

Background: Thyroid hormone (TH) is known to induce transcription of hepatic gluconeogenic genes.
Results: TH stimulates FoxO1 deacetylation to increase PCK1 and G6PC transcription.
Conclusion: FoxO1 deacetylation by SirT1 is crucial for TH induction of its target genes, PCK1 and G6PC.
Significance: This study demonstrates a novel role of FoxO1 in TH-mediated transcription that may explain some of the pleiotropic actions by TH.
Keywords: FoxO, Gene Regulation, Gene Transcription, Gluconeogenesis, Phosphoenolpyruvate Carboxykinase, Sirt1, Thyroid Hormone
Abstract
Hepatic gluconeogenesis is a concerted process that integrates transcriptional regulation with hormonal signals. A major regulator is thyroid hormone (TH), which acts through its nuclear receptor (TR) to induce the expression of the hepatic gluconeogenic genes, phosphoenolpyruvate carboxykinase (PCK1) and glucose-6-phosphatase (G6PC). Forkhead transcription factor FoxO1 also is an important regulator of these genes; however, its functional interactions with TR are not known. Here, we report that TR-mediated transcriptional activation of PCK1 and G6PC in human hepatic cells and mouse liver was FoxO1-dependent and furthermore required FoxO1 deacetylation by the NAD+-dependent deacetylase, SirT1. siRNA knockdown of FoxO1 decreased, whereas overexpression of FoxO1 increased, TH-dependent transcriptional activation of PCK1 and G6PC in cultured hepatic cells. FoxO1 siRNA knockdown also decreased TH-mediated transcription in vivo. Additionally, TH was unable to induce FoxO1 deacetylation or hepatic PCK1 gene expression in TH receptor β-null (TRβ−/−) mice. Moreover, TH stimulated FoxO1 recruitment to the PCK1 and G6PC gene promoters in a SirT1-dependent manner. In summary, our results show that TH-dependent deacetylation of a second metabolically regulated transcription factor represents a novel mechanism for transcriptional integration of nuclear hormone action with cellular energy status.
Introduction
Thyroid hormone (TH)2 plays critical roles in the gene expression of a diverse range of cellular signaling pathways, including glucose homeostasis (1, 2). TH regulates target gene expression by binding to its nuclear receptors (TRs). Liganded TRs then bind to thyroid hormone response elements (TREs), which are typically located in the promoter regions of target genes, and recruit co-activator complexes that have histone acetyltransferase activity. Histone acetyltransferases such as CBP/p300 induce histone acetylation and formation of euchromatin, enabling recruitment of RNA polymerase complex I and general transcription factors that are crucial for transcriptional activation (3, 4). Unliganded TRs also bind to TREs; however, in contrast to liganded TRs, they recruit a complex containing co-repressors (e.g. nuclear co-repressor complex (Nco-R), silencing mediator for retinoid or thyroid hormone receptors (SMRT)) and histone deacetylases (HDACs) that decrease histone acetylation and repress target gene expression (2, 4).
Previously, TH was shown to induce transcription of the key gluconeogenic gene, phosphoenolpyruvate carboxykinase (PCK1), which catalyzes formation of phosphoenolpyruvate from pyruvate, an early and highly regulated step in gluconeogenesis. This transcriptional activation appears to be mediated through a TRE located in the promoter region (5). TH also induces transcriptional activation of glucose-6-phosphatase (G6PC) that catalyzes the final step in gluconeogenesis, the conversion of glucose 6-phosphate to glucose (6); however, a putative TRE has not yet been identified thus far. The transcription of both the PCK1 and G6PC genes are regulated by Forkhead transcription factor FoxO1, which binds directly to its response element binding sites (insulin response element; IRE) in the promoters of these genes (7–9). These interactions have been shown to be critically important for the regulation of hepatic glucose production (9–11).
FoxO1 undergoes post-translational modifications such as phosphorylation, acetylation, and ubiquitination that modulate its functional activities (12). FoxO1 is phosphorylated through insulin-dependent activation of AKT/protein kinase B leading to its rapid nuclear exclusion (12, 13). Furthermore, histone deacetylases such as SirT1 (a class III histone deacetylase) or HDAC3 (a class I histone deacetylase) can deacetylate FoxO1 to regulate its transcriptional activity in mammalian cells (14, 15). Recently, SirT1 has been shown to modulate TH effects on carnitinepalmitoyl transferase 1α (CPT1α) and pyruvate dehydrogenase kinase 4 (PCK4) in rat primary hepatocytes by deacetylating PGC1α (16). However, thus far, no functional interactions among Sirt1, FoxO1, and TRs in TH action have been described.
Although TH directly regulates transcription of PCK1 by liganded TR binding to a TRE located within the promoter region, it is not known whether other hormone-responsive transcription factors, such as FoxO1, can modulate TH-induced transcription. Similarly, TH regulation of G6PC transcription is not well understood and may result from regulation/activation of other transcription factors such as FoxO1. In this study, we show that TH induction of hepatic gluconeogenic genes is FoxO1-mediated and requires SirT1-dependent deacetylation of FoxO1. We also demonstrate that TH promotes FoxO1 recruitment to PCK1 and G6PC promoters in a SirT1-dependent manner. Thus, our findings demonstrate a novel role of FoxO1 in TH-mediated transcription that may explain some of the pleiotropic actions of TH.
EXPERIMENTAL PROCEDURES
Cell Culture and Maintenance
We used an isogenic HepG2 cell line that ectopically expresses TRβ1 (TRβ1-HepG2) (a kind gift from Prof. Martin L. Privalsky, University of California, Davis) which has been characterized previously (17). All HepG2 cell lines were cultured and maintained as described earlier (18). For T3 treatments, all cells were grown for at least 3 days in DMEM containing 10% Dowex-stripped FBS and 1× penicillin/streptomycin (normal T3-depleted DMEM) before adding T3 with or without other compounds at the indicated concentrations and durations in the respective figures. Primary mouse hepatocytes were isolated from male C57BL/6 mice (8–10 weeks old) using a standard two-step collagenase perfusion method and cultured in normal T3-depleted DMEM as mentioned above.
Animals
Male C57BL/6 mice (8–10 weeks old) were purchased and housed in hanging polycarbonate cages under a 12-h light/12-h dark cycle at 23 °C with food and water available ad libitum. All cages contained shelters and nesting material. Hypothyroidism and hyperthyroidism were induced and confirmed as described earlier (19). During the course of treatment, animals were monitored daily for their general health and weight. The TRβ-null (TRβ−/−) mice, which lack both TRβ1 and TRβ2 isoforms, were in a C57BL/6:129sv mixed background (20). TRβ−/− mice and wild-type (TRβ+/+) mice of the same strain were treated with TH (a mix of T3 = 6.5 μg/mouse and T4 = 7.8 μg/mouse) for 5 days (21).
RNA Isolation and RT-qPCR Analysis
Total RNA isolation and RT-qPCR were performed as described previously (18). β-Actin levels were taken for normalization, and -fold change was calculated using 2−ΔΔCt. SYBR Green optimized primers from Sigma-Aldrich (KiCqStartTM SYBR® Green primers: KSPQ12012; Sigma) were used for the target gene analysis unless otherwise noted. All the primer sequences are provided in Table 1.
