

Background: Etomidate induces anesthesia via intersubunit transmembrane sites in GABAA receptors.
Results: In receptors engineered with α-M1 domain cysteines, GABA accelerates modification. Etomidate inhibits modification at three positions.
Conclusion: Etomidate contacts a subdomain of α-M1 linked to channel gating, consistent with in silico model docking.
Significance: We identify new structures that both bind anesthetic and modulate channel gating through rearrangement.
Keywords: Allosteric Regulation, Anesthetics, GABA Receptors, Ion Channels, Nicotinic Acetylcholine Receptors, Sulfhydryl
Abstract
Etomidate is a potent general anesthetic that acts as an allosteric co-agonist at GABAA receptors. Photoreactive etomidate derivatives labeled αMet-236 in transmembrane domain M1, which structural models locate in the β+/α- subunit interface. Other nearby residues may also contribute to etomidate binding and/or transduction through rearrangement of the site. In human α1β2γ2L GABAA receptors, we applied the substituted cysteine accessibility method to α1-M1 domain residues extending from α1Gln-229 to α1Gln-242. We used electrophysiology to characterize each mutant's sensitivity to GABA and etomidate. We also measured rates of sulfhydryl modification by p-chloromercuribenzenesulfonate (pCMBS) with and without GABA and tested if etomidate blocks modification of pCMBS-accessible cysteines. Cys substitutions in the outer α1-M1 domain impaired GABA activation and variably affected etomidate sensitivity. In seven of eight residues where pCMBS modification was evident, rates of modification were accelerated by GABA co-application, indicating that channel activation increases water and/or pCMBS access. Etomidate reduced the rate of modification for cysteine substitutions at α1Met-236, α1Leu-232 and α1Thr-237. We infer that these residues, predicted to face β2-M3 or M2 domains, contribute to etomidate binding. Thus, etomidate interacts with a short segment of the outer α1-M1 helix within a subdomain that undergoes significant structural rearrangement during channel gating. Our results are consistent with in silico docking calculations in a homology model that orient the long axis of etomidate approximately orthogonal to the transmembrane axis.
Introduction
Ionotropic γ-aminobutyric acid type A (GABAA) receptors are important targets for many general anesthetics (1), but we lack a detailed understanding of the anesthetic binding sites. GABAA receptors are pentamers of homologous subunits, each containing a large extracellular N-terminal domain, four transmembrane domains (M1–M4), and a large intracellular M3-M4 linker. The most abundant GABAA receptor subtypes contain 2α, 2β, and 1γ subunits arranged β-α-β-α-γ when viewed counterclockwise from the extracellular space (Fig. 1A) (2, 3).
FIGURE 1.
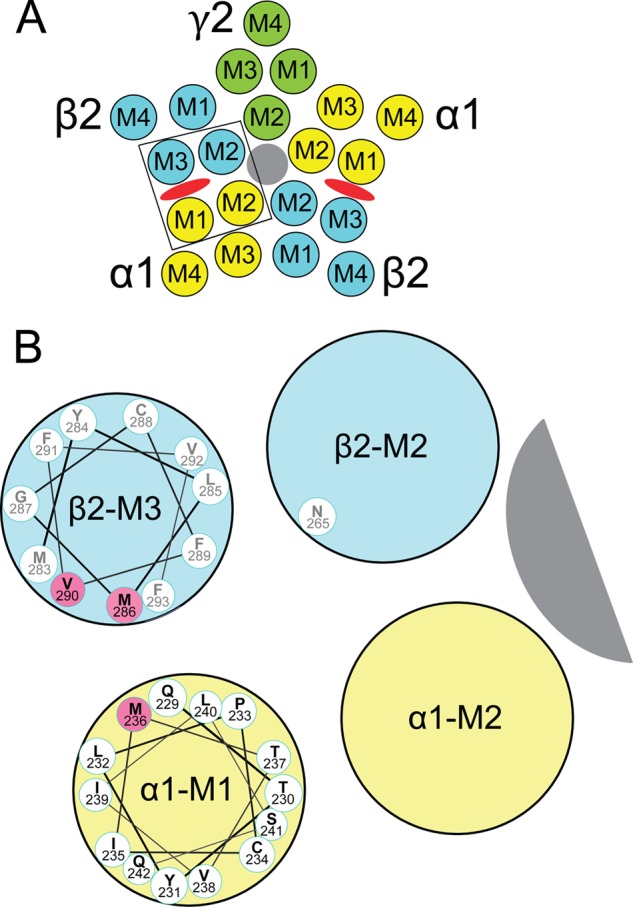
Transmembrane domains and amino acids forming etomidate sites in GABAA receptors. A, a diagram of a α1β2γ2L GABAA receptor cross-sectioned in the membrane plane and viewed from the extracellular space illustrates the arrangement of α1 (yellow), β2 (blue), and γ2 (green) subunits and the transmembrane domains (M1–M4) within each subunit. The chloride channel is shaded gray. Etomidate sites (red ovals) are located between α-M1 and β-M3 domains. B, an expanded diagram of one etomidate site (outlined in A), showing a helical wheel projection of the α1-M1 sequence from Gln-229 (nearest) through Gln-242. The orientation of the side chains is approximate and based on a homology model built from the structure of G. violaceous pentameric ion channels (33). GABAA receptor residues that are photolabeled by etomidate derivatives (13, 14), in both α-M1 and β-M3, are highlighted in pink. We also illustrate β2Asn-265 on β-M2, a residue where mutations influence etomidate sensitivity (37, 38).
Etomidate is a potent intravenous general anesthetic that produces its major effects through GABAA receptors (4, 5). Etomidate enhances receptor activation by GABA, increasing apparent affinity (reducing GABA EC50). At high concentrations, etomidate also directly activates GABAA receptors (6–9). These effects on human α1β2γ2L GABAA receptors are modeled by an equilibrium allosteric co-agonist scheme with two equivalent etomidate sites per receptor (10, 11).
Azietomidate and TDBzl-etomidate ((4-[3-(trifluoromethyl)-3H-diazirin-3-yl]benzyl 1-(1-phenylethyl)-1H-imidazole-5-carboxylate)) are photoreactive etomidate analogs (8, 12) that label purified GABAA receptors in detergent at αMet-236 in M1 and βMet-286 and βVal-290 in M3 (13, 14). Tryptophan mutations at α1Met-236 or β2Met-286 mimic etomidate effects (15), and a cysteine substitution at β2Met-286 is protected from covalent modification in the presence of etomidate (16). Based on x-ray crystal structures of pentameric ion channel homologs from Gloeobacter violaceus (GLIC)2 and Caenorhabditis elegans (GluCl) (17–19), GABAA receptor structural homology models (14, 20, 21) locate these transmembrane residues in interfacial clefts between β and α subunits (Fig. 1B). Disulfide trapping studies (22) further support this rotational orientation of α-M1 and β-M3 domains.
Although photolabeling has identified the general region where etomidate binds to GABAA receptors, complementary approaches are needed to more fully explore etomidate binding interactions in intact receptors. Receptor protein used for photolabeling was affinity-purified in detergent and may have undergone structural changes. The photosensitive diazirine moieties of azietomidate and TDBzl-etomidate are appended to one end of the stereoselective parent ligand and may fail to label some side chains in the site.
Given the identification of αMet-236 as a likely contact point, etomidate interactions with the α-M1 domain are of particular interest. Residues in α-M1 have previously been linked to GABAA receptor modulation by barbiturates (23, 24) and neurosteroids (25, 26) as well as gating transduction between extracellular GABA sites and the transmembrane channel (23, 27, 28). Cysteine substitution and modification studies (21, 22) have provided clues to some side chain orientations and evidence of rearrangements during gating for the pre-M1 and outer α-M1 domain from αIle-223 to αMet-236.
The aims of this study were to investigate the roles of α1-M1 domain residues in receptor gating, etomidate modulation, and etomidate binding interactions in functional α1β2γ2L GABAA receptors in native membranes. We applied the substituted cysteine accessibility method to a series of 13 single-residue cysteine-substituted mutations in the α1-M1 domain near α1Met-236. In each mutant channel, sensitivity to GABA and etomidate and the effects and rates of pCMBS modification with and without GABA were quantified electrophysiologically. We also tested proximity to bound etomidate at each accessible cysteine by testing if etomidate blocks sulfhydryl modification. Additional biochemical studies were performed on channels harboring the α1M236C mutation. Results were compared with in silico etomidate docking calculations in a homology model of the β2-M3/α1-M1 interfacial pocket.
MATERIALS AND METHODS
Animal Use
Female Xenopus laevis were housed in a veterinary-supervised environment in accordance with local and federal guidelines. Frogs were anesthetized by immersion in 0.2% tricaine (Sigma-Aldrich) prior to minilaparotomy to harvest oocytes.
Chemicals
R-(+)-Etomidate was obtained from Bedford Laboratories (Bedford, OH). The clinical preparation in 35% propylene glycol was diluted directly into buffer. Propylene glycol at the resulting concentrations has no effect on GABAA receptor function (10). Picrotoxin (PTX) was purchased from Sigma-Aldrich and dissolved (2 mm) in electrophysiology buffer. Alphaxalone was purchased from MP Biomedical (Solon, OH) and prepared as a stock solution in DMSO. p-Chloromercuribenzenesulfonic acid sodium salt (pCMBS), methyl methanethiosulfonate (MMTS), and 2-aminoethyl methanethiosulfonate hydrobromide (MTSEA) were purchased from Toronto Research Chemicals (North York, Canada). Salts and buffers were purchased from Sigma-Aldrich.
Molecular Biology
cDNAs for human GABAA receptor α1, β2, and γ2L subunits were cloned into pCDNA3.1 vectors (Invitrogen). Mutations in cDNA were created with oligonucleotide-directed mutagenesis using QuikChange kits (Agilent Technologies, Santa Clara, CA). Clones from each mutagenesis reaction were subjected to DNA sequencing through the entire cDNA region to confirm the presence of the mutation and absence of stray mutations.
