of shape. Nature 418, 729–730 [DOI] [PubMed] [Google Scholar]
- 13. Chiti F., Webster P., Taddei N., Clark A., Stefani M., Ramponi G., Dobson C. M. (1999) Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc. Natl. Acad. Sci. U.S.A. 96, 3590–3594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bouchard M., Zurdo J., Nettleton E. J., Dobson C. M., Robinson C. V. (2000) Formation of insulin amyloid fibrils followed by FTIR simultaneously with CD and electron microscopy. Protein Sci. 9, 1960–1967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Booth D. R., Sunde M., Bellotti V., Robinson C. V., Hutchinson W. L., Fraser P. E., Hawkins P. N., Dobson C. M., Radford S. E., Blake C. C., Pepys M. B. (1997) Instability, unfolding, and aggregation of human lysozyme variants underlying amyloid fibrillogenesis. Nature 385, 787–793 [DOI] [PubMed] [Google Scholar]
- 16. Chiti F., Taddei N., Bucciantini M., White P., Ramponi G., Dobson C. M. (2000) Mutational analysis of the propensity for amyloid formation by a globular protein. EMBO J. 19, 1441–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ohhashi Y., Ito K., Toyama B. H., Weissman J. S., Tanaka M. (2010) Differences in prion strain conformations result from non-native interactions in a nucleus. Nat. Chem. Biol. 6, 225–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yeh V., Broering J. M., Romanyuk A., Chen B., Chernoff Y. O., Bommarius A. S. (2010) The Hofmeister effect on amyloid formation using yeast prion protein. Protein Sci. 19, 47–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Walters R. H., Jacobson K. H., Pedersen J. A., Murphy R. M. (2012) Elongationkinetics of polyglutamine peptide fibrils: a quartz crystal microbalance with dissipation study. J. Mol. Biol. 421, 329–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chi E. Y., Krishnan S., Randolph T. W., Carpenter J. F. (2003) Physical stability of proteins in aqueous solution: Mechanism and driving forces in nonnative protein aggregation. Pharm. Res. 20, 1325–1336 [DOI] [PubMed] [Google Scholar]
- 21. Fink A. L. (1998) Protein aggregation: folding aggregates, inclusion bodies, and amyloid. Fold. Des. 3, R9–R23 [DOI] [PubMed] [Google Scholar]
- 22. Rubin J., San Miguel A., Bommarius A. S., Behrens S. H. (2010) Correlating aggregation kinetics and stationary diffusion in protein-sodium salt systems observed with dynamic light scattering. J. Phys. Chem. B. 114, 4383–4387 [DOI] [PubMed] [Google Scholar]
- 23. Bruce K. L., Chernoff Y. O. (2011) Sequence specificity and fidelity of prion transmission in yeast. Semin. Cell Dev. Biol. 22, 444–451 [DOI] [PubMed] [Google Scholar]
- 24. Chen B., Newnam G. P., Chernoff Y. O. (2007) Prion species barrier between the closely related yeast proteins is detected despite coaggregation. Proc. Natl. Acad. Sci. U.S.A. 104, 2791–2796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen B., Bruce K. L., Newnam G. P., Gyoneva S., Romanyuk A. V., Chernoff Y. O. (2010) Genetic and epigenetic control of the efficiency and fidelity of cross-species prion transmission. Mol. Microbiol. 76, 1483–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ross E. D., Toombs J. A. (2010) The effects of amino acid composition on yeast prion formation and prion domain interactions. Prion 4, 60–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Toombs J. A., Petri M., Paul K. R., Kan G. Y., Ben-Hur A., Ross E. D. (2012) De novo design of synthetic prion domains. Proc. Natl. Acad. Sci. U.S.A. 109, 6519–6524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tessier P. M., Lindquist S. (2007) Prion recognition elements govern nucleation, strain specificity, and species barriers. Nature 447, 556–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chien P., Weissman J. S., DePace A. H. (2004) Emerging principles of conformation based prion inheritance. Annu. Rev. Biochem. 73, 617–656 [DOI] [PubMed] [Google Scholar]
- 30. Caughey B., Lansbury P. T. (2003) Protofibrils, pores, fibrils, and neurodegeneration: Separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 26, 267–298 [DOI] [PubMed] [Google Scholar]
- 31. Tessier P. M., Lindquist S. (2009) Unraveling infectious structures, strain variants, and species barriers for the yeast prion [PSI+]. Nat. Struct. Mol. Biol. 16, 598–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tuite M. F., Cox B. S. (2006) The [PSI+] prion of yeast: A problem of inheritance. Methods 39, 9–22 [DOI] [PubMed] [Google Scholar]
- 33. Derkatch I. L., Chernoff Y. O., Kushnirov V. V., Inge-Vechtomov S. G., Liebman S. W. (1996) Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae. Genetics 144, 1375–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. King C. Y., Diaz-Avalos R. (2004) Protein-only transmission of three yeast prion strains. Nature 428, 319–323 [DOI] [PubMed] [Google Scholar]
- 35. Toyama B. H., Weissman J. S. (2011) Amyloid structure: Conformational diversity and consequences. Annu. Rev. Biochem. 80, 557–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Uptain S. M., Lindquist S. (2002) Prions as protein-based genetic elements. Annu. Rev. Microbiol. 56, 703–741 [DOI] [PubMed] [Google Scholar]
- 37. Tanaka M., Chien P., Naber N., Cooke R., Weissman J. S. (2004) Conformational variations in an infectious protein determine prion strain differences. Nature 428, 323–328 [DOI] [PubMed] [Google Scholar]