TABLE 1.
Primers used for qPCR analysis
| Gene name | Forward primer sequence | Reverse primer sequence |
|---|---|---|
| hPCK1 | CAGCGACGGGGGCGTTTACT | GGGCACAAGGTTCCCCATCCTCT |
| *mPCK1 | AATATGACAACTGTTGGCTG | AATGCTTTCTCAAAGTCCTC |
| *hG6PC | ACTGTGCATACATGTTCATC | TGAATGTTTTGACCTAGTGC |
| *mG6PC | TTCAAGTGGATTCTGTTTGG | AGATAGCAAGAGTAGAAGTGAC |
| *hSirT1 | AAGGAAAACTACTTCGCAAC | GGAACCATGACACTGAATTATC |
| *hFoxO1 | AACTACAGCCAAAATCACTG | ATGACAGGATTTCAACACAC |
| *hACTb | GACGACATGGAGAAAATCTG | ATGATCTGGGTCATCTTCTC |
| *mACTb | GATGTATGAAGGCTTTGGTC | TGTGCACTTTTATTGGTCTC |
| Promoter primers | ||
| hPCK1-TRE (−315 to −117) | CCCCGGCCAACCTTGTCCTT | TGCAACACGCCCCAGGGAGA |
| hPCK1-IRE (−426 to −170) | GGGTGCATCCTTCCCATGAA | GACTTCGAGCCCTCAACCAA |
| hG6PC-IRE (−237 to −70) | CATTGGCCCTGCTGAGTACA | AACCCAGCCCTGATCTTTGG |
* KiCqStart™ SYBR Green Primers (KSPQ12012) from Sigma-Aldrich optimized for SYBR Green dye used in qPCR analysis.
Western Blotting
Cultured cells or 50 mg of liver tissues were lysed using CelLytic M mammalian cell lysis/extraction reagent (C2978; Sigma-Aldrich). An aliquot was removed, and protein concentrations were measured using the BCA Kit (Bio-Rad). Western blotting was performed using standard protocol. Laemmli sample buffer (composition: 250 mmol/liter Tris, pH 7.4, 2% w/v sodium dodecyl sulfate, 25% v/v glycerol, 10% v/v 2-mercaptoethanol, and 0.01% w/v bromphenol blue) was added to the protein samples which were then heated to 100 °C for 5 min. These denatured samples were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred immediately onto polyvinylidene difluoride membranes (Bio-Rad) using 1×Towbin buffer (25 mmol/liter Tris, pH 8.8, 192 mmol/liter glycine, 15% v/v methanol). Membranes were blocked in 5% milk and subsequently were incubated in 1% w/v bovine serum albumin in TBST (1× TBS with 0.1% Tween 20) with specific antibodies overnight at 4 °C. Primary antibodies against acetylated FoxO1 (Ac-FKHR (D-19); SC-49437; which detects acetylation of lysine residues at 259, 262, and 271), FoxO1 (FKHR (H-128)X; SC-11350), SirT1 (clone 10E4; 04-1557; Millipore), β-actin (C4; SC-47778), and GAPDH (ab9485; Abcam) were used in this study. After overnight incubation in primary antibodies, membranes were washed three times in TBST and subsequently incubated with species-appropriate, peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology) for 1 h. Blots were washed three times with TBST and once with TBS without Tween and developed using an enhanced chemiluminescence system (GE Healthcare). Images were captured on the Gel-Doc system (Bio-Rad), and densitometry analysis was performed using ImageJ software (National Institutes of Health). The integrated density of target protein was normalized with indicated internal controls (β-actin or GAPDH), and the mean was plotted in graphs.
In Vitro Overexpression of SirT1 and FoxO1
To overexpress SirT1 in TRβ-HepG2 cells, FLAG-tagged human SirT1 and its deacetylase domain mutant (H363Y), cloned in pECE (Addgene plasmids 1791 and 1792, respectively) was used. To overexpress FoxO1 in TRβ-HepG2 cells, FLAG-tagged human FoxO1 cloned in pcDNA3.1 (Addgene plasmid 13507) was used. SirT1 and FoxO1 constructs were described previously (22, 23) and purchased from Addgene (nonprofit plasmid repository). Transfections were carried out in TRβ1-HepG2 cells in 12-well plates using 1 μg of the above indicated plasmids or empty pECE/pcDNA vector as control along with Lipofectamine 2000 (Invitrogen) following the reverse transfection protocol as provided by the manufacturer. After 48 h of transfection, cells were subjected to T3 (0.1 μm) treatment in normal T3-depleted medium as described above. After 24 h of treatment total RNA or protein was isolated for further analysis.
In Vitro Gene Silencing Using siRNA
Stealth siRNA duplex oligoribonucleotides targeting SirT1 (set of three siRNAs) or FoxO1 (set of three siRNAs) from Invitrogen were used. Transfections were carried out in HepG2 cells in a 6-well plate using 10 nm concentrations of the above indicated siRNAs or negative control siRNA with Lipofectamine RNAiMAX (Invitrogen) following the reverse transfection protocol as provided by the manufacturer. 48 h after transfection, T3 (0.1 μm) treatment was done for 24 h. Total RNA or protein was isolated for further analysis. For chromatin immunoprecipitation analysis in siRNA knockdown (KD) conditions, TRβ1-HepG2 cells were transfected in 150-mm culture dishes using Lipofectamine RNAiMAX with SirT1 siRNAs according to the manufacturer's protocol.
Pharmacological Inhibition of SirT1 in Vivo
Ex527 (6-chloro-2,3,4,9-tetrahydro-1H-carbazole-1-carboxamide; Sigma) (intraperitoneal 0.8 mg/day/100 g of body weight, for 3 days) was used to inhibit SirT1 activity in vivo (24). After the first injection of Ex527, mice were injected subcutaneously with T3 (10 μg/kg of body weight/day for 2 days) along with Ex527. After 3 days, animals were euthanized, and the liver tissues were subjected to Western blot analysis.
In Vivo Genetic Knockdown of FoxO1 Preferentially in the Liver Using siRNA
T3 treatment and siStable in vivo siRNA for mouse FoxO1 (GAGCGUGCCCCUACUUCAAGUU) or siGenome nontargeting siRNA (Dharmacon) was injected (40 μg/mice, for 3 days) using hydrodynamic tail vein injection to knock down FoxO1 preferentially in liver as described earlier (18). After the first injection of Ex527, mice were injected subcutaneously with T3 (10 μg/kg of body weight/day for 2 days) along with tail vein injection of FoxO1 siRNA. After 3 days, animals were euthanized, and the liver tissues were harvested and then subjected to RT-qPCR or Western blot analysis. FoxO1 KD was confirmed at both mRNA and protein levels.
Chromatin Immunoprecipitation (ChIP)-qPCR Analysis
The ChIP assays were performed using the EZ-Magna ChIP G-Chromatin Immunoprecipitation kit (17-610; Millipore) according to the manufacturer's protocol. TRβ1-HepG2 cells were transfected in 150-mm culture dishes with siRNAs as mentioned above or cultured in normal T3-depleted medium for 3 days in 150-mm dishes prior to the T3 treatment.