Oocyte Electrophysiology
Messenger RNA synthesis and Xenopus oocyte expression were performed as described previously (15). Experiments were performed at room temperature (21–23 °C) in ND96 buffer (96 mm NaCl, 2 mm KCl, 0.8 mm MgCl2, 1.8 mm CaCl2, 5 mm HEPES, pH 7.5). Peak current responses to varying GABA concentrations (range from 0.1 μm to 30 mm), alone or co-applied with 3.2 μm etomidate, were measured in Xenopus oocytes (n ≥ 3) using two-microelectrode voltage clamp electrophysiology, as described previously (29). GABA applications were from 5 to 20 s, depending on the time to reach steady-state peak current. Normalizing GABA responses at maximal GABA (1–30 mm) were recorded every second or third sweep. Picrotoxin-sensitive leak was measured using 2 mm PTX, followed by >5-min washout and a maximal GABA response test. Etomidate (3–10 μm) or alphaxalone (2 μm) was used as a gating enhancer together with maximal GABA to estimate GABA efficacy. Oocyte currents were low pass-filtered at 1 kHz (model OC-725B, Warner Instruments, Hamden, CT), digitized at 1–2 kHz (Digidata 1200, Molecular Devices, Sunnyvale, CA), and recorded digitally (pClamp 7, Molecular Devices).
Cysteine Modification with pCMBS and Etomidate Protection in Xenopus Oocytes
Maximally GABA-activated receptors were used for control (reference) pCMBS modification, and etomidate concentrations in protection studies were at least 2 times EC50 for direct activation (30–100 μm), establishing occupation of >90% of etomidate sites. The pCMBS concentration used for each mutant channel was chosen so that control modification was complete after 40–60 s of pCMBS exposure. Oocytes were repetitively stimulated with GABA pulses every 5 min until at least three sequential current responses were constant (±5%). For modification, oocytes were exposed to pCMBS (alone, with GABA, or with GABA plus etomidate) for 5, 10, or 20 s followed by a 5-min ND96 wash. A GABA response test, when appropriate, was repeated prior to the next pCMBS modification, and up to 15 (but usually fewer than 10) modification test cycles were repeated in each oocyte. For modification rate analysis, peak currents were normalized to a pre- or postmodification control and plotted against cumulative pCMBS exposure in units of s × mm. Normalized data were fitted to single exponential functions to determine the apparent modification rate constant in M−1 s−1.
Electrophysiology in HEK293 Cell Membrane Patches
HEK293 cell maintenance and transfection for functional studies were performed as described previously (15). Current recordings from excised outside-out membrane patches were performed at −50 mV and room temperature (21–23 °C) as described by Scheller and Forman (30). Currents were stimulated using pulses of GABA delivered via a multibarrel superfusion pipette coupled to piezo-electric elements that switched solutions in under 1 ms. Currents were filtered at 5 kHz and digitized at 10 kHz for off-line analysis.
HEK Cell Membrane Preparation for Binding Assays
HEK293T cells at 30–40% confluence were co-transfected using CaPO4 precipitation (31) with cDNAs encoding wild-type or mutant α1, β2, and γ2L. At 48 h after transfection, cells were harvested and centrifuged at 1500 × g for 10 min. The cell pellet was washed twice in PBS and again pelleted. The pellet was homogenized at 11,000 rpm for 30 s in PBS using an Ultra-Turrax T25 homogenizer (Janke & Kunkel, Staufen, Germany). The homogenate was centrifuged for 30 min at 25,000 × g at 4 °C. The membranes were washed and repelleted twice and then resuspended in PBS.
MMTS/MTSEA Reaction and Etomidate Protection in Membranes
Aliquots of membrane suspension were incubated for 30 min at 4 °C with a sulfhydryl-modifying reagent (MMTS or MTSEA) or with buffer as a control. For protection experiments, membranes were preincubated for 10 min with 200 μm etomidate, which remained present during MTSEA incubation. To remove sulfhydryl reagents (and etomidate), the membrane pellets (in both the MTSEA and the control groups) were resuspended with PBS buffer and repelleted four times (25,000 × g, 30 min, 4 °C).
[3H]Flunitrazepam Binding Assay in Membranes
Membrane suspensions were diluted into assay buffer (10 mm phosphate buffer (pH 7.4, 135 mm KCl, and 1 mm EDTA; 0.5 ml final volume) with 1 nm [3H]flunitrazepam (85.2 Ci/mmol; PerkinElmer Life Sciences) and incubated for 1 h at 4 °C. Nonspecific binding was measured by including 7.5 μm flurazepam. Etomidate modulation was tested at 0.1–100 μm. Multiple (n = 6) samples were suction-filtered on GF/B glass fiber filters. Filters were washed two times with 3 ml of assay buffer and transferred to scintillation vials with 2.5 ml of scintillation fluid (Ecolume, ICN Pharmaceuticals, Aurora, OH) for counting.
Electrophysiological Data Analysis
Analyses for agonist concentration responses, etomidate-induced left shift, and allosteric co-agonist model fitting followed our approach described elsewhere (15, 32). Peak GABA-stimulated currents were normalized to maximal GABA responses, and GABA concentration-response data for individual oocytes in the absence and presence of etomidate were fitted with logistic functions using non-linear least squares (Graphpad Prism version 5),
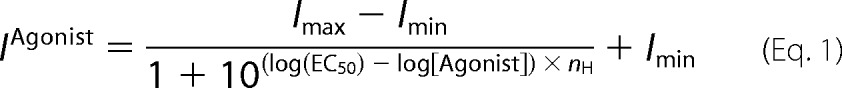 |
where EC50 is the half-maximal activating concentration and nH is Hill slope.
EC50 shift ratios were calculated from the difference in log(GABA EC50) values (Δlog(EC50)) measured in the presence of 3.2 μm etomidate versus control. Etomidate-dependent direct activation of receptors was analyzed similarly.
PTX-sensitive leak currents (IPTX) normalized to ImaxGABA were used to estimate basal open probability (Po). GABA efficacy was estimated based on enhancement of maximal GABA responses by etomidate or alphaxalone (32).
Estimated Po was calculated using average IPTX/ImaxGABA and IGABA+Enhancer/ImaxGABA values.
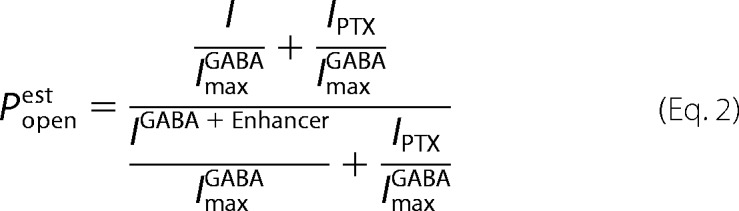 |
Non-linear least squares fits to a Monod-Wyman-Changeux (MWC) co-agonist mechanism with two equivalent sites each for GABA and etomidate (Equation 3) used estimated Po data (Equation 2) from GABA concentration responses with and without etomidate and etomidate direct activation, with both [GABA] and [etomidate] ([ETO]) specified as independent variables.
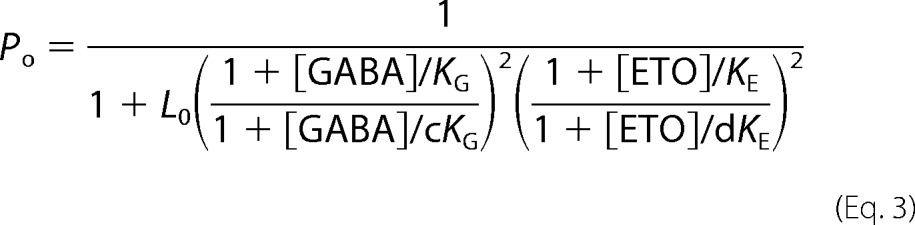 |
L0 in Equation 3 is a dimensionless basal equilibrium gating variable, approximately Po−1. KG and KE are equilibrium dissociation constants for GABA and etomidate binding to inactive receptors, and c and d are dimensionless parameters representing the respective ratios of dissociation constants in open versus inactive receptors. The agonist efficacies of GABA and etomidate are inversely related to c2 and d2, respectively.
Analysis of patch macrocurrents for activation, desensitization, and deactivation kinetics was performed as described previously (32) using non-linear least squares fits to Equation 4 and F-tests at p = 0.99 (Clampfit version 8.0, Molecular Devices) to choose the best number of exponents.
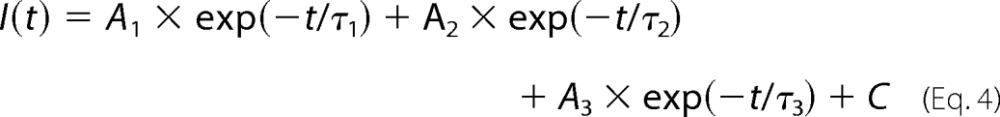 |
All activation traces were best fit with a single exponent, whereas desensitization and deactivation were consistently fitted with two exponents.
Molecular Modeling
A model of the α1β3γ2 GABAA receptor was constructed using the structure of the pentameric ion channel homolog GLIC from Protein Data Bank entry 3P50 as described (33). β2 peptide sequences were substituted into the β3 structures, requiring no insertions or deletions and only 26 amino acid replacements, seven of which were in the transmembrane domain (two in M3 and five in M4). No substitutions were near the etomidate binding sites. The model was placed within a simulated membrane force field and minimized with the photoreactive etomidate analog azietomidate placed in the etomidate binding interfaces, consistent with photolabeling results (13, 14). Using C-DOCKER in the Discovery Studio modeling software package (Accelrys Inc., San Diego, CA), R-(+)-etomidate (183-Å3 molecular volume, 213-Å3 Connolly surface volume) was docked within an 11-Å radius sphere in each binding site, solving for the 100 lowest energy solutions starting with 50 different initial orientations and 50 different molecular dynamics-altered etomidate configurations. Interaction energies for all 200 solutions (100 for each site) were between −36 and −39 kcal/mol. As controls, etomidate docking was performed at the other three model intersubunit transmembrane cavities.
