

Background: RASSF6 is a proapoptotic protein related to the Hippo pathway.
Results: RASSF6 interacts with MDM2 and stabilizes p53.
Conclusion: RASSF6 induces apoptosis and cell cycle arrest via p53.
Significance: Our work supports the importance of the C-terminal RASSF-MDM2-p53 axis.
Keywords: Apoptosis, Checkpoint Control, DNA Damage, p53, Tumor Suppressor Gene, Hippo Pathway, RASSF
Abstract
Ras association domain family (RASSF) 6 is a member of the C-terminal RASSF proteins such as RASSF1A and RASSF3. RASSF6 is involved in apoptosis in various cells under miscellaneous conditions, but it remains to be clarified how RASSF6 exerts tumor-suppressive roles. We reported previously that RASSF3 facilitates the degradation of MDM2, a major E3 ligase of p53, and stabilizes p53 to function as a tumor suppressor. In this study, we demonstrate that RASSF6 overexpression induces G1/S arrest in p53-positive cells. Its depletion prevents UV- and VP-16-induced apoptosis and G1/S arrest in HCT116 and U2OS cells. RASSF6-induced apoptosis partially depends on p53. RASSF6 binds MDM2 and facilitates its ubiquitination. RASSF6 depletion blocks the increase of p53 in response to UV exposure and up-regulation of p53 target genes. RASSF6 depletion delays DNA repair in UV- and VP-16-treated cells and increases polyploid cells after VP-16 treatment. These findings indicate that RASSF6 stabilizes p53, regulates apoptosis and the cell cycle, and functions as a tumor suppressor. Together with the previous reports regarding RASSF1A and RASSF3, the stabilization of p53 may be the common function of the C-terminal RASSF proteins.
Introduction
RASSF2 proteins comprise 10 members (RASSF1 to RASSF10) (1–4). RASSF1 to RASSF6 harbor Ras association (RA) domains and Salvador/RASSF/Hippo domains in the C-terminal region. RASSF1A, a splicing variant of RASSF1, is a well established tumor suppressor. RASSF6 expression is lost in various human cancers (5). It is epigenetically inactivated in childhood acute lymphocytic leukemia, and its decreased expression is proposed to be a prognostic marker of a worse outcome in gastric cancers (6, 7). These findings suggest that RASSF6 functions as a tumor suppressor in human cancers. However, our understanding, at the mechanistic level, of the tumor suppressive role of RASSF6 is limited. When RASSF6 is exogenously expressed, it induces apoptosis in various cells in caspase-dependent and caspase-independent manners (5, 8, 9). RASSF6 is implicated in tumor necrosis factor α-induced apoptosis in HeLa cells, in okadaic acid-induced apoptosis in rat hepatocytes, and in sorbitol-induced apoptosis in human renal proximal tubular epithelial cells. Considering its proapoptotic property, it is speculated that RASSF6 may kill cells with oncogenic gene mutations and contribute to the eradication of cancer cells. However, the molecular mechanism for RASSF6 to cause apoptosis is not fully clarified. The Hippo pathway is a tumor-suppressive signaling pathway that comprises mammalian Ste20-like (MST) 1 and 2 kinases and large tumor suppressor (LATS) 1 and 2 kinases (10–12). When the Hippo pathway is activated, LATS1 and 2 kinases phosphorylate Yes-associated protein (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ). The phosphorylated YAP and TAZ are recruited from the nucleus to the cytoplasm so that YAP- and TAZ-mediated gene transcriptions are shut down. RASSF6 interacts with MST kinases but induces apoptosis independently of the Hippo pathway (13). The implication of modulator of apoptosis (MOAP) 1, which activates Bax, is demonstrated, but MOAP1 depletion does not completely block RASSF6-induced apoptosis, suggesting that other molecules also work downstream of RASSF6.
The previous studies demonstrate that p53 plays a role in cell cycle regulation by RASSF1A and RASSF5 (also called Nore1) (14, 15). We reported that RASSF3 induces apoptosis and regulates the cell cycle via p53 (16). RASSF1A loss enhances p53-mediated tumor predisposition (17). Restricted activation of p53 because of RASSF1A mutation at the ataxia telangiectasia mutated kinase phosphorylation site exacerbates soft tissue sarcoma (18). Our study also indicates that RASSF3 function depends on p53, although it also plays a tumor suppressive role in p53-compromised cells (16). Here we address the question whether and how p53 is involved in the tumor-suppressive role of RASSF6. In a previous study, RASSF6 suppressed the growth of p53-positive A549 cells but not of p53-defective H1299 cells (5). This finding is supportive of the fact that p53 is required for RASSF6 functions, but it is not yet conclusive. In this study, we examined whether RASSF6 regulates the cell cycle as other RASSF proteins do, whether p53 is indispensable for the tumor-suppressive role of RASSF6, and how RASSF6 suppression potentially leads to oncogenesis at the cell level.
EXPERIMENTAL PROCEDURES
Construction of Expression Vectors
pClneoFH-RASSF6b, pClneoMyc-RASSF6b, pClneoGFP-RASSF6b, pClneoFLAG-MDM2, pCGN HA-ubiquitin (Ubc), pClneoFH, pClneoGFP, and pBudCE2 have been described previously (13, 16). NheI/EcoRI fragments from pEGFPC2 (Clontech) and pClneoFH were subcloned into the same sites of pBudCE2 to generate pBudCGFP and pBudCFLAG. pBudCGFP-SUMO-1 and pBudCFLAG-CIN85 were constructed using pBudCGFP and pBudCFLAG. pClneomCherry was constructed by inserting mCherry into the NheI/EcoRI sites of pClneo. pClneomCherry-MDM2, pClneoGFP-p53, and pClneoFLAG-p53 were generated by using pClneomCherry, pClneoGFP, and pClneoFLAG.