Immunoprecipitation was performed with antibodies against control rabbit IgG (sc2025; Santa Cruz Biotechnology) and anti-acetylated histone H3 (anti-histone H3 antibody, ChIP grade; ab47915; Abcam), anti-acetylated histone H4 (anti-acetyl-histone H4 antibody, 06-598; Millipore), and anti-FoxO1 (FKHR antibody (H-128)X; sc-11350 X; Santa Cruz Biotechnologies). Eluted DNA was purified with QIAamp DNA Mini Kit (Qiagen). 2 μl of immune-precipitated DNA (1% input DNA) was used with a QuantiFast SYBR Green PCR kit (Qiagen) for 40 cycles of qPCR using Rotor-Gene®Q qPCR machine (Qiagen). Primers and regions used to amplify PCK1 and G6PC promoter are given in Table 1.
Statistical Analysis
Individual culture experiments were performed in triplicate and repeated at least three times independently using matched controls; the data were pooled. Results are expressed as mean ± S.D. for all in vitro experiments and mean ± S.E. for all in vivo experiments. The statistical significance of differences (p < 0.05) was assessed by a two-tailed Student's t test.
Ethics Statement and Study Approval
All mice were maintained according to the Guide for the Care and Use of Laboratory Animals (National Institutes of Health publication 1.0.0. revised 2011), and experiments were approved by the IACUCs at the University of Pennsylvania, the National Cancer Institute, and the Duke-NUS Graduate Medical School.
RESULTS
FoxO1 Is Necessary for T3-dependent PCK1 and G6PC Gene Expression
T3 regulation of PCK1 and G6PC was confirmed in a HepG2 cell line that ectopically expressed hTRβ1 (TRβ1-HepG2) (Fig. 1A) (17). The pattern of gene induction closely mimicked induction of these genes by T3 in mouse primary hepatocytes (Fig. 1B) (6, 25). Moreover, T3 rapidly induced PCK1 and G6PC transcription in a time- and dose-dependent manner in TRβ1-HepG2 cells (Fig. 1C).
FIGURE 1.
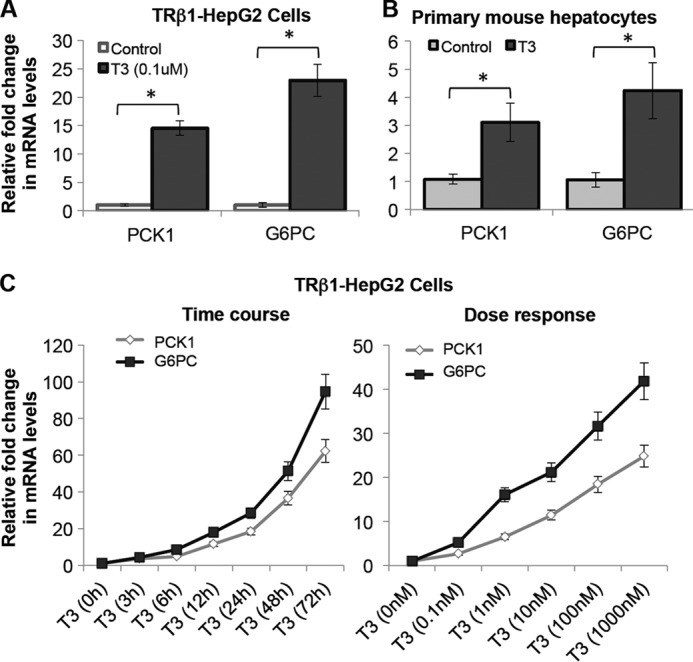
T3 induced mRNA expression of PCK1 and G6PC in human hepatic cells ectopically expressing TRβ1 (TRβ1-HepG2) and in primary mouse hepatocytes. A and B, cells were treated with or without T3 (at 0.1 μm concentration) for 24 h. Total mRNA was collected, and RT-qPCR analysis was done as indicated under “Experimental Procedures.” The figure shows mRNA expression of PCK1 and G6PC in HepG2 cells ectopically expressed TRβ1 (A) and primary mouse hepatocytes (B). C, T3 significantly increased PCK1 and G6PC mRNA expression in a time-dependent manner (at 0.1 μm concentration), as well as dose-dependent manner (for 24 h) in TRβ1-HepG2 cells. Cells were treated with or without T3 at the indicated time points or concentrations and maintained in normal T3-depleted medium as indicated under “Experimental Procedures.” β-Actin was used as normalization control (n = 3; *, p < 0.05). Error bars represent mean ± S.D.
To assess the role of FoxO1 in T3-induced transcription of PCK1 and G6PC genes, we performed loss-of-function as well as gain-of-function experiments (Fig. 2, A and B). We observed that FoxO1 siRNA KD abolished T3 induction of PCK1 and G6PC mRNAs in these HepG2 cells (Fig. 2C). We next overexpressed FoxO1 in TRβ1-HepG2 cells and found that it synergistically increased T3 induction of PCK1 mRNA by 53-fold (p < 0.001) and G6PC mRNA by 471-fold (p < 0.001) compared with empty vector control (Fig. 2D). These results demonstrated that FoxO1 was essential for TH induction of key gluconeogenic genes.
FIGURE 2.
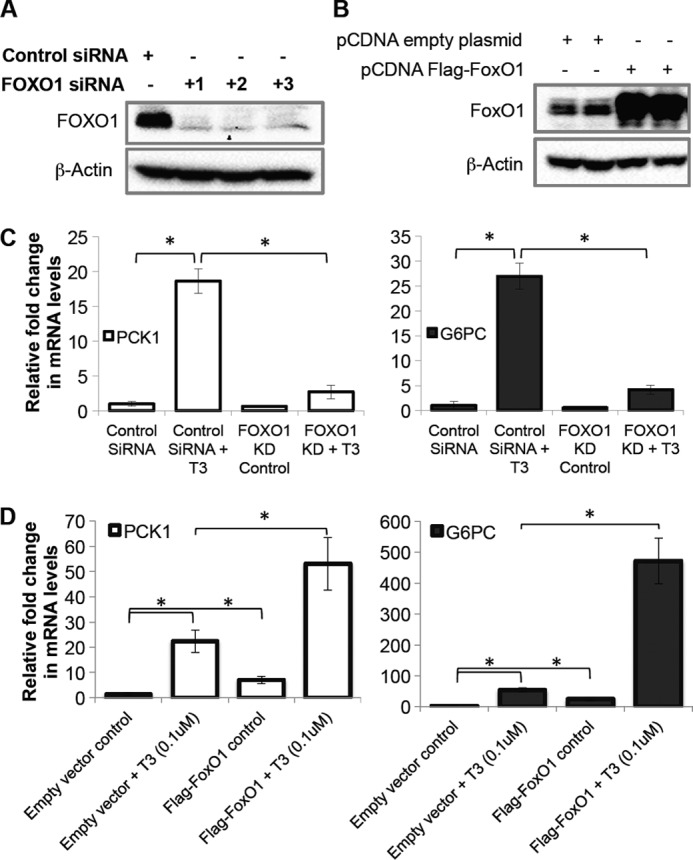
FoxO1 knockdown significantly reduced T3 induction of PCK1 and G6PC gene expression in TRβ1-HepG2 cells. A, FoxO1 was knocked down using three independent siRNAs in TRβ1-HepG2 cells, and KD was confirmed at the protein level. Cells were cultured with control siRNA or FoxO1 siRNA for 48 h followed by replacement of medium with T3-depleted medium as indicated under “Experimental Procedures.” Protein was harvested, and KD was confirmed by Western blotting. B, to overexpress FoxO1 in TRβ1-HepG2 cells, FLAG-tagged human FoxO1 cloned in pcDNA3.1 (Addgene plasmid 13507) was transfected and confirmed at protein level. Cells were treated with empty vector as control or FoxO1-vector for 48 h followed by replacement of medium with normal medium as indicated under “Experimental Procedures.” Protein was collected, and FoxO1 expression was confirmed by Western blotting. C, FoxO1 KD using siRNA significantly decreased T3 induced PCK1 and G6PC expression. After 48 h FoxO1 siRNA treatment cells were treated with T3 (0.1 μm) for 24 h, total mRNA was isolated, and RT-qPCR analysis was performed as indicated under “Experimental Procedures.” D, FoxO1 overexpression significantly increased T3-induced PCK1 and G6PC expression. 48 h after transfection cells were treated with T3 (0.1 μm) for 24 h, total mRNA was isolated, and RT-qPCR analysis was performed as indicated under “Experimental Procedures.” β-Actin was used as normalization control (n = 3; *, p < 0.05). Error bars represent mean ± S.D.