Statistical Analysis
Results are reported as mean ± S.D. unless otherwise noted. Wild-type and all mutant group comparisons for log(EC50) and Δlog(EC50) were performed using one-way ANOVA with Dunnett's post hoc multiple comparisons test in GraphPad Prism. Within the mutant group, statistical comparisons of pCMBS modification rates measured under three conditions were performed using ANOVA with Tukey's post hoc test. Etomidate-dependent [3H]flunitrazepam binding data were analyzed using two-way ANOVA with post hoc multiple comparisons test. Pairwise comparisons were performed using Student's t tests. Statistical significance was inferred at p < 0.05.
RESULTS
Functional Characterization of Wild-type and α1M236Cβ2γ2L GABAA Receptors
GABA- and Etomidate-dependent Channel Gating
Initial studies focused on the α1Met-236 residue identified in photolabeling studies (13, 14). GABA- and etomidate-dependent gating, spontaneous activation, and macrocurrent rapid kinetics were characterized in α1M236Cβ2γ2L channels expressed in Xenopus oocytes or HEK293 cells and compared with α1β2γ2L (wild type). Results of oocyte studies are summarized in Table 1. Functional characteristics of α1β2γ2L receptors in oocytes were similar to those reported in earlier studies (10, 15, 32). Wild-type GABA EC50 value averaged 37 μm, and the addition of 3.2 μm etomidate reduced GABA EC50 18-fold to about 2 μm. Etomidate directly activated wild-type channels with an average EC50 of 30 μm, and maximum etomidate activation averaged 42% of the maximal GABA response. In oocytes expressing α1M236Cβ2γ2L GABAA receptors, GABA EC50 averaged about 10-fold higher (390 μm) than that in wild-type (Fig. 2A (solid symbols) and Table 1). In the presence of etomidate, α1M236Cβ2γ2L receptor currents elicited with maximal GABA (10–30 mm) increased about 4-fold, indicating a low agonist efficacy for GABA. GABA EC50 was reduced about 5-fold (Fig. 2A, open symbols). Etomidate alone was a potent and efficacious agonist of α1M236Cβ2γ2L receptors, inducing maximal currents that were 4-fold larger than those elicited by 10–30 mm GABA (Fig. 2B). Rapid application of GABA (10 mm) to voltage-clamped outside-out patches from HEK293 cells expressing α1M236Cβ2γ2L receptors elicited currents with activation, desensitization, and deactivation phases similar to those of wild-type receptors activated with 1 mm GABA (Fig. 2C and Table 2). With reduced GABA potency and efficacy, accelerated deactivation and slower desensitization might be expected if desensitization proceeds only from open states. However, GABA-bound pre-open states may desensitize at rates comparable with those of open states (34). Also, GABA efficacy in this mutant may differ in HEK293 cells and Xenopus oocytes.
TABLE 1.
GABA and etomidate sensitivities of wild-type and mutated GABAA receptors expressed in Xenopus oocytes
EC50 values and confidence intervals were calculated from the average and S.D. of fitted log(EC50) values from individual oocytes (n > 3). GABA efficacy was calculated from the ratio of maximal GABA response to maximal GABA plus an enhancing drug (etomidate or alphaxalone). Etomidate efficacy is expressed as the ratio of maximal etomidate current to the maximal GABA current. Comparisons with wild type were performed using one-way ANOVA with Dunnett's post hoc test. CI, confidence interval.
| Receptor type | GABA EC50 (95% CI) | GABA efficacy (n) | Etomidate EC50 (95% CI) | Etomidate efficacy (n) | GABA EC50 ratio (95% CI) |
|---|---|---|---|---|---|
| μm | μm | ||||
| Wild type | 36 (24–55) | 0.80 ± 0.03 (6) | 30 (14–66) | 0.42 ± 0.10 (6) | 0.059 (0.043–0.082) |
| α1Q229C | 610 (500–750)a | 0.65 ± 0.02 (6)b | 40 (17–93) | 0.24 ± 0.14 (3) | 0.29 (0.19–0.45)a |
| α1T230C | 78 (57–105)b | 0.90 ± 0.04 (4) | 12 (7.1–19) | 0.68 ± 0.18 (3) | 0.021 (0.012–0.036)c |
| α1Y231C | 69 (40–119) | 0.63 ± 0.06 (4)b | 10 (8.7–12) | 0.99 ± 0.08 (3) | 0.012 (0.005–0.030)a |
| α1L232C | 77 (56–107)b | 0.95 ± 0.03 (5) | 20 (13–32) | 0.68 ± 0.23 (4) | 0.060 (0.038–0.094) |
| α1P233C | 63 (52–76) | 0.32 ± 0.06 (5)c | 15 (12–19) | 3.0 ± 0.95 (3)a | 0.069 (0.040–0.118) |
| α1I235C | 96 (65–142)a | 0.85 ± 0.03 (5) | 3.0 (1.3–6.7)a | 0.83 ± 0.24 (3) | 0.142 (0.072–0.279) |
| α1M236C | 360 (260–510)a | 0.24 ± 0.06 (6)c | 19 (13–27) | 4.0 ± 0.86 (3)a | 0.186 (0.097–0.356)a |
| α1T237C | 43 (32–58) | 0.78 ± 0.12 (4) | 31 (24–39) | 0.9 ± 0.26 (3) | 0.078 (0.024–0.259) |
| α1V238C | 51 (38–67) | 0.76 ± 0.03 (6) | 20 (13–31) | 0.9 ± 0.15 (3) | 0.122 (0.068–0.220) |
| α1I239C | 46 (39–53) | 1.00 ± 0.02 (3) | 32 (18–57) | 0.31 ± 0.10 (3) | 0.084 (0.070–0.101) |
| α1L240C | 28 (20–40) | 0.91 ± 0.02 (4) | 16 (8.9–30) | 0.72 ± 0.12 (4) | 0.114 (0.072–0.179) |
| α1S241C | 38 (31–48) | 0.72 ± 0.03 (4) | 13 (7.3–23) | 0.62 ± 0.20 (3) | 0.028 (0.016–0.042)c |
| α1Q242C | 66 (46–93) | 0.89 ± 0.04 (4) | 36 (14–96) | 0.38 ± 0.11 (3) | 0.067 (0.037–0.121) |
a p < 0.001.
b p < 0.05.
c p < 0.01.
FIGURE 2.

Electrophysiological characterization of α1M236Cβ2γ2L GABAA receptors. A, GABA concentration response in oocytes. Data points are mean ± S.D. (error bars) (n > 3) peak currents normalized to maximal GABA (10 mm) responses. Lines overlaying points represent nonlinear least squares fits to Hill equations (Equation 1). Solid symbols, GABA alone; EC50 = 320 μm (95% confidence interval, 290–410 μm); nH = 0.75 ± 0.075. Open symbols, GABA plus 3.2 μm etomidate; EC50 = 61 μm (95% confidence interval, 56–77 μm); nH = 0.83 ± 0.071; maximum response = 3.7 ± 0.10. B, etomidate agonism concentration response in oocytes. Data points are mean ± S.D. (n > 3) peak currents normalized to maximal GABA (10 mm) responses. The line represents a fitted Hill equation. EC50 = 18 μm (95% confidence interval, 14–29 μm); nH = 1.7 ± 0.52; maximum response = 4.2 ± 0.63. C, current sweep recorded from an HEK293 cell patch during a 1-s pulse of 10 mm GABA. The white bar indicates GABA application. Average rates of activation, desensitization, and deactivation are summarized in Table 2. D, spontaneous channel gating current in an oocyte. The small outward current during PTX application is due to inhibition of active channels. Current elicited with 10 mm GABA in the same cell is also displayed. Average spontaneous activity is 1.8 ± 0.28% of maximal GABA response. E, estimation of maximal GABA efficacy in an oocyte. GABA (10 mm; white bar) alone elicits a current that is enhanced severalfold with co-application of alphaxalone (2 μm; black bar). Average results for GABA efficacy are reported in Table 1. F, estimation of etomidate agonist efficacy in an oocyte. Etomidate (100 μm; black bar) elicits a maximal current that is only modestly enhanced with co-application of GABA (3 mm; white bar). Average results for etomidate efficacy are reported in Table 1. G, allosteric co-agonist modeling of GABA and etomidate activation. Estimated Po was calculated using Equation 2 from data in A and B (same symbols used for each data set). Equation 3 was fitted to estimated Po using nonlinear least squares with both [GABA] and [etomidate] as input variables. Lines represent the fitted model. Fitted model parameters are reported in Table 3.
TABLE 2.