Antibodies and Reagents
Rabbit anti-RASSF6 antibody has been described previously (9, 13). The other antibodies and reagents were obtained from commercial sources as follows: mouse anti-Myc (9E10, ATCC); mouse anti-FLAG M2 (catalog no. F3165), mouse anti-α-tubulin (catalog no. T9026), Hoechst 33342, thymidine, and propidium iodide (PI) (Sigma-Aldrich); mouse anti-GFP (catalog no. sc-9996) and mouse anti-MDM2 (catalog no. sc-965) (Santa Cruz Biotechnology); mouse anti-HA (Roche); mouse anti-p53 (clone DO-1) (Leica); mouse anti-cytochrome c (clone 6H2.B4), mouse anti-cytochrome c (clone 7H8.2C12) (BD Biosciences); rabbit anti-acetyl-p53 (Lys-373) (catalog no. 06-916), mouse anti-actin (clone C4), and donkey fluorescein-isothiocyanate-conjugated and donkey rhodamine-conjugated secondary antibody (Millipore); rabbit anti-mCherry (catalog no. 5993-100) (BioVision); peroxidase-conjugated secondary antibodies (ICN Cappel); etoposide (VP-16) and doxorubicin hydrochloride (Wako Chemicals); tetramethylrhodamine methyl ester perchlorate (Molecular Probes); mouse anti-voltage-dependent anion channel (catalog no. 529532), mouse anti-phosphorylated histone γ-H2AX (JBW301), and MG132 (Calbiochem); and Nutlin-3 (Cayman Chemical Co.).
Cell Cultures, Transfection, and RNA Interferences
HEK293FT, H1299, U2S, TIG3, HCT116 p53+/+, and p53−/− cells were cultured in DMEM containing 10% FBS, 10 mm Hepes-NaOH (pH 7.4), 100 units/ml of penicillin, and 100 mg/liter of streptomycin under 5% CO2 at 37 °C. HCT116 p53+/+ and p53−/− cells were gifts from Dr. Bert Vogelstein. DNA and siRNA transfections were performed using Lipofectamine 2000 (Invitrogen) and Lipofectamine RNAiMAX (Invitrogen). dsRNAs were purchased from Thermo Fisher Scientific and Ambion. p53 (catalog no. 7157) and p73 (catalog no. 7161) were purchased from Dharmacon. Human MOAP1 (catalog no. 130306) was purchased from Ambion. Human RASSF6 (catalog nos. 217240 #1, S46640 #2, and S46639 #3) and human MDM2 (catalog no. s8629) were from Ambion. siGENOME non-targeting siRNA pool #1 (Thermo Fisher Scientific) and Silencer Select negative control no.2 (Ambion) were used as negative controls.
Quantitative RT-PCR
Quantitative RT-PCR analysis was performed using SYBR Green (Roche) and the DNA Engine Opticon system (Bio-Rad) as described previously (13). The primers for RASSF6, MOAP-1, p53, MDM2, PUMA, p21, and actin have been described previously (13, 16). The primers for p73, Bax, and Btg2 were as follows: human p73, 5′-cccaccactttgaggtcact-3′ and 5′-ggcgatctggcagtagagtt-3′; human Bax, 5′-atgttttctgacggcaacttc-3′ and 5′-atcagttccggcaccttg-3′; and human Btg2, 5′-gcgagcagaggcttaaggt-3′ and 5′-gggaaaccagtggtgtttgta-3′.
Senescence
Senescence was evaluated using a senescence β-galactosidase staining kit (Cell Signaling Technology).
Immunofluorescence
Immunofluorescence was performed as described previously (8).
Immunoprecipitation
Myc-RASSF6 and FLAG-MDM2 were expressed in HEK293FT cells. Immunoprecipitation was performed as described previously (13). To detect the interaction of endogenous RASSF6 with MDM2, Wistar rat (male, 8 weeks old) liver was lysed in buffer (25 mm Tris-HCl (pH 7.4), 100 mm NaCl, 1% (w/v) Triton X-100, 1 mg/liter 4-amidinophenylmethanesulfonyl fluoride, 10 mg/l leupeptin, 10 mg/l pepstatin A, 10 μm MG132, and 1 mm EDTA). The lysates were centrifuged at 20,000 × g for 15 min at 4 °C. The supernatant was incubated with RASSF6 antibody or MDM2 antibody fixed on protein G-Sepharose 4 fast-flow beads (GE Healthcare). The beads were washed three times with buffer (25 mm Tris-HCl (pH 7.4), 100 mm NaCl, and 1% (w/v) Triton X-100). The precipitates were analyzed by SDS-PAGE and immunoblotting.
Ultraviolet Light Exposure
Cells were exposed to ultraviolet light using the UV cross-linker CL-100 (UVP, an Analytik Jena Co.).
Apoptosis Detection Assays
Cell survival and cell death were analyzed as described previously (8, 13, 16). In brief, control GFP or GFP-RASSF6 was expressed in U2OS, HCT116 p53+/+, and HCT116 p53−/− cells. GFP-positive cells were gated and analyzed by flow cytometry (FACSCalibur, BD Biosciences). Mitochondrial membrane permeability was measured by 200 nm TMRM. DNA content was determined by PI staining to analyze sub-G1 population. FACS data were analyzed by CellQuest software (BD Biosciences).
Subcellular Fractionation
Cells were grown at 4 × 106 in a 100-mm plate and irradiated with 25 J/m2 UV light. 24 h later, cells were washed with PBS, harvested by scraping on ice, and pelleted by centrifugation at 2,000 × g for 2 min. Cell pellets were resuspended in 200 μl of buffer A (20 mm HEPES-KOH (pH 7.5), 10 mm KCl, 1.5 mm MgCl2, 1 mm sodium EDTA, 1 mm sodium EGTA, 1 mm dithiothreitol, and 250 mm sucrose) with a mixture of protease inhibitors (1 mg/liter 4-amidinophenylmethanesulfonyl fluoride, 10 mg/liter leupeptin, and 10 mg/liter pepstatin A). Cell suspensions were passed through a 26-gauge needle eight times. Unbroken cells, large plasma membrane pieces, and nuclei were removed by centrifugation at 1,000 × g for 10 min at 4 °C. The resulting supernatant was centrifuged at 10,000 × g for 20 min at 4 °C. The pellet fraction (mitochondria) was washed with buffer A and then solubilized and boiled in 50 μl of SDS sample buffer. The supernatant was centrifuged at 100,000 × g for 1 h at 4 °C. The second supernatant was mixed with 5× volume of acetone, stored at −80 °C overnight, and centrifuged at 10,000 × g for 20 min at 4 °C. The pellet fraction (cytosol) was solubilized and boiled in 50 μl of SDS sample buffer.