T3 Increases FoxO1 Deacetylation, Which Is SirT1-dependent
Deacetylation is critical for FoxO1 transcriptional activity (22, 26). T3 significantly decreased acetylation of FoxO1 in TRβ1-HepG2 cells in a time-dependent manner (Fig. 3A). Similar results were also observed in primary mouse hepatocytes (Fig. 3B). Interestingly, TH induction of FoxO1 deacetylation was abrogated when SirT1 was knocked down (Fig. 3C). To confirm the SirT1 effect on T3-induced deacetylation of FoxO1, we performed SirT1 KD using three independent siRNAs and overexpression of FLAG-tagged hSirT1 or mutant SirT1 that lacked deacetylase activity (Fig. 4, A and B). SirT1 siRNA KD significantly reduced T3 induction of PCK1 and G6PC mRNAs in these HepG2 cells (Fig. 4C). Overexpression of hSirT1 led to higher basal as well as T3-induced PCK1 and G6PC gene expression (Fig. 4D). The putative SirT1 activator, resveratrol (25–50 μm), also significantly increased T3-mediated induction of PCK1 and G6PC mRNA expression in a dose-dependent manner (data not shown). Furthermore, overexpression of mutant SirT1 was unable to increase T3-dependent induction of these genes (Fig. 4D), confirming the requirement of SirT1 deacetylase activity in FoxO1-mediated transcription.
FIGURE 3.
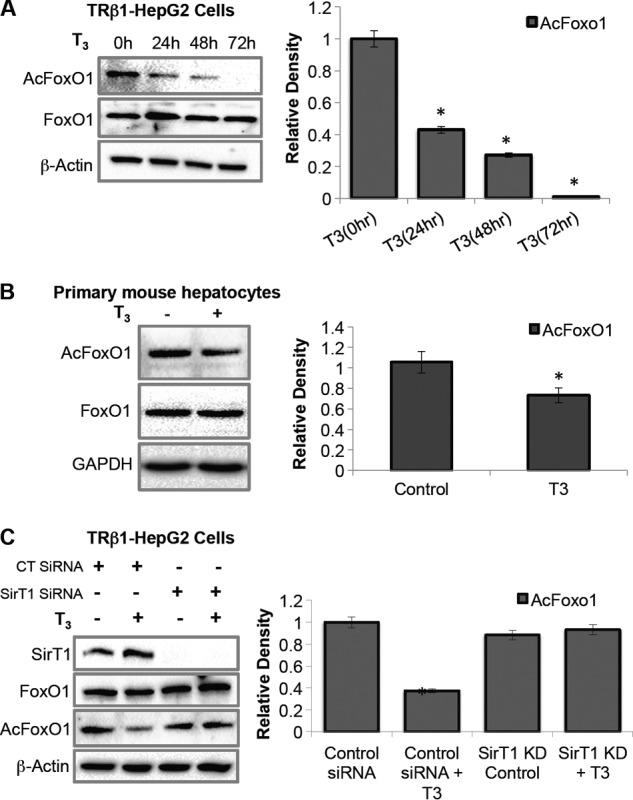
T3 induced deacetylation of FoxO1 in a SirT1-dependent manner. A, T3 decreased acetylation of FoxO1 in a time-dependent manner. TRβ1-HepG2 cells were cultured with or without T3 (0.1 μm) for the indicated time points. Protein was isolated, and Western blotting was performed to observe FoxO1 deacetylation. B, primary mouse hepatocytes were isolated using a standard two-step collagenase perfusion method and cultured in DMEM containing 10% Dowex-stripped FBS and 1× penicillin/streptomycin with or without T3 (0.1 μm) treatment for 16 h. Protein was isolated, and Western blotting was performed to observe FoxO1 deacetylation. C, SirT1 KD significantly reduced T3-dependent deacetylation of FoxO1. Cells were cultured with control siRNA or SirT1 siRNA for 48 h followed by T3 treatment (0.1 μm) for 24 h. Protein was isolated, and Western blotting was done to observe FoxO1 deacetylation. Densitometric values of AcFoxO1 and FoxO1 were normalized with β-actin, then the ratio (AcFoxO1/FoxO1) was plotted as relative density (n = 3; *, p < 0.05). Error bars represent mean ± S.D.
FIGURE 4.