Wild-type and mutant GABAA receptor rates of activation, desensitization, and deactivation
Rates for mutant receptors did not significantly differ from those for wild type. amp, amplitude.
| Receptor | Maximal activation rate | Fast desensitization rate (s−1) (amp (%)) | Slow desensitization rate (s−1) (amp (%)) | Fast deactivation rate (s−1) (amp (%)) | Slow deactivation rate (s−1) (amp (%)) |
|---|---|---|---|---|---|
| s−1 | |||||
| α1β2γ2L | 3000 ± 1200 (n = 4) | 20 ± 13 (30 ± 11) | 1.0 ± 0.45 (70 ± 11) | 50 ± 18 (60 ± 17) | 6 ± 3.5 (40 ± 17) |
| α1M236Cβ2γ2L | 2700 ± 1000 (n = 3) | 28 ± 12 (30 ± 13) | 1.1 ± 0.45 (70 ± 13) | 60 ± 16 (40 ± 23) | 10 ± 5 (60 ± 23) |
Voltage-clamped oocytes (n = 3) expressing α1M236Cβ2γ2L receptors displayed small picrotoxin-sensitive leak currents in the absence of GABA (Fig. 2D) that were 1–2% of that elicited with 10 mm GABA (0.3–0.5% of maximal currents elicited with 100 μm etomidate). Oocytes expressing wild-type receptors displayed no picrotoxin-sensitive leak currents. GABA and etomidate efficacies (maximal fraction of receptors activated) were assessed using single-sweep experiments, where receptors were initially activated with one of these agonists, followed by the addition of a co-agonist. Maximal α1M236Cβ2γ2L currents elicited by 10 mm GABA increased about 4-fold (300% over base line) with the addition of 2 μm alphaxalone (Fig. 2E), whereas currents elicited by 100 μm etomidate were enhanced only 4–5% with the addition of 3 mm GABA (Fig. 2F). Assuming that both of these combinations of co-agonists open all activatable receptors, these results indicate that the efficacies of GABA and etomidate are ∼0.24 and 0.96, respectively.
Monod-Wyman-Changeux Model Analysis
A two-state MWC allosteric co-agonist model accounts for the gating effects of both GABA and etomidate in oocytes (10) and provides a mechanistic framework for interpretation of the functional effects of mutations (15, 16, 32). A non-linear least squares fit to this model (Equation 3) was performed using estimates of [GABA] and [etomidate]-dependent Po derived from α1M236Cβ2γ2L oocyte data corrected for spontaneous channel activity and rescaled for GABA efficacy (Equation 2).
Model analysis of pooled α1M236Cβ2γ2L receptor data was constrained by an L0 value (200) derived from the spontaneous gating measurement. The resulting fit and parameters are shown in Fig. 2G and compared with model parameters fitted to wild-type data (Table 3). The four free fitted parameters were well constrained by the data sets. MWC model analysis indicates that the M236C mutation dramatically reduced GABA efficacy (Table 3; c for α1M236Cβ2γ2L is 70-fold larger than for wild-type) while only slightly reducing etomidate efficacy (d for α1M236Cβ2γ2L does not significantly differ from the wild-type value). The relatively small GABA EC50 shift with etomidate is mostly due to the low efficacy of GABA, whereas spontaneous receptor activity accounts for efficacious etomidate agonism. A similar sensitivity to etomidate agonism was observed previously in α1M236Wβ2γ2L receptors, which also display spontaneous gating (15).
TABLE 3.
Equilibrium co-agonist model parameters for wild-type and α1M236Cβ2γ2L GABAA receptors
Results are from nonlinear least squares fits to Equation 3, which describes an allosteric two-state equilibrium mechanism with two classes of agonist sites (one for GABA and one for etomidate), each with two equivalent sites (10). L0 is a dimensionless basal equilibrium gating variable, inversely related to spontaneous activity. For wild-type receptors, spontaneous activity was undetectable, and we constrained L0 to a previous estimate (25,000) for wild type (32). KG and KE are equilibrium dissociation constants for GABA and etomidate binding to inactive states, and c and d are dimensionless parameters representing the respective ratios of binding constants in active versus inactive states. The agonist efficacies of GABA and etomidate are inversely related to c2 and d2, respectively.
| Receptor | L0 | KG | c | KE | d |
|---|---|---|---|---|---|
| μm | μm | ||||
| α1β2γ2L | 25,000 | 70 ± 22 | 0.0019 ± 0.00038 | 40 ± 14 | 0.0076 ± 0.0010 |
| α1M236Cβ2γ2L | 200a | 100 ± 19 | 0.135 ± 0.0070a | 70 ± 22 | 0.014 ± 0.0046 |
a Differs from wild-type value at p < 0.01.
pCMBS Modification and Etomidate Protection
Application of pCMBS (up to 2 mm for 60 s, with or without 1 mm GABA) to oocytes expressing wild-type GABAA receptors produced no detectable changes in electrophysiological properties, including spontaneous activation and responses to low and high GABA (data not shown). Applying pCMBS to oocytes expressing α1M236Cβ2γ2L receptors irreversibly increased leak currents and reduced 10 mm GABA current responses, suggesting that modification activates and enhances desensitization. In the absence of GABA, pCMBS concentrations over 1 mm were needed to produce functional changes within 60 s, and the apparent rate of modification at room temperature was low (16 m−1 s−1; Table 4). Co-application of 10 mm GABA with pCMBS increased the rate of modification over 10-fold to 210 m−1 s−1 (Fig. 3A). Because α1M236Cβ2γ2L channels display spontaneous activity and were readily activated by etomidate, we used a maximally GABA-activated receptor condition (10 mm GABA) for control studies of pCMBS modification to compare with modification with GABA plus etomidate. For protection, we used 30 μm etomidate, based on the fitted allosteric model (Table 3), which indicated an etomidate dissociation constant for GABA-activated α1M236Cβ2γ2L receptors (KE × d) near 1 μm. Co-application of 30 μm etomidate and 10 mm GABA reduced the apparent pCMBS modification rate about 10-fold compared with GABA alone (Fig. 3, B and C, and Table 4).
TABLE 4.
Apparent modification rates in cysteine-substituted GABAA receptors
| Receptor type | pCMBS alone | pCMBS + GABA | pCMBS + GABA + etomidate | Effect of modification | Rate ratio +GABA/−GABA | Rate ratio GABA+ ETO/GABA |
|---|---|---|---|---|---|---|
| Wild-type | NEa (4) | NEb (4) | ND | NE | ||
| α1Q229C | 540 ± 130 (4) | 7000 ± 1600 (4) | 22,700 ± 5200 (3) | ↑ low GABA response | 13 ± 4.3c | 3.2 ± 1.03d |
| α1T230C | 3000 ± 550 (3) | 520,000 ± 115,000 (3) | 960,000 ± 167,000 (3) | ↑ low GABA response | 170 ± 50d | 1.8 ± 0.5d |
| α1Y231C | NE (3) | NE (3) | ND | NE | ||
| α1L232C | 70 ± 18 (6) | 150 ± 37 (7) | 40 ± 5 (7) | ↓ GABA response | 2.1 ± 0.75e | 0.32 ± 0.16e,f |
| α1P233C | NE (3) | NE (3) | ND | NE | ||
| α1I235C | NE (3) | 20 ± 7.8 (3) | 90 ± 6.4 (3) | ↑ low GABA response | > 2d | 4.5 ± 1.8c |
| α1M236C | 16 ± 4.6 (4) | 210 ± 80 (6) | 20 ± 17 (6) | ↓ high GABA response | 13 ± 6.2c | 0.1 ± 0.09e,f |
| α1T237C | 140 ± 92 (3) | 1200 ± 360 (3) | 260 ± 84 (3) | ↓ GABA response | 8.9 ± 6.5d | 0.22 ± 0.09c,f |
| α1V238C | NE (3) | NE (3) | ND | NE | ||
| α1I239C | NE (3) | NE (3) | ND | NE | ||
| α1L240C | 10 ± 5.1 (4) | 26 ± 8.2 (7) | 29 ± 2.4 (5) | ↓ GABA response | 2.7 ± 1.7d | 1.14 ± 0.37 |
| α1S241C | 60 ± 15 (3) | 40 ± 19 (3) | 70 ± 23 (3) | ↑ low GABA response | 0.65 ± 0.35 | 1.72 ± 0.99 |
| α1Q242C | NE (3) | NE (3) | ND | NE |
Rates are expressed in m−1 s−1 (mean ± S.D.) (n). Ratios were calculated from means with propogation of S.D. Statistical significance was determined using Student's t test.
a NE, no effect of modification.
b ND, experiment was not done.
c p < 0.01.
d p < 0.05.
e p < 0.001.
f Protection was inferred when modification rate ratios for GABA + etomidate/GABA were significantly less than 1.0.
FIGURE 3.

Cysteine modification and etomidate protection in heterologously expressed α1M236Cβ2γ2L GABAA receptors. A, traces represent sequential measurements of maximal current elicited with 10 mm GABA (black bars) in an oocyte expressing α1M236Cβ2γ2L GABAA receptors. Arrows, 10-s exposure to 10 mm GABA plus 500 μm pCMBS, followed by wash. Basal leak current increases and maximal GABA current diminishes with incremental exposure to pCMBS. B, traces are sequential maximal current tests (10 mm GABA; black bars) from another oocyte expressing α1M236Cβ2γ2L GABAA receptors. Arrows, 10-s exposure to 10 mm GABA plus 1 mm pCMBS plus 30 μm etomidate, followed by wash. C, rate analyses of peak current data from A and B are shown, plotted against cumulative [pCMBS] × exposure time. Lines represent nonlinear least squares fits to single exponential functions. The fitted time constants are 6.3 s × mm (control; solid symbols) and 51 s × mm (+ etomidate; open symbols). The second order rate constants are 157 and 20 m−1 s−1, respectively. D, data points represent mean ± S.E. (error bars) (n = 6) measurements of membrane-bound [3H]flunitrazepam, normalized to control (100%). Data were analyzed by ANOVA with Bonferroni post-tests. Etomidate at concentrations up to 30 μm positively modulates flunitrazepam binding to untreated membranes from HEK293 cells expressing α1M236Cβ2γ2L receptors (solid squares). Membranes pre-exposed to 2.5 mm MTSEA (solid circles) show significantly reduced etomidate modulation (*, p < 0.05; ***, p < 0.001). In membranes exposed to MTSEA in the presence of 200 μm etomidate (open circles), subsequent maximal etomidate modulation (at 10 μm) is indistinguishable from that in untreated membranes but significantly different from MTSEA-treated membranes (†, p < 0.05).