Cell Cycle Analysis
HCT116 p53+/+ and U2OS cells were transfected with control pBudGFP-SUMO-1 or pClneoGFP-RASSF6. To evaluate the effect of GFP-RASSF6, 6 h after transfection, a thymidine block was started by culturing cells in DMEM containing 10% FBS and 2 mm thymidine. 20 h later, cells were washed, incubated in fresh DMEM containing 10% FBS, and fixed at the indicated time points. GFP-positive cells were gated and analyzed. For the FACS analysis, the cells were washed with PBS, fixed with 70% (v/v) ethanol in PBS overnight at −20 °C, washed with PBS, and stained for 15 min with 10 mg/liter PI in PBS containing 100 mg/liter RNaseA. Each time, at least 5 × 103 GFP-positive cells were analyzed. For the knockdown experiments, the cells were transfected with control dsRNA or RASSF6 dsRNA, cultured in serum-free medium for 48 h to block the cell cycle, exposed to 10 J/m2 UV light, and released from serum starvation.
Ubiquitination Assay
Various tagged RASSF6 and MDM2 were expressed with HA-Ubc in HEK293FT cells. The cells were treated with 10 μm MG132 for 6 h, and immunoprecipitation was performed as described above.
Statistical Analysis
Statistical analyses were performed with Student's t test for comparison between two samples and analysis of variance with Bonferroni's post hoc test for multiple comparisons using GraphPad Prism software (GraphPad Software). Data expressed as percentages were subjected to arcsine square root transformation before analysis of variance.
RESULTS
RASSF6 Is Involved in UV light- and VP-16-induced Apoptosis and UV light-induced G1/S Arrest
We first tested whether RASSF6 is involved in UV light-induced apoptosis and cell cycle arrest in HCT116 cells. The exposure to 25 J/m2 of UV light induced cell death and cytochrome c release and increased the sub-G1 population in HCT116 cells (Fig. 1, A, C, and D, No treatment and 25 J/m2 UV). In this study, we used three dsRNAs to knock down human RASSF6 for U2OS cells and HCT116 cells. All of them effectively suppressed RASSF6 (Fig. 1B). RASSF6 knockdown blocked cytochrome c release and attenuated the increase of the sub-G1 population (Fig. 1, A, C, and D, siCont and siRASSF6). We confirmed the decrease of the sub-G1 population by RASSF6 knockdown using additional dsRNAs (dsRNA#2 and #3) (data not shown). RASSF6 knockdown decreased the VP-16-induced sub-G1 population (Fig. 1E). We also evaluated UV light-induced cytochrome c release in the subcellular fractionation (Fig. 1F, white and black arrows). The exposure to 10 J/m2 UV light caused G1/S arrest in HCT116 cells (Fig. 2A, a white arrow). RASSF6 knockdown overrode UV light-induced cell cycle arrest (Fig. 2A, black arrow). RASSF6 knockdown by the different dsRNA showed a similar effect (data not shown). However, when HCT116 cells were exposed to 25 J/m2 UV light, RASSF6 knockdown had no effect on G1/S arrest (data not shown). When we exogenously overexpressed RASSF6 in HCT116 and U2OS cells, it caused G1/S arrest (Fig. 2B, black arrows). These data suggest that overexpressed RASSF6 induces not only apoptosis but also cell cycle arrest and that RASSF6 at the endogenous level is necessary for UV light- and VP-16-induced apoptosis and UV light-induced G1/S arrest.
FIGURE 1.

RASSF6 is involved in UV light-induced and VP-16-induced apoptosis in HCT116 cells. A–D, HCT116 cells were transfected with control (siCont) or RASSF6-targeted (siRASSF6#1) dsRNA. 72 h later, the cells were exposed to 25 J/m2 UV light and cultured for 12 h. A, representative phase contrast images. Scale bar = 100 μm. B, validation of three dsRNAs for RASSF6. The validities of knockdowns of RASSF6 (Ambion 217240 (#1), Ambion S46640 (#2), and Ambion S46639 (#3)) were confirmed by quantitative RT-PCR in HCT116 or U2OS cells. Error bars indicate S.D. of three independent experiments. **, p < 0.01. C, cells were stained with PI, and DNA contents were analyzed using FACS. RASSF6 knockdown reduced the sub-G1 population. Error bars indicate S.D. of three independent experiments. **, p < 0.01. D, in control cells, UV light exposure induced cytochrome c release (white arrows) and nuclear condensation (white arrowheads). Scale bar = 20 μm. E, HCT116 cells were transfected with control dsRNA or RASSF6 dsRNA (siRASSF6#2). 72 h after transfection, the cells were exposed to 10 μM VP-16 and cultured for 24 h. The cells were stained with PI, and DNA contents were analyzed with FACS. The histogram demonstrates the summary of three independent experiments. F, HCT116 cells were transfected with control dsRNA or RASSF6 dsRNA (siRASSF6#2). 72 h after transfection, the cells were exposed to 25 J/m2 UV light. The subcellular fractionation was performed 24 h later. Voltage-dependent anion channel (VDAC) and α-tubulin were used as markers for the mitochondrial and the cytosolic fractions, respectively. UV exposure induced the cytochrome release to the cytosolic fraction (white arrow), but RASSF6 knockdown reduced it (black arrow).
FIGURE 2.

RASSF6 is involved in UV light-induced G1/S arrest in HCT116 cells, and GFP-RASSF6 expression induces G1/S arrest in HCT116 and U2OS cells. A, HCT116 cells were transfected with control dsRNA (siCont) or RASSF6 dsRNA (siRASSF6#2). The cells were cultured in serum-free medium for 48 h to block the cell cycle, exposed to 10 J/m2 UV light, and released from serum starvation. UV light induced G1/S arrest (white arrow). RASSF6 depletion overrode this arrest (black arrow). B, HCT116 cells and U2OS cells were transfected with pBudCGFP-SUMO1 (GFP cont) or pClneoGFP-RASSF6 (GFP-RASSF6). A single thymidine block was performed. GFP-positive cells were gated and analyzed. GFP-RASSF6 induced G1/S arrest (arrows).