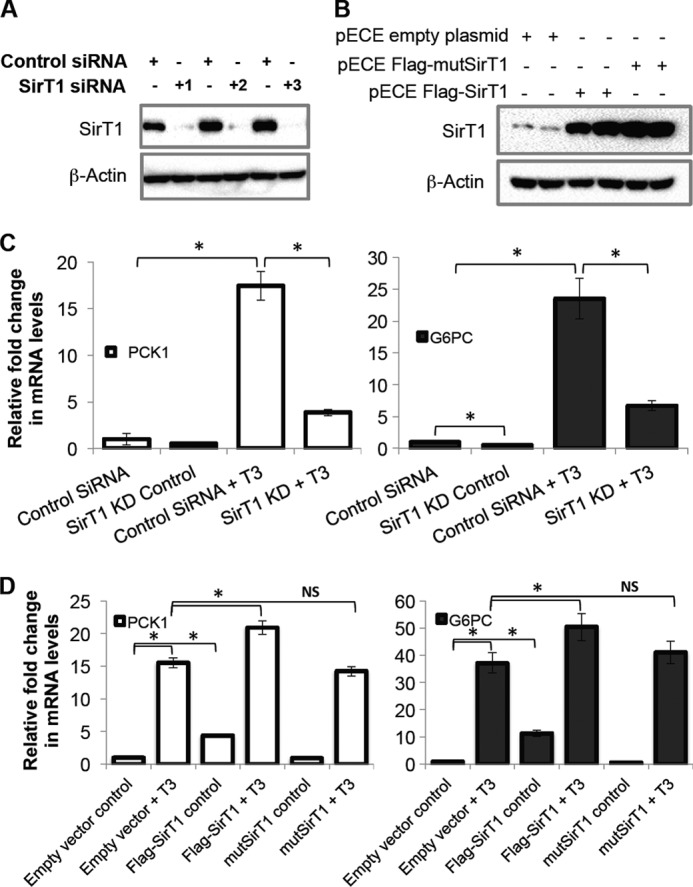
T3 induced PCK1 and G6PC mRNA expression in a SirT1-dependent manner. A, SirT1 was knocked down using three independent siRNAs in TRβ1-HepG2 cells and confirmed at the protein level. Cells were cultured with control siRNA or SirT1siRNA for 48 h followed by replacement of medium with T3-depleted medium as indicated under “Experimental Procedures.” Protein was harvested, and SirT1 KD was confirmed by Western blotting. B, to overexpress SirT1 in TRβ1-HepG2 cells, FLAG-tagged human SirT1 and its deacetylase domain mutant (H363Y), cloned in pECE (Addgene plasmids 1791 and 1792, respectively) were transfected and confirmed at the protein level. Cells were treated with empty vector as control or SirT1 vectors for 48 followed by replacement of medium with normal medium as indicated under “Experimental Procedures.” Protein was isolated, and SirT1 expression was confirmed by Western blotting. C, SirT1 deficiency significantly reduced T3-dependent increase in PCK1 and G6PC expression. Cells were treated with control siRNA or SirT1 siRNA for 48 h followed by T3 treatment (0.1 μm) for 24 h. 48 h after transfection cells were treated with T3 (0.1 μm) for 24 h, total mRNA was collected, and RT-qPCR analysis was performed as indicated under “Experimental Procedures.” D, SirT1 overexpression significantly increased T3-induced PCK1 and G6PC expression. After 48 h of transfection cells were treated with T3 (0.1 μm) for 24 h, total mRNA was collected, and RT-qPCR analysis was performed as indicated under “Experimental Procedures.” β-Actin was used as normalization control (n = 3; *, p < 0.05). Error bars represent mean ± S.D.
We next examined TH regulation of PCK1 and G6PC mRNA and its association with FoxO1 deacetylation in vivo. TH-induced hepatic PCK1 and G6PC mRNA levels (Fig. 5A) in parallel with hepatic FoxO1 deacetylation in hyperthyroid mice (HyperTH) (Fig. 5B). Furthermore, to confirm that TH-induced deacetylation is SirT1-dependent, SirT1 inhibitor Ex527 (24) was administered in vivo. Ex527 administration significantly blocked TH-mediated deacetylation of FoxO1 (Fig. 5C).
FIGURE 5.
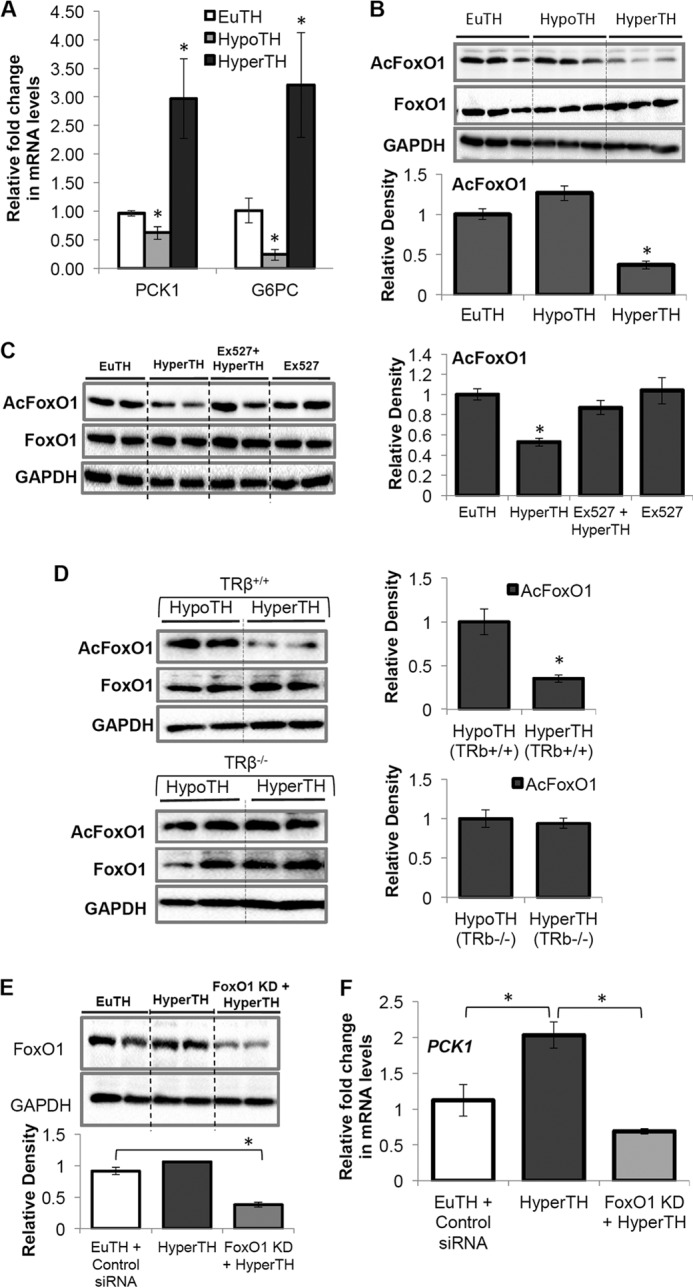
T3 increased PCK1, G6PC gene expression, and SirT1-dependent deacetylation of FoxO1 in mice which also required TRβ. A, hypothyroidism and hyperthyroidism in male C57BL/6 mice (8–10 weeks old) were induced and confirmed as described under “Experimental Procedures.” B, after 14 h of TH injection, liver tissues were harvested, and total mRNA/protein was extracted. RT-qPCR and Western blot analysis were performed on EuTH, HypoTH, and HyperTH mice liver tissues as described under “Experimental Procedures.” An increase in PCK1 and G6PC expression in hyperthyroid mouse liver is significantly corroborated by FoxO1 deacetylation. C, Ex527 (0.8 mg/day/100 g of body weight, injected intraperitoneally for 3 days) was used to inhibit SirT1 activity in vivo. After the first injection of Ex527, mice were injected subcutaneously with T3 (10 μg/kg of body weight/day for 2 days) along with Ex527. After 3 days, animals were euthanized, and the liver tissues were subjected to Western blot analysis as indicated under “Experimental Procedures.” Ex527-treated hyperthyroid mouse liver tissues showed a significant increase in FoxO1 acetylation. D, inhibition of TH-induced FoxO1 deacetylation in TRβ-null (TRβ−/−) mouse liver tissues showed that TRβ is essential for TH-dependent FoxO1 deacetylation. A description of TRβ−/− mice is as indicated under “Experimental Procedures.” E and F, in vivo FoxO1 KD using hydrodynamic tail vein injection, as indicated under “Experimental Procedures,” significantly inhibited induction in PCK1 mRNA in HyperTH mouse livers when compared with EuTH. After TH treatment in FoxO1 KD mice, liver tissues were harvested, and total mRNA/protein was extracted. RT-qPCR and Western blot analysis were performed on EuTH and HyperTH mice liver tissues as described under “Experimental Procedures.” Densitometric values of FoxO1 were normalized with GAPDH and plotted as relative density. β-Actin was used as normalization control in RT-qPCR analysis (n = 5; *, p < 0.05). Error bars represent mean ± S.D.