Etomidate protection of α1M236C from sulfhydryl modification was also assessed using a biochemical assay in HEK293 cell membranes. [3H]Flunitrazepam binding to both wild-type (data not shown) and α1M236Cβ2γ2L receptors (Fig. 3D) was modulated by etomidate. Exposure of α1M236Cβ2γ2L receptors (but not α1β2γ2L) to the modifying reagent MTSEA significantly reduced etomidate modulation. Co-application of 200 μm etomidate during MTSEA treatment blocked the modification effect, preserving modulation. Exposure of α1M236Cβ2γ2L receptors to MMTS, which creates a smaller adduct than MTSEA, did not alter [3H]flunitrazepam binding modulation (data not shown).
Cysteine Substitution Scan of α1-M1 Residues
GABA- and Etomidate-dependent Gating
The molecular length of etomidate in an extended configuration is ∼12 Å, about the distance of two full turns along an α-helical axis. We therefore characterized single residue cysteine mutants along the α1-M1 domain from α1Q229 to α1Q242, spanning four helical turns with the photolabeled α1Met-236 residue in the middle of this region. All cysteine-substituted receptors were characterized using a two-microelectrode voltage clamp in Xenopus oocytes to assess GABA EC50, GABA response modulation by 3.2 μm etomidate, and etomidate direct activation. Results are summarized in Table 1 and Fig. 4.
FIGURE 4.

GABA sensitivity and etomidate modulation in α1-M1 cysteine substituted GABAA receptors. A, bars represent mean ± S.D. (error bars) GABA EC50 plotted on a log scale, averaged from multiple individual measurements in separate oocytes (n ≥ 4). B, bars represent mean ± S.D. GABA EC50 shift ratio in the presence versus absence of 3.2 μm etomidate, averaged from multiple individual measurements in separate oocytes (n ≥ 4) and plotted on a log scale. Large leftward shifts (low shift ratio) indicate high etomidate sensitivity. Mutant results were compared with wild type by ANOVA with Dunnett's post hoc test. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Five mutant channels in this series, α1Q229C, α1T230C, α1L232C, α1I235C, and α1M236C, displayed GABA EC50 values that were significantly (one-way ANOVA; p < 0.05) higher than the wild-type value (Table 1 and Fig. 4A). No mutants were characterized by GABA EC50 values lower than wild type. GABA efficacy was significantly reduced in four mutant channels: α1Q229C, α1Y231C, α1P233C, and α1M236C. Thus, cysteine substitutions at all seven α1-M1 residues from Gln-229 to Met-236 (excluding Cys-234) increased GABA EC50 and/or reduced GABA efficacy, whereas mutations intracellular to Met-236 did not significantly affect GABA EC50 or efficacy (Table 1).
In the presence of 3.2 μm etomidate, five mutant channels exhibited GABA EC50 shifts that significantly differed (one-way ANOVA, p < 0.05) from the wild-type value (Table 1 and Fig. 4B). Receptors with α1Q229C and α1M236C mutations showed etomidate shifts smaller than that in wild type, and α1T230C, α1Y231C, and α1S241C were associated with larger shifts. All α1-M1 domain cysteine-substituted mutant receptors were directly activated by etomidate (Table 1). Etomidate direct activation EC50 values varied from 3 μm (α1I235Cβ2γ2L) to 40 μm (α1Q229Cβ2γ2L), and the maximal relative efficacy of etomidate activation, normalized to maximal GABA efficacy, varied from about 0.24 in α1Q229Cβ2γ2L channels up to about 4.0 in both α1P233Cβ2γ2L and α1M236Cβ2γ2L receptors. Whereas maximum etomidate agonist efficacy relative to GABA was significantly increased by P233C and M236C mutations, the estimated normalized etomidate efficacy for the mutants (calculated 95% confidence intervals for GABA efficacy × etomidate efficacy, from Table 1) did not significantly differ from wild type. In other words, high relative etomidate efficacy simply reflected low GABA efficacy with P233C and M236C mutants. Overall, six mutations, all but one extracellular to Thr-237, altered etomidate sensitivity as assessed by direct activation EC50 or EC50 shifts (Table 1).
pCMBS Modification and Etomidate Protection
In the resting closed state (no GABA), exposure to pCMBS up to 1 mm for 60 s produced no functional changes in receptors containing α1Y231C, α1P233C, α1I235C, α1V238C, αI239C, and αQ242C mutations. For the other seven cysteine-substituted mutant channels, evidence of pCMBS modification in the absence of GABA was observed with apparent rates ranging from 10 to 3000 m−1 s−1 (Table 4). Along the transmembrane axis, closed state aqueous accessibility extended from Gln-229 to Ser-241. Modification of cysteine substitutions at the most extracellular residues we studied, Q229C and T230C, was much faster than at more internal residues, with L240C slowest. A helical wheel projection based on the homology model (Fig. 5A) locates all closed state accessible residues within one hemi-face of α-M1, projecting toward β2-M3 and the intersubunit space or toward α-M2. None are predicted to face lipid in the model.
FIGURE 5.

Effects of GABA and etomidate on modification rates of pCMBS-accessible α1-M1 cysteine substituted GABAA receptors. A, a helical wheel projection of α1-M1 similar to that in Fig. 1B shows pCMBS-modified residues as colored circles; green, modification enhanced by etomidate; magenta, modification reduced by etomidate. The heavy outline around Ile-235 indicates modification only in the presence of GABA. B, bars represent log ratios (mean ± S.D. (error bars)) of pCMBS modification rates measured in the presence of maximally activating GABA (>10 × EC50) to the rate without GABA (n ≥ 3 for each condition). *, rates in the presence versus absence of GABA differ significantly with p < 0.05. †, a ratio for I235C could not be calculated because no modification was evident without GABA. C, bars represent log ratios (mean ± S.D.) of pCMBS modification rates measured in the presence of maximally activating GABA plus etomidate (30–100 μm) to rates in the presence of GABA alone (n ≥ 3 for each condition). Black bars, reduced rates; white bars, increased rates. *, rates in the presence versus absence of etomidate differ significantly with p < 0.05.
With the addition of GABA at >10 × EC50, pCMBS modification was observed in eight mutant receptors: the seven that were modified in the resting state plus α1I235Cβ2γ2L. Rates of pCMBS modification in the presence of GABA varied from 20 to 520,000 m−1 s−1 (Table 4). GABA significantly increased pCMBS modification rates at all modifiable α1-M1 cysteine-substituted sites, except α1Ser-241. The ratio of modification rates in the presence versus absence of GABA varied from 2-fold at α1L232C and α1L240C up to 170-fold at α1T230C (Fig. 5B). A rate ratio could not be calculated for α1I235C receptors, which displayed no modification in the absence of GABA.
The addition of etomidate significantly increased the apparent rate of pCMBS modification in three cysteine-substituted receptors at α1Gln-229, α1Thr-230, and α1Ile-235 (Fig. 5C and Table 4). Fig. 6, A and B, illustrates the enhancement of α1Q229C modification by both GABA and etomidate. As observed for α1M236C, etomidate reduced apparent rates of pCMBS modification at α1L232C and α1T237C (Table 4 and Fig. 5C). In GABA-activated α1L232Cβ2γ2L receptors, 30 μm etomidate reduced the pCMBS modification rate about 3-fold (Fig. 6, C and D). In α1T237Cβ2γ2L receptors, 100 μm etomidate reduced the pCMBS modification rate nearly 5-fold (Fig. 6, E and F).
FIGURE 6.

Etomidate effects on cysteine modification in GABAA receptors with α1Q229C, α1L232C, and α1T237C mutations. Left-hand panels display current traces from oocytes expressing mutant GABAA receptors. Currents were activated with GABA, indicated by solid black bars above each trace. Downward arrows indicate exposures to pCMBS alone or with GABA or with GABA plus etomidate, followed by washout. Modification conditions (P, pCMBS; G, GABA; E, etomidate) for each set of traces are indicated in micromolar above each set of arrows. Right-hand panels show normalized peak current data from the traces shown on the left plotted against cumulative pCMBS exposure (s × mm). Lines through plotted symbols represent least squares fits to single exponentials. A, α1Q229Cβ2γ2L channels. Currents were stimulated with 50 μm GABA (EC10). B, solid squares, modification with pCMBS alone (fitted rate = 520 m−1 s−1); open squares, modification with pCMBS plus GABA (fitted rate = 3280 m−1 s−1); crossed squares, modification with pCMBS plus GABA and etomidate (fitted rate = 6500 m−1 s−1). Inset, rate analyses are shown with a magnified time base. C, α1L232Cβ2γ2L channels. Currents were stimulated with 1 mm GABA (EC100). D, solid triangles, modification with pCMBS plus GABA (fitted rate = 170 m−1 s−1); open triangles, modification with pCMBS plus GABA and etomidate (fitted rate = 34 m−1 s−1). E, α1T237β2γ2L channels. Currents were stimulated with 1 mm GABA (EC100). F, solid circles, modification with pCMBS plus GABA (fitted rate = 1450 m−1 s−1); open circles, modification with pCMBS plus GABA and etomidate (fitted rate = 210 m−1 s−1).