RASSF6 Knockdown Has No Effect on H2O2-induced and Low-dose Doxorubicin-induced Senescence
We also tested whether or not RASSF6 plays a role in senescence. The treatment of 150 μm H2O2 and 1 μm doxorubicin induced senescence in human fibroblast TIG3 cells (Fig. 3). RASSF6 knockdown had no remarkable effect on senescence in both cases.
FIGURE 3.
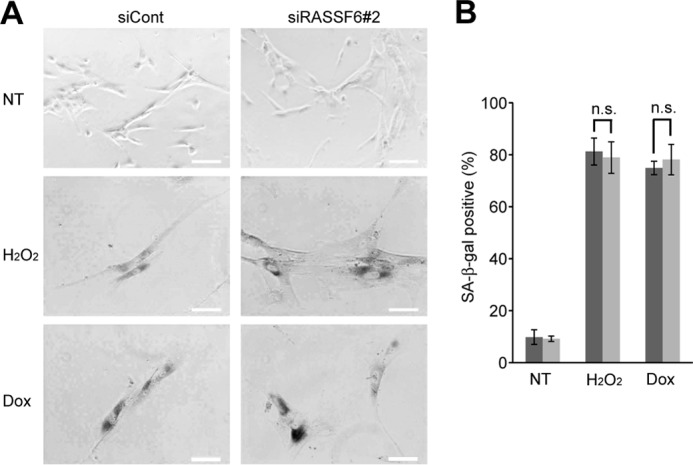
RASSF6 knockdown does not affect the senescence induced by H2O2 and doxorubicin. TIG3 cells were transfected with control dsRNA (siCont) or RASSF6 dsRNA (siRASSF6#2). 72 h after transfection, the cells were treated with 150 μM H2O2 or 1 μM doxorubicin (Dox) for 1 h. The senescent β-galactosidase was detected on day 6. The numbers of senescent β-galactosidase-positive cells per 100 cells were counted from three independent preparations. n.s., not significant; NT, no treatment.
RASSF6-induced Apoptosis Is p53-dependent
p53 is involved in cell cycle regulation by RASSF1A and Nore1A (14, 15). We reported that RASSF3 regulates apoptosis and the cell cycle via p53 (16). We wanted to know whether p53 is also involved in RASSF6-induced apoptosis. Consistent with a previous report (5), we observed that p53-deficient H1299 cells expressing RASSF6 remain viable (data not shown). To further confirm the requirement of p53 for RASSF6-induced apoptosis, we expressed RASSF6 in isogenic HCT116 p53+/+ and p53−/− cells. 48 h after transfection, HCT116 p53+/+ cells expressing FLAG-RASSF6 exhibited nuclear condensation (Fig. 4A, arrows), whereas HCT116 p53−/− cells with FLAG-RASSF6 did not (arrowheads). The quantitative studies of DNA content and mitochondrial membrane permeability using FACS indicated that RASSF6 induced apoptosis even in HCT116 p53−/− cells (Fig. 4, B and C, gray columns) but that RASSF6 caused apoptosis more efficiently in HCT116 p53+/+ cells (white columns). These findings support the fact that p53 is involved in RASSF6-induced apoptosis but that RASSF6-induced apoptosis does not solely depend on p53. Because p73 functions downstream of RASSF1A (19), we suppressed p73 in HCT116 cells, but p73 knockdown did not affect RASSF6-induced apoptosis in contrast to p53 knockdown (Fig. 5, A–D). We reported previously that MOAP1 knockdown suppresses RASSF6-induced apoptosis in HeLa cells (13). Because p53 is compromised in HeLa cells, we hypothesized that MOAP1 may function independently of p53. However, MOAP1 knockdown in HCT116 p53−/− cells did not reduce apoptosis (Fig. 5E). This result implies that the MOAP1 pathway functions in the same pathway as p53.
FIGURE 4.

p53 is involved in RASSF6-induced apoptosis. A, HCT116 p53+/+ and p53−/− cells were transfected with control pBudCFLAG-CIN85 or pClneoFH-RASSF6. At the indicated time points, the cells were fixed and stained. 48 h later, HCT116 p53+/+ cells exhibited nuclear condensation (arrows). HCT116 p53−/− cells expressing FLAG-RASSF6 remained viable (arrowheads). Scale bars = 10 μm. We observed 50 FLAG-CIN85- or FLAG-RASSF6-expressing cells from HCT116 p53+/+ and p53−/− cells, and the ratios of the cells with the nuclear condensation were evaluated. The data were obtained from three independent preparations. B and C, control GFP or GFP-RASSF6 was expressed in HCT116 p53+/+ and p53−/− cells. The cells were stained with PI to determine DNA content (B) or loaded with TMRM to evaluate mitochondrial membrane permeability (C). GFP-positive cells were gated and analyzed. Error bars indicate S.D. of three independent experiments. **, p < 0.01.
FIGURE 5.

p73 is not involved in RASSF6-induced apoptosis. HCT116 cells were transfected with control dsRNA (siCont), p53 dsRNA (sip53) or p73 dsRNA (sip73); replated 48 h later; and cultured overnight. A, the cells expressing control FLAG-CIN85 or FLAG-RASSF6 were analyzed as in Fig. 2. Arrows and arrowheads show the RASSF6-expressing cells with and without nuclear apoptosis. Error bars indicate S.D. of three independent experiments. **, p < 0.01; n.s., not significant. B, the validities of knockdown of p73 (catalog no. 7161, Dharmacon) and MOAP1 (catalog no. 130306, Ambion) were evaluated by quantitative RT-PCR, whereas those of p53 (catalog no. 7157, Dharmacon) and MDM2 (catalog no. S8629, Ambion) were evaluated by immunoblotting. **, p < 0.01. C–E, the sub-G1 population and mitochondrial permeability were analyzed in cells expressing control GFP or GFP-RASSF6. p73 knockdown had no effect. MOAP1 knockdown did not reduce apoptosis in HCT116 p53−/− cells.