Moreover, to determine whether FoxO1 deacetylation was TRβ-dependent, we examined hepatic FoxO1 deacetylation in TRβ-null (TRβ−/−) mouse livers under HypoTH and HyperTH conditions (Fig. 5D). In HyperTH condition, WT (TRβ+/+) mice displayed significant deacetylation of FoxO1 compared with HypoTH (TRβ+/+) livers, whereas this effect of TH was lost in TRβ−/− mice (Fig. 5D).
Next, to confirm that FoxO1 is required for TH-induced transcription activation of key gluconeogenic genes, we performed hydrodynamic tail vein injection of FoxO1 siRNA to knock down FoxO1 preferentially in the liver (Fig. 5E). We observed that FoxO1 knockdown significantly reduced PCK1 expression in HyperTH mouse livers compared with euthyroid (EuTH) (Fig. 5F). Taken together, these in vitro and in vivo data showed that TH induction of gluconeogenic gene expression was mediated through FoxO1 deacetylation that was dependent upon both SirT1 and TRβ expression.
T3 Induction of PCK1 and G6PC mRNA Expression Requires FoxO1 Recruitment to Their Promoters in a SirT1-dependent Manner
From our preceding data, TH induction of PCK1 and G6PC transcription was SirT1-dependent because pharmacological stimulation and overexpression of SirT1 increased, and knockdown of SirT1 decreased, TH-induced PCK1 and G6PC transcription. To further confirm FoxO1 requirement for TH action, we performed ChIP-qPCR to analyze FoxO1 binding to the IRE located on the PCK1 and G6PC promoters, which has been shown earlier to be necessary for FoxO1-mediated transcription (9, 27). ChIP-qPCR analyses showed that T3 significantly increased FoxO1 binding to the IRE regions of PCK1 (−426 to −170) and G6PC (−237 to −70) promoters (Fig. 6A). Furthermore, FoxO1 recruitment to these target gene promoters was SirT1-dependent as SirT1 knockdown blocked T3-mediated FoxO1 binding to both promoters. Taken together, these results demonstrated that TH-induced FoxO1 recruitment to PCK1 and G6PC promoters was SirT1-dependent.
FIGURE 6.
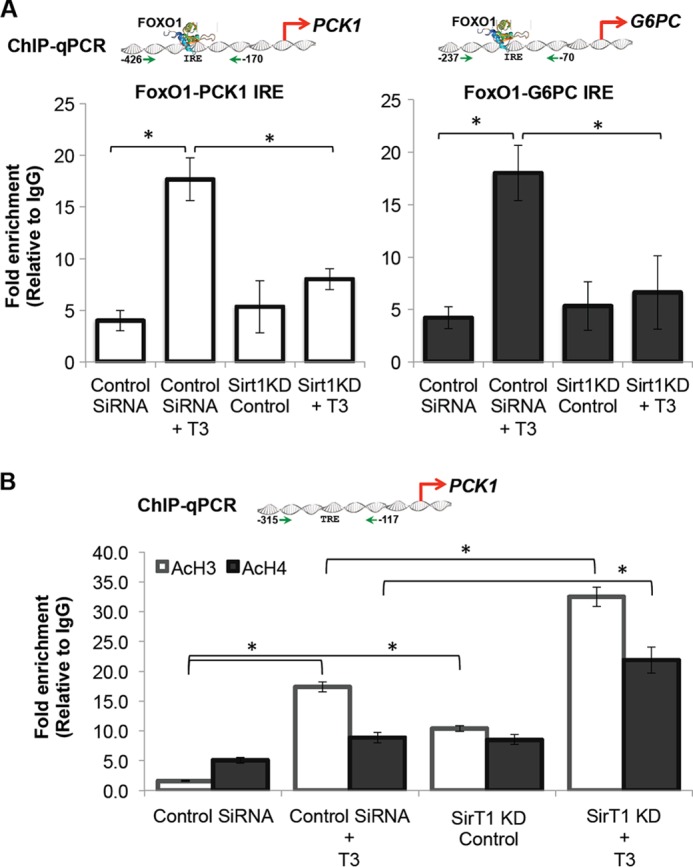
T3 induced FoxO1 recruitment to PCK1 and G6PC promoters in a SirT1-dependent manner. A, ChIP-qPCR analysis revealed that T3 increased FoxO1 recruitment on PCK1 and G6PC promoters at the IRE region (−426 to −170 for PCK1 and −237 to −70 for G6PC gene promoter), which is known for FoxO1 binding in relation to gluconeogenesis. SirT1 deficiency significantly reduced T3-dependent binding of FoxO1. Cells were cultured with control siRNA or SirT1 siRNA for 48 h followed by replacement of medium with normal medium as indicated under “Experimental Procedures.” After 72 h of transfection, cells were treated with T3 for 1 h to analyze FoxO1 recruitment. B, ChIP-qPCR analysis of TRE on PCK1 promoter demonstrates that T3 (0.1 μm) rapidly increased acetylation of histone H3 and H4 in this region. HDAC activity of SirT1 was also confirmed as evident from increased basal acetylation of histone H3 and H4 during SirT1 KD in TRβ-HepG2 cells. During SirT1 KD, T3 (0.1 μm) further increased acetylation of these histones, which is significant. 2 μl of immunoprecipitated DNA (1% input DNA) was used for qPCR analysis as indicated under “Experimental Procedures” (n = 3; *, p < 0.05). Error bars represent mean ± S.D.
Moreover, we also analyzed histone H3 and H4 acetylation (AcH3, AcH4) in the region encompassing the TRE (−315 to −117) of the PCK1 promoter (28) using ChIP-qPCR. In agreement with previous studies, we found that T3 rapidly increased histone H3 and H4 acetylation near the TRE on PCK1 gene promoter (Fig. 6B). We found that histone H3 and H4 acetylation was increased at this promoter by the knockdown of SirT1 in TRβ-HepG2 cells and even further increased by T3 treatment after SirT1 knockdown in these cells. Although this change in histone acetylation is expected with an HDAC knockdown, it presents an apparent paradox because loss of SirT1 impairs T3-induced transcription of these genes. One possible explanation for this observation could be that although increased histone acetylation may be permissive for transcriptional activity, net transcription of these genes is dependent on FoxO1 recruitment as a principal enhancer. In this scenario, increased deacetylation of FoxO1 by SirT1 may override its histone deacetylation activity and increase net transcription by T3. The further increase in histone acetylation by T3 when SirT1 is knocked down suggests that there may be increased histone acetyltransferase activity induced by T3 that does not result in a productive transcriptional activation. We were not able to analyze G6PC-TRE because no TRE has been identified thus far.
DISCUSSION
Although TH is known to induce the expression of the gluconeogenic genes PCK1 and G6PC, little is known about the mechanism of induction. Because PCK1 and G6PC also are transcriptional targets for FoxO1 (9, 11), we examined whether FoxO1 could co-regulate PCK1 and G6PC transcription by TH. Surprisingly, we found that FoxO1 plays a key role in TH induction of PCK1 and G6PC gene transcription in vitro and in vivo. FoxO1 gain-of-function experiments in vitro significantly increased PCK1 and G6PC gene transcription, and genetic knockdown of FoxO1 using siRNA in vitro and in vivo significantly abolished the effect of T3 (Figs. 2 and 5).