Etomidate Docking to a Molecular Structure Homology Model
The energy-minimized α1β2γ2L homology model (Fig. 7) is based on the crystal structure of GLIC, a prokaryotic homopentameric ion channel that is thought to represent either an open or desensitized channel state (17, 18, 35, 36). The orientations of α1-M1 residues in the model are similar to those in a α1β2γ2L homology model (21) based on GluCl (19). R-(+)-Etomidate docked between α1-M1 and β2-M3 transmembrane domains adopts an L-shape when viewed along the transmembrane axis, with the plane of the benzene ring forming the short leg and approximately orthogonal to the plane of the imidazole ring and ester linkage, which together form the long leg (Fig. 7). In the 100 lowest energy docking orientations in the binding sites at GABAA receptor β2-M3/α1-M1 interfaces, the etomidate molecule is oriented with the benzene ring located near β2-M2. In the lowest energy poise, the non-branching nitrogen in the imidazole group is located 3.0 Å from the amide nitrogen of β2N265 (M2–15′), a residue where mutations affect etomidate sensitivity (37, 38). The ester leaving group (ethanol) projects outward from the ion channel toward the lipid-protein interface between α1-M1 and β2-M3. The C2 ethanol carbon is 4.6 Å from the α carbon of β2Met-286, and the ester carbonyl oxygen is 3.5 Å from both α1Pro-233 and α1Met-236 and 4 Å from β2Phe-289. In α1-M1, etomidate makes contact with part of the water-accessible surface of Ile-228, Leu-232, Pro-233, Met-236, Thr-237, and Leu-240 side chains. Control etomidate docking calculations in β-M1/α-M3, β-M1/γ-M3, and γ-M1/α-M3 subunit interfaces produced similar energetically stable interactions.
FIGURE 7.

Etomidate docked within an α1β2γ2L GABAA receptor homology model contacts α1Leu-232, α1Met-236, and α1Thr-237 in the M1 domain. A–C, multiple views of a homology model of GABAA receptor built on the structure of pentameric GLIC (Protein Data Bank entry 3P50), showing α-helices as cylinders and β-sheets as ribbons with subunits α1 (yellow), β2 (blue), and γ2L (green). The GABAA receptor-specific ligands GABA (blue), a benzodiazepine (magenta), and etomidate (red) are shown within their intersubunit binding sites as Connolly surfaces. A, the model viewed parallel to the membrane. The extent of the α1-M1 helix examined in this study is colored brown. B, a view from the extracellular space of the extracellular domains. C, a view from the extracellular space of the transmembrane domains. D–F, enlarged views of one etomidate site. Shown in stick format are residues protected by etomidate from pCMBS modification after cysteine substitution, α1Leu-232 (red), α1Met-236 (purple), and α1Thr-237 (green). Also shown in stick format color-coded by atom type (gray, carbon; red, oxygen; blue, nitrogen; gold, sulfur) are residues of interest in the β2 subunit and etomidate docked at its lowest energy orientation. A Connolly surface of the composite of 100 most stable docked poises surrounds the etomidate molecule. D, a view from within the membrane. E, a view from the β2 subunit. F, a view from the synaptic end of the transmembrane domain.
DISCUSSION
Our main findings are that cysteine substitution at most residues in the outer GABAA receptor α-M1 domain impair GABA agonism, whereas effects on etomidate modulation vary. GABA accelerates pCMBS modification at most accessible positions. Etomidate protection from modification indicates proximity to three (two newly identified) residues in this region.
The Outer α1-M1 Domain Is Linked to GABA Agonism and the Channel State
Cysteine substitutions in α1-M1 from Gln-229 through Met-236 (excluding Cys-234) consistently impair GABA agonism, increasing GABA EC50, reducing GABA efficacy, or both (Table 1). Five of these seven cysteine substitutions are modifiable with pCMBS, and modification accelerates with application of GABA (Table 4). In contrast, α1-M1 residues intracellular to Met-236 do not significantly affect GABA sensitivity and are relatively inaccessible to pCMBS. Modification by pCMBS signifies accessibility via an aqueous pathway (39). Thus, water-accessible α1-M1 residues are largely extracellular to Thr-237, consistent with structural homology models in which the space between transmembrane domains narrows intracellularly. Accelerated pCMBS modification by GABA suggests increased mobility or aqueous access. Bali and Akabas (21) have also examined substituted cysteine accessibility in α1-M1 from Gly-224 to Met-236. At residues examined in both studies (Gln-229 to Met-236), the pattern of GABA-dependent pCMBS modification at Q229C, T230C, L232C, and M236C was similar. Contrasting with our results, Bali and Akabas reported lower GABA EC50 values and higher pCMBS modification rates and reported that Y231C mutations abolish channel function. These differences may be due to their rat “Cys-light” channel background, lacking native transmembrane cysteines, including α1Cys-234. Importantly, our main conclusions are unaffected by these differences.
A variety of data support the hypothesis that GABA activation expands the space around α1-M1, increasing both water and side chain mobility to a depth near α1Leu-240. Cysteine substitutions could impair GABA-induced gating by reducing side chain volume and increasing water in transmembrane pockets, increasing entropic energy costs. Cysteine, being smaller than most of the residues we mutated, does not occlude etomidate binding, which displaces water and reduces entropy, stabilizing open channel states. Tryptophan substitution at α1Met-236 or β2Met-286 enhances gating in the same manner while occluding the etomidate site (15). A similar mechanism was proposed by Jenkins et al. (40) for volatile anesthetic actions at intrasubunit pockets. Bali et al. (22) observed GABA-dependent disulfide formation between β2M286C and α1Q229C and suggested that these residues approach each other during gating. However, this intersubunit disulfide bond stabilizes a non-conducting receptor state. GABA-dependent cross-linking could also result from increased side chain or backbone mobility. Delineating transmembrane domain motions during channel gating and desensitization remains an important experimental challenge.
Outer α-M1 Domain Linkage to Etomidate Modulation
Several outer α-M1 domain mutations also affected etomidate sensitivity with a pattern distinct from that for GABA sensitivity (Fig. 4). Two cysteine substitutions, Q229C and M236C, reduce etomidate-induced left-shift, whereas T230C, Y231C, and S241C increased modulation. MWC allosteric analysis indicates that the α1M236C mutation insignificantly affects etomidate allosteric efficacy (Table 3, d). Thus, the small GABA EC50 shift in α1M236Cβ2γ2L receptors reflects only a portion of etomidate modulation; the remainder is evident as increased GABA efficacy (Fig. 1A). The divergence of cysteine mutant effects on orthosteric and allosteric agonists rules out isolated effects on channel gating (L0 in MWC models). Considering evidence that etomidate contacts α-M1 domain residues (discussed below), we speculate that its movements differ when gating is triggered by GABA alone versus with etomidate bound, altering open state stability and channel gating modes.
Etomidate Binds Next to a Short Segment of α1-M1
Our pCMBS modification experiments identify M236C, L232C, and T237C as etomidate-protected residues (Fig. 5). Parallel experiments based on [3H]flunitrazepam binding (Fig. 3D) further support etomidate protection of α1M236C. Based on MWC affinity parameters for activated α1M236Cβ2γ2L receptors (KE × d ≈ 1 μm), etomidate site occupation was predicted to exceed 96% at 30 μm. Etomidate slowed pCMBS modification at M236C about 10-fold (Fig. 5C). However, GABA alone (reference modification conditions) activated only about 24% of receptors, whereas co-application of etomidate with GABA (protection conditions) probably activated nearly 100% of receptors (Fig. 2, E and F). Supporting this interpretation, alphaxalone, which enhances GABA activation without blocking etomidate binding (41), accelerated pCMBS modification of α1M236C about 3-fold over the GABA-only reference rate (data not shown). Thus, etomidate probably reduced pCMBS modification rates much more than 10-fold in GABA-activated α1M236Cβ2γ2L receptors. We reported similar protection in GABA-activated α1β2M286Cγ2L receptors (16).
We used 30–100 μm etomidate, concentrations associated with direct receptor agonism, to achieve high etomidate site occupation in protection experiments. Importantly, etomidate agonism is mediated by the same sites that produce modulation by low (general anesthetic) concentrations (10). Despite high predicted site occupancy, etomidate protection at L232C and T237C was less complete than at M236C, suggesting less steric proximity. An alternative protection mechanism whereby etomidate blocks the aqueous pCMBS access pathway is unlikely, because access to L240C and S241C, furthest from the membrane surface, was preserved with etomidate bound. The possibility that etomidate indirectly induces movement of protected residues to become pCMBS-inaccessible also exists. However, a steric protection mechanism is also supported by comparison with Q229C, T230C, and I235C. In receptors with these mutations, etomidate increased pCMBS modification rates beyond those with GABA alone (Table 4 and Fig. 5), paralleling electrophysiological evidence that etomidate increased channel activation. Notably, etomidate also enhanced channel gating in the three protected Cys-mutant receptors (Table 1).
Combining current results, other protection studies, and photolabeling (13, 14, 16) identifies three α1-M1 residues (αLeu-232, αMet-236, and αThr-237) and two β2-M3 residues (βMet-286 and βVal-290) that form etomidate binding sites. Sensitivity to volatile anesthetics is also affected by mutations at αLeu-232 and βMet-286 (40, 42). Mutations at β2Met-286 also affect propofol sensitivity (43), and propofol protects β2M286C from modification (44). Both propofol and isoflurane inhibit azietomidate photolabeling of GABAA receptors (45). Thus, the α1-M1/β2-M3 interface apparently contains overlapping sites for volatile anesthetics, propofol and etomidate. Hosie and Smart (25) also identified α1T237 and α1Q242 as determinants for neurosteroid sensitivity, although neurosteroids do not displace etomidate (41). Our cysteine protection results argue against alphaxalone contact with α1Met-236. Similar studies may help specify which residues contact other anesthetics.
Our results suggest that etomidate interactions are limited to 1.5 helical turns of α1-M1, about 8 Å along the transmembrane axis. The long leg of etomidate fits into a right cylinder of diameter 7.4 Å and length 11.9 Å, sufficiently long to contact residues on two or three sequential helical turns. Thus, our results are most consistent with etomidate binding with its long axis approximately perpendicular (90 ± 45°) to the transmembrane axis.