RASSF6 Is Necessary for UV-induced Stabilization of p53
RASSF1A and RASSF3 both interact with MDM2 and facilitate self-ubiquitination and the subsequent degradation, resulting in the stabilization of p53 (14, 16). We hypothesized that RASSF6 drives apoptosis via p53 through a similar mechanism. RASSF6 knockdown did not reduce the basal expression of p53 in HCT116 cells (Fig. 6A, 0 h). However, RASSF6 knockdown blocked the UV light-induced increase of p53 (Fig. 6A, UV, 4 h to 12 h). MDM2 expression was enhanced in RASSF6-depleted cells (Fig. 6A, top panel). MG132 treatment and the additional knockdown of MDM2 cancelled the effect of RASSF6 knockdown (Fig. 6, B and C, white and black arrows). RASSF6 knockdown facilitated the degradation of endogenous p53 in cycloheximide-treated HCT116 cells (Fig. 6D, center panel). MDM2 decrease was slightly slacked in RASSF6-depleted cells (Fig. 6D, top panel, arrowhead). Nutlin-3 treatment remarkably enhanced p53 expression in cycloheximide-treated HCT116 cells (Fig. 6E, third lane). RASSF6 knockdown did not decrease p53 expression under this condition (Fig. 6E, fourth lane). As a reverse experiment, we expressed FLAG-RASSF6 and GFP-p53 in HCT116 cells. The expression of GFP-p53 was enhanced by coexpression of RASSF6 (Fig. 6F).
FIGURE 6.

RASSF6 depletion blocks UV light-induced p53 increase via MDM2. HCT116 cells were transfected with control dsRNA (siCont) or RASSF6 dsRNA (siRASSF6#1). A, B, and C, 72 h after transfection, cells were irradiated with 25 J/m2 UV light and harvested at the indicated time points. A, RASSF6 depletion blocked p53 increase at 4, 8, and 12 h. IB, immunoblot. B, cells were treated with 10 μM MG132. C, MDM2 was additionally knocked down. MG132 treatment and MDM2 depletion recovered the UV light-induced p53 increase under RASSF6 depletion (white and black arrows). D and E, HCT116 cells were transfected with control dsRNA or RASSF6 dsRNA (siRASSF6#2). D, 72 h after transfection, the cells were treated with 50 μg/ml cycloheximide. The cells were harvested at the indicated time points. The cell lysates were immunoblotted with anti-MDM2, anti-p53, and anti-α-tubulin antibodies. RASSF6 knockdown facilitated the degradation of p53. E, 72 h after transfection, cells were treated with (+) or without (−) 10 μM Nutrin-3 for 18 h. Cells were treated with 50 μg/ml cycloheximide for 4 h. Cell lysates were immunoblotted with anti-p53 and anti-α-tubulin antibodies. F, RASSF6 coexpression enhanced p53 expression. HCT116 cells were transfected with pClneoGFP-p53 alone or with pClneoFH-RASSF6. Cell lysates were immunoblotted with anti-GFP, anti-FLAG, and anti-α-tubulin antibodies. α-Tubulin was used as a loading control.
RASSF6 Interacts with MDM2 and Augments Self-ubiquitination
The above findings suggest that p53 undergoes degradation by MDM2 in RASSF6-depleted cells. An immunoprecipitation experiment using rat liver indicated that RASSF6 and MDM2 interact with each other (Fig. 7A). The interaction was confirmed in heterologous HEK293FT cells (Fig. 7B). MDM2 ubiquitination was remarkably enhanced in the presence of RASSF6 (Fig. 7C). Conversely, MDM2 reduced expression of RASSF6 (Fig. 7D). These data indicate that RASSF6, like RASSF1A and RASSF3, interacts with MDM2 to facilitate its degradation and to stabilize p53 and that MDM2 reciprocally suppresses RASSF6 expression. p53 is acetylated at seven lysine residues in the carboxyl-terminal region and one lysine in the DNA-binding domain. The acetylation stabilizes p53. We examined the acetylation at lysine 373 as a representative one, but could not detect change by coexpression of RASSF6 (data not shown).
FIGURE 7.

RASSF6 interacts with MDM2 and enhances its ubiquitination. A, coimmunoprecipitation of RASSF6 and MDM2 from rat liver. RASSF6 (left panel) or MDM2 (right panel) was immunoprecipitated (IP) from rat liver. The immunoprecipitates were immunoblotted (IB) with anti-MDM2 and anti-RASSF6 antibodies. Cont, control. B, Myc-RASSF6 was coimmunoprecipitated with FLAG-MDM2 by anti-FLAG-M2 affinity gel from MG132-treated HEK293FT cells. The immunoprecipitates were immunoblotted with anti-Myc and anti-FLAG antibodies. C, FLAG-MDM2 and HA-Ubc were expressed with control GFP or GFP-RASSF6 in MG132-treated HEK293FT cells. The immunoprecipitation was done with anti-FLAG M2 affinity gel. The input and the immunoprecipitate were immunoblotted with anti-FLAG, anti-GFP, and anti-HA antibodies as indicated. RASSF6 enhanced the ubiquitination of MDM2. D, MDM2 coexpression reduces RASSF6 expression. HCT116 cells were transfected with pClneoFH-RASSF6 alone or with the indicated amounts of pClneomCherry-MDM2. The cell lysates were immunoblotted with anti-FLAG, anti-mCherry, and anti-α-tubulin antibodies. α-Tubulin was used as a loading control. RASSF6 (arrowhead) decreased as MDM2 increased (arrow).
RASSF6 Regulates p53 Target Genes
We next evaluated p53 target gene transcriptions in quantitative RT-PCR. 10 J/m2 UV light exposure increased the mRNA of Bax, PUMA, p21, and Btg2 in HCT116 cells (Fig. 8, siCont). RASSF6 depletion decreased all of these gene transcriptions (Fig. 8, siRASSF6). As described above, RASSF6 knockdown did not affect G1/S arrest in cells exposed to 25 J/m2 UV light. Consistently, RASSF6 depletion reduced Bax and PUMA transcriptions in 25 J/m2 UV light-exposed HCT116 cells, whereas it had no effect on p21 and Btg2 transcriptions (data not shown). It suggests that the RASSF6-independent mechanism more significantly contributes to p21 and Btg2 up-regulation under this condition.