FoxO1 transcription factors are tightly regulated by post-translational modifications; in particular, FoxO1 deacetylation facilitates nuclear retention and DNA binding (29, 30). Because SirT1 is known to deacetylate FoxO1 (12, 14, 31), we examined whether TH induction of PCK1 and G6PC gene transcription involved FoxO1 deacetylation by SirT1. Interestingly, TH increased deacetylation of FoxO1 via SirT1 in vitro and in vivo (Figs. 3 and 5, B and C). Moreover, pharmacological activation of SirT1 by resveratrol and/or genetic loss-of-function and gain-of-function experiments showed that SirT1 is important for full induction of PCK1 and G6PC transcription by TH (Fig. 4). Surprisingly, we found that these effects were not regulated by these increases in histone H3 and H4 acetylation (Fig. 6B) on the PCK1 promoter because transcription decreased both in the absence or presence of T3 when SirT1 was knocked down in TRβ1-HepG2 cells. Instead, our data strongly suggest that TH induction of SirT-mediated FoxO1 deacetylation stimulated transcription of PCK1 and G6PC target genes by increasing FoxO1 binding to their gene promoters (Fig. 6A). Because both TRβ and SirT1 are primarily nuclear proteins,3 it is likely that FoxO1 deacetylation occurs in the nucleus. Moreover, TRβ is required for TH induction of FoxO1 deacetylation because TH is unable to deacetylate FoxO1 in TRβ-null mice (Fig. 5C).
Recently, Thakran et al. (16) showed that SirT1 deacetylation of co-activator PGC1α may mediate TH induction of CPT1α mRNA in the liver. Our findings provide an even greater understanding of TH-mediated transcription by showing that SirT1 plays a critical role in TH-mediated transcription of gluconeogenic genes in addition to those involved in fatty acid β oxidation; thus, SirT1 may be a central regulator of TH metabolic action in the liver. Additionally, FoxO1 is a major target of SirT1, and its deacetylation is necessary for the TH transcriptional effects on PCK1 and G6PC transcription. Besides SirT1, other HDACs (e.g. HDAC3, HDAC4, and HDAC5) previously have been shown to deacetylate FoxO transcription factors (15), so it is possible that TH or other hormones may stimulate their activities to regulate target genes. Further studies will need to be performed to determine whether this is the case.
Whereas a TRE has been described in the PCK1 promoter, no TRE has been identified thus far in G6PC. Thus, our findings raise the possibility that TR binding to target genes may not be absolutely required to activate the transcription of some TH-regulated target genes. TH may be able to indirectly regulate transcription of target genes without TREs by activating other transcription factors such as FoxO1. These results challenge the common assumption that genes induced early by TH are regulated directly by TR binding to TREs of target genes whereas genes induced late are due to TR-mediated transcription of secondary transcription factors. Indeed, our findings with G6PC suggest that the latter may not need to occur for TH to indirectly regulate the transcription of certain target genes and could even apply to the actions of other nuclear hormone receptors. Comparison of TR binding by genomic ChIP-Seq analyses with genomic microarray data should help determine whether this occurs.
In summary, our study shows that FoxO1 is crucial for TH induction of the key gluconeogenic genes, PCK1 and G6PC. We demonstrate that TH induction of PCK1 and G6PC gene transcription occurs by a novel mechanism that involves SirT1-dependent deacetylation of FoxO1, leading to its binding to their gene promoters and activating their transcription. This elegant example of co-regulation of gene target transcription via nuclear hormone receptor activation of another transcription factor reveals a mechanism by which a hormone can increase the number of target genes that it can regulate while concomitantly allowing its actions to be modulated by cellular metabolic and energy conditions.
Acknowledgments
We thank Prof. Mitchell A. Lazar (Division of Endocrinology, Diabetes, and Metabolism, Department of Medicine, The Institute for Diabetes, Obesity, and Metabolism, Perelman School of Medicine at the University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania) and Prof. Christopher B. Newgard (Sarah W. Stedman Nutrition and Metabolism Center, Departments of Medicine and Pharmacology and Cancer Biology, Duke University Medical Center, Durham, NC) for critical reading and suggestions. We also thank Dr. Chui Sun Yap, Dr. M. M. Siddique, Laura Martinez, Benjamin Livingston Farah (Cardiovascular and Metabolic Disorders Program, Duke-NUS Graduate Medical School), and Cyrielle Billon (Institut de Génomique Fonctionnelle de Lyon, Université de Lyon, Université Lyon 1, CNRS, Ecole Normale Supérieure de Lyon), for helpful advice and constructive comments; and Prof. Martin L. Privalsky for the kind gift of HepG2 cells ectopically expressing TRβ1, used in this study.
This work was supported by Duke-NUS Graduate Medical School Faculty Funds (to P. M. Y.) sponsored by the Ministry of Health, Ministry of Education and Ministry of Trade, Singapore, and A*StaR Singapore; Singapore National Medical Research Council (NMRC) Early Development Grant (EDG)/1044/2011; Singapore NMRC Clinical Investigator Research Grant 1340/2012; and NMRC/CIRG/1340/2012 grants. This work also was supported by an American Diabetes Association mentored research fellowship (to S. H. Y. and M. A. Lazar).
B. K. Singh and P. M. Yen, unpublished results.
- TH
- thyroid hormone
- EuTH
- euthyroid
- FoxO1
- Forkhead box O1
- G6PC
- glucose-6-phosphatase (G6Pase)
- HDAC
- histone deacetylase
- HyperTH
- hyperthyroid
- HypoTH
- hypothyroid
- IRE
- insulin response element
- KD
- knockdown
- PCK1
- phosphoenolpyruvate carboxykinase (PEPCK)
- PGC1α
- peroxisome proliferator-activated receptor-γ co-activator-1α
- qPCR
- quantitative PCR
- T3
- triiodothyronine
- TR
- thyroid hormone receptor
- TRE
- thyroid hormone response element
- CPT1α
- carnitine palmitoyltransferase 1α.