Comparison with Structural Homology Models and Crystallized Receptors
In silico docking of etomidate to the α-M1/β-M3 cavity in a structural homology model (Fig. 7) placed etomidate adjacent to all three protected residues (Leu-232, Met-236, and Thr-237) with its long axis oriented approximately orthogonal to the transmembrane axis. Thus, cysteine protection results agree with the predictions of this and previous models of the etomidate site (13, 14). In the model, etomidate also contacts Ile-228, Pro-233, and Leu-240. We did not include Ile-228 in our study, and α1P233Cβ2γ2L receptors were unaffected by exposure to pCMBS, precluding protection tests. The P233C mutation also did not alter etomidate sensitivity (Table 1 and Fig. 4B). Thus, α1P233 could be an etomidate contact point with a “silent” cysteine mutation. The L240C mutation produced no change in etomidate sensitivity and was modified by pCMBS but not protected. The model places Leu-240 farther from etomidate than the protected residues, and a cysteine mutation would further increase this separation. Assessing other Pro-233 and Leu-240 mutations might provide additional information about etomidate interactions.
High resolution structural evidence locating allosteric agonists in transmembrane intersubunit sites is emerging from several pentameric ligand-gated ion channel models. Co-crystals of C. elegans GluCl channels locate ivermectin (19) in sites that are structural homologs of etomidate sites in GABAA receptors. Ivermectin is much larger than etomidate and makes extensive contacts with GluCl M1 domains. Co-crystallization of propofol and desflurane with GLIC (46) identified intrasubunit transmembrane sites, although cysteine accessibility studies by Ghosh et al. (47) suggest that propofol inhibition is mediated by intersubunit sites. Recent crystal data from positively modulated GLIC-F238A mutants also locate anesthetics and alcohols in intersubunit cavities (48). Co-crystallization of Erwinia chrysanthemi (ELIC) channels with bromoform also identified an intersubunit site deep in the membrane (near residues corresponding to GABAA α1Gln-242 and α1Trp-246), along with other sites (49). Thus, both prokaryotic and eukaryotic pentameric ion channels harbor multiple types of anesthetic binding sites, linked to both enhancing and inhibitory anesthetic effects (50).
In our current study, cysteine mutations at residues predicted to project into the intrasubunit helical bundle pocket displayed weaker effects on GABA and etomidate sensitivity than at residues facing the intersubunit site. This suggests that intersubunit transmembrane pockets in GABAA receptors influence channel gating more than neighboring intrasubunit transmembrane pockets. Accordingly, although we and others (21) find evidence for variable α-M1 side chain movements with gating, suggesting local secondary structure changes, our data also point to important quaternary rearrangements (at subunit interfaces) linked to gating.
Despite agreement between model docking and protection results, the limitations of in silico docking to current homology models are evident from control computations showing that etomidate docks with similar stability to all five transmembrane subunit interfaces. This is probably because the model was built on a homomeric template. Nonetheless, recent photolabeling studies demonstrate that R-(+)-etomidate binds to β-M3/α-M1 interfaces with greater than 100-fold higher affinity than to α-M3/β-M1 or γ-M3/β-M1 interfaces that are selectively photolabeled by a barbiturate derivative (33). More broadly, links between crystallized receptor structures and functional states remain tenuous, and co-crystal structures reveal no significant rearrangements in anesthetic binding cavities (46, 48), highlighting the value of complementary approaches.
Can Mutant Function Predict Anesthetic Contact?
We thoroughly characterized cysteine mutant channel functions yet found no phenotypic “fingerprint” for residues contacting etomidate. Considering the three protected α1-M1 mutants we identified and β2Met-286 (16), two (β2Met-286 and α1Met-236) reduce etomidate left shift, and three (β2Met-286, α1Met-236, and α1Leu-232) reduce GABA sensitivity. However, in α1Q229Cβ2γ2L, a mutant with functional characteristics similar to α1M236Cβ2γ2L and β2M286Cβ2γ2L, etomidate enhances rather than inhibits pCMBS modification. MWC allosteric analysis, which derives independent parameters for etomidate binding and efficacy, also fails to unambiguously identify etomidate binding site mutations. MWC analysis of α1M236C indicates minimal changes from wild type in both etomidate affinity and efficacy, whereas cysteine protection and photolabeling clearly place this residue in the etomidate binding site. Functional analysis of multiple mutations at candidate residues might improve inferences regarding ligand interactions. MWC analyses of α1M236W, β2M286W, and β2M286C mutations (15, 16) identified dominant effects on the etomidate efficacy parameter (d), which reflects binding to the high affinity activated state. Our current protection results were obtained in GABA-bound receptors and thus reflect interactions with open and/or desensitized states. Although β2M286C is protected by etomidate in resting (closed) state receptors (16), the low affinity of this state makes its study more challenging.
In summary, we find that cysteine substitutions at outer α1-M1 residues (Gln-229 through αMet-236) reduce GABA sensitivity and variably affect etomidate sensitivity. Accessibility to pCMBS was evident in α1-M1 residues predicted to face the intersubunit space or the α1-M2 domain and few others. GABA enhanced modification at most of these positions, indicating gating-coupled rearrangements at the subdomain level. Etomidate inhibited modification of L232C, M236C, and T237C, indicating contact with a limited portion of α-M1 at the subunit-subunit interface. These results agree with in silico model docking calculations, suggesting that the long axis of etomidate lies perpendicular to the α1-M1 domain.
Acknowledgments
We thank Aiping Liu, Youssef Jounaidi, and Alex Stern (all at Massachusetts General Hospital) for technical help and Professors Keith W. Miller (Massachusetts General Hospital), Douglas E. Raines (Massachusetts General Hospital), and Jonathan B. Cohen (Harvard Medical School) for helpful comments on the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants R01GM089745 and P01GM58448.
- GLIC
- G. violaceus ion channel
- ELIC
- E. chrysanthemi ion channel
- MMTS
- methyl methanethiosulfonate
- MTSEA
- 2-aminoethyl methanethiosulfonate
- MWC
- Monod-Wyman-Changeux
- pCMBS
- para-chloromercuribenzenesulfonate
- PTX
- picrotoxin
- ANOVA
- analysis of variance.
REFERENCES
- 1. Franks N. P. (2008) General anaesthesia. From molecular targets to neuronal pathways of sleep and arousal. Nat. Rev. Neurosci. 9, 370–386 [DOI] [PubMed] [Google Scholar]
- 2. Olsen R. W., Sieghart W. (2008) International Union of Pharmacology. LXX. Subtypes of γ-aminobutyric acid(A) receptors. Classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol. Rev. 60, 243–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baumann S. W., Baur R., Sigel E. (2002) Forced subunit assembly in α1β2γ2 GABAA receptors. Insight into the absolute arrangement. J. Biol. Chem. 277, 46020–46025 [DOI] [PubMed] [Google Scholar]
- 4. Jurd R., Arras M., Lambert S., Drexler B., Siegwart R., Crestani F., Zaugg M., Vogt K. E., Ledermann B., Antkowiak B., Rudolph U. (2003) General anesthetic actions in vivo strongly attenuated by a point mutation in the GABAA receptor β3 subunit. FASEB J. 17, 250–252 [DOI] [PubMed] [Google Scholar]
- 5. Reynolds D. S., Rosahl T. W., Cirone J., O'Meara G. F., Haythornthwaite A., Newman R. J., Myers J., Sur C., Howell O., Rutter A. R., Atack J., Macaulay A. J., Hadingham K. L., Hutson P. H., Belelli D., Lambert J. J., Dawson G. R., McKernan R., Whiting P. J., Wafford K. A. (2003) Sedation and anesthesia mediated by distinct GABAA receptor isoforms. J. Neurosci. 23, 8608–8617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang J., Uchida I. (1996) Mechanisms of etomidate potentiation of GABAA receptor-gated currents in cultured postnatal hippocampal neurons. Neuroscience 73, 69–78 [DOI] [PubMed] [Google Scholar]
- 7. Hill-Venning C., Belelli D., Peters J. A., Lambert J. J. (1997) Subunit-dependent interaction of the general anaesthetic etomidate with the γ-aminobutyric acid type A receptor. Br. J. Pharmacol. 120, 749–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Husain S. S., Ziebell M. R., Ruesch D., Hong F., Arevalo E., Kosterlitz J. A., Olsen R. W., Forman S. A., Cohen J. B., Miller K. W. (2003) 2-(3-Methyl-3H-diaziren-3-yl)ethyl 1-(1-phenylethyl)-1H-imidazole-5-carboxylate. A derivative of the stereoselective general anesthetic etomidate for photolabeling ligand-gated ion channels. J. Med. Chem. 46, 1257–1265 [DOI] [PubMed] [Google Scholar]
- 9. Tomlin S. L., Jenkins A., Lieb W. R., Franks N. P. (1998) Stereoselective effects of etomidate optical isomers on γ-aminobutyric acid type A receptors and animals. Anesthesiology 88, 708–717 [DOI] [PubMed] [Google Scholar]
- 10. Rüsch D., Zhong H., Forman S. A. (2004) Gating allosterism at a single class of etomidate sites on α1β2γ2L GABAA receptors accounts for both direct activation and agonist modulation. J. Biol. Chem. 279, 20982–20992 [DOI] [PubMed] [Google Scholar]
- 11. Guitchounts G., Stewart D. S., Forman S. A. (2012) Two etomidate sites in α1β2γ2 γ-aminobutyric acid type A receptors contribute equally and noncooperatively to modulation of channel gating. Anesthesiology 116, 1235–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Husain S. S., Nirthanan S., Ruesch D., Solt K., Cheng Q., Li G. D., Arevalo E., Olsen R. W., Raines D. E., Forman S. A., Cohen J. B., Miller K. W. (2006) Synthesis of trifluoromethylaryl diazirine and benzophenone derivatives of etomidate that are potent general anesthetics and effective photolabels for probing sites on ligand-gated ion channels. J. Med. Chem. 49, 4818–4825 [DOI] [PubMed] [Google Scholar]