FIGURE 8.
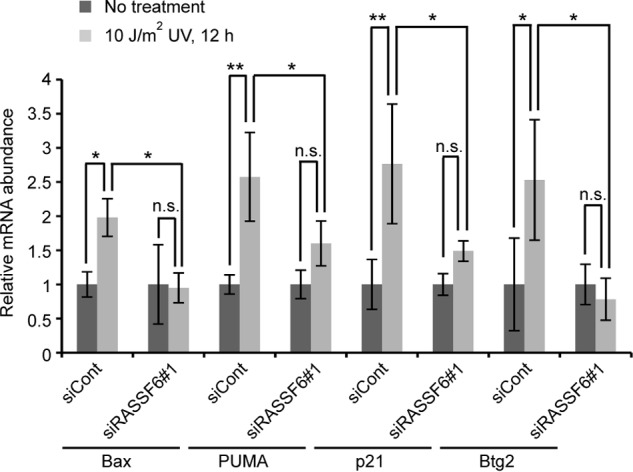
RASSF6 depletion blocks the increase of p53 target genes. HCT116 cells were irradiated with 10 J/m2 UV light and harvested at 12 h. Quantitative RT-PCR was performed. Error bars indicate S.D. of three independent experiments. *, p < 0.05; **, p < 0.01; n.s., not significant.
Depletion of RASSF6 Gives Birth to Polyploid Cells
In the final set of experiments, we tested whether and how RASSF6 exerts tumor-suppressive roles under DNA damage. The exposure to 25 J/m2 UV light and to 50 μm of VP-16-generated γ-H2A-positive cells (Fig. 9). In control cells, γ-H2AX signals significantly decreased after 6 h. However, the signals remained in RASSF6-depleted cells at 24 h. RASSF6 depletion itself did not increase polyploid cells but increased polyploid cells after VP-16 treatment (Fig. 10). These findings support the fact that RASSF6 functions as a tumor suppressor.
FIGURE 9.
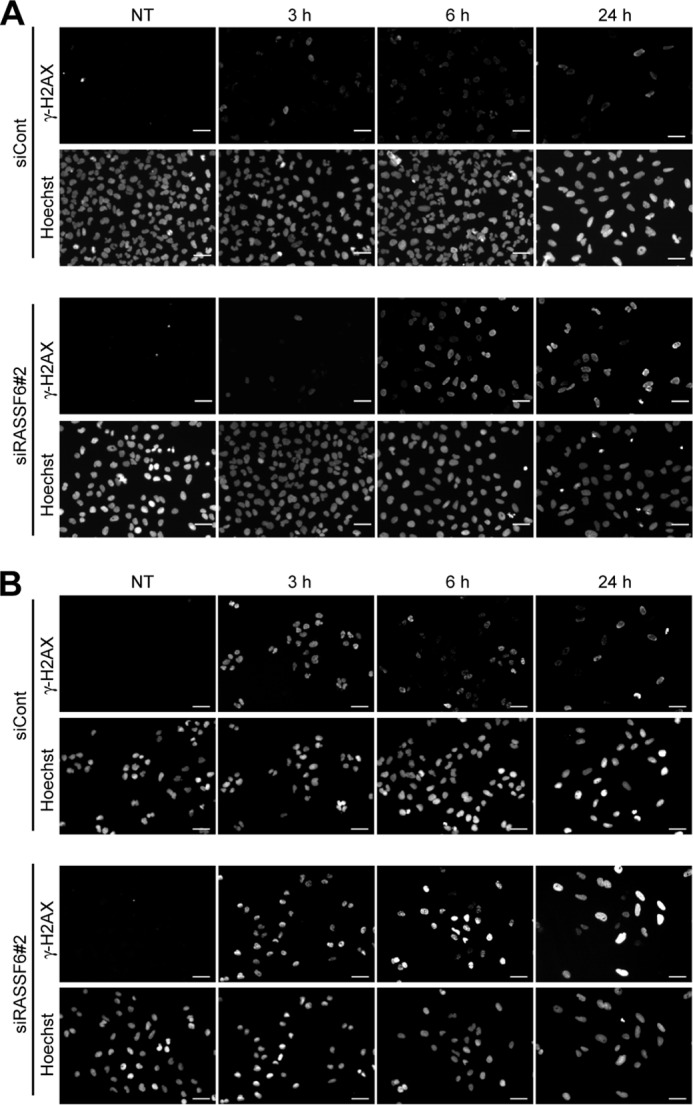
RASSF6 knockdown impairs DNA repair in UV light-exposed and VP-16-treated HCT116 cells. HCT116 cells were transfected with control dsRNA (siCont) or RASSF6 dsRNA (siRASSF6#2). 72 h after transfection, the cells were exposed to 25 J/m2 UV light (A) or to 50 μM VP-16 for 3 h (B). The cells were fixed immediately or cultured for 3, 6, or 24 h. γ-H2AX was immunostained. The signals were detected at 3 h in both control cells and RASSF6-depleted cells. In control cells, the signals disappeared at 24 h, whereas the signals remained detectable in RASSF6-depleted cells. NT, no treatment.
FIGURE 10.

RASSF6 depletion increases polyploid cells after VP-16 treatment. U2OS cells were transfected with control dsRNA (siCont) or RASSF6 dsRNA (siRASSF6#2). The cells were replated at 4 x 105 /100-mm plate 48 h later, cultured overnight, treated with 50 μM VP-16 for 3 h, and washed with fresh medium. The cells were cultured for 48 and 96 h before flow cytometric profiling. Three independent experiments were performed. A, representative flow cytometric profiles. B, quantitative analysis. Error bars indicate S.D. of three independent experiments. **, p < 0.01; n.s., not significant. White columns, no treatment. Gray columns, VP-16 treatment. NT, no treatment.