- 38. Tanaka M., Collins S. R., Toyama B. H., Weissman J. S. (2006) The physical basis of how prion conformations determine strain phenotypes. Nature 442, 585–589 [DOI] [PubMed] [Google Scholar]
- 39. Toyama B. H., Kelly M. J. S., Gross J. D., Weissman J. S. (2007) The structural basis of yeast prion strain variants. Nature 449, 233–237 [DOI] [PubMed] [Google Scholar]
- 40. Verges K. J., Smith M. H., Toyama B. H., Weissman J. S. (2011) Strain conformation, primary structure, and the propagation of the yeast prion [PSI+]. Nat. Struct. Mol. Biol. 18, 493–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Munishkina L. A., Henriques J., Uversky V. N., Fink A. L. (2004) Role of protein-water interactions and electrostatics in α-synuclein fibril formation. Biochemistry 43, 3289–3300 [DOI] [PubMed] [Google Scholar]
- 42. Jain S., Udgaonkar J. B. (2010) Salt-induced modulation of the pathway of amyloid fibril formation by the mouse prion protein. Biochemistry 49, 7615–7624 [DOI] [PubMed] [Google Scholar]
- 43. Diaz-Espinoza R., Mukherjee A., Soto C. (2012) Kosmotropic anions promote conversion of recombinant prion protein into a PrPSc-like misfolded form. PLoS One 7, e31678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lodderstedt G., Sachs R., Faust J., Bordusa F., Kühn U., Golbik R., Kerth A., Wahle E., Balbach J., Schwarz E. (2008) Hofmeister salts and potential therapeutic compounds accelerate in vitro fibril formation of the N-terminal domain of PABPN1 containing a disease-causing alanine extension. Biochemistry 47, 2181–2189 [DOI] [PubMed] [Google Scholar]
- 45. Allen K. D., Wegrzyn R. D., Chernova T. A., Müller S., Newnam G. P., Winslett P. A., Wittich K. B., Wilkinson K. D., Chernoff Y. O. (2005) Hsp70 chaperones as modulators of prion life cycle: Novel effects of Ssa and Ssb on the Saccharomyces cerevisiae prion [PSI+]. Genetics 169, 1227–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Halfmann R., Lindquist S. (2008) Screening for amyloid aggregation by semi-denaturing detergent-agarose gel electrophoresis. J. Vis. Exp. pii: 838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bailleul P. A., Newnam G. P., Steenbergen J. N., Chernoff Y. O. (1999) Genetic study of interactions between the cytoskeletal assembly protein Sla1 and prion-forming domain of the release factor Sup35 (eRF3) in Saccharomyces cerevisiae. Genetics 153, 81–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kushnirov V. V., Alexandrov I. M., Mitkevich O. V., Shkundina I. S., Ter-Avanesyan M. D. (2006) Purification and analysis of prion and amyloid aggregates. Methods 39, 50–55 [DOI] [PubMed] [Google Scholar]
- 49. Wolfe L. S., Calabrese M. F., Nath A., Blaho D. V., Miranker A. D., Xiong Y. (2010) Protein-induced photophysical changes to the amyloid indicator dye thioflavin T. Proc. Natl. Acad. Sci. U.S.A. 107, 16863–16868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hellstrand E., Boland B., Walsh D. M., Linse S. (2010) Amyloid β-protein aggregation produces highly reproducible kinetic data and occurs by a two-phase process. ACS Chem. Neurosci. 1, 13–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nielsen L., Khurana R., Coats A., Frokjaer S., Brange J., Vyas S., Uversky V. N., Fink A. L. (2001) Effect of environmental factors on the kinetics of insulin fibril formation: Elucidation of the molecular mechanism. Biochemistry 40, 6036–6046 [DOI] [PubMed] [Google Scholar]
- 52. Sorci M., Grassucci R. A., Hahn I., Frank J., Belfort G. (2009) Time-dependent insulin oligomer reaction pathway prior to fibril formation: cooling and seeding. Proteins 77, 62–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim A. Y., Berg J. C. (2000) Fractal aggregation: Scaling of fractal dimension with stability ratio. Langmuir 16, 2101–2104 [Google Scholar]
- 54. Chernoff Y. O., Uptain S. M., Lindquist S. L. (2002) Analysis of prion factors in yeast. Methods Enzymol. 351, 499–538 [DOI] [PubMed] [Google Scholar]
- 55. Peula-Garcia J. M., Ortega-Vinuesa J. L., Bastos-Gonzalez D. (2010) Inversion of Hofmeister series by changing the surface of colloidal particles from hydrophobic to hydrophilic. J. Phys. Chem. C. 114, 11133–11139 [Google Scholar]
- 56. Pegram L. M., Wendorff T., Erdmann R., Shkel I., Bellissimo D., Felitsky D. J., Record M. T. (2010) Why Hofmeister effects of many salts favor protein folding but not DNA helix formation. Proc. Natl. Acad. Sci. U.S.A. 107, 7716–7721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pegram L. M., Record M. T. (2008) Thermodynamic origin of Hofmeister ion effects. J. Phys. Chem. B. 112, 9428–9436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Asakura S., Oosawa F. (1954) On Interaction between two bodies immersed in a solution of macromolecules. J. Chem. Phys. 22, 1255–1256 [Google Scholar]
- 59. Baynes B. M., Trout B. L. (2004) Rational design of solution additives for the prevention of protein aggregation. Biophys. J. 87, 1631–1639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Glover J. R., Kowal A. S., Schirmer E. C., Patino M. M., Liu J. J., Lindquist S. (1997) Self-seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion-like factor of S. cerevisiae. Cell 89, 811–819 [DOI] [PubMed] [Google Scholar]
- 61. DePace A. H., Weissman J. S. (2002) Origins and kinetic consequences of diversity in Sup35 yeast prion fibers. Nat. Struct. Biol. 9, 389–396 [DOI] [PubMed] [Google Scholar]