- 13. Li G. D., Chiara D. C., Sawyer G. W., Husain S. S., Olsen R. W., Cohen J. B. (2006) Identification of a GABAA receptor anesthetic binding site at subunit interfaces by photolabeling with an etomidate analog. J. Neurosci. 26, 11599–11605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chiara D. C., Dostalova Z., Jayakar S. S., Zhou X., Miller K. W., Cohen J. B. (2012) Mapping general anesthetic binding site(s) in human α1β3 γ-aminobutyric acid type A receptors with [3H]TDBzl-etomidate, a photoreactive etomidate analogue. Biochemistry 51, 836–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stewart D., Desai R., Cheng Q., Liu A., Forman S. A. (2008) Tryptophan mutations at azi-etomidate photo-incorporation sites on α1 or β2 subunits enhance GABAA receptor gating and reduce etomidate modulation. Mol. Pharmacol. 74, 1687–1695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stewart D. S., Hotta M., Desai R., Forman S. A. (2013) State-dependent etomidate occupancy of its allosteric agonist sites measured in a cysteine-substituted GABAA receptor. Mol. Pharmacol. 83, 1200–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bocquet N., Nury H., Baaden M., Le Poupon C., Changeux J. P., Delarue M., Corringer P. J. (2009) X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature 457, 111–114 [DOI] [PubMed] [Google Scholar]
- 18. Hilf R. J., Dutzler R. (2009) Structure of a potentially open state of a proton-activated pentameric ligand-gated ion channel. Nature 457, 115–118 [DOI] [PubMed] [Google Scholar]
- 19. Hibbs R. E., Gouaux E. (2011) Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature 474, 54–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jansen M., Akabas M. H. (2006) State-dependent cross-linking of the M2 and M3 segments. Functional basis for the alignment of GABAA and acetylcholine receptor M3 segments. J. Neurosci. 26, 4492–4499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bali M., Akabas M. H. (2012) Gating-induced conformational rearrangement of the γ-aminobutyric acid type A receptor β-α subunit interface in the membrane-spanning domain. J. Biol. Chem. 287, 27762–27770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bali M., Jansen M., Akabas M. H. (2009) GABA-induced intersubunit conformational movement in the GABAA receptor α 1M1-β 2M3 transmembrane subunit interface. Experimental basis for homology modeling of an intravenous anesthetic binding site. J. Neurosci. 29, 3083–3092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Greenfield L. J., Jr., Zaman S. H., Sutherland M. L., Lummis S. C., Niemeyer M. I., Barnard E. A., Macdonald R. L. (2002) Mutation of the GABAA receptor M1 transmembrane proline increases GABA affinity and reduces barbiturate enhancement. Neuropharmacology 42, 502–521 [DOI] [PubMed] [Google Scholar]
- 24. Carlson B. X., Engblom A. C., Kristiansen U., Schousboe A., Olsen R. W. (2000) A single glycine residue at the entrance to the first membrane-spanning domain of the γ-aminobutyric acid type A receptor β2 subunit affects allosteric sensitivity to GABA and anesthetics. Mol. Pharmacol. 57, 474–484 [DOI] [PubMed] [Google Scholar]
- 25. Hosie A. M., Wilkins M. E., da Silva H. M., Smart T. G. (2006) Endogenous neurosteroids regulate GABAA receptors through two discrete transmembrane sites. Nature 444, 486–489 [DOI] [PubMed] [Google Scholar]
- 26. Akk G., Li P., Bracamontes J., Reichert D. E., Covey D. F., Steinbach J. H. (2008) Mutations of the GABA-A receptor alpha1 subunit M1 domain reveal unexpected complexity for modulation by neuroactive steroids. Mol. Pharmacol. 74, 614–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Keramidas A., Kash T. L., Harrison N. L. (2006) The pre-M1 segment of the α1 subunit is a transduction element in the activation of the GABAA receptor. J. Physiol. 575, 11–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mercado J., Czajkowski C. (2006) Charged residues in the α1 and β2 pre-M1 regions involved in GABAA receptor activation. J. Neurosci. 26, 2031–2040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rüsch D., Forman S. A. (2005) Classic benzodiazepines modulate the open-close equilibrium in α1β2γ2L γ-aminobutyric acid type A receptors. Anesthesiology 102, 783–792 [DOI] [PubMed] [Google Scholar]
- 30. Scheller M., Forman S. A. (2002) Coupled and uncoupled gating and desensitization effects by pore domain mutations in GABAA receptors. J. Neurosci. 22, 8411–8421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Graham F. L., van der Eb A. J. (1973) A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology 52, 456–467 [DOI] [PubMed] [Google Scholar]
- 32. Desai R., Ruesch D., Forman S. A. (2009) γ-amino butyric acid type A receptor mutations at β2N265 alter etomidate efficacy while preserving basal and agonist-dependent activity. Anesthesiology 111, 774–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chiara D. C., Jayakar S. S., Zhou X., Zhang X., Savechenkov P. Y., Bruzik K. S., Miller K. W., Cohen J. B. (2013) Specificity of intersubunit general anesthetic-binding sites in the transmembrane domain of the human α1β3γ2 γ-aminobutyric acid type A (GABAA) receptor. J. Biol. Chem. 288, 19343–19357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Burkat P. M., Yang J., Gingrich K. J. (2001) Dominant gating governing transient GABAA receptor activity. A first latency and Po/o analysis. J. Neurosci. 21, 7026–7036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gonzalez-Gutierrez G., Grosman C. (2010) Bridging the gap between structural models of nicotinic receptor superfamily ion channels and their corresponding functional states. J. Mol. Biol. 403, 693–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Parikh R. B., Bali M., Akabas M. H. (2011) Structure of the M2 transmembrane segment of GLIC, a prokaryotic Cys loop receptor homologue from Gloeobacter violaceus, probed by substituted cysteine accessibility. J. Biol. Chem. 286, 14098–14109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Belelli D., Lambert J. J., Peters J. A., Wafford K., Whiting P. J. (1997) The interaction of the general anesthetic etomidate with the γ-aminobutyric acid type A receptor is influenced by a single amino acid. Proc. Natl. Acad. Sci. U.S.A. 94, 11031–11036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Siegwart R., Krähenbühl K., Lambert S., Rudolph U. (2003) Mutational analysis of molecular requirements for the actions of general anaesthetics at the γ-aminobutyric acidA receptor subtype, α1β2γ2. BMC Pharmacol. 3, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Karlin A., Akabas M. H. (1998) Substituted-cysteine accessibility method. Methods Enzymol. 293, 123–145 [DOI] [PubMed] [Google Scholar]
- 40. Jenkins A., Greenblatt E. P., Faulkner H. J., Bertaccini E., Light A., Lin A., Andreasen A., Viner A., Trudell J. R., Harrison N. L. (2001) Evidence for a common binding cavity for three general anesthetics within the GABAA receptor. J. Neurosci. 21, RC136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li G. D., Chiara D. C., Cohen J. B., Olsen R. W. (2009) Neurosteroids allosterically modulate binding of the anesthetic etomidate to γ-aminobutyric acid type A receptors. J. Biol. Chem. 284, 11771–11775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Krasowski M. D., Finn S. E., Ye Q., Harrison N. L. (1998) Trichloroethanol modulation of recombinant GABAA, glycine and GABA rho 1 receptors. J. Pharmacol. Exp. Ther. 284, 934–942 [PubMed] [Google Scholar]
- 43. Krasowski M. D., Nishikawa K., Nikolaeva N., Lin A., Harrison N. L. (2001) Methionine 286 in transmembrane domain 3 of the GABAA receptor beta subunit controls a binding cavity for propofol and other alkylphenol general anesthetics. Neuropharmacology 41, 952–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bali M., Akabas M. H. (2004) Defining the propofol binding site location on the GABAA receptor. Mol. Pharmacol. 65, 68–76 [DOI] [PubMed] [Google Scholar]
- 45. Li G. D., Chiara D. C., Cohen J. B., Olsen R. W. (2010) Numerous classes of general anesthetics inhibit etomidate binding to γ-aminobutyric acid type A (GABAA) receptors. J. Biol. Chem. 285, 8615–8620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nury H., Van Renterghem C., Weng Y., Tran A., Baaden M., Dufresne V., Changeux J. P., Sonner J. M., Delarue M., Corringer P. J. (2011) X-ray structures of general anaesthetics bound to a pentameric ligand-gated ion channel. Nature 469, 428–431 [DOI] [PubMed] [Google Scholar]
- 47. Ghosh B., Satyshur K. A., Czajkowski C. (2013) Propofol binding to the resting state of the Gloeobacter violaceus ligand-gated ion channel (GLIC) induces structural changes in the inter- and intrasubunit transmembrane domain (TMD) cavities. J. Biol. Chem. 288, 17420–17431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sauguet L., Howard R. J., Malherbe L., Lee U. S., Corringer P. J., Harris R. A., Delarue M. (2013) Structural basis for potentiation by alcohols and anaesthetics in a ligand-gated ion channel. Nat. Commun. 4, 1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Spurny R., Billen B., Howard R. J., Brams M., Debaveye S., Price K. L., Weston D. A., Strelkov S. V., Tytgat J., Bertrand S., Bertrand D., Lummis S. C., Ulens C. (2013) Multisite binding of a general anesthetic to the prokaryotic pentameric Erwinia chrysanthemi ligand-gated ion channel (ELIC). J. Biol. Chem. 288, 8355–8364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Forman S. A., Miller K. W. (2011) Anesthetic sites and allosteric mechanisms of action on Cys-loop ligand-gated ion channels. Can J. Anaesth. 58, 191–205 [DOI] [PMC free article] [PubMed] [Google Scholar]