DISCUSSION
Mammals have six C-terminal RASSFs (RASSF1 to RASSF6) (1–4). They have the RA domain in the middle region and the Salvador/RASSF/Hippo domain in the C-terminal region. RASSF1A is the one studied most extensively. RASSF1A is frequently suppressed in human cancers. The knockout mice are susceptible to oncogenesis. On the basis of these findings, RASSF1A is regarded as a tumor suppressor. The molecular mechanism underlying the tumor-suppressive function of RASSF1A is well studied. RASSF1A regulates the cell cycle and is involved in Ras-induced and ataxia telangiectasia mutated kinase-related apoptosis (20–23). Other C-terminal RASSFs are also epigenetically inactivated in human cancers. By analogy with RASSF1A, these RASSFs are considered to be tumor suppressors and reported to regulate the cell cycle and apoptosis. Although these RASSFs have many common characters, they also diverge in several points. First, the subcellular localizations are different. Because of the limitation of available antibodies, the subcellular localizations of endogenous RASSF proteins are not conclusively determined, but when expressed exogenously, RASSF1A is associated with microtubules but RASSF3 or RASSF6 is not. Second, RASSF1A and RASSF2 activate MST2 to induce apoptosis through the Hippo pathway, whereas RASSF6 inhibits MST1/2 and induces apoptosis independently of the Hippo pathway (13, 24–27). Third, although all RASSF proteins have the Ras association domain, each RASSF protein has a different affinity for Ras proteins (28). Because more than one RASSF protein is expressed in the same cell, RASSF proteins may cooperate to function as tumor suppressors. It is important to characterize each RASSF protein and to know whether and how they compensate each other when some RASSF proteins are suppressed.
In this study, we demonstrated that RASSF6 is involved in UV light- and VP-16-induced apoptosis and that p53 knockdown attenuates RASSF6-induced apoptosis. RASSF6 induces apoptosis more remarkably in HCT116 p53+/+ cells than in HCT116 p53−/− cells. However, p53 knockdown does not completely abolish RASSF6-induced apoptosis, and RASSF6 induces apoptosis in HCT116 p53−/− cells to some extent. These findings suggest that RASSF6-induced apoptosis does not depend solely on p53. Although p73 is implicated in RASSF1A-induced apoptosis, p73 knockdown had no effect on RASSF6-induced apoptosis. We reported previously that MOAP1 is involved in RASSF6-induced apoptosis in HeLa cells. Because p53 is compromised in HeLa cells, we tested the idea that MOAP1 plays a role in RASSF6-induced apoptosis independently of p53. Contrary to our expectations, MOAP1 knockdown does not decrease apoptosis in HCT116 p53−/− cells. This finding implicates that MOAP1 functions in the same pathway as p53 in the context of RASSF6-induced apoptosis. We need to clarify in a future study how RASSF6 induces apoptosis independently of p53.
We have also shown that RASSF6 is involved in p53-mediated G1/S arrest. Mechanistically, RASSF6 interacts with MDM2 and promotes its self-ubiquitination. Conversely, RASSF6 undergoes degradation by MDM2. UV light exposure reduces MDM2 expression in the presence of RASSF6 and increases p53 expression. These findings suggest some mechanism that determines whether RASSF6 or MDM2 is more readily degraded and that DNA damage shifts the balance between RASSF6 and MDM2. In RASSF6-depleted cells, MDM2 expression is maintained even after UV light exposure, and p53 expression is not up-regulated. Therefore, it is predicted that the G1/S checkpoint does not work properly. Indeed, RASSF6 depletion impairs DNA repair in UV light-exposed or VP-16-treated cells and eventually increases polyploid cells. All of these findings support that RASSF6 functions as a tumor suppressor through MDM2 and p53. RASSF1A and RASSF3 also interact with MDM2, facilitate its protein degradation, and stabilize p53 (14, 16). Although further studies of other RASSF proteins are necessary, the interaction with MDM2 and the stabilization of p53 under DNA damage may be the common feature for the C-terminal RASSF proteins.
Acknowledgments
We thank Kiyotsugu Yoshida (Jikei University School of Medicine), Bert Vogelstein (Johns Hopkins University), and Ken-ichi Nakahama (Tokyo Medical and Dental University) for materials and advice.
This work was supported by Ministry of Education, Sports, Science, and Technology Grant 17081008 and by Japan Society for the Promotion of Science (JSPS) Grants 22790275 and 22590267.
- RASSF
- Ras association domain family
- RA
- Ras association
- MST
- mammalian Ste20-like
- MOAP
- modulator of apoptosis
- PI
- propidium iodide
- TAZ
- transcriptional co-activator with PDZ-binding motif
- TMRM
- tetramethylrhodamine methyl ester perchorate
- YAP
- Yes-associated protein.