REFERENCES
- 1. Aub J. C., Bauer W., Heath C., Ropes M. (1929) Studies of calcium and phosphorus metabolism. III. The effects of the thyroid hormone and thyroid disease. J. Clin. Invest. 7, 97–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yen P. M., Ando S., Feng X., Liu Y., Maruvada P., Xia X. (2006) Thyroid hormone action at the cellular, genomic and target gene levels. Mol. Cell. Endocrinol. 246, 121–127 [DOI] [PubMed] [Google Scholar]
- 3. Zhang J., Lazar M. A. (2000) The mechanism of action of thyroid hormones. Annu. Rev. Physiol. 62, 439–466 [DOI] [PubMed] [Google Scholar]
- 4. Yen P. M. (2001) Physiological and molecular basis of thyroid hormone action. Physiol. Rev. 81, 1097–1142 [DOI] [PubMed] [Google Scholar]
- 5. Yang J., Reshef L., Cassuto H., Aleman G., Hanson R. W. (2009) Aspects of the control of phosphoenolpyruvate carboxykinase gene transcription. J. Biol. Chem. 284, 27031–27035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Feng X., Jiang Y., Meltzer P., Yen P. M. (2000) Thyroid hormone regulation of hepatic genes in vivo detected by complementary DNA microarray. Mol. Endocrinol. 14, 947–955 [DOI] [PubMed] [Google Scholar]
- 7. Durham S. K., Suwanichkul A., Scheimann A. O., Yee D., Jackson J. G., Barr F. G., Powell D. R. (1999) FKHR binds the insulin response element in the insulin-like growth factor binding protein-1 promoter. Endocrinology 140, 3140–3146 [DOI] [PubMed] [Google Scholar]
- 8. Gross D. N., van den Heuvel A. P., Birnbaum M. J. (2008) The role of FoxO in the regulation of metabolism. Oncogene 27, 2320–2336 [DOI] [PubMed] [Google Scholar]
- 9. Yabaluri N., Bashyam M. D. (2010) Hormonal regulation of gluconeogenic gene transcription in the liver. J. Biosci. 35, 473–484 [DOI] [PubMed] [Google Scholar]
- 10. Matsumoto M., Pocai A., Rossetti L., Depinho R. A., Accili D. (2007) Impaired regulation of hepatic glucose production in mice lacking the Forkhead transcription factor FoxO1 in liver. Cell Metab. 6, 208–216 [DOI] [PubMed] [Google Scholar]
- 11. Gross D. N., Wan M., Birnbaum M. J. (2009) The role of FoxO in the regulation of metabolism. Curr. Diab. Rep. 9, 208–214 [DOI] [PubMed] [Google Scholar]
- 12. van der Horst A., Burgering B. M. (2007) Stressing the role of FoxO proteins in lifespan and disease. Nat. Rev. Mol. Cell Biol. 8, 440–450 [DOI] [PubMed] [Google Scholar]
- 13. Schilling M. M., Oeser J. K., Boustead J. N., Flemming B. P., O'Brien R. M. (2006) Gluconeogenesis: re-evaluating the FoxO1-PGC-1α connection. Nature 443, E10-E11 [DOI] [PubMed] [Google Scholar]
- 14. Frescas D., Valenti L., Accili D. (2005) Nuclear trapping of the Forkhead transcription factor FoxO1 via Sirt-dependent deacetylation promotes expression of glucogenetic genes. J. Biol. Chem. 280, 20589–20595 [DOI] [PubMed] [Google Scholar]
- 15. Mihaylova M. M., Vasquez D. S., Ravnskjaer K., Denechaud P. D., Yu R. T., Alvarez J. G., Downes M., Evans R. M., Montminy M., Shaw R. J. (2011) Class IIa histone deacetylases are hormone-activated regulators of FoxO and mammalian glucose homeostasis. Cell 145, 607–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thakran S., Sharma P., Attia R. R., Hori R. T., Deng X., Elam M. B., Park E. A. (2013) Role of sirtuin 1 in the regulation of hepatic gene expression by thyroid hormone. J. Biol. Chem. 288, 807–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chan I. H., Privalsky M. L. (2009) Isoform-specific transcriptional activity of overlapping target genes that respond to thyroid hormone receptors α1 and β1. Mol. Endocrinol. 23, 1758–1775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sinha R. A., You S. H., Zhou J., Siddique M. M., Bay B. H., Zhu X., Privalsky M. L., Cheng S. Y., Stevens R. D., Summers S. A., Newgard C. B., Lazar M. A., Yen P. M. (2012) Thyroid hormone stimulates hepatic lipid catabolism via activation of autophagy. J. Clin. Invest. 122, 2428–2438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. You S. H., Liao X., Weiss R. E., Lazar M. A. (2010) The interaction between nuclear receptor corepressor and histone deacetylase 3 regulates both positive and negative thyroid hormone action in vivo. Mol. Endocrinol. 24, 1359–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gauthier K., Chassande O., Plateroti M., Roux J. P., Legrand C., Pain B., Rousset B., Weiss R., Trouillas J., Samarut J. (1999) Different functions for the thyroid hormone receptors TRα and TRβ in the control of thyroid hormone production and post-natal development. EMBO J. 18, 623–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gauthier K., Billon C., Bissler M., Beylot M., Lobaccaro J. M., Vanacker J. M., Samarut J. (2010) Thyroid hormone receptor β (TRβ) and liver X receptor (LXR) regulate carbohydrate-response element-binding protein (ChREBP) expression in a tissue-selective manner. J. Biol. Chem. 285, 28156–28163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brunet A., Sweeney L. B., Sturgill J. F., Chua K. F., Greer P. L., Lin Y., Tran H., Ross S. E., Mostoslavsky R., Cohen H. Y., Hu L. S., Cheng H. L., Jedrychowski M. P., Gygi S. P., Sinclair D. A., Alt F. W., Greenberg M. E. (2004) Stress-dependent regulation of FoxO transcription factors by the SIRT1 deacetylase. Science 303, 2011–2015 [DOI] [PubMed] [Google Scholar]
- 23. Tang E. D., Nuñez G., Barr F. G., Guan K. L. (1999) Negative regulation of the Forkhead transcription factor FKHR by Akt. J. Biol. Chem. 274, 16741–16746 [DOI] [PubMed] [Google Scholar]
- 24. Nie Y., Erion D. M., Yuan Z., Dietrich M., Shulman G. I., Horvath T. L., Gao Q. (2009) STAT3 inhibition of gluconeogenesis is down-regulated by SirT1. Nat. Cell Biol. 11, 492–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hanson R. W., Reshef L. (1997) Regulation of phosphoenolpyruvate carboxykinase (GTP) gene expression. Annu. Rev. Biochem. 66, 581–611 [DOI] [PubMed] [Google Scholar]
- 26. Matsuzaki H., Daitoku H., Hatta M., Aoyama H., Yoshimochi K., Fukamizu A. (2005) Acetylation of FoxO1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 102, 11278–11283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Webb A. E., Brunet A. (2013) FoxO flips the longevity SWItch. Nat. Cell Biol. 15, 444–446 [DOI] [PubMed] [Google Scholar]
- 28. Park E. A., Jerden D. C., Bahouth S. W. (1995) Regulation of phosphoenolpyruvate carboxykinase gene transcription by thyroid hormone involves two distinct binding sites in the promoter. Biochem. J. 309, 913–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang X. J., Seto E. (2008) Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol. Cell 31, 449–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nasrin N., Ogg S., Cahill C. M., Biggs W., Nui S., Dore J., Calvo D., Shi Y., Ruvkun G., Alexander-Bridges M. C. (2000) DAF-16 recruits the CREB-binding protein coactivator complex to the insulin-like growth factor binding protein 1 promoter in HepG2 cells. Proc. Natl. Acad. Sci. U.S.A. 97, 10412–10417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang K., Li L., Qi Y., Zhu X., Gan B., DePinho R. A., Averitt T., Guo S. (2012) Hepatic suppression of FoxO1 and FoxO3 causes hypoglycemia and hyperlipidemia in mice. Endocrinology 153, 631–646 [DOI] [PMC free article] [PubMed] [Google Scholar]