REFERENCES
- 1. Avruch J., Xavier R., Bardeesy N., Zhang X. F., Praskova M., Zhou D., Xia F. (2009) Rassf family of tumor suppressor polypeptides. J. Biol. Chem. 284, 11001–11005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Richter A. M., Pfeifer G. P., Dammann R. H. (2009) The RASSF proteins in cancer. From epigenetic silencing to functional characterization. Biochim. Biophys. Acta 1796, 114–128 [DOI] [PubMed] [Google Scholar]
- 3. Sherwood V., Recino A., Jeffries A., Ward A., Chalmers A. D. (2010) The N-terminal RASSF family. A new group of Ras-association-domain-containing proteins, with emerging links to cancer formation. Biochem. J. 425, 303–311 [DOI] [PubMed] [Google Scholar]
- 4. Underhill-Day N., Hill V., Latif F. (2011) N-terminal RASSF family. RASSF7-RASSF10. Epigenetics 6, 284–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Allen N. P., Donninger H., Vos M. D., Eckfeld K., Hesson L., Gordon L., Birrer M. J., Latif F., Clark G. J. (2007) RASSF6 is a novel member of the RASSF family of tumor suppressors. Oncogene 26, 6203–6211 [DOI] [PubMed] [Google Scholar]
- 6. Hesson L. B., Dunwell T. L., Cooper W. N., Catchpoole D., Brini A. T., Chiaramonte R., Griffiths M., Chalmers A. D., Maher E. R., Latif F. (2009) The novel RASSF6 and RASSF10 candidate tumour suppressor genes are frequently epigenetically inactivated in childhood leukaemias. Mol. Cancer 8, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wen Y., Wang Q., Zhou C., Yan D., Qiu G., Yang C., Tang H., Peng Z. (2011) Decreased expression of RASSF6 is a novel independent prognostic marker of a worse outcome in gastric cancer patients after curative surgery. Ann. Surg. Oncol. 18, 3858–3867 [DOI] [PubMed] [Google Scholar]
- 8. Ikeda M., Hirabayashi S., Fujiwara N., Mori H., Kawata A., Iida J., Bao Y., Sato Y., Iida T., Sugimura H., Hata Y. (2007) Ras-association domain family protein 6 induces apoptosis via both caspase-dependent and caspase-independent pathways. Exp. Cell Res. 313, 1484–1495 [DOI] [PubMed] [Google Scholar]
- 9. Withanage K., Nakagawa K., Ikeda M., Kurihara H., Kudo T., Yang Z., Sakane A., Sasaki T., Hata Y. (2012) Expression of RASSF6 in kidney and the implication of RASSF6 and the Hippo pathway in the sorbitol-induced apoptosis in renal proximal tubular epithelial cells. J. Biochem. 152, 111–119 [DOI] [PubMed] [Google Scholar]
- 10. Pan D. (2010) The Hippo signaling pathway in development and cancer. Dev. Cell 19, 491–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bao Y., Hata Y., Ikeda M., Withanage K. (2011) Mammalian Hippo pathway. From development to cancer and beyond. J. Biochem. 149, 361–379 [DOI] [PubMed] [Google Scholar]
- 12. Zhao B., Tumaneng K., Guan K. L. (2011) The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat. Cell Biol. 13, 877–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ikeda M., Kawata A., Nishikawa M., Tateishi Y., Yamaguchi M., Nakagawa K., Hirabayashi S., Bao Y., Hidaka S., Hirata Y., Hata Y. (2009) Hippo pathway-dependent and -independent roles of RASSF6. Sci. Signal. 2, ra59. [DOI] [PubMed] [Google Scholar]
- 14. Song M. S., Song S. J., Kim S. Y., Oh H. J., Lim D. S. (2008) The tumour suppressor RASSF1A promotes MDM2 self-ubiquitination by disrupting the MDM2-DAXX-HAUSP complex. EMBO J. 27, 1863–1874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Calvisi D. F., Donninger H., Vos M. D., Birrer M. J., Gordon L., Leaner V., Clark G. J. (2009) NORE1A tumor suppressor candidate modulates p21CIP1 via p53. Cancer Res. 69, 4629–4637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kudo T., Ikeda M., Nishikawa M., Yang Z., Ohno K., Nakagawa K., Hata Y. (2012) The RASSF3 candidate tumor suppressor induces apoptosis and G1-S cell-cycle arrest via p53. Cancer Res. 72, 2901–2911 [DOI] [PubMed] [Google Scholar]
- 17. Tommasi S., Besaratinia A., Wilczynski S. P., Pfeifer G. P. (2011) Loss of Rassf1a enhances p53-mediated tumor predisposition and accelerates progression to aneuploidy. Oncogene 30, 690–700 [DOI] [PubMed] [Google Scholar]
- 18. Yee K. S., Grochola L., Hamilton G., Grawenda A., Bond E. E., Taubert H., Wurl P., Bond G. L., O'Neill E. (2012) A RASSF1A polymorphism restricts p53/p73 activation and associates with poor survival and accelerated age of onset of soft tissue sarcoma. Cancer Res. 72, 2206–2217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Matallanas D., Romano D., Yee K., Meissl K., Kucerova L., Piazzolla D., Baccarini M., Vass J. K., Kolch W., O'Neill E. (2007) RASSF1A elicits apoptosis through an MST2 pathway directing proapoptotic transcription by the p73 tumor suppressor protein. Mol. Cell 27, 962–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Donninger H., Vos M. D., Clark G. J. (2007) The RASSF1A tumor suppressor. J. Cell Sci. 120, 3163–3172 [DOI] [PubMed] [Google Scholar]
- 21. Hamilton G., Yee K. S., Scrace S., O'Neill E. (2009) ATM regulates a RASSF1A-dependent DNA damage response. Curr. Biol. 19, 2020–2025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Khokhlatchev A., Rabizadeh S., Xavier R., Nedwidek M., Chen T., Zhang X. F., Seed B., Avruch J. (2002) Identification of a novel Ras-regulated proapoptotic pathway. Curr. Biol. 12, 253–265 [DOI] [PubMed] [Google Scholar]
- 23. Matallanas D., Romano D., Al-Mulla F., O'Neill E., Al-Ali W., Crespo P., Doyle B., Nixon C., Sansom O., Drosten M., Barbacid M., Kolch W. (2011) Mutant K-Ras activation of the proapoptotic MST2 pathway is antagonized by wild-type K-Ras. Mol. Cell 44, 893–906 [DOI] [PubMed] [Google Scholar]
- 24. Praskova M., Khoklatchev A., Ortiz-Vega S., Avruch J. (2004) Regulation of the MST1 kinase by autophosphorylation, by the growth inhibitory proteins, RASSF1 and NORE1, and by Ras. Biochem. J. 381, 453–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guo C., Tommasi S., Liu L., Yee J. K., Dammann R., Pfeifer G. P. (2007) RASSF1A is part of a complex similar to the Drosophila Hippo/Salvador/Lats tumor-suppressor network. Curr. Biol. 17, 700–705 [DOI] [PubMed] [Google Scholar]
- 26. Cooper W. N., Hesson L. B., Matallanas D., Dallol A., von Kriegsheim A., Ward R., Kolch W., Latif F. (2009) RASSF2 associates with and stabilizes the proapoptotic kinase MST2. Oncogene 28, 2988–2998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Song H., Oh S., Oh H. J., Lim D. S. (2010) Role of the tumor suppressor RASSF2 in regulation of MST1 kinase activity. Biochem. Biophys. Res. Commun. 391, 969–973 [DOI] [PubMed] [Google Scholar]
- 28. Chan J. J., Flatters D., Rodrigues-Lima F., Yan J., Thalassinos K., Katan M. (2013) Comparative analysis of interactions of RASSF1–10. Adv. Biol. Regul. 53, 190–201 [DOI] [PMC free article] [PubMed] [Google Scholar]